Unlocking the Ringed Architecture of Natural Products: From Biosynthetic Logic to Drug Discovery Innovation
This article provides a comprehensive exploration of ring systems and molecular frameworks in natural products, tailored for researchers and drug development professionals.
Unlocking the Ringed Architecture of Natural Products: From Biosynthetic Logic to Drug Discovery Innovation
Abstract
This article provides a comprehensive exploration of ring systems and molecular frameworks in natural products, tailored for researchers and drug development professionals. It bridges the gap between the foundational structural diversity found in nature and the cutting-edge methodologies used to exploit it for therapeutic design. The scope encompasses an analysis of privileged natural product scaffolds, advances in synthetic and computational strategies for ring manipulation (including skeletal editing and biomimetic synthesis), and the critical evaluation of these frameworks within the drug discovery pipeline. By synthesizing insights from cheminformatics, synthetic chemistry, and AI-driven design, the article aims to equip scientists with a holistic understanding of how natural product-inspired ring systems can be optimized, validated, and leveraged to navigate chemical space and develop novel bioactive entities.
The Structural Diversity and Biosynthetic Origins of Natural Product Ring Systems
The structural cores of bioactive molecules are predominantly defined by their ring systems. These systems establish molecular shape, dictate the spatial orientation of substituents, and critically influence key pharmacokinetic and pharmacodynamic properties [1]. Within this broad chemical space, privileged scaffolds—recurrent molecular frameworks capable of providing ligands for diverse biological targets—hold particular significance for drug discovery. Natural products (NPs) serve as a primary evolutionary source of such scaffolds, encoding biologically relevant information through frameworks honed by biosynthetic pathways [2].
A comprehensive analysis of 38,662 ring systems from natural products reveals their exceptional structural diversity and unique physicochemical profiles [3]. Despite this diversity, a stark disconnect exists between NP chemical space and contemporary drug design: only about 2% of NP ring systems are present in approved drugs [3]. This discrepancy underscores a vast reservoir of unexplored, biologically validated chemotypes. Concurrently, analyses of medicinal chemistry literature demonstrate a conservative reliance on established ring motifs, with approximately 67% of clinical trial compounds incorporating known drug ring systems [1]. This highlights the critical challenge and opportunity in drug discovery: to systematically bridge NP-inspired chemical space with synthetic feasibility, thereby unlocking novel privileged scaffolds for therapeutic development.
Table 1: Prevalence of Key Ring System Characteristics in Natural Products vs. Synthetic Compounds [3] [1]
| Characteristic | Natural Products (NPs) | Synthetic Compounds (SCs) / Drugs | Implication for Scaffold Privilege |
|---|---|---|---|
| Representation in Approved Drugs | ~2% of NP ring systems are present [3] | Majority based on known motifs [1] | Vast untapped potential in NPs. |
| 3D Shape/Electrostatic Coverage | ~50% have analogous shape/electrostatics in SCs [3] | SC libraries capture half of NP 3D space. | NP-inspired design can access validated geometries. |
| Frequency Distribution | High diversity, long-tail distribution [3]. | Power-law distribution; few very common rings [1]. | Privileged scaffolds often reside in "frequent" clusters. |
| Stereochemical Complexity | Generally high [3]. | Often lower [1]. | Complexity may enhance selectivity and binding [2]. |
Strategic Design of Natural Product-Inspired Scaffolds
The translation of NP-derived ring systems into novel privileged scaffolds requires deliberate synthetic strategy. Approaches exist along a continuum, from closely mimicking NP frameworks to creating entirely novel architectures inspired by NP fragments [2].
- Biology-Oriented Synthesis (BIOS) and Function-Oriented Synthesis (FOS) start from a bioactive NP core, aiming to retain or improve its biological function through analogue synthesis [2] [4].
- Pseudo-Natural Products (PNPs) represent a powerful fragment-based strategy. This involves the synthesis of novel scaffolds by combining two or more biosynthetically unrelated NP fragments, creating chemotypes not found in nature but retaining NP-like properties [2] [5]. A study combining fragments from quinine, sinomenine, and griseofulvin with chromanone or indole fragments yielded 244 PNPs with high chemical diversity and distinct bioactivity profiles [5].
- Complexity-to-Diversity (CtD) and Ring Distortion strategies begin with a complex NP and apply reactions that dramatically alter the core scaffold, often via ring expansion, contraction, or cleavage, to access new polycyclic systems [2] [6]. A seminal example involves the diversification of steroids via C–H oxidation followed by ring expansion to synthesize medium-sized rings (7–11 membered), an underexplored yet privileged chemical space [6].
Table 2: Core Strategies for Privileged Scaffold Discovery from Natural Products
| Strategy | Core Principle | Key Advantage | Example Outcome |
|---|---|---|---|
| Biology-Oriented Synthesis (BIOS) | Modification of a known bioactive NP scaffold [2]. | Retains biological relevance of the original NP framework. | Discovery of novel Hedgehog pathway inhibitors from a macrolactone scaffold [4]. |
| Pseudo-Natural Product (PNP) | Combinatorial fusion of unrelated NP fragments [2] [5]. | Generates unprecedented scaffolds with NP-like properties. | A 244-member library with diverse phenotypic bioactivities [5]. |
| Complexity-to-Diversity (CtD/Ring Distortion) | Drastic skeletal remodeling of a complex NP starting material [2] [6]. | Accesses highly novel and strained ring systems from readily available NPs. | Synthesis of polycyclic scaffolds with medium-sized rings from steroids [6]. |
| Hybrid Design | Combination of primary NP scaffold with secondary privileged motifs [7]. | Integrates target "bait" with diversity elements for selectivity. | Identification of a spirooxepinoindole privileged scaffold for sterol transport proteins [7]. |
Diagram 1: The Design-Evaluate Cycle for Discovering Privileged Scaffolds from Natural Products. This workflow outlines the strategic transition from a complex natural product to an identified privileged scaffold, incorporating iterative feedback from structure-activity relationship (SAR) analysis [2] [7] [4].
Experimental Methodologies for Scaffold Diversification and Evaluation
The discovery of privileged scaffolds necessitates robust synthetic and analytical methodologies. Key experimental workflows involve scaffold diversification followed by rigorous biological and biophysical evaluation.
Protocol 1: Ring Distortion and Expansion to Access Medium-Sized Rings [6] This protocol details a two-phase strategy to diversify polycyclic natural products (e.g., steroids) into scaffolds containing medium-sized rings.
- Phase 1: C-H Oxidation. Employ site-selective C-H oxidation to install functional handles. For allylic oxidations, use an electrochemical cell with carbon felt electrodes, a Pt counter electrode, and a solvent mixture of acetic acid and water with NaHCO₃ as electrolyte. Alternatively, use metal-mediated oxidations (e.g., with Cu or Cr catalysts) for benzylic or other C-H bonds.
- Intermediate Purification. Isolate the ketone or alcohol oxidation product via standard flash chromatography.
- Phase 2: Ring Expansion. Subject the oxidized intermediate to ring-expanding reactions.
- For Schmidt Reaction: Treat a ketone with sodium azide in a mixture of trifluoroacetic acid and dichloromethane at 0°C to RT to form a lactam.
- For Formal [2+2] Cycloaddition/Fragmentation: React a β-keto ester with dimethyl acetylenedicarboxylate (DMAD) in refluxing xylenes, followed by treatment with acetic acid and HCl to yield ring-expanded anhydrides.
- For Beckmann Rearrangement: Convert a ketone to its oxime, then treat with Lewis acid (e.g., TiCl₄) or under Beckmann conditions (e.g., PCl₅) to form the expanded lactam.
- Characterization: Confirm structures using NMR, HRMS, and, where possible, X-ray crystallography to assign novel stereochemistry and ring conformations.
Protocol 2: Synthesis and Evaluation of a Pseudo-Natural Product (PNP) Library [7] [5] This protocol outlines the generation of a cholic acid-inspired PNP collection for inhibitor discovery.
- Primary Scaffold Synthesis. Synthesize the core cis-decalone scaffold from cholic acid derivatives in multi-gram scale (6 steps, ~41% yield) [7].
- Scaffold Fusion (PNP Formation). Employ one-pot fusion reactions from the core ketone.
- Fischer Indole Synthesis: React ketone with substituted phenylhydrazine using tosylic acid (TsOH) in refluxing ethanol.
- Friedländer Quinoline Synthesis: React ketone with o-aminoacetophenone derivatives under solvent-free microwave irradiation.
- Hantzsch Thiazole Synthesis: React an α-bromoketone derivative with thioamides in refluxing ethanol.
- Ring Distortion (CtD Phase). Further diversify selected PNPs.
- Witkop Oxidation: Treat an indole-fused PNP with sodium periodate (NaIO₄) to perform oxidative cleavage to a ketolactam.
- Camps Cyclization: Treat the ketolactam with base (e.g., NaOH) to induce ring contraction to a quinolone.
- Biological Evaluation.
- Primary Screening: Use a fluorescence polarization (FP) assay monitoring displacement of a fluorescent sterol probe from the target protein.
- Hit Validation: Confirm binding via differential scanning fluorimetry (DSF), monitoring protein thermal stabilization.
- Functional Assay: Measure inhibition of intracellular cholesterol transport in a cell-based assay.
Diagram 2: Ring Expansion via C-H Functionalization. This two-phase synthetic workflow converts common polycyclic natural product cores into novel scaffolds containing medium-sized rings, an underexplored class of privileged structures [6].
The Scientist's Toolkit: Essential Reagents and Materials
| Reagent/Material | Function in Protocol | Key Application |
|---|---|---|
| Sodium Azide (NaN₃) | Nitrogen source for the Schmidt reaction [6]. | One-step conversion of ketones to ring-expanded lactams. |
| Dimethyl Acetylenedicarboxylate (DMAD) | Dienophile for formal [2+2] cycloaddition [6]. | Two-carbon ring expansion of β-keto esters to anhydrides. |
| Tosylate Salt of Phenylhydrazine | Substrate for Fischer indole synthesis [7]. | Robust formation of edge-fused indole rings from ketones. |
| 2-Hydroxyacetophenones | Substrates for the Kabbe condensation [5]. | Forms spirocyclic chromanone fragments fused to NP cores. |
| Sodium Periodate (NaIO₄) | Oxidizing agent for Witkop oxidation [7]. | Cleaves indole rings to synthetically versatile ketolactams. |
| Fluorescent Sterol Probe (e.g., DHEA-BODIPY) | Tracer for Fluorescence Polarization (FP) assays [7]. | Enables high-throughput screening for sterol transport protein inhibitors. |
Computational and Analytical Frameworks
Modern cheminformatic tools are indispensable for analyzing ring system diversity, predicting scaffold privilege, and navigating NP chemical space.
Computational Analysis of Ring Systems: Studies utilize large-scale cheminformatic analyses of databases like COCONUT and ChEMBL to profile ring systems [3]. Key steps include:
- Data Curation: Standardizing structures, considering stereochemistry, and defining ring systems as isolated cyclic systems.
- Descriptor Calculation: Generating 3D conformers and calculating shape (e.g., principal moments of inertia) and electrostatic potential descriptors.
- Similarity Mapping: Using metrics like ET_combo scores to quantify the coverage of NP ring system shape/electrostatics by synthetic compound libraries [3].
Prediction of Bioactive Categories: The AgreementPred framework aids in annotating NPs by predicting their pharmacological categories [8].
- Multi-Representation Similarity: Calculates structural similarity between an unannotated NP and a labeled database using 22 different molecular representations (e.g., ECFP4, AP, PHFP fingerprints).
- Data Fusion & Agreement Scoring: Aggregates similarity results and assigns an "agreement score" to predicted category labels, filtering for high-precision predictions. This method achieved a recall of 0.74 and precision of 0.55 for predicting across 1,520 categories [8].
- Application: This framework can prioritize NP-derived scaffolds for therapeutic areas where they are predicted to be active, guiding library design.
Privileged scaffolds derived from natural product ring systems represent a cornerstone for future drug discovery, offering a unique blend of biological validation and chemical novelty. The path forward lies in the systematic integration of advanced synthetic strategies (like PNP design and CtD), robust computational prediction tools (like AgreementPred), and high-content biological screening. This integrated approach will enable researchers to more effectively mine the vast, untapped chemical space of natural products, moving beyond the mere 2% of NP ring systems currently reflected in drugs [3]. By doing so, the field can overcome the inherent conservatism in scaffold selection and deliver novel, privileged molecular frameworks to address unmet therapeutic needs.
Within the intricate architectures of natural products and therapeutic agents, ring systems serve as the fundamental scaffolds that define molecular destiny. These cyclic frameworks are not mere structural curiosities but are the primary determinants of three-dimensional shape, conformational rigidity, and biochemical function [1]. In the realm of drug discovery, the prevalence of rings is overwhelming: 95.1% of small-molecule drugs approved by the FDA over the past two decades contain at least one ring system [1]. This predominance underscores a central thesis in natural products research: biological activity and bioavailability are inextricably linked to the topological and electronic properties encoded within these cyclic structures. Natural products, evolutionarily optimized for interaction with biological macromolecules, provide a rich library of privileged ring topologies. These scaffolds, characterized by specific ring sizes, patterns of fusion, and strategic heteroatom incorporation, dictate key pharmacodynamic and pharmacokinetic properties—from binding affinity and target selectivity to metabolic stability and cellular permeability [1]. This whitepaper provides a technical guide for researchers, deconstructing how the core variables of ring size, fusion, and heteroatom identity govern molecular shape and properties, thereby offering a rational framework for the design of next-generation bioactive compounds inspired by nature's blueprint.
The Determinant of Spatial Occupation: Ring Size
Ring size is a primary variable that controls the spatial footprint and conformational flexibility of a molecule. It directly influences the molecule's ability to present functional groups in three-dimensional space for optimal target interaction.
Energetics, Strain, and Conformational Freedom
The stability and geometry of a ring are governed by torsional strain, angle strain, and steric interactions, which vary predictably with size. Small rings (3-4 members) are highly strained and rigid, while common 5- and 6-membered rings are typically low in strain. Medium (7-13 members) and large (≥14 members) rings contend with transannular strain and complex conformational dynamics but offer unique spatial arrangements for binding extended, shallow protein interfaces [9].
Systematic Studies and Biological Implications
The profound impact of ring size on biological activity is elegantly demonstrated in synthetic studies of natural product-inspired macrocycles. Research on cyclic oligomeric depsipeptide (COD) analogs of ent-verticilide, targeting the cardiac ryanodine receptor (RyR2), revealed a sharp "Goldilocks" zone for activity [9]. As summarized in Table 1, only the 18- and 24-membered rings showed potent sub-micromolar inhibition, while smaller (6-, 12-membered) and larger (30-, 36-membered) analogs were completely inactive. This indicates that a specific spatial presentation of side chains, achievable only within a narrow range of ring sizes, is critical for target engagement [9].
Table 1: Impact of Macrocycle Ring Size on Biological Activity and Properties [9]
| Ring Size (Membered) | RyR2 Inhibition (Ca²⁺ Spark Frequency) | Postulated Conformational State | Relative Lipophilicity (AlogP Trend) |
|---|---|---|---|
| 6 | Inactive | Highly rigid, deviated sidechain presentation | Lowest |
| 12 | Inactive | Rigid, insufficient interaction surface | Low |
| 18 | Potently Active | Optimal balance of pre-organization and flexibility | Moderate |
| 24 | Potently Active | Flexible, accessible binding conformation | High |
| 30 | Inactive | Excessively flexible, "floppy" | Higher |
| 36 | Inactive | Excessively flexible, poor conformational population | Highest |
Similarly, in materials science, the ring size of C3-symmetrical dehydrobenzoannulene (DBA) derivatives dictates supramolecular assembly and optical properties. A [12]DBA ring formed a stable molecular glass, while the larger [18]DBA favored crystalline π-dimer assemblies due to stronger intermolecular interactions [10].
Diagram 1: Rational Design Logic: From Ring Topology to Biological Outcome. This workflow illustrates how the three core topological variables (size, fusion, heteroatoms) govern physical consequences that converge to define key molecular properties and ultimate biological function.
Experimental Protocol: Synthesis and Evaluation of Ring-Size Analogs
The following protocol, adapted from research on cyclic depsipeptides, outlines a systematic approach to studying ring-size effects [9].
- Objective: To synthesize a homologous series of macrocycles varying only in ring size and evaluate their biological activity.
- Materials: Protected linear depsipeptide precursor units, coupling reagents (e.g., HATU, DIC), catalysts for cyclization (e.g., high-dilution conditions with PyBOP), deprotection reagents (e.g., TFA for Boc removal, H₂/Pd-C for benzyl esters), anhydrous solvents (DMF, DCM, THF).
- Linear Precursor Synthesis: Iteratively couple protected monomer units via standard peptide coupling chemistry. The length of the linear precursor dictates the final ring size.
- Macrocyclization: Under high-dilution conditions (≈1 mM), activate the terminal carboxylic acid of the linear precursor and initiate cyclization by nucleophilic attack of the terminal amine or alcohol. Precise control of temperature, concentration, and additive use is critical to favor intramolecular cyclization over oligomerization.
- Deprotection & Purification: Remove all protecting groups using conditions orthogonal to the macrocycle stability. Purify the crude product via preparative HPLC.
- Conformational & Biological Analysis:
- Conformational Analysis: Use NMR (e.g., ROESY) to identify through-space correlations and define predominant conformers in solution. Computational modeling (MD simulations) can supplement this.
- Biological Assay: In the cited study, compounds were tested on permeabilized cardiomyocytes. Cells were incubated with compound (e.g., 25 µM), and RyR2 activity was quantified by measuring spontaneous Ca²⁺ spark frequency via fluorescence microscopy [9].
- Data Interpretation: Correlate activity data with ring size and conformational models to identify the optimal spatial geometry for target interaction.
The Architecture of Complexity: Ring Fusion and Polycyclization
The fusion of two or more rings creates polycyclic systems that introduce profound rigidity, define stereochemical complexity, and create unique topological landscapes.
Conformational Locking and Stereochemistry
Fused ring systems dramatically limit conformational mobility. The classic example is decalin (bicyclo[4.4.0]decane). trans-Decalin, with both bridgehead hydrogens on opposite faces, is conformationally locked in a favorable diequatorial, chair-chair conformation and cannot ring-flip due to geometric constraints [11] [12]. cis-Decalin can undergo ring inversion, but its "tent-like" shape introduces destabilizing gauche interactions, making it ~2.7 kcal/mol less stable than its trans counterpart [12]. This locking effect is leveraged in steroid frameworks (like cholesterol and sex hormones) to maintain a precise, bioactive conformation [11].
Topology and Shape in Drug Design
Fusion patterns create distinct molecular shapes—linear, angular, or globular—that are matched to binding pockets. Furthermore, fusion generates topological complexity that can be essential for activity. For instance, the β-lactam ring fused to a thiazolidine ring in penicillin creates a highly strained, reactive system crucial for its mechanism of action as an acylating agent [13].
Diagram 2: Structural and Conformational Consequences of Ring Fusion Patterns. Different fusion geometries lead to distinct three-dimensional shapes and degrees of conformational freedom, directly impacting molecular properties.
Experimental Protocol: Conformational Analysis of Fused Ring Systems
- Objective: To determine the preferred conformation and energy difference between isomers of a fused ring system (e.g., cis- vs. trans-decalin).
- Materials: Pure samples of isomers, deuterated NMR solvent (e.g., CDCl₃), X-ray crystallography equipment (if suitable crystals can be grown), computational chemistry software.
- NMR Spectroscopy:
- Acquire ¹H and ¹³C NMR spectra at room temperature.
- For cis-decalin, the ability to ring-flip leads to averaged signals at room temperature. To freeze the conformation and observe distinct axial/equatorial protons, acquire Variable-Temperature (VT) NMR spectra at low temperature (e.g., -90°C in a toluene-d₈ solvent mixture) [12].
- For trans-decalin, signals are consistent at all temperatures due to conformational locking.
- Use coupling constants (J-values) and Nuclear Overhauser Effect (NOE/ROESY) measurements to confirm the spatial proximity of protons and assign stereochemistry.
- X-ray Crystallography: Grow single crystals of each isomer. The crystal structure provides definitive proof of the ring junction stereochemistry and the exact chair conformations of the cyclohexane rings.
- Computational Energy Minimization: Perform molecular mechanics (MM2, MMFF) or density functional theory (DFT) calculations to model the lowest energy conformation and calculate the relative steric energy difference between isomers.
- Data Interpretation: Synthesize data from all techniques. NMR shows dynamic behavior, crystallography provides a static snapshot, and computation offers energetic rationale. The combined data validates the conformational models and explains stability differences.
The Electronic and Functional Modulators: Heteroatom Incorporation
The replacement of carbon atoms within a ring with heteroatoms (most commonly N, O, S) is a powerful strategy for modulating electronic distribution, polarity, and intermolecular interactions.
Prevalence and Functional Roles
Heterocycles are ubiquitous in medicinal chemistry and biology. Approximately 59% of U.S. FDA-approved drugs contain a nitrogen heterocycle [14]. Their functions are diverse [15] [1] [14]:
- Hydrogen Bonding: Serve as hydrogen bond acceptors (e.g., pyridine N, carbonyl O) or donors/acceptors (e.g., pyrrole NH, imidazole).
- Polarity & Solubility: Increase aqueous solubility relative to their carbocyclic analogs.
- Metal Coordination: Act as ligands for metal ions in enzyme active sites (e.g., histidine in zinc fingers, porphyrins in heme).
- Bioisosteric Replacement: Mimic the geometry and electronic properties of other functional groups (e.g., a phenyl ring can be replaced by pyridyl, thiophene, or furan rings to fine-tune properties).
Table 2: Key Heterocycles in Bioactive Natural Products and Drugs [15] [14]
| Heterocycle (Ring Size) | Example Heteroatoms | Key Natural Product/Drug Examples | Primary Biological Role/Function |
|---|---|---|---|
| Pyridine (6) | N | Vitamin B3 (niacin), nicotine | Hydrogen bond acceptor, weak base. |
| Imidazole (5) | 2N | Amino acid histidine, antifungal drugs (e.g., ketoconazole) | Hydrogen bond donor/acceptor, metal ligand, key in enzyme catalysis. |
| Thiazole (5) | N, S | Vitamin B1 (thiamine), penicillin antibiotics | Essential for covalent reaction mechanism (penicillin), cofactor. |
| Pyrimidine (6) | 2N | DNA/RNA bases (cytosine, thymine, uracil) | Hydrogen bonding for base-pairing, component of genetic code. |
| Purine (Fused 5+6) | 4N | DNA/RNA bases (adenine, guanine), caffeine | Hydrogen bonding for base-pairing, cellular signaling (cAMP). |
| Indole (Fused Benzene+Pyrrole) | N | Amino acid tryptophan, serotonin, reserpine (alkaloid) | Hydrophobic interactions, neurotransmitter activity. |
| Morpholine (6) | O, N | Synthetic building block in drugs (e.g., gefitinib) | Solubilizing moiety, hydrogen bond acceptor. |
Impact on Aromaticity and Reactivity
Incorporating heteroatoms into aromatic rings (heteroaromatics) alters the electron density distribution. For example, pyridine is π-deficient with the nitrogen withdrawing electron density, making it susceptible to nucleophilic attack. Conversely, pyrrole is π-excessive, with the nitrogen lone pair contributing to the aromatic sextet, making it prone to electrophilic substitution [13]. This directly influences the chemical reactivity and metabolic fate of the molecule.
Diagram 3: The Multifunctional Roles of Heteroatoms in Ring Systems. Introducing heteroatoms influences electronic structure, intermolecular interaction capacity, and acid-base properties, which collectively fine-tune biological performance.
Experimental Protocol: Synthesis of a Representative Heterocycle (Paal-Knorr Pyrrole Synthesis)
- Objective: To synthesize a substituted pyrrole, a fundamental π-excessive nitrogen heterocycle.
- Principle: The Paal-Knorr synthesis involves the condensation of a 1,4-diketone with a primary amine or ammonia under acid catalysis [13].
- Materials: 2,5-hexanedione (acetonylacetone), aniline (or other primary amine), acetic acid (AcOH) or p-toluenesulfonic acid (p-TsOH) as catalyst, toluene or ethanol as solvent, standard extraction and purification glassware.
- Procedure:
- In a round-bottom flask equipped with a reflux condenser, dissolve the 1,4-diketone (10 mmol) and aniline (10 mmol) in 20 mL of toluene.
- Add a catalytic amount of p-TsOH (0.1 mmol).
- Reflux the reaction mixture for 4-16 hours, monitoring by TLC.
- After completion, cool the mixture and wash with aqueous sodium bicarbonate to remove the acid catalyst.
- Dry the organic layer over anhydrous magnesium sulfate, filter, and concentrate under reduced pressure.
- Purify the crude product by column chromatography (silica gel, hexanes/ethyl acetate) to yield the pure 2,5-dimethyl-1-phenylpyrrole.
- Characterization: Confirm the structure by ¹H NMR (look for the characteristic pyrrole NH signal ~8-10 ppm and aromatic protons), ¹³C NMR, and mass spectrometry.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Ring System Research [11] [12] [9]
| Category | Reagent/Material | Function in Ring System Research | Example Application |
|---|---|---|---|
| Synthesis & Cyclization | HATU / PyBOP | Peptide coupling reagents for amide bond formation. | Macrocyclization of linear depsipeptide precursors [9]. |
| p-Toluenesulfonic Acid (p-TsOH) | Acid catalyst for cyclization and condensation reactions. | Paal-Knorr pyrrole synthesis [13]. | |
| High-Dilution Apparatus | Ensures slow addition of linear precursor to favor intramolecular cyclization over intermolecular oligomerization. | Synthesis of medium/large ring macrocycles [9]. | |
| Conformational & Structural Analysis | Deuterated Solvents for VT-NMR (e.g., Toluene-d₈) | Low-temperature NMR solvent for "freezing out" conformational exchange. | Observing distinct axial/equatorial protons in cis-decalin [12]. |
| Crystallography Reagents | Solvents for slow vapor diffusion or layering to grow single crystals. | Determining absolute stereochemistry of fused ring junctions [12]. | |
| Functionalization & Assay | m-Chloroperoxybenzoic Acid (mCPBA) | Electrophilic oxygen source for epoxidation of alkenes. | Probing stereoselectivity on rigid fused ring scaffolds (e.g., alkenyl-decalins) [12]. |
| Fluorescent Dyes & Microscopy Buffers | For labeling and visualizing biological activity in cellular assays. | Measuring intracellular calcium spark frequency in RyR2 inhibition assays [9]. |
The strategic manipulation of ring size, fusion, and heteroatom identity constitutes the core of rational molecular design in natural products research and drug discovery. As demonstrated, ring size controls the spatial canvas, fusion dictates architectural rigidity and shape, and heteroatoms install the electronic and interactive functionalities. The conservation of privileged ring systems across the chemical space of drugs—with common motifs like benzene, piperidine, pyridine, and imidazole dominating—testifies to their validated biological compatibility [1]. The future lies not only in exploring novel ring systems accessible through advanced synthesis but also in the deeper computational understanding of how these topological features dictate molecular properties within biological systems [16]. By mastering the principles outlined in this guide, researchers can more effectively decode the lessons embedded in natural product architectures and harness ring topology as a powerful, predictable tool for crafting the next generation of bioactive molecules.
Within the expansive universe of natural products, molecular frameworks and ring systems are fundamental determinants of biological activity, physicochemical properties, and synthetic accessibility. This examination positions the cyclopropane ring—the smallest possible carbocycle—as a critical case study within this broader thesis [17]. Despite, or perhaps because of, its extreme ring strain and structural simplicity, the cyclopropane motif is a disproportionately powerful actor in medicinal chemistry and natural product biosynthesis [18]. It is a ubiquitous feature in a remarkable array of complex terpenoids, alkaloids, steroids, and fatty acids, where it often serves as a key pharmacophore responsible for significant and diverse biological effects [19] [20]. This whitepaper provides an in-depth technical analysis of the cyclopropane ring, detailing its unique physicochemical properties, its strategic role in drug design, and the advanced synthetic methodologies that enable its study and application. By exploring this "small but mighty" motif, we illuminate the profound impact that specific, strained ring systems can exert on the field of natural products research and therapeutic development.
Biological Significance and Activity Profiles
The cyclopropane ring is not a mere structural curiosity; its incorporation into complex molecular architectures confers a wide spectrum of potent biological activities. Found in natural products derived from terrestrial plants, marine organisms, and microorganisms, these strained rings are key contributors to drug discovery campaigns [19].
Table 1: Bioactive Natural Products Featuring Cyclopropane Motifs
| Natural Product Class | Example Compounds | Reported Biological Activities | Source Organism |
|---|---|---|---|
| Terpenoids | Curacin A, Illudalane sesquiterpenes | Cytotoxic, Anticancer, Antiviral, Immunosuppressive | Marine Cyanobacteria, Fungi [19] [21] |
| Alkaloids | Mirabazines, Cyclopropane-containing indoles | Anti-HIV, Antimicrobial, Cytotoxic | Marine Sponges, Plants [19] |
| Steroids | Withanolides (modified) | Cytotoxic | Plants [19] |
| Fatty Acids | Sterculic acid, Malvalic acid | Enzyme inhibition (e.g., Δ9-desaturase) | Plants [19] |
The biological profile extends beyond natural products into designed therapeutics. The strategic fusion of a cyclopropane ring to a core scaffold is a recognized strategy in medicinal chemistry to improve drug-like properties. As illustrated in the diagram below, this single modification can simultaneously influence multiple pharmacokinetic and pharmacodynamic parameters, leading to enhanced therapeutic potential [18].
Diagram Title: Multifunctional Impact of Fused Cyclopropanes in Drug Design
Chemical Properties and Synthesis Fundamentals
The extraordinary effects of the cyclopropane ring stem from its unique and strained chemical geometry. With internal C-C-C bond angles forced to approximately 60°—a severe deviation from the ideal tetrahedral angle of 109.5°—the ring possesses a high strain energy of 27-28 kcal/mol [18] [21]. This strain has several critical consequences:
- Bent Bond Geometry: The C-C bonds exhibit significant "banana" or bent character, with greater p-orbital character, making them behave similarly to π-bonds in their reactivity.
- Altered C-H Bonds: The C-H bonds have higher s-character (≈33%), making them stronger and less acidic, which contributes to enhanced metabolic stability against oxidative enzymes like cytochrome P450s [18].
- Structural Rigidity and 3D Shape: The ring imposes planarity and rigidity, preventing free rotation and presenting substituents in well-defined spatial orientations. This is invaluable for pre-organizing a molecule for optimal target binding [18].
The synthesis of cyclopropanes is a cornerstone of organic methodology. Classical approaches can be broadly categorized as [2+1] cycloadditions or 1,3-cyclizations [20] [22].
Table 2: Core Methodologies for Cyclopropane Synthesis
| Method Name | Key Reagents/Conditions | Mechanistic Class | Typical Substrate Scope | Key Stereochemical Notes |
|---|---|---|---|---|
| Simmons-Smith | CH₂I₂, Zn(Cu) (or related carbenoids) | Carbenoid addition to alkene [21] [22] | Electron-rich alkenes, often directed by allylic alcohols | Stereospecific (syn addition); diastereoselective with chiral directing groups. |
| Metal-Catalyzed Diazo Decomposition | Diazo compound (e.g., N₂CHCO₂Et), Rh₂(OAc)₄, Cu(acac)₂ | Metal-carbene formation & addition [21] [22] | Broad; works well with acceptor-substituted alkenes (e.g., enones). | Highly enantioselective versions exist with chiral metal complexes (e.g., Rh, Co). |
| Michael-Initiated Ring Closure (MIRC) | Sulfur Ylide (e.g., (CH₃)₂S(O)=CH₂) + α,β-unsaturated carbonyl | Nucleophilic addition then intramolecular substitution [22] | Excellent for electron-deficient alkenes (Michael acceptors). | Can proceed with high diastereoselectivity using chiral substrates, nucleophiles, or catalysts. |
| Kulinkovich Reaction | Ti(OiPr)₄, Grignard Reagent (EtMgBr), Ester | Titanium-mediated coupling [22] | Esters or amides to form hydroxy- or amino-cyclopropanes. | Provides a distinct route to functionalized cyclopropanes. |
Experimental Protocols and Recent Methodological Advances
Recent years have witnessed significant innovation in cyclopropane synthesis, focusing on safety, selectivity, and applicability to complex molecules. Below are detailed protocols for two contemporary, high-impact methods.
This method provides a safe, practical alternative to traditional carbene pathways by using a radical mechanism initiated by visible light.
- Objective: To convert alkenes into cyclopropanes using methylene precursors without hazardous diazo compounds or metal carbenoids.
- Materials: Alkene substrate, methylene precursor (e.g., diethyl bromomalonate, 19 variants demonstrated), 9,10-dicyanoanthracene (DCA, photocatalyst), dimethylformamide (DMF, solvent), oxygen (from air, 10% in atmosphere).
- Procedure:
- In a vial equipped with a stir bar, combine the alkene (1.0 equiv), the methylene precursor (2.0-3.0 equiv), and DCA (2 mol%).
- Add dry DMF to achieve a substrate concentration of approximately 0.1 M.
- Seal the vial and purge the headspace with an O₂/N₂ mixture (10% O₂) or simply perform the reaction open to air for simplicity.
- Irradiate the reaction mixture with blue LEDs (456 nm) while stirring vigorously at room temperature for 12-24 hours.
- Monitor reaction completion by TLC or LCMS.
- Upon completion, dilute the mixture with water and extract with ethyl acetate (3x). Combine the organic layers, dry over anhydrous Na₂SO₄, filter, and concentrate in vacuo.
- Purify the crude product by flash chromatography on silica gel.
- Key Advantages: Avoids explosive diazo compounds; uses stable, commercial reagents; tolerant of ambient oxygen and a wide range of functional groups; successful on complex substrates like estrone and penicillin derivatives [23].
This protocol leverages engineered biocatalysis for the highly stereoselective synthesis of cyclopropanes, followed by chemical diversification.
- Objective: To perform an enantioselective cyclopropanation of vinylarenes with diazoketones using an engineered myoglobin (Mb) variant.
- Materials: Styrene or vinylarene substrate, α-aryl diazoketone carbene donor, Engineered sperm whale myoglobin variant Mb(H64G,V68A) (expressed and purified), Sodium borate buffer (100 mM, pH 9.0), Sodium dithionite (reducing agent).
- Procedure:
- In an anaerobic glovebox, prepare a solution of the Mb(H64G,V68A) catalyst (0.1-0.2 mol%) in Na-borate buffer (pH 9.0).
- Add a slight excess of solid sodium dithionite to the protein solution to reduce the heme iron to the active Fe(II) state. Incubate for 10 minutes.
- Add the vinylarene substrate (20 mM final concentration) and the diazoketone reagent (20 mM final concentration) from stock solutions in a minimal amount of DMSO (<2% v/v final).
- Seal the reaction vessel and incubate at 25°C with gentle shaking for 6-16 hours.
- Quench the reaction by extracting with ethyl acetate (3x). Combine the organic extracts, dry (Na₂SO₄), and concentrate.
- Analyze the product by chiral HPLC or SFC to determine enantiomeric excess (typically >99% ee) [24].
- The resulting cyclopropyl ketone can be diversified via downstream chemical reactions (e.g., reduction, olefination, Grignard addition) to create a library of chiral cyclopropane scaffolds.
- Key Advantages: Exceptional enantioselectivity (>99% ee) and diastereoselectivity (>99% de); broad substrate scope for both coupling partners; green, aqueous conditions; provides chiral building blocks not easily accessible by traditional chemical catalysis [24].
The workflow for creating diverse chiral libraries via this chemoenzymatic approach is outlined below.
Diagram Title: Chemoenzymatic Workflow for Chiral Cyclopropane Library Synthesis
Applications in Therapeutics and Case Studies
The strategic value of the cyclopropane ring is exemplified by its application in modern drug discovery, where it is used to solve specific pharmacological challenges.
Table 3: Therapeutic Case Studies of Fused-Cyclopropane Motifs [18]
| Therapeutic Target / Drug | Role of Cyclopropane Motif | Outcome Achieved | Development Stage |
|---|---|---|---|
| SARS-CoV-2 Main Protease (Mᵖʳᵒ) Inhibitor (Nirmatrelvir) | Replaced a tert-leucine amide with a fused cyclopropyl amide. | Enhanced permeability and oral bioavailability, while maintaining potency against the viral protease. | Approved (Paxlovid) [18] |
| Protein Kinase B (Akt) Inhibitor (NTQ1062) | Installed a fused cyclopropane in the solvent-exposed region. | Significantly improved metabolic stability (reduced clearance) and in vivo exposure in mice compared to the predecessor. | Clinical Stage (NCT06172322) [18] |
| β-Secretase 1 (BACE-1) Inhibitor for Alzheimer's | Incorporated a fused cyclopropane to constrain conformation. | Reduced hERG channel liability (cardiotoxicity risk) while maintaining target potency and improving brain exposure. | Preclinical/Lead Optimization [18] |
| Complement Factor D (FD) Inhibitor | Fused cyclopropane formed part of a macrocyclic constraint. | Achieved exquisite target selectivity over related serine proteases (kallikrein), crucial for minimizing side effects. | Preclinical [18] |
The Scientist's Toolkit: Research Reagent Solutions
Working with cyclopropanes requires specialized reagents and building blocks. The following table details key materials for synthesis and analysis in this field.
Table 4: Essential Research Reagents for Cyclopropane Chemistry
| Reagent / Material | Function / Purpose | Application Notes |
|---|---|---|
| Diiodomethane (CH₂I₂) & Zinc-Copper Couple | Core reagents for the Simmons-Smith cyclopropanation [21]. | The classic system for methylene transfer. Zn/Cu couple activates CH₂I₂ to form the iodomethylzinc iodide carbenoid. Handle in anhydrous ether under inert atmosphere. |
| Ethyl Diazoacetate (EDA) & Rh₂(OAc)₄ | Standard carbene donor and catalyst for metal-catalyzed cyclopropanation [21] [22]. | EDA is a hazardous, potentially explosive liquid. Must be handled with extreme caution in dilute solutions, using appropriate shielding. Rh₂(OAc)₄ is a robust, commonly used catalyst. |
| Dimethylsulfoxonium Methylide (Corey-Chaykovsky Reagent) | Sulfur ylide for MIRC reactions with enones [22]. | Typically generated in situ from trimethylsulfoxonium iodide and a strong base (e.g., NaH). Provides access to donor-acceptor cyclopropanes. |
| Chiral Bisoxazoline (Box) Ligands | Ligands for enantioselective metal-catalyzed cyclopropanations (with Cu, Rh, etc.) [22]. | Induce asymmetry in the metal-carbene intermediate, leading to high enantiomeric excess in the product. Available in a variety of chiral backbones. |
| Engineered Myoglobin (e.g., Mb(H64G,V68A)) | Biocatalyst for enantioselective cyclopropanation with diazo reagents [24]. | Requires protein expression and purification. Offers an alternative to synthetic chiral catalysts with exceptional selectivity in aqueous buffers. |
| 9,10-Dicyanoanthracene (DCA) | Photocatalyst for visible-light-mediated radical cyclopropanation [23]. | Organic, metal-free catalyst. Used in the Giri protocol with methylene precursors and oxygen. Operates under mild blue light irradiation. |
| (R)- or (S)-Limonene Oxide | Chiral pool starting material for the synthesis of enantiopure cyclopropane building blocks. | A readily available, inexpensive terpene that can be elaborated into complex chiral cyclopropane structures found in natural products. |
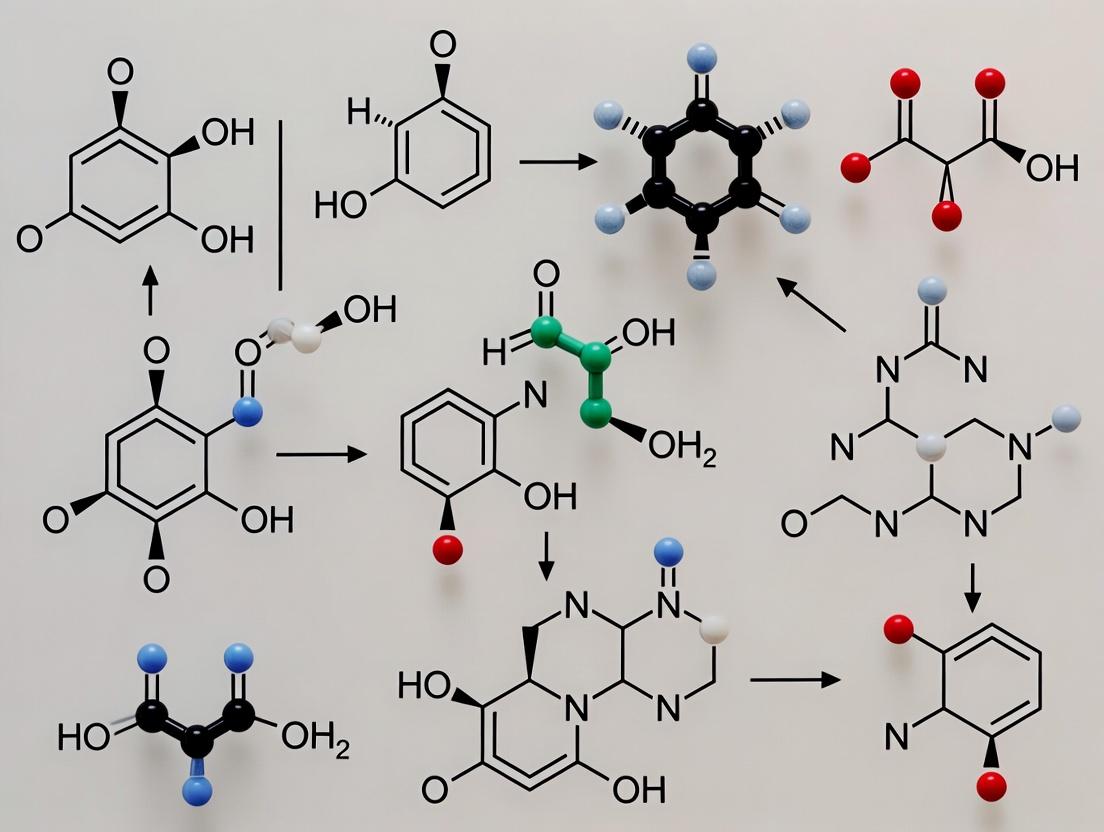
The structural architecture of bioactive small molecules, particularly those derived from nature, is predominantly defined by their ring systems. These cyclic frameworks form the core scaffolds of most clinically approved drugs and are responsible for conferring essential physicochemical properties, three-dimensional shape, and precise biological targeting capabilities [3]. Within the broader thesis of natural products research, understanding biosynthetic pathways transcends mere metabolic mapping; it reveals the fundamental chemical and evolutionary logic that nature employs to construct these critical ring systems. This logic operates on a principle of economy and diversification: a limited set of core, scaffold-generating chemical transformations acts on primary metabolic building blocks to create key intermediates. These intermediates are then expansively diversified through tailoring reactions into vast families of structurally related natural products [25]. The recent convergence of synthetic biology, advanced genomics, and computational cheminformatics has transformed this understanding from a descriptive science into a predictive and engineering discipline [26] [27] [28]. This whitepaper delves into the natural logic of ring construction, detailing the experimental and computational methodologies that now allow researchers to reverse-engineer, reprogram, and harness these biosynthetic blueprints for the discovery and rational design of novel molecular frameworks.
The Foundational Chemical Logic of Scaffold Assembly
The biosynthesis of natural product (NP) ring systems is not an endless catalog of unique reactions but is instead built upon a concise repertoire of highly efficient, scaffold-generating chemical themes. These transformations convert readily available precursors from primary metabolism into stereochemically defined cyclic or polycyclic cores [25].
Core Scaffold-Generating Chemistries: A limited set of enzyme-catalyzed reactions is responsible for the initial ring-forming steps across the major classes of NPs. The following table summarizes these key transformations, their substrates, and representative molecular outcomes [25].
Table 1: Key Scaffold-Generating Chemistries in Natural Product Biosynthesis [25]
| Chemistry Type | Substrate(s) | Representative Enzyme | Ring-Containing Scaffold Formed |
|---|---|---|---|
| Amine-aldehyde condensation (intermolecular, e.g., Pictet-Spengler) | Tryptamine + secologanin | Strictosidine synthase (STR1) | Strictosidine (tetracyclic indole alkaloid precursor) |
| Amine-aldehyde condensation (intramolecular) | N-methylputrescine | Copper amine oxidase (CuAO) | N-methyl-Δ¹-pyrrolinium (monocyclic tropane precursor) |
| Aryl-CoA acylation (Type III Polyketide Synthase) | Coumaroyl-CoA + Malonyl-CoA | Chalcone synthase (CHS) | Naringenin chalcone (tricyclic flavonoid precursor) |
| C–C Radical Coupling | Coniferyl alcohol radicals | Laccase/Dirigent protein complex | Pinoresinol (dicyclic lignan scaffold) |
| Terpene Cyclization | Farnesyl pyrophosphate (FPP) | Cadinene synthase (COS) | δ-Cadinene (bicyclic sesquiterpene) |
This logic of scaffold generation followed by diversification is a powerful paradigm. For instance, the Pictet-Spenglerase-catalyzed condensation creates the strictosidine scaffold, which is subsequently tailored by over a dozen different downstream enzymes (e.g., oxidoreductases, methyltransferases, glucosidases) to yield more than 3,000 distinct monoterpene indole alkaloids [25]. Similarly, the chalcone scaffold from CHS is the common precursor to all flavonoids, with downstream enzymes like isomerases, hydroxylases, and reductases generating the diversity of this massive NP class.
Statistical Landscape of NP Ring Systems: The functional importance of ring systems is underscored by cheminformatic analysis. A comprehensive study of 38,662 NP ring systems revealed their structural centrality but also a significant unexplored potential [3].
Table 2: Cheminformatic Analysis of Natural Product Ring Systems [3]
| Metric | Finding | Implication for Drug Discovery |
|---|---|---|
| Coverage in Approved Drugs | Only ~2% of known NP ring systems are present in approved drugs. | Vast untapped reservoir of novel, biologically pre-validated scaffolds. |
| Representation in Screening Libraries | ~50% of NP ring systems have analogs with similar 3D shape/electrostatics in typical screening compound sets. | Commercial libraries capture some NP-like chemical space, but a significant portion remains unique to nature. |
| Structural Diversity | High density of stereochemistry, bridged ring systems, and complex polycycles not commonly found in synthetic libraries. | NPs explore regions of chemical shape and complexity that are challenging for traditional synthetic chemistry. |
This data positions biosynthetic pathway investigation as the most direct route to access and engineer this underrepresented chemical space for drug development.
Diagram 1: Chemical logic of scaffold diversification from a common precursor.
Experimental Methodologies for Pathway Refactoring and Engineering
The experimental interrogation and manipulation of biosynthetic gene clusters (BGCs) are fundamental to testing hypotheses about ring-construction logic. Recent methodological advances enable the precise cloning, refactoring, and heterologous expression of BGCs to activate silent pathways and produce novel analogs [26] [27].
Hierarchical Golden Gate Assembly for BGC Engineering: Traditional cloning methods like Transformation-Associated Recombination (TAR) can be error-prone for complex, repetitive BGCs. A robust alternative is a hierarchical Golden Gate Assembly (GGA) strategy, which uses Type IIS restriction enzymes for scarless, multi-fragment assembly in a defined order [26].
- Protocol: De Novo Assembly and Refactoring of a Polyketide BGC [26]
- BGC Selection and Domestication: Select a target BGC (e.g., the 23-kb actinorhodin (act) cluster). In silico "domesticate" the sequence by silently mutating all internal recognition sites for the chosen Type IIS enzymes (e.g., BsaI, PaqCI) within coding regions and modifying sites in non-coding regions.
- Fragment Preparation: Design the BGC to be assembled from ~2 kb fragments. Amplify and clone each domesticated fragment into a dedicated entry vector (e.g., pKan) for stability and sequence verification.
- Hierarchical Assembly:
- Primary Assembly: Combine 6-10 entry plasmids in a single GGA reaction with BsaI-HFv2 and T4 DNA ligase, assembling them into an intermediate vector (e.g., pAmp-RFP-BsaI). This step achieves near 100% efficiency for up to six fragments.
- Secondary Assembly: Perform a second GGA reaction using PaqCI on 2-3 intermediate plasmids to assemble the full-length BGC into the final expression vector (e.g., pPAP-RFP-PaqCI).
- Pathway Refactoring: To interrogate logic, systematically inactivate individual genes within the refactored cluster via promoter deletions or insertions during the assembly process.
- Heterologous Expression: Transfer the assembled BGCs into an optimized, BGC-free heterologous host (e.g., Streptomyces coelicolor M1152) via conjugation.
- Metabolite Analysis: Culture strains and analyze metabolite production using HPLC-MS. Employ molecular networking (e.g., via GNPS) to compare the metabolic profiles of mutant strains to the wild-type pathway, identifying novel shunt products that reveal pathway logic and flexibility [26].
This platform approach enables the parallel construction of numerous pathway variants. For the act cluster, generating 23 mutant derivatives in a single experiment revealed that only 9 genes were essential for actinorhodin production, while inactivation of others led to significant pathway rewiring and the production of previously unidentified molecules [26].
Table 3: Performance Metrics of Hierarchical GGA Platform [26]
| Assembly Parameter | One-Pot GGA | Hierarchical GGA |
|---|---|---|
| Number of Fragments Assembled | 12 fragments + vector | 6-10 fragments per step |
| Assembly Efficiency | <20% correct clones | ~100% correct clones |
| Transformation Efficiency | Baseline | >10-fold higher |
| Suitability for High-Throughput Engineering | Low | High |
Diagram 2: Workflow for BGC refactoring via hierarchical Golden Gate Assembly.
The Scientist's Toolkit: Research Reagent Solutions Table 4: Essential Research Reagents for BGC Engineering Experiments [26]
| Reagent/Material | Function in Experiment | Specific Example / Notes |
|---|---|---|
| Type IIS Restriction Enzymes | Enable scarless, directional assembly of DNA fragments with unique 4-bp overhangs. | BsaI-HFv2 (for primary assembly), PaqCI (for secondary assembly). |
| T4 DNA Ligase | Ligates the cohesive ends generated by Type IIS digestion in the same reaction mix. | Used in the GGA master mix concurrently with restriction enzymes. |
| Entry & Destination Vectors | Provide stable propagation for fragments and final assembly, with selectable markers and cassette removal sites. | pKan (entry), pAmp-RFP-BsaI (intermediate), pPAP-RFP-PaqCI (destination). |
| Chemically Competent E. coli | For high-efficiency transformation and propagation of assembled constructs. | Essential after each GGA step to recover plasmids. |
| Optimized Heterologous Host | A genetically tractable host devoid of competing endogenous pathways for clean metabolite production. | Streptomyces coelicolor M1152 (Δact, Δred, etc.) [26]. |
| Conjugation Donor Strain | Facilitates the transfer of large, non-mobilizable BGC vectors from E. coli to the actinobacterial host. | E. coli ET12567/pUZ8002. |
Computational Tools for Pathway Prediction and Design
The vastness of genomic and chemical space necessitates computational tools to predict the logic of uncharacterized BGCs and design new pathways. This integrates biological big data, retrosynthetic analysis, and enzyme engineering [28].
Biological Big Data Resources: Computational pathway design relies on comprehensive, curated databases spanning compounds, reactions, and enzymes [28].
Table 5: Key Computational Resources for Biosynthetic Pathway Design [28]
| Data Category | Representative Databases | Primary Utility in Ring Logic Analysis |
|---|---|---|
| Compound Information | PubChem, ChEBI, COCONUT, NPAtlas | Provides structures of known NP scaffolds and analogs for comparison [28]. |
| Reaction/Pathway Information | KEGG, MetaCyc, Rhea, BKMS-react | Catalogs known enzymatic transformations, especially ring-forming and tailoring reactions [28]. |
| Enzyme Information | BRENDA, UniProt, PDB, AlphaFold DB | Offers functional data, sequences, and 3D structures to infer or engineer substrate specificity [28]. |
Retrosynthetic Pathway Prediction: Retrosynthesis algorithms work backwards from a target ring system (scaffold) to identify plausible biological precursors and enzyme-catalyzed steps. Tools leverage reaction rules mined from the above databases to propose pathways that can be tested experimentally [28].
Enzyme Discovery and Engineering: When a predicted pathway requires a novel or promiscuous enzyme activity, computational tools are used to:
- Identify Candidate Enzymes: Mine genomic databases for enzymes with similarity to those catalyzing known, analogous reactions.
- Model Substrate Binding: Use protein structures (from PDB or AlphaFold) for docking studies to predict if an enzyme might accept a non-native substrate.
- Design Mutants: Propose active site mutations to alter or broaden substrate specificity, a key step in repurposing tailoring enzymes to act on novel scaffolds [27] [28].
Diagram 3: Computational cycle for designing biosynthetic pathways to target molecules.
Applications and Future Outlook: Engineering Novel Ring Systems
The integrated application of the described experimental and computational methodologies moves the field from observation to creation, directly impacting drug discovery.
Combinatorial Biosynthesis and Scaffold Diversification: By swapping domains in polyketide synthases (PKS) or non-ribosomal peptide synthetases (NRPS), or by expressing tailoring enzymes from different pathways in a heterologous host, researchers can generate "unnatural natural products." This approach has been used to produce novel analogs of antibiotics, antifungals, and anticancer agents with improved properties [27]. For example, reprogramming the avermectin PKS led to the commercial production of doramectin, a potent anthelmintic with a tailored ring system [27].
Activating Silent BGCs for Novel Scaffold Discovery: Many BGCs are not expressed under laboratory conditions. Heterologous expression in a well-characterized host, often after refactoring with strong promoters, can activate these silent pathways, leading to the discovery of entirely new ring-containing scaffolds with potentially novel bioactivities [26] [27].
Synthetic Biology for Scaffold Production: For complex plant-derived ring systems (e.g., alkaloids, terpenes), complete biosynthetic pathways are being reconstructed in microbial hosts like yeast and E. coli. This not only enables sustainable production but also provides a plug-and-play platform for engineering. By modulating the expression of tailoring enzymes, libraries of differentially decorated scaffolds can be generated for structure-activity relationship studies [27] [25].
The future of understanding and utilizing the natural logic of ring construction lies in deepening the integration of computational predictions with high-throughput automated strain construction and screening. As algorithms better predict the outcome of pathway rewiring and enzyme engineering, the Design-Build-Test-Learn cycle will accelerate, systematically unlocking the blueprint of biosynthetic pathways to build the next generation of bioactive molecular frameworks.
The structural core of most bioactive small molecules is defined by their ring systems, which determine molecular shape, rigidity, and key pharmacodynamic properties [3]. In drug discovery, the exploration of chemical space—the vast, multidimensional universe of possible molecules—is fundamentally an exploration of ring systems and molecular frameworks [1]. Natural products (NPs), honed by evolution for biological interaction, represent a privileged subspace of chemical diversity, rich in complex and stereochemically dense scaffolds [3]. Conversely, synthetic compound libraries, built for accessibility and modularity, often explore different regions of chemical space [29].
This analysis is framed within a critical thesis: that the unique ring systems and frameworks prevalent in natural products occupy distinct and underutilized regions of biologically relevant chemical space (BioReCS) compared to common synthetic libraries [30]. Mapping this uniqueness is not merely an academic exercise; it is a strategic imperative for reinvigorating scaffold-based drug discovery. This guide provides a technical roadmap for conducting such analyses, detailing methodologies for framework extraction, comparative cheminformatics, and advanced mapping techniques to quantify and visualize the structural distinctiveness of natural product architectures.
Foundational Concepts: Frameworks, Ring Systems, and Chemical Space
A precise lexicon is essential for reproducible analysis. In this context, a molecular framework (or scaffold) is typically derived by removing all side chain atoms, leaving only the ring systems and the linkers that connect them. A ring system refers to a single ring or a set of fused or spiro-connected rings, excluding any linker atoms [1]. Chemical space is a conceptual multidimensional space where each dimension corresponds to a molecular property or descriptor, and each compound occupies a specific coordinate [31]. The Biologically Relevant Chemical Space (BioReCS) is the subspace populated by molecules exhibiting bioactivity, which includes but is far more constrained than the total theoretical chemical space [30].
The analytical focus on ring systems is justified by their overwhelming prevalence: over 99.5% of bioactive molecules in databases like ChEMBL contain at least one ring system, and they are the primary determinants of a molecule's global properties [1].
Table 1: Key Definitions for Chemical Space and Framework Analysis
| Term | Definition | Analytical Significance |
|---|---|---|
| Molecular Framework/Scaffold | The core structure remaining after removal of all side chains and functional groups, retaining ring systems and connecting atoms. | Enables abstraction of molecules to their core architectures for diversity analysis and scaffold hopping. |
| Ring System | A single cyclic structure or multiple rings connected via fusion (shared bonds) or spiro junctions (shared single atom). | The fundamental building block of molecular shape and rigidity; the primary unit for comparative cheminformatics [3]. |
| Chemical Space | A multi-dimensional space defined by molecular descriptors where each compound is represented as a point. | Provides a visual and quantitative model for comparing large compound collections [31]. |
| Biologically Relevant Chemical Space (BioReCS) | The subspace of chemical space populated by molecules with documented biological activity [30]. | The target region for drug discovery; mapping NP frameworks here highlights their validated bioactivity potential. |
Quantitative Landscape: Diversity and Coverage of Ring Systems
Comprehensive analyses reveal a stark dichotomy between the structural wealth of NPs and the conservative, focused diversity of synthetic libraries. A landmark study analyzing 38,662 ring systems from NPs found an immense structural and stereochemical diversity far exceeding that of typical synthetic compounds (SCs) [3]. Notably, only about 2% of NP ring systems are directly present in approved drugs, indicating a vast reservoir of unexplored chemotypes [3].
However, approximately 50% of NP ring systems have a close counterpart in synthetic libraries when considering similar 3D shape and electrostatic properties, suggesting that synthetic chemistry has, to some degree, converged on biologically relevant shapes, albeit with different atomic compositions [3]. The frequency distribution of ring systems follows a power law: a small number of very simple systems (e.g., benzene, pyridine) are ubiquitous, while a "long tail" consists of thousands of unique, complex systems appearing only once or a few times [1]. This long tail is disproportionately populated by NPs.
Table 2: Comparative Analysis of Ring Systems in Natural Products vs. Synthetic Compounds [3]
| Metric | Natural Product Ring Systems | Synthetic Compound Ring Systems | Implication |
|---|---|---|---|
| Total Unique Systems Analyzed | 38,662 | From large screening libraries | NPs offer orders of magnitude more unique ring system templates. |
| Coverage in Approved Drugs | ~2% | Higher percentage (common systems) | Vast majority of NP ring systems are pharmaceutically underexplored. |
| 3D Shape/Electrostatic Coverage | ~50% have a similar counterpart in SCs | Covers half of NP shape space | Synthetic libraries mimic many NP shapes but with different atoms/scaffolds. |
| Stereochemical Complexity | High prevalence of chiral centers and unique stereoisomers. | Generally lower complexity. | NP frameworks encode richer 3D information critical for selective binding. |
| Representative Common Systems | Complex fused and bridged systems (e.g., pentacyclic triterpenes). | Simple mono- and bicyclic aromatics (e.g., benzene, indole). | Core structural preferences differ fundamentally. |
Fragment-based analysis corroborates this divergence. A 2025 study generated fragment libraries from over 695,000 non-redundant NPs (COCONUT database) and compared them to the CRAFT library of novel synthetic fragments [29] [32]. The NP-derived library contained 2.58 million fragments, showcasing an explosive combinatorial diversity stemming from their complex cores, while the synthetic CRAFT library contained 1,214 carefully curated fragments [32]. The chemical space covered by these libraries is distinct, with NP fragments exploring more stereochemistry and sp3-carbon richness.
Table 3: Fragment Library Comparison: Natural Product vs. Synthetic Sources [29] [32]
| Library | Source Database | Number of Source Compounds | Number of Fragments Generated | Key Characteristics |
|---|---|---|---|---|
| NP Fragment Library | COCONUT (Curated NP database) | >695,133 unique NPs | 2,583,127 | Extreme diversity, high stereochemical and 3D complexity. |
| LANaP Fragment Library | Latin America Natural Product Database | 13,578 unique NPs | 74,193 | Regionally sourced biodiversity, unique chemotypes. |
| CRAFT Library | Designed synthetic & NP-derived chemicals | N/A (designed library) | 1,214 | Focus on novel heterocyclic scaffolds, high synthetic accessibility. |
Experimental & Computational Methodologies
Protocol 1: Cheminformatic Analysis of Ring System Diversity and Coverage
This protocol outlines the steps to compare ring systems between NP and synthetic libraries [3].
Data Curation and Standardization:
- Source NP Databases: Use comprehensive, curated databases such as COCONUT or LOTUS [3] [8]. For synthetic compounds, use vendor catalogs (e.g., Enamine, MolPort) or screening libraries like ChEMBL.
- Standardization: Apply consistent rules for neutralization, tautomerization, and removal of salts using toolkits like RDKit or OpenBabel. Crucially, retain stereochemical information.
Ring System Perception and Extraction:
- Apply an algorithm (e.g., the Murcko framework decomposition or the RDKit
GetSymmSSSRfunction) to decompose each molecule into its constituent ring systems [1]. - Store each unique ring system as a canonical SMILES string, with and without stereochemical indicators, to create two separate sets: a stereo-aware and a stereo-agnostic inventory.
- Apply an algorithm (e.g., the Murcko framework decomposition or the RDKit
Descriptor Calculation and 3D Shape Analysis:
- Generate 3D conformers for each unique ring system using a conformer generation algorithm (e.g., ETKDG in RDKit).
- Calculate 3D molecular shape descriptors (e.g., via Ultra-Fast Shape Recognition, USR, or its electrostatic variant, ECFP) and 2D topological fingerprints (e.g., Morgan fingerprints).
Coverage Analysis:
- For each NP ring system, search for the most similar synthetic ring system using a combined shape-electrostatic similarity score (e.g., ET_combo) [3].
- Set a similarity threshold (e.g., ET_combo ≥ 0.8) to define "coverage." Calculate the percentage of NP ring systems covered by synthetic systems.
Visualization and Clustering:
- Use dimensionality reduction techniques like t-SNE or UMAP on the fingerprint descriptors to project the high-dimensional data into 2D.
- Color-code points by source (NP vs. synthetic) to create a chemical space map illustrating regions of overlap and uniqueness.
Figure 1: Workflow for Cheminformatic Ring System Analysis. This diagram outlines the computational pipeline for extracting, comparing, and visualizing ring systems from natural product and synthetic compound libraries [3].
Protocol 2: Multi-Representation Similarity for Framework Categorization (AgreementPred)
This protocol uses the AgreementPred framework to recommend pharmacological categories for unannotated NP frameworks by fusing similarity searches across multiple molecular representations [8].
Construct Annotated Training Set:
- Compile a set of drugs and NPs with known pharmacological annotations from sources like PubChem, which integrates ATC (Anatomical Therapeutic Chemical) and MeSH (Medical Subject Headings) codes [8].
- Extract all unique textual category labels (e.g., "Enzyme Inhibitors," "Antineoplastic Agents") to serve as prediction targets.
Generate Multiple Molecular Representations:
- For each compound in the training set and for each unannotated NP framework, calculate a diverse set of 22+ molecular fingerprints and descriptors. This includes 2D fingerprints (e.g., ECFP4, Atom Pair, MACCS keys), 3D shape descriptors, and learned neural network embeddings [8].
Similarity Search and Result Fusion:
- For an unannotated NP framework, perform a similarity search (e.g., Tanimoto similarity) against the annotated training set using each of the 22+ representations independently.
- For each representation, retrieve the top-k most similar annotated compounds and their associated category labels.
Calculate Agreement Scores and Filter Predictions:
- Fuse the results by counting how many different representations "agree" on recommending a specific category label for the query framework.
- Calculate an Agreement Score for each recommended category:
(Number of Representations Recommending the Category) / (Total Number of Representations). - Filter predictions by setting an Agreement Score threshold (e.g., >0.1). A category recommended by many diverse representations is considered a high-confidence prediction [8].
Figure 2: The AgreementPred Framework for Category Prediction. This process uses multiple structural representations to achieve robust pharmacological category recommendations for unannotated natural product frameworks [8].
Protocol 3: 3D Structure-Aware Molecular Optimization (3DToMolo)
This protocol leverages a diffusion model to optimize a lead molecule's properties while preserving a desired NP-derived core framework, using textual and 3D structural guidance [33].
Problem Definition and Input:
- Input: A starting molecule
M_0(e.g., a simplified NP derivative) with its 2D graph and 3D conformer. - Constraint: A specific substructure (the NP framework) to be preserved throughout optimization.
- Goal: A textual prompt
ydescribing desired properties (e.g., "increase solubility," "reduce logP," "maintain kinase binding").
- Input: A starting molecule
Forward Diffusion Process:
- The model gradually adds noise to the 3D coordinates and atom types of
M_0over many stepst, following a stochastic differential equation (SDE) [33]. - This process creates a noisy, intermediate representation
M_tthat progressively obscures atomic details but retains the overall semantic shape and topology.
- The model gradually adds noise to the 3D coordinates and atom types of
Conditional Denoising (Optimization) Process:
- A text encoder (e.g., a lightweight language model) processes the textual prompt
yinto a feature vector. - An SE(3)-equivariant graph neural network is trained to denoise
M_tback towards a valid molecule. Crucially, its denoising direction is guided by the text feature vector and the constraint to preserve the defined core substructure [33]. - By reversing the diffusion process from a noisy state under this multi-modal guidance, the model generates a new molecule
M_1that aligns better with the text prompt while keeping the NP framework intact.
- A text encoder (e.g., a lightweight language model) processes the textual prompt
Figure 3: 3D-Aware Molecular Optimization with the 3DToMolo Framework. This AI-driven process optimizes lead molecules based on textual property goals while preserving a fixed natural product core substructure in 3D space [33].
Table 4: Key Research Reagent Solutions for Chemical Space Analysis
| Item / Resource | Type | Function in Analysis | Example / Source |
|---|---|---|---|
| Curated NP Databases | Data | Provide standardized, high-quality structural data for NP ring system extraction. | COCONUT [29], LANaPDB [32], LOTUS [8] |
| Synthetic Compound Libraries | Data | Provide reference chemical space for comparison and coverage analysis. | CRAFT [32], Enamine REAL, ChEMBL [30] |
| Cheminformatics Toolkits | Software | Enable molecule standardization, ring perception, descriptor calculation, and fingerprinting. | RDKit, OpenBabel, scikit-learn [3] |
| Molecular Representation Libraries | Software/Algorithm | Provide diverse descriptors for multi-perspective similarity analysis. | RDKit fingerprints, Shape- & Electrostatic-Descriptors (e.g., USR), MAP4 fingerprint [30] [8] |
| Similarity & Clustering Algorithms | Software/Algorithm | Quantify molecular similarity and group compounds in chemical space. | Tanimoto coefficient, t-SNE, UMAP, hierarchical clustering [31] |
| Category Annotation Sources | Data | Provide pharmacological labels for training predictive models. | PubChem ATC/MeSH annotations [8] |
| Generative AI Frameworks | Software/Model | Enable property-driven optimization of molecules while preserving core scaffolds. | 3DToMolo [33], Chemeleon [34] |
Future Directions and Integrative Approaches
The frontier of chemical space analysis is being reshaped by generative artificial intelligence and multi-modal data integration. Tools like Chemeleon, which uses text-guided diffusion models to generate crystal structures, demonstrate the potential of language to steer exploration in vast compositional spaces [34]. Translating this to NP frameworks, future methods will allow researchers to navigate BioReCS using prompts like "generate novel variants of the indole alkaloid scaffold with improved metabolic stability."
Furthermore, the concept of BioReCS is expanding to include historically underexplored regions such as metallodrugs, macrocycles, and protein-protein interaction inhibitors [30]. Integrating the unique frameworks of NPs with these non-traditional chemotypes represents a powerful strategy for addressing challenging biological targets. The continuous development of universal molecular descriptors, capable of encoding information from small molecules to peptides and inorganic complexes, will be crucial for mapping these unified chemical spaces [30].
Systematic chemical space analysis confirms that natural product frameworks occupy a region of structural and stereochemical diversity that is both unique and highly relevant to biology. While synthetic libraries provide broad coverage of simple, accessible chemotypes, the complex, three-dimensional architectures characteristic of NPs remain a distinctive and invaluable resource. The methodologies detailed here—from foundational cheminformatics to advanced, AI-driven optimization—provide researchers with a toolkit to quantify this uniqueness, mine it for novel scaffolds, and intelligently hybridize it with synthetic approaches. Embracing the complexity of NP ring systems is not a step backward into natural product isolation, but a leap forward into a new era of informed, diversity-driven molecular design.
From Nature-Inspired to Lab-Created: Synthetic and Computational Strategies for Ring Systems
Within the grand thesis of exploring ring systems and molecular frameworks in natural products research, biomimetic synthesis represents a paradigm-shifting philosophy. It moves beyond merely recreating natural product structures to emulating the efficiency and logic of their biosynthetic pathways in living organisms [35]. This approach is particularly transformative for assembling complex ring systems—the core architectural motifs that define the bioactivity of countless therapeutics [17]. Traditional stepwise synthesis often struggles with the thermodynamic and kinetic hurdles of constructing medium-sized (8-11 membered) and macrocyclic rings, facing issues like transannular strain and unfavorable entropic factors [36]. Biomimetic synthesis addresses these challenges by drawing inspiration from nature's own catalysts and step-economical processes, such as polyene cyclizations and oxidative couplings [37]. By integrating this approach, researchers can populate underexplored regions of chemical space with novel, biologically relevant ring systems, directly advancing the core objective of the broader thesis: to understand, catalog, and exploit molecular frameworks for drug discovery [36] [17].
Strategic Approaches to Biomimetic Ring Assembly
The biomimetic synthesis of complex ring systems employs strategies directly inspired by biosynthetic machinery. Three cornerstone approaches have enabled the efficient construction of intricate molecular frameworks.
Biomimetic Polyene Cyclization: This strategy mimics the enzymatic conversion of linear oligoprenyl diphosphates into stereochemically dense polycyclic terpenoids and steroids. In nature, acid-initiated cascade reactions form multiple carbon-carbon bonds and rings in a single operation. Synthetic chemists emulate this using Brønsted or Lewis acids to trigger controlled, stepwise cyclizations of polyene substrates. This approach has been pivotal for synthesizing the core ring systems of steroids like progesterone and complex terpenoid alkaloids, providing critical insights into achieving high levels of stereoselective control that mirror enzymatic precision [37].
Oxidative Dearomatization-Ring Expansion (ODRE) Sequences: Inspired by phenolic oxidative coupling in nature, this powerful tactic builds medium-sized rings from smaller, aromatic precursors [36]. The process begins with the oxidative dearomatization of a phenol to a highly reactive cyclohexadienone. This intermediate then undergoes a nucleophile-triggered ring expansion, cleaving a strategic bond to form a larger ring, often followed by rearomatization. This ODRE sequence is a versatile tool for constructing benzannulated medium-ring ethers, lactones, and biaryl systems—common motifs in bioactive natural products that are challenging to access via direct cyclization [36].
Biomimetic Diels-Alder Cycloadditions: Many natural [4+2] cycloadditions are hypothesized in biosynthesis. The biomimetic version utilizes electron-rich dienes and dienophiles under thermal or Lewis acid-catalyzed conditions to rapidly assemble six-membered rings embedded within larger, polycyclic architectures. This strategy is celebrated for its atom economy and ability to install multiple stereocenters simultaneously. It has been successfully deployed in the total synthesis of highly complex targets like FR182877, showcasing its power for constructing intricate, bridged ring systems [37].
Table 1: Core Biomimetic Strategies for Ring Assembly
| Strategy | Biosynthetic Inspiration | Key Reactive Intermediate | Typical Ring Systems Formed | Primary Advantage |
|---|---|---|---|---|
| Polyene Cyclization | Terpene/steroid biosynthesis | Carbocation cascade | Fused 6-, 5-, and 3-membered rings (e.g., steroid cores) | Rapid construction of multiple rings and stereocenters |
| ODRE Sequence | Phenolic oxidative coupling | Cyclohexadienone | Benzannulated 8-11 membered rings (medium-ring ethers, lactones) | Access to strained medium-sized rings from stable aromatics |
| Diels-Alder Cycloaddition | Proposed enzymatic [4+2] cyclizations | N/A (concerted pericyclic) | 6-membered rings within polycyclic frameworks | High atom economy and stereochemical control |
Detailed Experimental Protocols
Translating biomimetic strategies into practice requires carefully designed experimental protocols. The following detailed methodologies are foundational to the field.
Protocol for Oxidative Dearomatization-Ring Expansion (ODRE)
This protocol outlines the synthesis of a benzannulated 9-membered aryl ether via a tandem oxidative dearomatization-ring expansion reaction, adapted from Tan et al. [36].
Reagents:
- Phenolic substrate (e.g., 2-(4-hydroxybenzyl)-1,4-dimethoxybenzene), 1.0 equiv.
- Iodobenzene diacetate (PIDA), 1.1 equiv.
- Trifluoroethanol (TFE), as nucleophile and solvent.
- Boron trifluoride diethyl etherate (BF₃·OEt₂), 0.2 equiv.
- Anhydrous dichloromethane (DCM), distilled.
- Saturated aqueous sodium thiosulfate (Na₂S₂O₃) solution.
- Saturated aqueous sodium bicarbonate (NaHCO₃) solution.
- Anhydrous magnesium sulfate (MgSO₄).
- Silica gel for flash chromatography.
Procedure:
- Setup: Under a nitrogen atmosphere, charge a flame-dried round-bottom flask with the phenolic substrate (1.0 mmol) and dissolve in anhydrous DCM (0.1 M concentration). Cool the solution to -40°C using an acetonitrile/dry ice bath.
- Oxidation: Add trifluoroethanol (10.0 equiv.) followed by dropwise addition of BF₃·OEt₂ (0.2 equiv.). Stir for 5 minutes.
- Dearomatization & Expansion: In one portion, add solid iodobenzene diacetate (1.1 equiv.). Maintain the temperature at -40°C and monitor the reaction by thin-layer chromatography (TLC). The reaction is typically complete within 2-4 hours.
- Quenching: Slowly add a saturated aqueous Na₂S₂O₃ solution (5 mL per mmol substrate) to quench excess oxidant. Warm the mixture to room temperature.
- Extraction: Transfer the mixture to a separatory funnel. Dilute with DCM (20 mL) and wash sequentially with saturated NaHCO₃ solution (10 mL) and brine (10 mL).
- Work-up: Dry the combined organic layers over anhydrous MgSO₄, filter, and concentrate under reduced pressure using a rotary evaporator.
- Purification: Purify the crude residue by flash column chromatography on silica gel (gradient elution: hexanes to 20% ethyl acetate in hexanes) to obtain the ring-expanded 9-membered benzannulated ether.
Protocol for Electrochemical Dehydrogenative Ring Expansion
This protocol describes an electrochemical method for synthesizing medium-ring lactams via amidyl radical formation and C–C bond cleavage, based on the work of Liu et al. [36].
Reagents:
- Benzocyclic ketone (e.g., 1-tetralone), 1.0 equiv.
- Primary amide (e.g., acetamide), 2.0 equiv.
- Tetrabutylammonium hexafluorophosphate (Bu₄NPF₆), 0.1 M as supporting electrolyte.
- Anhydrous acetonitrile (MeCN), distilled.
- Platinum plate electrodes (anode and cathode).
- Constant current power supply.
- Saturated aqueous ammonium chloride (NH₄Cl) solution.
- Ethyl acetate for extraction.
- Anhydrous sodium sulfate (Na₂SO₄).
Procedure:
- Electrochemical Cell Setup: In an undivided electrochemical cell equipped with two platinum plate electrodes (approx. 1 cm²), combine the benzocyclic ketone (0.5 mmol), primary amide (1.0 mmol), and Bu₄NPF₆ (0.1 mmol) in anhydrous MeCN (10 mL). Stir until a homogeneous solution is achieved.
- Electrolysis: Connect the cell to a constant current power supply. Apply a constant current of 5 mA and perform the electrolysis at room temperature for 8 hours. Monitor the reaction progress by TLC or LC-MS.
- Reaction Completion & Quench: Once starting material is consumed, disconnect the power supply. Pour the reaction mixture into a separatory funnel containing saturated NH₄Cl solution (20 mL).
- Extraction and Isolation: Extract the aqueous mixture with ethyl acetate (3 x 15 mL). Combine the organic extracts and wash with brine (20 mL). Dry the organic phase over anhydrous Na₂SO₄, filter, and concentrate.
- Purification: Purify the crude product via flash chromatography (silica gel, eluting with 15-30% ethyl acetate in petroleum ether) to afford the medium-ring (8-11 membered) lactam product.
Protocol for Biomimetic Aldol Reaction
This protocol outlines an asymmetric biomimetic aldol reaction catalyzed by a chiral pyridoxal derivative, enabling efficient synthesis of chiral β-hydroxy-α-amino acid derivatives, as reported by Liang et al. [38].
Reagents:
- Glycine ethyl ester hydrochloride, 1.0 equiv.
- Aldehyde substrate, 1.2 equiv.
- Chiral pyridoxal catalyst (e.g., (S)-4-tert-butyl-2-(2-hydroxybenzylidene)imidazolidin-5-one), 0.1 equiv.
- Triethylamine (Et₃N), 2.0 equiv.
- Molecular sieves (4Å), powdered and activated.
- Anhydrous methanol (MeOH).
- Anhydrous dichloromethane (DCM).
- 1M aqueous hydrochloric acid (HCl) solution.
- Saturated aqueous sodium bicarbonate (NaHCO₃) solution.
- Brine.
Procedure:
- Activation: In a flame-dried Schlenk flask, add glycine ethyl ester hydrochloride (1.0 mmol), the chiral pyridoxal catalyst (0.1 mmol), and powdered 4Å molecular sieves (100 mg). Evacuate and backfill with nitrogen three times.
- Reaction Mixture: Under nitrogen, add anhydrous MeOH (5 mL) and DCM (5 mL). Cool the mixture to 0°C in an ice bath.
- Deprotonation: Add triethylamine (2.0 mmol) dropwise and stir for 15 minutes to generate the free glycine ester and the active catalyst complex.
- Aldol Addition: Add the aldehyde substrate (1.2 mmol) dropwise. Remove the ice bath and allow the reaction to warm to room temperature. Stir for 24-48 hours, monitoring by TLC.
- Work-up: Filter the reaction mixture through a pad of Celite to remove molecular sieves, washing thoroughly with DCM. Transfer the filtrate to a separatory funnel.
- Acid-Base Extraction: Wash the organic solution with 1M HCl (10 mL), then with saturated NaHCO₃ (10 mL), and finally with brine (10 mL). Dry the organic layer over anhydrous Na₂SO₄.
- Isolation: Filter, concentrate, and purify the residue by flash chromatography (silica gel, hexanes/ethyl acetate gradient) to yield the enantiomerically enriched β-hydroxy-α-amino ester.
Table 2: Representative Experimental Outcomes from Biomimetic Protocols
| Protocol | Starting Material Class | Key Reaction Conditions | Typical Product Ring Size | Reported Yield Range | Primary Challenge |
|---|---|---|---|---|---|
| ODRE Sequence [36] | Polycyclic Phenol | PIDA, BF₃·OEt₂, -40°C | 8-11 membered benzannulated rings | 45-75% | Controlling regioselectivity of nucleophile attack |
| Electrochemical Expansion [36] | Benzocyclic Ketone + Amide | Constant Current (5 mA), Pt electrodes, rt | 8-11 membered lactams | 60-82% | Optimization of current density and electrode material |
| Biomimetic Aldol [38] | Glycinate + Aldehyde | Chiral Pyridoxal (10 mol%), 0°C to rt | Forms linear precursor for heterocycles | 80-95%, >90% ee | Sensitivity to moisture and substrate scope limitations |
Diagram 1: Conceptual Workflow for Biomimetic Synthesis of Ring Systems.
Diagram 2: Mechanism of the Oxidative Dearomatization-Ring Expansion (ODRE) Reaction.
Data Presentation and Analysis
The success of biomimetic strategies is quantified by their ability to efficiently generate complex ring systems with high fidelity. The following data, synthesized from recent literature, demonstrates the scope and efficiency of these approaches. A critical analysis of ring system diversity in drug databases shows that while a limited set of simple aromatic (SA) ring systems dominates known bioactive molecules, there exists a vast unexplored space of complex and medium-sized rings that are prime targets for biomimetic synthesis [17].
Table 3: Analysis of Ring Systems in Bioactive Molecules vs. Natural Products
| Metric | Data from Known Drugs / Bioactive Molecules [17] | Implication for Biomimetic Synthesis |
|---|---|---|
| Number of Distinct Simple Aromatic (SA) Ring Systems | ~780 systems found in 150,000 bioactive molecules. | Highlights the high value of a relatively small set of privileged frameworks. |
| Coverage of Chemical Space | SA systems cover a significant portion of current drug space but represent a limited chemical subspace. | A vast territory of complex, bridged, and medium-sized rings from natural products remains underexploited. |
| Frequency of Medium-Sized Rings (8-11 membered) | Notably underrepresented in screening libraries and top-selling drugs. | Biomimetic synthesis (e.g., ODRE, electrochemical expansion) is specifically tasked with populating this gap. |
| Strategy for Discovery | Bioisosteric replacement and scaffold hopping within known SA systems. | Biomimetic synthesis enables de novo generation of novel, biologically pre-validated ring systems from natural product blueprints. |
The Scientist's Toolkit: Key Reagents & Materials
Executing advanced biomimetic syntheses requires a specialized set of reagents, catalysts, and analytical tools.
Table 4: Essential Research Reagent Solutions for Biomimetic Ring Synthesis
| Reagent/Material | Function in Biomimetic Synthesis | Example Use Case |
|---|---|---|
| Hypervalent Iodine Reagents (e.g., PIDA, PIFA) | Mild, selective oxidants for dearomatization. Mimic the single-electron oxidation function of certain enzymes. | Generating the key cyclohexadienone intermediate in ODRE sequences [36]. |
| Chiral Pyridoxal / Pyridoxamine Derivatives | Biomimetic asymmetric catalysis. Mimics the function of pyridoxal phosphate (PLP)-dependent enzymes in amino acid metabolism. | Catalyzing enantioselective biomimetic aldol reactions of glycine derivatives [38]. |
| Electrochemical Cell Setup (Pt electrodes, supporting electrolyte) | Provides a controlled, "green" source of electrons or holes to generate reactive intermediates. Mimics redox processes in biological systems. | Generating amidyl radicals for dehydrogenative ring expansion to medium-ring lactams [36]. |
| Lewis Acids (BF₃·OEt₂, SnCl₄) | Activates substrates and controls stereochemistry in polycyclization events. Mimics the action of metal ions in terpene cyclase enzymes. | Initiating and directing the stereochemical outcome of biomimetic polyene cyclizations [37]. |
| Polyprenyl / Polyene Chain Precursors | Linear substrates designed to fold and cyclize in a specific conformation, emulating the natural polyisoprene building blocks. | Serving as starting materials for biomimetic syntheses of terpenoids and steroids via cation cascades [37]. |
Biomimetic synthesis stands as a cornerstone methodology for advancing the thesis of ring systems and molecular frameworks in natural products research. By emulating nature's inherent efficiency—through strategies like polyene cyclization, ODRE sequences, and biomimetic cycloadditions—it provides reliable access to complex and strained ring architectures that defy conventional synthesis [36] [37]. This approach does more than replicate structures; it validates biosynthetic hypotheses and unlocks efficient routes to novel chemical space, particularly the underexplored domain of medium-sized rings [36] [17]. The future of this field lies in deeper integration with bioinformatics for pathway prediction, artificial intelligence for reaction optimization, and the continued development of bio-inspired catalysts that operate with enzymatic precision under mild conditions [38] [37]. As these tools mature, biomimetic synthesis will increasingly serve as the primary engine for discovering and constructing the next generation of bioactive molecular frameworks, solidifying its critical role in the evolution of natural products-based drug discovery.
Abstract Ring-expanding skeletal editing represents a paradigm shift in molecular design, enabling the direct, late-stage insertion of atoms into the core frameworks of complex molecules. This in-depth technical guide details the mechanisms, methodologies, and applications of this transformative approach, with a specific focus on its role in diversifying natural product-derived scaffolds for drug discovery. We provide a comprehensive analysis of carbon and heteroatom insertion strategies, supported by quantitative data, detailed experimental protocols for key transformations, and an exploration of integrated computational workflows. This whitepoon underscores how skeletal editing, by facilitating efficient navigation of chemical space around privileged ring systems, accelerates the discovery of novel bioactive compounds.
The structural core of bioactive molecules, particularly natural products (NPs) and their synthetic analogues, is predominantly defined by their ring systems [39]. These carbo- and heterocyclic frameworks dictate fundamental molecular properties—including three-dimensional shape, conformational rigidity, solubility, and metabolic stability—which in turn govern biological function [39]. For decades, diversifying these core structures to explore structure-activity relationships (SAR) or improve drug-like properties necessitated laborious de novo synthesis or peripheral functionalization, both of which have significant limitations in step economy and the degree of scaffold change achievable [39] [40].
The emerging discipline of skeletal editing addresses this bottleneck by enabling precise, atom-level “surgery” on molecular frameworks [40]. This guide focuses on ring-expanding skeletal editing, a subset defined as the insertion of one or more atoms into a cyclic system, thereby increasing its ring size [41] [39]. This strategy is especially powerful for late-stage diversification, where complex, densely functionalized intermediates—such as advanced NP derivatives—can be directly transformed into novel analogues with altered ring architectures [42] [43]. Such transformations provide efficient access to underrepresented chemical space, including medium-sized rings (8-11 members), which are challenging to synthesize by conventional cyclization but are prevalent in bioactive NPs [44].
Within the broader thesis of NP research, ring-expanding editing serves as a critical tool for Complexity-to-Diversity (CtD) strategies. CtD uses the inherent complexity of NPs as a starting point, applying distortion reactions—including ring expansion, cleavage, and fusion—to rapidly generate diverse, NP-like compound libraries for phenotypic and target-based screening [42]. This approach leverages biologically relevant, pre-validated scaffolds to populate chemical space with molecules of high therapeutic potential [44].
Mechanisms and Strategies of Ring-Expanding Skeletal Editing
Ring-expansion editing is fundamentally categorized by the nature of the atom inserted and the mechanistic pathway employed. The most developed strategies involve the insertion of single carbon or nitrogen atoms, often via the generation of reactive intermediates that undergo controlled rearrangement or cycloaddition [39] [40].
Table 1: Core Strategies for Single-Atom Ring-Expanding Skeletal Editing
| Strategy | Atom Inserted | Key Reactive Intermediate/Reagent | Typical Product Ring Size Change | Common Substrate Classes |
|---|---|---|---|---|
| Classical Rearrangement [41] [39] | None (C/N exchange) | Oxime, nitrene | n → n (atom transmutation) | Pyridines, quinolines |
| Carbene/Carbenoid Insertion [40] | C | Dichlorocarbene, metal-carbenes | n → n+1 | Pyrroles, indoles (Ciamician-Dennstedt) |
| Homologation with Diazo Compounds [39] | C | α-Diazo esters, rhodium carbenoids | n → n+1 (ketone ring expansion) | Cyclic ketones, benzocyclobutenones |
| Nitrene Insertion [41] | N | Organic azides, metal-nitrenes | n → n+1 | Saturated carbocycles, arenes |
| Photoredox-Mediated Radical Insertion [43] | Functionalized C | α-Iodonium diazo reagents, atomic carbon equivalents | n → n+1 | Indenes, indoles |
2.1 Carbon Atom Insertion This is the most prevalent and widely studied approach. A landmark historical method is the Ciamician-Dennstedt rearrangement, where a dichlorocarbene (generated from chloroform under basic conditions) adds to a pyrrole ring, leading to ring expansion to a pyridine via a cyclopropanation-fragmentation-aromatization sequence [40]. Modern advancements have focused on safer, more controllable carbene precursors and catalysts. A powerful contemporary method involves the homologation of cyclic ketones using diazo compounds. For instance, the enantioselective ring expansion of cyclic ketones with α-alkyl α-diazo esters, catalyzed by chiral scandium(III) complexes, provides access to chiral β-keto esters with excellent stereocontrol [39]. Furthermore, rhodium-catalyzed formal (4+1) cycloadditions using styrenes as carbenoid equivalents offer a safer alternative to diazo compounds for inserting a carbon unit into strained systems like benzocyclobutenones [39]. Recently, photoredox catalysis has unlocked new pathways for diversity-generating carbon insertion. For example, an α-iodonium diazo-based reagent acts as a “carbynyl radical equivalent,” enabling the ring expansion of indenes to naphthalenes while simultaneously installing a versatile functional group (e.g., ester, ketone, cyano) from the reagent pool [43].
2.2 Nitrogen and Other Heteroatom Insertion The insertion of nitrogen into carbocyclic rings is a direct method for generating nitrogen-containing heterocycles, which are ubiquitous in pharmaceuticals. This is often achieved via nitrene insertion, where a metal-nitrene species, typically generated from an organic azide, inserts into a C–C bond [41]. While powerful, controlling the regioselectivity and avoiding over-reaction remains a key challenge. The transmutation of ring atoms, such as converting a carbon in a pyridine to another atom, is also a form of skeletal editing, though it does not change ring size [41] [40].
Application in Natural Product Diversification and Drug Discovery
Ring-expanding skeletal editing is uniquely positioned to address central challenges in modern drug discovery, particularly in diversifying NP-inspired chemical space and accessing novel, drug-like architectures.
3.1 Enabling Complexity-to-Diversity (CtD) and Pseudo-Natural Product Synthesis The CtD strategy uses the complex core of a NP as a launchpad for generating diverse libraries [42]. Ring-expansion is a cornerstone reaction in this paradigm. For instance, applying ring-expansion edits to a single, complex NP scaffold can generate a family of analogues with varying ring sizes and functionalities, rapidly exploring SAR and potentially improving properties like selectivity or metabolic stability [42] [44]. Similarly, in pseudo-natural product synthesis, fragments from biosynthetically unrelated NPs are combined; skeletal editing can then be used to further reshape and diversify these novel hybrid cores, accessing unprecedented chemical space with biological relevance [44].
3.2 Synthesis of Challenging Medium-Sized Rings Medium-sized rings (8-11 members) are highly represented in bioactive NPs but are notoriously difficult to synthesize via direct end-to-end cyclization due to transannular strain and entropic penalties [44]. Ring-expansion of smaller, less-strained cyclic precursors provides a thermodynamically favored route to these valuable structures. Strategies include oxidative cleavage-driven expansion of bicyclic systems and biomimetic dearomatization-expansion sequences, which efficiently construct benzannulated medium rings found in many NPs [44].
3.3 Impact on Drug Discovery Pipelines The direct late-stage diversification of lead compounds accelerates hit-to-lead and lead optimization campaigns. By allowing medicinal chemists to alter the core scaffold of a promising molecule without total re-synthesis, these methods compress the Design-Make-Test-Analyze (DMTA) cycle [45]. This aligns with the broader 2025 trend of integrating innovative chemistry with AI-driven design and predictive analytics to mitigate attrition risk and shorten development timelines [45] [46].
Detailed Experimental Protocols for Key Transformations
Protocol 1: Photoredox-Mediated, Diversity-Oriented Ring Expansion of Indenes [43] Objective: To convert indene scaffolds to functionalized naphthalenes via insertion of a functionalized carbon atom.
- Reaction Setup: In a dried Schlenk tube under an inert atmosphere (N₂ or Ar), combine the indene substrate (1.0 equiv, 0.1 mmol) and the α-iodonium diazo reagent (e.g., ethyl 2-diazo-2-(iodonium)acetate, 1.2 equiv). Add dry, degassed dichloromethane (DCM, 2 mL).
- Catalyst Addition: Add the photocatalyst (e.g., fac-Ir(ppy)₃, 2 mol%) and a weak base (e.g., K₂HPO₄, 2.0 equiv).
- Irradiation: Seal the tube and place it in a blue LED photoreactor (456 nm). Stir the reaction mixture at room temperature for 12-16 hours.
- Monitoring: Monitor reaction progress by TLC or LC-MS.
- Work-up: After completion, quench the reaction by direct filtration through a short plug of silica gel, eluting with DCM. Concentrate the filtrate under reduced pressure.
- Purification: Purify the crude residue by flash column chromatography on silica gel (hexanes/ethyl acetate gradient) to obtain the ring-expanded naphthalene product. Key Note: The α-iodonium diazo reagent serves as a carbynyl radical equivalent. The functional group (R) on the reagent is incorporated into the final product, allowing for diversification by simply changing the reagent [43].
Protocol 2: Rhodium-Catalyzed Formal (4+1) Cycloaddition for Carbon Insertion [39] Objective: One-carbon ring expansion of benzocyclobutenones to 1,1,3-trisubstituted 2-indanones.
- Reaction Setup: Charge an oven-dried microwave vial with benzocyclobutenone (1.0 equiv, 0.05 mmol), the styrene derivative (as the carbene precursor, 2.0 equiv), and a stir bar.
- Catalyst System: Add the cationic rhodium catalyst precursor [Rh(C₂H₄)₂Cl]₂ (2.5 mol%) and the ligand 1,2-bis(diphenylphosphino)ethane (dppe, 5.5 mol%). Use dichloroethane (DCE, 1.0 M) as solvent.
- Reaction Execution: Seal the vial and heat the mixture to 80°C with vigorous stirring for 12 hours.
- Monitoring: Analyze an aliquot by ¹H NMR or LC-MS to confirm consumption of the starting material.
- Work-up and Purification: Cool the reaction to room temperature. Concentrate directly under vacuum. Purify the crude product via preparative thin-layer chromatography (PTLC) or flash chromatography to yield the 2-indanone product. Mechanistic Insight: DFT calculations indicate the reaction proceeds via rhodium-styrene complex formation, carbene formation, cyclopropanation with the four-membered ring, and a kinetically favored β-hydride elimination/reductive elimination sequence to afford the (4+1) product selectively over (4+2) [39].
Integration with Computational and AI-Driven Workflows
The fusion of skeletal editing with advanced computational methods creates a powerful, iterative discovery engine.
5.1 Molecular Representation and AI-Guided Design Effective molecular representation—translating chemical structures into computable data—is foundational for AI applications [47]. Beyond traditional SMILES strings or fingerprints, graph-based representations like Graph Neural Networks (GNNs) treat molecules as graphs with atoms as nodes and bonds as edges, naturally capturing structural relationships critical for predicting the outcomes of complex skeletal edits [47]. These AI models, trained on reaction databases, can predict feasible editing pathways, recommend optimal reagents, and forecast the properties of the resulting molecules.
5.2 Enabling Scaffold Hopping and Generative Chemistry AI-driven scaffold hopping aims to identify core structures with similar bioactivity but distinct frameworks from a known lead [47]. Skeletal editing provides the experimental toolbox to execute such ambitious hops. Conversely, generative AI models (e.g., variational autoencoders, transformers) can propose novel, synthetically accessible ring-expanded structures within desired property space. Platforms like Exscientia’s and Insilico Medicine’s integrate such generative design with automated synthesis planning, rapidly proposing and prioritizing ring-editing strategies for lead optimization [46] [48]. This integration is exemplified by AI-platforms compressing early discovery timelines from years to months [46].
5.3 Predictive Modeling and Simulation Density Functional Theory (DFT) calculations are indispensable for elucidating reaction mechanisms, identifying key intermediates (e.g., metal-carbene or nitrene species), and understanding regioselectivity in skeletal editing transformations [39]. These insights guide the rational design of new catalysts and reagents. Furthermore, molecular docking and free-energy perturbation calculations can predict the binding affinity and selectivity of ring-expanded analogues before synthesis, prioritizing the most promising targets for experimental exploration [45] [46].
The following diagram illustrates the synergistic, closed-loop workflow integrating computational design, skeletal editing synthesis, and biological testing.
AI-Driven Skeletal Editing Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Ring-Expanding Skeletal Editing
| Reagent/Material | Function in Skeletal Editing | Example Application & Key Note |
|---|---|---|
| α-Diazo Esters (e.g., Ethyl diazoacetate) | Source of metal-carbene for C–H or C–C bond insertion; one-carbon homologation agent. | Ring expansion of cyclic ketones [39]. Handle with extreme care: diazo compounds are potentially explosive and toxic. |
| Organic Azides (e.g., TsN₃) | Precursor to metal-nitrene species upon activation, enabling N-atom insertion into C–C bonds. | Synthesis of N-heterocycles from saturated carbocycles [41]. |
| Dihalomethanes (e.g., CHCl₃, CHBr₃) | Source of dihalocarbenes (:CX₂) under strong base conditions (e.g., NaOH). | Classic Ciamician-Dennstedt ring expansion of pyrroles/indoles [40]. |
| α-Iodonium Diazo Reagents [43] | Act as functionalized “carbynyl radical equivalents” under photoredox conditions. | Diversity-generating ring expansion of indenes to naphthalenes. Allows R-group diversification. |
| Styrene Derivatives | Serve as safe carbenoid equivalents in transition metal catalysis. | Rh-catalyzed (4+1) cycloaddition with benzocyclobutenones [39]. |
| Chiral Sc(III) N,N′-Dioxide Complexes | Asymmetric Lewis acid catalysts for enantioselective transformations. | Enantioselective ring expansion of ketones with diazo compounds [39]. |
| Photoredox Catalysts (e.g., fac-Ir(ppy)₃) | Mediate single-electron transfer processes under mild visible light irradiation. | Generate radical intermediates for novel insertion pathways [43]. |
| Cationic Rhodium Catalysts (e.g., [Rh(C₂H₄)₂Cl]₂) | Form highly reactive electrophilic metal-carbene complexes from alkenes or other precursors. | Facilitate cycloaddition and insertion reactions for ring expansion [39]. |
Ring-expanding skeletal editing has evolved from a conceptual curiosity to a practical and powerful toolkit for the late-stage diversification of complex molecular frameworks, particularly those derived from natural products. By enabling precise, single-atom changes to core ring systems, it offers unparalleled efficiency in scaffold hopping and exploring complex, biologically relevant chemical spaces, such as medium-sized rings.
The future of this field lies in the deepening integration with computational and AI platforms. As molecular representation and generative models become more sophisticated, they will not only predict the outcomes of known edits but also invent novel, synthetically feasible ring-expansion transformations in silico. The closing of the “robotic synthesis-AI design” loop, where AI proposes edits and automated platforms execute them, will further accelerate discovery cycles [46] [48]. Key challenges remain, including expanding the substrate scope to increasingly complex polyfunctional molecules, improving stereocontrol, and developing milder, more sustainable reaction conditions. Addressing these challenges will solidify ring-expanding skeletal editing as an indispensable component of modern molecular innovation, directly contributing to the accelerated discovery of next-generation therapeutics.
Multicomponent Reactions (MCRs) as a Power Tool for Rapid Generation of Novel Medicinal Rings
The architectural backbone of the vast majority of bioactive molecules and approved drugs is composed of ring systems [1]. These structures are not merely passive frameworks; they define molecular shape, enforce crucial three-dimensional conformations that enhance target binding, and directly influence fundamental pharmacokinetic properties such as solubility, metabolic stability, and membrane permeability [1]. The pursuit of novel ring systems is therefore a core objective in drug discovery, driven by the need to escape the "flatland" of overused aromatic scaffolds, address undruggable targets, and circumvent existing intellectual property [49].
Historically, the discovery of novel ring systems has been bottlenecked by traditional linear synthesis, which is often step-intensive, low-yielding, and poorly suited for rapid library generation [49]. Within this context, Multicomponent Reactions (MCRs) have emerged as a transformative, convergent synthetic strategy. MCRs are defined as one-pot processes where three or more starting materials combine to form a single product that incorporates most of their atoms [49] [50]. This paradigm offers unparalleled efficiency through high atom- and step-economy, operational simplicity, and the rapid generation of structural complexity and diversity from simple, commercially available building blocks [49] [51].
This technical guide explores the pivotal role of MCRs in expanding the accessible chemical space of medicinally relevant ring systems. By focusing on recent advances (post-2019), we detail specific methodologies for constructing privileged and novel heterocycles, provide experimental protocols, and analyze the unique chemical space MCR-derived scaffolds occupy. Furthermore, we frame this discussion within the broader inspiration drawn from natural products—a traditional source of bioactive ring systems—and demonstrate how MCRs provide an efficient synthetic counterpart for exploring and diversifying these complex molecular frameworks [52].
MCRs for the Construction of Nitrogen-Containing Heterocycles
Nitrogen heterocycles are omnipresent in pharmaceuticals and natural alkaloids, serving as critical hydrogen-bond donors/acceptors [49]. MCRs provide streamlined access to both saturated and aromatic N-heterocycles with high diversity.
Table 1: Recent MCRs for the Synthesis of Nitrogen Heterocycles
| Target Ring System | MCR Components (Example) | Key Conditions/Catalyst | Medicinal Chemistry Value | Reference |
|---|---|---|---|---|
| Tetrahydropyridines (THPs) | F-masked sulfonamide allene, alkene, TMSCN (cyanide source) | [Cu(CH₃CN)₄]PF₆, BOX ligand, fluorobenzene, 60°C | Introduces rare C3/C5 substituents; high 3D character; post-MCR diversification via nitrile [49]. | [49] |
| Benzofuran-Fused Piperidines | Electron-rich benzofuran, primary amine, formaldehyde | Acetic acid, 80°C, double Mannich-type C–H functionalization | Fused, rigid polycyclic systems mimicking natural product frameworks; late-stage functionalization of drug amines [49]. | [49] |
| Piperidine-Fused Indoles (γ-Tetrahydrocarbolines) | 2-Methyl indole, amino acid ester, formaldehyde | DMF, 100°C | Core scaffold of numerous bioactive alkaloids; access to diverse stereochemistry from chiral amino acids [49]. | [49] |
| Oxoindole-β-Lactams | Isatin, β- or γ-amino acid, isocyanide | MeOH, rt (Ugi-4C-3CR) | Dual pharmacophore; evaluated as potent butyrylcholinesterase inhibitors for Alzheimer's disease [50]. | [50] |
Experimental Protocol: Synthesis of Polysubstituted 1,2,5,6-Tetrahydropyridines via Copper-Catalyzed Radical Cascade [49]
- Reaction Setup: In an oven-dried Schlenk tube under an inert atmosphere (N₂ or Ar), combine the F-masked benzene-sulfonamide allene 1 (1.0 equiv, 0.2 mmol), alkene 2 (1.2 equiv), and the bisoxazoline (BOX) ligand (10 mol%).
- Catalyst Addition: Add tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(CH₃CN)₄]PF₆, 10 mol%) and anhydrous fluorobenzene (2 mL).
- Nucleophile Addition: Cool the mixture to 0°C, then add trimethylsilyl cyanide (TMSCN, 2.0 equiv) via syringe.
- Reaction Execution: Remove the ice bath and stir the reaction mixture at 60°C for 12-24 hours, monitoring by TLC or LC-MS.
- Work-up: After completion, cool to room temperature and dilute with ethyl acetate (10 mL). Wash sequentially with saturated aqueous NaHCO₃ solution and brine. Dry the organic layer over anhydrous Na₂SO₄, filter, and concentrate under reduced pressure.
- Purification: Purify the crude residue by flash column chromatography on silica gel (eluent: hexane/ethyl acetate) to afford the desired tetrahydropyridine product 4.
MCRs for Other Medicinally Relevant Ring Systems
Beyond nitrogen heterocycles, MCRs efficiently construct O- and S-containing rings, as well as complex fused and spirocyclic systems often found in natural products.
MCRs in Natural Product-Inspired Synthesis: The power of MCRs is exemplified in concise syntheses of complex alkaloids. A multiple MCR strategy was employed to synthesize "tubugi" analogs of the potent antimitotic agent tubulysin D, combining Ugi, Passerini, and further couplings to assemble the tetrapeptide in a highly convergent manner, yielding compounds with sub-nanomolar cytotoxicity [52]. Similarly, the challenging pentacyclic scaffold of the alkaloid luotonin A was assembled in a single step from commercial materials (isatoic anhydride, propargylamine, aniline, glyoxal) via a Yb(OTf)₃-catalyzed, one-pot MCR involving an aza-Diels-Alder cyclization [52].
Scaffold Hopping and Library Design: MCRs are ideal for scaffold hopping—generating novel core structures with retained bioactivity [47]. The Ugi reaction, followed by post-condensation modifications like cyclization (e.g., Ugi-Deprotection-Cyclization, UDC), is a premier tool for this purpose [50]. For instance, Ugi azide reactions with benzofuran-pyrazole aldehydes have generated hybrid scaffolds combining three pharmacophores (benzofuran, tetrazole, pyrazole) for multitarget Alzheimer's therapy [50].
Analysis of Chemical Space and Scaffold Novelty
Cheminformatic analysis confirms that MCRs access novel and desirable regions of chemical space. A 2025 review analyzed scaffolds from recent MCRs and compared them to rings found in approved drugs and clinical candidates [49]. Key findings are summarized below:
Table 2: Chemical Space Analysis of MCR-Derived Scaffolds vs. Approved Drugs [49]
| Property | MCR-Derived Scaffolds | Approved Drugs / Clinical Candidates | Implication for Drug Discovery |
|---|---|---|---|
| Molecular Complexity | Higher (more globular, 3D) | Lower (more planar) | Better potential for targeting complex protein interfaces. |
| Fraction of sp³-Hybridized Carbons (Fsp³) | Generally higher | Generally lower | Increased 3D character often correlates with improved solubility and clinical success. |
| Ring Systems per Molecule | Often contain novel, fused, or bridged rings | Predominantly simple, common rings (e.g., benzene, piperidine) | Access to unprecedented intellectual property and bioactivity. |
| Synthetic Accessibility | High (by design, from commercial blocks) | Variable | Enables rapid hit-to-lead optimization and library synthesis. |
This analysis underscores that MCRs are not just efficient but strategically valuable, populating the under-explored, complex regions of chemical space that are increasingly sought after in modern drug discovery.
Experimental Workflow and Computational Integration
The modern application of MCRs in medicinal chemistry is enhanced by integrated experimental and computational workflows.
Diagram Title: Integrated Workflow for MCR-Based Drug Discovery
The Scientist's Toolkit: Key Research Reagent Solutions
- F-Masked Sulfonamide Allenes: Serve as versatile bifunctional components in radical MCRs, providing both the initiating N-radical and the allene acceptor for cyclization [49].
- Tetrakis(acetonitrile)copper(I) Hexafluorophosphate ([Cu(CH₃CN)₄]PF₆): A stable, soluble Cu(I) source used in conjunction with chiral bisoxazoline (BOX) ligands to catalyze enantioselective radical cyclizations for stereocontrolled heterocycle formation [49].
- Isocyanides (R-NC): The quintessential MCR reactant, central to Ugi, Passerini, and Groebke-Blackburn-Bienaymé reactions. Their unique divalent carbon allows sequential addition with nucleophiles and electrophiles to assemble diverse α-functionalized amides and heterocycles [50].
- Trimethylsilyl Cyanide (TMSCN): A safe, handleable source of cyanide anion used as a C1 building block nucleophile in MCRs to introduce nitrile functionalities, which are versatile handles for further derivatization (e.g., hydrolysis to acids, reduction to amines) [49].
Case Studies in Drug Discovery Applications
MCRs have directly contributed to lead identification and optimization programs across therapeutic areas.
Central Nervous System (CNS) Drug Discovery: MCRs are extensively used to generate libraries targeting CNS diseases. For example, Ugi and Passerini reactions have been employed to create multi-target-directed ligands for Alzheimer's disease, combining cholinesterase inhibition with anti-aggregation or antioxidant properties in a single molecule [50]. Similarly, MCR-derived compounds are being explored as ligands for serotonin and dopamine receptors relevant to schizophrenia and depression [50].
Anticancer Agent Development: The synthesis of tubulysin analogs via multiple MCRs is a prime example of efficiently generating complex, potent cytotoxic agents [52]. Furthermore, MCRs facilitate the generation of analogs of natural products like luotonin A and rigidins, enabling thorough structure-activity relationship studies to optimize their antitumor and antiproliferative activities [52].
The future of MCRs in medicinal chemistry is intrinsically linked to technological integration. Artificial Intelligence and Automated Discovery: Computational algorithms can now design novel, mechanistically distinct MCR networks by analyzing vast arrays of potential substrate combinations and reaction pathways, moving MCR discovery from serendipity to prediction [51]. Advanced Molecular Representation: Graph neural networks and language models trained on SMILES strings or molecular graphs enable better navigation of chemical space, facilitating scaffold hopping from MCR-derived hits to truly novel isofunctional cores [47].
In conclusion, Multicomponent Reactions represent a powerful and efficient engine for populating medicinal chemistry's next generation of novel ring systems. By enabling the rapid, one-pot assembly of complex, three-dimensional, and diverse scaffolds from simple inputs, MCRs directly address the critical need for new molecular frameworks in drug discovery. When integrated with modern computational design, AI-powered planning, and natural product-inspired logic, MCRs evolve from a synthetic tool into a central strategy for pioneering the unexplored frontiers of bioactive chemical space.
The structural core of most small-molecule drugs is formed by a ring system, with a significant proportion of these frameworks tracing their origins to natural products (NPs) [3]. These naturally occurring ring systems represent an evolutionary-optimized library of chemical scaffolds, pre-validated by biological systems for interaction with protein targets. However, a comprehensive cheminformatic analysis reveals a striking underutilization: only approximately 2% of the ring systems observed in natural products are present in approved drugs [3]. This vast, untapped reservoir of chemical scaffolds presents both a monumental opportunity and a significant challenge for drug discovery. The challenge lies in navigating this structural diversity to identify novel frameworks that retain desirable bioactivity while improving upon pharmacokinetic profiles, synthetic accessibility, or intellectual property (IP) landscapes.
Computational scaffold hopping emerges as the pivotal strategy to address this challenge. Originally defined by Schneider et al. in 1999, scaffold hopping aims to identify or generate novel molecular core structures (scaffolds) that maintain similar biological activity to a known reference compound [47] [53]. This is not a simple task; it requires a delicate balance between structural novelty and biofunctional equivalence, often moving beyond the traditional "similarity-property principle" which posits that similar structures confer similar properties [53]. The process is fundamentally dependent on how molecules are represented computationally—how their complex, three-dimensional structures are translated into a format that algorithms can process and compare.
Recent advancements in artificial intelligence (AI) and machine learning (ML) have dramatically transformed the capabilities of scaffold hopping. Modern AI-driven methods, leveraging deep learning (DL) models such as graph neural networks (GNNs) and transformers, can now learn continuous, high-dimensional molecular representations directly from data [47]. These representations capture intricate structural and functional relationships that rule-based, traditional methods often miss. This technical guide explores the convergence of sophisticated molecular representation methods and AI-driven generative models, framing them within the critical context of natural product research to unlock new bioactive frameworks from nature's blueprint.
Foundations of Molecular Representation
Molecular representation is the cornerstone of computational chemistry, serving as the critical bridge between a chemical structure and its numerical interpretation by an algorithm [47]. An effective representation must encode information pertinent to the task at hand, whether it be predicting biological activity, optimizing physicochemical properties, or, as in this case, identifying functionally equivalent but structurally distinct scaffolds.
Traditional Representation Methods
Traditional methods rely on explicit, rule-based feature extraction or linear string notations.
- String-Based Representations: The Simplified Molecular-Input Line-Entry System (SMILES) is the most prevalent, encoding molecular graphs as compact strings of characters representing atoms, bonds, and branching [47]. While human-readable and efficient, SMILES has inherent limitations in capturing molecular complexity and can suffer from syntactic ambiguity (multiple valid SMILES for one structure).
- Molecular Descriptors and Fingerprints: These are numerical representations encoding specific chemical information. Descriptors quantify physicochemical properties (e.g., molecular weight, logP, polar surface area). Fingerprints, such as Extended-Connectivity Fingerprints (ECFPs), encode the presence of molecular substructures as bit strings and are extensively used for similarity searching and quantitative structure-activity relationship (QSAR) modeling [47].
These traditional representations have powered early virtual screening and QSAR but are limited by their reliance on pre-defined features. They often struggle to capture the nuanced, non-linear relationships between distant structural changes and biological function, a key requirement for effective scaffold hopping [47].
Modern AI-Driven Representation Learning
The advent of deep learning has shifted the paradigm from manual feature engineering to data-driven representation learning. These methods automatically derive informative features directly from raw molecular data.
- Graph-Based Representations: Molecules are natively represented as graphs (atoms as nodes, bonds as edges). Graph Neural Networks (GNNs) operate directly on this structure, passing and transforming information along bonds to learn embeddings that capture both local atomic environments and global topology [47]. This makes them exceptionally well-suited for tasks involving structural changes to a core scaffold.
- Language Model-Based Representations: Inspired by natural language processing (NLP), models like Transformers treat SMILES or SELFIES strings as a chemical language [47]. By learning the contextual relationships between molecular "tokens," these models build powerful representations useful for generation and property prediction.
- 3D and Multimodal Representations: Advanced models integrate multiple data views. For example, the DeepHop model for scaffold hopping uses a multimodal transformer that integrates 2D graph structure, 3D molecular conformation (via a spatial GNN), and protein target sequence information [54]. This is crucial because bioactive molecules interact with their targets through 3D shape and electrostatics, not just 2D connectivity.
Table 1: Comparison of Key Molecular Representation Methods for Scaffold Hopping
| Method Type | Core Principle | Key Advantages for Scaffold Hopping | Primary Limitations | Example Applications/Tools |
|---|---|---|---|---|
| Traditional (ECFP) [47] | Hashed substructure patterns encoded in a fixed-length bit string. | Computationally cheap, interpretable, excellent for fast similarity search. | Limited expressivity; cannot generate novel structures; misses 3D shape. | Virtual screening, QSAR models (e.g., FP-ADMET) [55]. |
| Graph Neural Networks [47] | Learns node/edge embeddings by message-passing on the molecular graph. | Captures inherent topological structure; powerful for property prediction. | Requires careful architecture design; 2D graph may not fully represent 3D bioactivity. | Property prediction (DMPNN), molecular optimization. |
| Language Models (Transformer) [47] | Treats SMILES as sequence; learns contextual token embeddings via self-attention. | Excellent for de novo generation of valid SMILES strings. | SMILES syntax non-uniqueness can confuse models; 2D-centric. | De novo molecule design, SMILES-based translation. |
| Multimodal (3D-Aware) [54] | Combines multiple representations (e.g., 2D graph + 3D conformer + protein info). | Directly models the 3D interaction paradigm; targets specific proteins. | Computationally intensive; requires 3D conformer generation or data. | Target-specific scaffold hopping (e.g., DeepHop model) [54]. |
Scaffold Hopping: Concepts, Classification, and AI-Driven Evolution
Definition and Strategic Importance
Scaffold hopping is the deliberate modification of a molecule's central core structure to generate novel chemotypes with retained or improved bioactivity [47] [53]. Its strategic value in drug discovery is multifaceted:
- Overcoming Liabilities: Replacing a scaffold can address toxicity, metabolic instability, or poor pharmacokinetics inherent to an original lead series.
- Expanding IP Space: Novel scaffolds form the basis for new patent estates, essential for developing best-in-class therapies.
- Exploring Chemical Space: It enables systematic exploration of regions of chemical space around a validated pharmacophore, potentially leading to superior drugs.
The process is deeply connected to the study of natural products. As NP-derived ring systems are vastly underexploited, scaffold hopping provides the methodology to use these complex NPs as inspiration, "hopping" to synthetically tractable, novel frameworks that retain the key bioactivity determinants [3].
Classification of Hopping Approaches
Scaffold hops can be categorized by the degree and nature of structural change [53]:
- Heterocycle Replacements (1° Hop): Swapping or replacing heteroatoms within a ring (e.g., carbon for nitrogen). This is a small step but can significantly alter electronic properties and patentability (e.g., Sildenafil to Vardenafil) [53].
- Ring Opening or Closure (2° Hop): Breaking or forming rings to change saturation, flexibility, and shape. A classic example is the ring-opening transformation from the rigid morphine to the more flexible tramadol [53].
- Peptidomimetics: Replacing peptide backbones with non-peptide moieties to enhance metabolic stability and oral bioavailability.
- Topology-Based Hopping (3° Hop): The most dramatic change, involving a complete overhaul of the scaffold's connectivity while preserving the spatial arrangement of key pharmacophoric elements. This offers the highest novelty but is the most challenging [53].
Table 2: Classification of Scaffold Hopping Approaches with Natural Product Context
| Hop Category | Degree of Change | Structural Novelty | Success Rate Consideration | Natural Product (NP) Relevance | Example (NP-Derived Context) |
|---|---|---|---|---|---|
| Heterocycle Replacement [53] | Low (1°) | Low | High | Common in NP analog synthesis to modulate polarity, solubility, and binding interactions. | Modification of alkaloid cores (e.g., pyridine to benzene isosteres). |
| Ring Opening/Closure [53] | Medium (2°) | Medium | Medium | Crucial for simplifying complex polycyclic NP scaffolds or rigidifying flexible NP chains for potency. | Morphine (fused polycycle) → Tramadol (opened chain) [53]. |
| Peptidomimetics | Medium-High | Medium-High | Medium | Direct application to cyclic peptide NPs (e.g., cyclosporine) to create orally available non-peptide analogs. | Design of small-molecule mimics of macrocyclic peptide pharmacophores. |
| Topology-Based Hop [53] | High (3°) | High | Lower (but increasing with AI) | AI can use NP pharmacophore as a 3D blueprint to generate topologically novel, synthetically accessible scaffolds. | Using the 3D shape/electrostatics of a complex terpenoid to generate a novel aromatic scaffold. |
The AI Revolution in Scaffold Hopping
Traditional computational methods for scaffold hopping relied on searching pre-existing databases using 2D or 3D similarity metrics [54]. These methods are inherently limited by the scope of the database. AI, particularly deep generative models, has reformulated scaffold hopping from a search problem to a generation problem [54].
Modern AI models like DeepHop are trained on curated pairs of molecules that demonstrate a successful "hop": increased bioactivity, high 3D similarity, but low 2D scaffold similarity [54]. By learning this complex mapping, the model can then propose novel, generated scaffolds for a new input molecule, effectively exploring the vast, unenumerated chemical space (estimated at 10^60 drug-like molecules) [54]. This data-driven approach can identify non-intuitive hops beyond human medicinal chemistry intuition or predefined rules.
Diagram: AI-Driven Multimodal Scaffold Hopping Workflow (Based on the DeepHop architecture [54]) This illustrates how a modern AI model integrates multiple molecular and target representations to generate novel, target-aware scaffolds.
Technical Methodologies and Experimental Protocols
Implementing an AI-driven scaffold hopping pipeline involves several key stages, from data curation to model validation. The following protocol outlines the methodology based on state-of-the-art practices exemplified by models like DeepHop [54].
Data Curation and Preparation for Supervised Learning
Effective models require high-quality training data of successful scaffold hop pairs.
- Source Raw Bioactivity Data: Extract bioactivity data (IC50, Ki, Kd) for a target family of interest from curated public databases like ChEMBL [54]. Kinases are a common starting point due to abundant data and high therapeutic relevance.
- Preprocess Molecules: Standardize structures using toolkits like RDKit. Steps include: removal of salts and isotopes, neutralization of charges, and normalization of tautomers.
- Construct Scaffold-Hop Pairs: This is the critical step. Identify pairs of molecules (
(X, Y)) for a shared targetZthat meet strict criteria mimicking a successful hop:- Bioactivity Improvement:
pChEMBL(Y) - pChEMBL(X) >= 1.0(i.e., a 10-fold increase in potency) [54]. - 2D Dissimilarity: The Tanimoto similarity of their Bemis-Murcko scaffolds (core frameworks) is
≤ 0.6[54]. - 3D Similarity: The shape/electrostatic similarity (e.g., ComboScore from ROCS) of their bioactive conformations is
≥ 0.6[54].
- Bioactivity Improvement:
- Build a Validation QSAR Model: Train a separate, high-accuracy deep learning model (e.g., a Multi-Task DNN) on all bioactivity data for the target family. This model acts as a virtual profiler to rapidly predict the activity of newly generated molecules during the hopping process [54].
Model Architecture and Training
The core architecture is a multimodal conditional generative model.
- Input Encoding:
- Encode the reference molecule
Xas a 2D molecular graph (processed by a GNN) and its 3D conformation (processed by a spatial GNN) [54]. - Encode the target protein
Zsequence via a protein language model (e.g., Transformer).
- Encode the reference molecule
- Multimodal Fusion: The encoded representations are fused in a central transformer module. This module learns the complex relationships between the 2D/3D structure of
Xand the targetZ[54]. - Conditional Generation: The fused representation conditions a molecular decoder (e.g., a SMILES-based transformer decoder or a graph decoder). The decoder is trained to generate the SMILES string or graph of the hopped molecule
Y[54]. - Training Objective: The model is trained to maximize the likelihood of the true hopped molecule
Ygiven the inputs(X, Z)and the learned representations.
Evaluation and Validation
Generated molecules must be rigorously assessed.
- Computational Filters:
- Validity & Drug-likeness: Ensure the generated SMILES corresponds to a valid, synthetically accessible (SA Score) structure with drug-like properties.
- 2D/3D Similarity: Verify the hop was successful (low 2D scaffold similarity, high 3D similarity to
X). - Predicted Activity: Use the pre-trained QSAR model to predict
pChEMBLforYagainst targetZ. A successful hop should show improved or equipotent activity.
- Expert Review & Prioritization: The top-ranked generated structures should be reviewed by medicinal chemists for synthetic feasibility, novelty, and potential off-target effects based on scaffold.
- Experimental Validation: The ultimate step involves the chemical synthesis and in vitro biological testing of prioritized compounds to confirm the computational predictions—closing the AI-design cycle.
Diagram: Experimental Protocol for AI-Driven Scaffold Hopping This workflow outlines the end-to-end process from data preparation to experimental validation.
Table 3: Research Reagent Solutions for Computational Scaffold Hopping
| Category | Tool/Resource | Primary Function | Key Utility in Scaffold Hopping | Reference/Origin |
|---|---|---|---|---|
| Core Cheminformatics | RDKit | Open-source toolkit for cheminformatics and ML. | Molecule standardization, fingerprint generation, scaffold extraction (Bemis-Murcko), descriptor calculation, and basic property prediction. | Widely used standard [54]. |
| Deep Learning Frameworks | PyTorch / TensorFlow | Open-source libraries for building and training deep neural networks. | Implementation of custom GNNs, transformers, and multimodal architectures for molecular representation and generation. | Industry standards. |
| Specialized ML for Chemistry | DeepChem | Open-source library for deep learning in drug discovery and quantum chemistry. | Provides high-level APIs for building graph models, training on chemical datasets, and performing hyperparameter tuning. | [56] |
| 3D Conformation & Alignment | OpenEye ROCS | Tool for rapid shape-based superposition and comparison of molecules. | Critical for calculating 3D similarity scores (ShapeTanimoto, ComboScore) to evaluate and constrain scaffold hops. | Cited in 3D hopping methods [54]. |
| Generative Model Platforms | GT4SD (Generative Toolkit for Scientific Discovery) | Framework for developing, training, and deploying generative models for molecules and materials. | Access to and development of state-of-the-art generative models applicable to scaffold hopping. | Emerging platform. |
| Bioactivity Data | ChEMBL Database | Manually curated database of bioactive molecules with drug-like properties. | Primary source for extracting target-annotated bioactivity data to train and validate scaffold hopping models. | Used in major studies [54]. |
| Target Information | Protein Data Bank (PDB) | Repository of 3D structural data for biological macromolecules. | Source of target protein structures for structure-based analysis and for informing 3D pharmacophore constraints. | [56] |
| Synthetic Accessibility | SA Score | Algorithm to estimate the ease of synthesizing a molecule based on fragment contributions and complexity. | Filter for generated scaffolds to prioritize those within realistic synthesis scope for medicinal chemists. | Common post-filter. |
The convergence of sophisticated AI-driven molecular representation and generative modeling has fundamentally expanded the horizons of scaffold hopping. By moving beyond database lookup to true de novo generation conditioned on 3D shape and target information, these tools allow researchers to explore the chemical space surrounding a validated pharmacophore with unprecedented breadth and creativity [54]. This capability is especially potent when applied within the context of natural product research.
The vast structural diversity of natural product ring systems, of which over 98% remain unexploited in drugs, provides a rich library of biologically pre-validated starting points [3]. Computational scaffold hopping, powered by the AI methodologies described herein, offers a systematic pathway to mine this library. It enables the translation of complex, often synthetically challenging NP scaffolds into novel, patentable, and synthetically tractable frameworks that retain the essential bioactivity encoded by nature. This synergy between NP-inspired design and AI-powered execution represents a promising frontier for discovering the next generation of bioactive compounds.
This technical guide presents an integrated workflow for the design, synthesis, and evaluation of novel natural product (NP)-inspired ring systems, framed within the broader thesis that such frameworks constitute a privileged yet underexplored region of biologically relevant chemical space. Natural products and their derivatives account for a substantial proportion of approved drugs, with their unique ring systems forming the structural core essential for bioactivity [57]. However, only an estimated 2% of the tens of thousands of known NP ring systems are present in approved drugs, revealing a vast untapped resource [58] [57]. This whitepaper synthesizes contemporary strategies—including biology-oriented synthesis (BIOS), pseudo-natural product (PNP) design, diversity-oriented synthesis (DOS), and complexity-to-diversity (CtD) transformations—into a coherent, actionable pipeline. We detail computational tools for scaffold generation and analysis, practical synthetic methodologies with a focus on challenging medium-sized ring construction, and robust protocols for biological evaluation. By providing a unified framework that bridges cheminformatic analysis, synthetic chemistry, and screening, this guide aims to equip researchers with a systematic approach for expanding into novel, NP-inspired ring chemical space to accelerate drug discovery.
The structural core of most bioactive small molecules is defined by a ring system. These frameworks dictate molecular shape, conformational flexibility, and the spatial display of functional groups, making them indispensable for target recognition and binding [57]. Natural products, refined by evolution, are an unparalleled source of such privileged ring systems. Analyses of comprehensive databases like the Collection of Open Natural Products (COCONUT) reveal an extraordinary diversity of 38,662 unique ring systems within NPs, characterized by high three-dimensionality, stereochemical complexity, and a prevalence of oxygen-containing heterocycles [58] [57].
Despite this richness, the NP ring system universe remains poorly exploited in synthetic libraries. A seminal finding indicates that approximately 50% of NP ring systems have a representative with similar 3D shape and electrostatic properties in commercially available synthetic screening compounds, suggesting viable starting points for design [57]. Conversely, this also means half of NP-inspired chemical space is truly novel and inaccessible through conventional screening collections. This gap motivates the development of integrated strategies to deliberately design and access these unexplored regions. The overarching thesis of this field posits that by understanding, deconstructing, and recombining the principles encoded in NP ring systems, researchers can generate novel frameworks with enhanced biological relevance, improved synthetic tractability, and the potential for unprecedented bioactivities.
Core Strategic Approaches for Ring System Design and Diversification
Multiple complementary strategies have been developed to navigate NP-inspired chemical space. Their selection and integration depend on the project goals, ranging from closely mimicking a bioactive NP to generating entirely unprecedented scaffolds.
Biology-Oriented Synthesis (BIOS) starts with a validated, bioactive NP scaffold as the guiding structure. The core ring system is conserved, while peripheral regions are diversified to explore structure-activity relationships (SAR) and improve properties [2] [4]. This strategy leverages nature's pre-validation but may limit exploration to the immediate chemical vicinity of the parent NP.
Pseudo-Natural Product (PNP) Design involves the fragment-based combination of biosynthetically unrelated NP fragments into novel molecular frameworks not found in nature [2] [59]. This approach aims to retain the "biological relevance" of the constituent fragments while exploring new regions of chemical space. For example, indole and tropane alkaloid fragments have been fused to create "indotropanes," a novel chemotype with unique biological profiles [59].
Diversity-Oriented Synthesis (DOS) aims to generate high skeletal and stereochemical diversity from common starting materials using branching reaction pathways. When applied to NP-inspired design, it often employs complexity-generating reactions to build collections rich in sp3-hybridized centers and polycyclic systems reminiscent of NPs [2] [4].
Complexity-to-Diversity (CtD) and Ring Distortion begins with a complex NP itself and applies transformative, often ring-distorting, reactions to rapidly generate diverse derivatives from a single advanced intermediate. This can include ring cleavage, expansion, contraction, and rearrangements [2] [36].
Table 1: Comparison of Core Strategies for NP-Inspired Ring System Design
| Strategy | Guiding Principle | Typical Ring System Outcome | Key Advantage | Primary Challenge |
|---|---|---|---|---|
| Biology-Oriented Synthesis (BIOS) | Conserve a bioactive NP scaffold [4]. | Close analogues of known NP rings. | High probability of retained bioactivity. | Limited exploration of novel chemical space. |
| Pseudo-Natural Product (PNP) | Recombine unrelated NP fragments [2] [59]. | Novel fused/spiro ring systems. | Explores uncharted, yet biologically relevant space. | Design and synthesis of novel fusion patterns can be complex. |
| Diversity-Oriented Synthesis (DOS) | Maximize skeletal diversity from simple precursors [2]. | Broad array of novel, NP-like polycyclic systems. | Efficient exploration of a wide chemical area. | Risk of generating biologically irrelevant structures. |
| Complexity-to-Diversity (CtD) | Diversify a complex NP via ring distortion [36]. | Distorted, rearranged versions of the original NP core. | Rapid access to high complexity from pure NPs. | Heavily dependent on specific reactivity of the starting NP. |
An Integrated Workflow: From Computational Design to Biological Evaluation
A modern, efficient workflow for novel ring system development integrates computational design, synthetic execution, and biological assessment in an iterative cycle.
Phase 1: Computational Design & In Silico Analysis The process is initiated with cheminformatic analysis and generative design. Tools like the NIMO (Natural Product-Inspired Molecular Generative) model exemplify this approach [60]. NIMO uses transformer neural networks trained on motif sequences extracted from NPs. Its two models serve distinct purposes:
- NIMO-M: A generic model for de novo generation of molecules under multi-property constraints (e.g., drug-likeness, synthetic accessibility).
- NIMO-S: A scaffold-based model for lead optimization that expands a specified central core ring system [60].
This computational stage assesses novel designs for properties like synthetic accessibility score (SAS), natural product-likeness (NP-score), and coverage of underrepresented regions in ring-descriptor space [60] [17].
Phase 2: Synthetic Access & Library Construction Synthesizing designed ring systems, particularly medium-sized rings (8-11 members), is a core challenge due to unfavorable transannular interactions and entropic barriers [36]. Ring-expansion reactions of polycyclic precursors have emerged as a powerful solution, avoiding the high-dilution conditions of direct macrocyclization.
- Oxidative Dearomatization-Ring Expansion (ODRE): This biomimetic strategy uses oxidative dearomatization of phenolic substrates to create reactive intermediates that undergo ring expansion upon rearomatization, efficiently constructing benzannulated medium rings [36].
- Electrochemical Ring Expansion: A sustainable method using electrochemical oxidation to generate amidyl radicals, which induce C–C bond cleavage and expansion to form medium-sized lactams [36].
Phase 3: Biological Evaluation & Target Identification Synthesized libraries are profiled using phenotypic and target-based screens. High-content morphological profiling (e.g., Cell Painting) is particularly valuable for PNPs and novel scaffolds, as it provides an unbiased, multiparametric bioactivity fingerprint that can suggest a mechanism of action or identify unique phenotypes [59]. For target deconvolution, techniques like thermal proteome profiling (TPP) and quantitative proteomics are employed in tandem with computational target prediction [59].
Diagram Title: Integrated Workflow for NP-Inspired Ring System Design
Detailed Experimental Protocols & Methodologies
Protocol: Oxidative Dearomatization-Ring Expansion (ODRE) for Benzannulated Medium Rings
This biomimetic protocol is adapted from methodologies for synthesizing 8-11 membered benzannulated rings [36].
Principle: A phenolic substrate undergoes oxidative dearomatization to form a reactive cyclohexadienone intermediate. This intermediate undergoes a ring-expanding rearrangement upon rearomatization, cleaving a strategic C-C bond.
Materials:
- Substrate: Polycyclic phenol (e.g., a bicyclic phenol like compound 3 [36]).
- Oxidant: Hypervalent iodine reagent, e.g., Phenyliodine(III) diacetate (PIDA) or (Bis(trifluoroacetoxy)iodo)benzene (PIFA).
- Solvent: Anhydrous dichloromethane (DCM), acetonitrile (MeCN), or a mixture.
- Nucleophile: (If applicable) Alcohols, carboxylic acids, or electron-rich arenes to trap the expanded intermediate.
- Work-up & Purification: Standard aqueous work-up materials (e.g., saturated NaHCO₃, brine), silica gel for flash chromatography.
Procedure:
- Reaction Setup: In an oven-dried round-bottom flask under an inert atmosphere (N₂ or Ar), dissolve the phenolic substrate (1.0 equiv) in dry, degassed DCM (0.05-0.1 M concentration).
- Addition of Oxidant: Cool the solution to 0°C (ice-water bath). Add the hypervalent iodine oxidant (e.g., PIFA, 1.1-1.5 equiv) in one portion or as a solution in minimal DCM.
- Reaction Progression: Allow the reaction to warm to room temperature and stir. Monitor by TLC. The reaction is typically complete within 1-12 hours.
- Nucleophile Quench (Optional): If the protocol calls for a nucleophile, add it (1.5-3.0 equiv) after the oxidation step is complete (often after 30-60 min at 0°C). Stir until TLC indicates consumption of the intermediate.
- Work-up: Quench the reaction by adding a saturated aqueous solution of sodium thiosulfate (to reduce excess iodine) and saturated sodium bicarbonate. Extract the aqueous layer with DCM (3x). Combine the organic layers, dry over anhydrous Na₂SO₄, filter, and concentrate under reduced pressure.
- Purification: Purify the crude residue by flash chromatography on silica gel to isolate the ring-expanded product. Characterize using ¹H/¹³C NMR, HRMS, and IR spectroscopy.
Protocol: Metabolomic-Guided Library Analysis for Diversity Assessment
This protocol, inspired by fungal isolate studies, uses LC-MS to quantitatively assess the chemical diversity of a synthesized NP-inspired library [61].
Principle: Liquid Chromatography-Mass Spectrometry (LC-MS) data are processed to detect "chemical features" (unique m/z at a specific retention time). Feature accumulation curves are generated to determine how many compounds are needed to capture the scaffold diversity of the library.
Materials:
- Samples: Purified compounds from the synthesized library, dissolved in a suitable MS-compatible solvent (e.g., methanol, DMSO).
- LC-MS System: High-resolution LC-MS system (e.g., UHPLC coupled to Q-TOF or Orbitrap mass spectrometer).
- Software: Data processing software (e.g., MZmine, XCMS, proprietary vendor software).
- Columns: Reversed-phase C18 column for LC separation.
Procedure:
- Data Acquisition: Analyze all library compounds using a standardized LC-MS method. Use consistent gradients and MS settings (positive/negative ionization mode, broad mass range scan).
- Feature Detection: Process raw data files to perform peak picking, alignment, and deisotoping. The output is a list of all detected "chemical features" across all samples, defined by mass-to-charge ratio (m/z) and retention time (RT).
- Dereplication & Scaffold Binning: Using in-house or public databases (e.g., COCONUT, GNPS), annotate features where possible. Group features that share a common core ring system (scaffold) based on MS/MS fragmentation patterns or prior knowledge.
- Generate Accumulation Curve: Randomize the sample order. Sequentially, calculate the cumulative number of unique chemical features (or unique scaffolds) discovered as each new sample is added to the analysis.
- Analysis: Plot the accumulation curve. The point where the curve begins to plateau indicates the library size sufficient to capture most of the accessible diversity from that particular synthetic strategy or design principle. This data can inform decisions on optimal library size for future iterations [61].
Table 2: The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in Workflow | Key Application / Example |
|---|---|---|
| Hypervalent Iodine Reagents (PIDA, PIFA) | Oxidative dearomatization agent [36]. | Key oxidant in the ODRE ring-expansion reaction to form reactive cyclohexadienone intermediates. |
| Chiral Ligands (e.g., BOX, PyBOX, Quinoline-based) | Control stereochemistry in cycloadditions [59]. | Enabling enantioselective synthesis of novel spiro- or fused-ring systems in PNP synthesis (e.g., Cu-catalyzed 1,3-dipolar cycloadditions). |
| Solid-Phase Synthesis Resins | Enable parallel synthesis and purification [4]. | Used in DOS libraries to facilitate the synthesis of complex natural product-like frameworks (e.g., gemmacin antibiotic discovery). |
| Electrochemical Cell | Enables sustainable oxidation/reduction without chemical oxidants [36]. | Used in electrochemical ring-expansion reactions for medium-sized lactam/lactone synthesis. |
| LC-MS Metabolomics Platform | High-throughput chemical profiling and diversity analysis [61]. | Generating feature accumulation curves to quantify chemical diversity coverage in a synthesized library. |
| Cell Painting Assay Dyes | Multiparametric morphological profiling [59]. | Unbiased phenotypic screening to generate bioactivity fingerprints for novel PNPs and guide target identification. |
Data Presentation & Analysis of NP Ring System Space
Table 3: Statistical Overview of Natural Product Ring System Diversity [58] [57]
| Metric | Value | Implication for Design |
|---|---|---|
| Total Unique NP Ring Systems | 38,662 (from COCONUT) | Vast diversity available for inspiration. |
| NP Ring Systems in Approved Drugs | ~2% | Huge untapped potential; majority are novel starting points. |
| NPs with ≥1 Ring System | 94% | Confirms centrality of rings in NP structure. |
| Coverage by Synthetic Compounds | ~50% (by 3D shape/electrostatics) | Half of NP-like shape space is accessible via commercial compounds; half requires de novo synthesis. |
| Common Heteroatoms | Oxygen > Nitrogen | Designs favoring O-heterocycles may have higher NP-likeness. |
Table 4: Performance Comparison of Generative Models for NP-Inspired Scaffolds [60]
| Model (Type) | Validity (%) | Novelty (MOSES, %) | Synthetic Accessibility (SAS)↓ | Best Use Case |
|---|---|---|---|---|
| NIMO-M (Fragment-based) | 94.5 | 71.4 | 0.78 | De novo generation of novel, synthesizable NP-like motifs. |
| NIMO-S (Scaffold-based) | 99.3 | 89.0 | 0.91 | Optimizing/elaborating a known core ring system. |
| MCMG (SMILES-based) | 95.0 | 79.5 | 1.22 | General molecular generation under constraints. |
| FBMG (Fragment-based) | 42.9 | 99.9 | 0.94 | High novelty, but poor synthetic guidance and validity. |
↓ Lower SAS score indicates easier predicted synthesis. *Novelty relative to training set.*
Synthesis of Challenging Ring Systems: Focus on Medium-Sized Rings
The synthesis of 8- to 11-membered rings remains a significant hurdle due to transannular strain and entropic penalties. Ring-expansion strategies are critical to the workflow's success [36].
Key Ring-Expansion Tactics:
- C–C Bond Cleavage Expansion: Pre-formed polycyclic substrates contain a strained or cleavable bond. Oxidative cleavage (e.g., of an alkene bridge) directly yields a medium-sized ring [36].
- Tandem Oxidative Dearomatization: As detailed in Protocol 4.1, this method converts readily available phenolic precursors into complex benzannulated medium rings [36].
- Electrochemical Radical Expansion: An electrochemical setup generates radical intermediates (e.g., amidyl radicals) that trigger selective C–C bond cleavage and expansion, offering a green chemistry alternative [36].
Diagram Title: Ring-Expansion via Oxidative Dearomatization
The systematic exploration of natural product-inspired ring systems represents a frontier in drug discovery, grounded in the thesis that evolutionary selection has privileged certain molecular frameworks for biological interaction. This whitepaper has outlined an integrated workflow that moves beyond singular strategies, combining the target-focused logic of BIOS, the innovative scaffold generation of PNP design, the broad exploratory power of DOS, and the efficient diversification of CtD. Critical to this pipeline are modern computational tools like the NIMO generator for in silico design and robust synthetic methodologies, particularly ring-expansion reactions, to overcome the historical challenge of constructing medium-sized and novel polycyclic ring systems. By applying quantitative metrics for diversity assessment and employing unbiased phenotypic profiling for evaluation, researchers can iteratively refine their approach to populate the vast, uncharted regions of NP-like chemical space. The ultimate goal is to transform the immense structural diversity encoded in natural product ring systems into novel, synthetically tractable chemotypes that yield next-generation therapeutics and biological probes.
Balancing Innovation and Feasibility: Design Challenges for Complex Ring Systems
The quest for novel therapeutic agents exists in a state of tension—a novelty paradox. While the chemical space of potential ring systems is astronomically large, estimated at approximately 450,000 unique systems derived from billions of molecules, the practical world of drug development exhibits profound conservatism [62]. Rings are the architectural backbone of bioactive molecules, determining three-dimensional shape, pre-organizing substituents for target binding, and critically influencing pharmacokinetic and pharmacodynamic profiles [63]. In natural products research, which has historically been the most prolific source of novel molecular scaffolds, this paradox is particularly acute. Nature itself recycles and elaborates upon a finite set of core frameworks, yet produces astounding biological diversity.
This technical guide frames the novelty paradox within the broader thesis that molecular frameworks derived from natural products are not merely structures but evolutionary-validated solutions to biological interaction. The persistent reliance on known ring systems in drug candidates—67% of small molecules in clinical trials comprise only rings found in marketed drugs—is not a failure of imagination but a rational, risk-managed strategy [62]. This document provides an in-depth analysis of the quantitative evidence for this trend, deconstructs the multidimensional drivers of conservatism, identifies strategic inflection points that justify breaking the pattern, and provides actionable experimental and computational protocols for the principled exploration of novel chemical space.
Quantitative Landscape: The Empirical Basis of the Paradox
A data-driven analysis reveals the stark contrast between potential and practiced chemical novelty. The following tables synthesize key quantitative findings from large-scale analyses of clinical trial and approved drug databases.
Table 1: Utilization of Ring Systems in Drug Development Pipelines
| Metric | Clinical Trial Compounds | Marketed Drugs | Source / Note |
|---|---|---|---|
| Compounds with only known ring systems | 67% | ~70% (annual new approvals) | Mirrors drug approval trends [62] |
| Utilization of available ring system pool | 0.1% | Even more restricted | From ~450,000 unique systems [62] |
| Novel ring systems per novel drug | Often just one | Typically zero or one | Most novelty is in side-chains/combinations [62] |
| Predicted coverage of novel trial systems | ~50% by 3902 systems | N/A | From systematic 1-2 atom changes to known systems [62] |
Table 2: Analysis of the Antibacterial Pipeline (Illustrating a High-Need Field)
| Pipeline Category | Number of Candidates | Meeting WHO Innovation Criteria | Implication |
|---|---|---|---|
| All traditional antibacterial agents | 57 | 12 | Limited novelty [64] |
| Agents targeting WHO Priority Pathogens | 32 | 4 (target critical pathogens) | Acute unmet need [64] |
| New chemical classes (since 2017) | 2 (e.g., vaborbactam, lefamulin) | 2 | Extreme scarcity of novel scaffolds [64] |
| β-lactamase inhibitor combinations | Dominant proportion | 0 (analogues of existing classes) | "Buying time" vs. long-term solution [64] |
The data underscore a systemic pattern: the drug development funnel acts as a powerful filter for chemical novelty. While clinical trials show marginally more diversity than marketed drugs, the attrition of novel rings increases toward approval [62]. This funnel effect is economically rational but poses a fundamental threat to addressing novel biological targets and evolving resistance mechanisms, as starkly evidenced by the antibiotic pipeline [64] [65].
The Drivers of Conservatism: A Multifactorial Analysis
The reliance on known rings is a consequence of interconnected scientific, economic, and risk-based drivers.
De-risked Pharmacological and Synthetic Profiles
Known ring systems come with established structure-activity relationship (SAR) libraries, understood metabolic soft spots, and vetted synthetic routes. Their physicochemical properties—such as solubility, polarity, and three-dimensional shape—are navigable within a known "drug-like" space, often aligned with heuristic rules like the Rule of Five, though antibiotics frequently violate these norms [65]. This prior knowledge dramatically reduces the uncertainty in lead optimization, allowing teams to focus on improving potency and selectivity.
The Overwhelming Economics of Failure
Drug development is governed by Eroom's Law (the inverse of Moore's Law), where costs skyrocket as efficiency declines [66]. The financial impact of failure is catastrophic: a failed Phase 3 program costs between $600 million and $1.2 billion [67]. Up to 90% of drug programs collapse, predominantly due to flawed target biology rather than chemistry [67]. In this environment, introducing a novel, unproven ring system adds a layer of chemical risk to an already high-risk undertaking. For antibiotics, the economic case is weakest, with short treatment durations and low prices stifling investment in novel classes despite dire public health need [64].
Target Validation and Translational Gaps
A novel ring system is often unjustified when the biological target itself is uncertain. Failures frequently originate in the earliest phase: target identification [67]. Historical debacles—such as BACE inhibitors for Alzheimer's, CETP inhibitors for cardiovascular disease, and matrix metalloproteinase (MMP) inhibitors in oncology—exemplify "elegant execution against false assumptions" [67]. When the core disease-driving mechanism is misidentified, even perfect chemistry will fail. Known ring systems provide a stable variable in this otherwise high-risk equation.
Diagram 1: The Multifactorial Filter of Chemical Novelty. Economic, scientific, and biological risk factors collectively filter the vast potential chemical space into the narrow range of ring systems used in practice.
Strategic Inflection Points: When to Break the Pattern
Principled innovation is warranted at specific strategic inflection points where the value of novelty outweighs its inherent risk.
Confronting Established Mechanisms of Resistance
This is paramount in antimicrobial and anticancer therapy. When resistance arises from target mutation or enzymatic degradation of a core scaffold, incremental modification of known rings often fails. The WHO defines four innovation criteria to combat antimicrobial resistance: new chemical class, new target, new mode of action, and lack of cross-resistance [65]. A novel ring system is frequently required to meet the first and last criteria. For example, the discovery of Halicin via deep learning revealed a structurally novel compound with a unique mode of action and no pre-existing cross-resistance, demonstrating the value of escaping known chemical space [65].
Engaging Novel or "Undruggable" Biological Targets
When targeting a novel protein class (e.g., a new enzyme family or a protein-protein interaction interface), existing ring libraries may be inadequate. The shape and electronic complementarity required might demand a novel molecular framework. Natural products, with their vast and evolved scaffold diversity, often provide starting points for such challenges.
Overcoming Insurmountable ADMET Hurdles
If all optimized leads within a known chemical series possess a fatal flaw—such as mechanism-based toxicity, irreversible metabolism, or an inability to penetrate a key barrier (e.g., the Gram-negative bacterial outer membrane or the blood-brain barrier)—a scaffold hop to a novel ring system may be the only path forward [65].
Diagram 2: Strategic Inflection Points for Novel Ring Exploration. Specific high-need scenarios justify accepting the increased risk of novel scaffold development.
Experimental & Computational Protocols for Principled Exploration
Protocol: AI-Driven Virtual Screening for Novel Bioactive Scaffolds
This protocol leverages deep learning to explore vast chemical spaces for novel ring systems with predicted activity [65] [66].
- Model Training: Assemble a high-quality dataset of compounds with whole-cell or target-based activity data against the pathogen or target of interest. Encode molecules using a graph-based representation (e.g., Directed-Message Passing Neural Network, D-MPNN) where nodes are atoms and edges are bonds [65].
- Virtual Library Construction: Generate or access a virtual library encompassing novel ring systems, such as those from enumerated fused rings (e.g., ~570,000 systems) [62] or "dark chemical matter" [65]. Filter for synthetic accessibility using a scoring model.
- Prediction & Prioritization: Use the trained model to screen the virtual library. Prioritize hits that are (a) structurally distant from known bioactive rings (via Tanimoto distance on Morgan fingerprints), (b) predicted to be active, and (c) synthetically tractable.
- Experimental Validation: Synthesize or procure top-ranked novel scaffolds. Validate activity in a primary assay (e.g., minimum inhibitory concentration (MIC) for antibiotics). Confirm the mechanism of action is novel (e.g., via whole-genome sequencing of resistant mutants or a targeted protein-binding assay).
Diagram 3: AI-Driven Workflow for Novel Ring System Discovery. A computational-experimental pipeline for identifying and validating novel bioactive scaffolds.
Protocol: Target-Driven Synthesis Based on Natural Product Frameworks
This protocol uses natural product frameworks as inspiration for synthesizing novel, simplified analogues targeting a specific protein.
- Target Analysis & Pharmacophore Mapping: Determine the 3D structure of the target protein (e.g., via X-ray crystallography or AlphaFold). Identify key binding pockets and define a pharmacophore model (hydrogen bond donors/acceptors, hydrophobic regions, electrostatic constraints).
- Natural Product Database Mining: Search databases (e.g., NPASS, COCONUT) for natural product scaffolds that could spatially and electronically satisfy the pharmacophore. Prioritize under-explored or synthetically challenging frameworks.
- Scaffold Simplification & Retrosynthesis: Design a simplified, synthetically accessible core ring system that retains the critical spatial orientation of functional groups needed for target binding. Perform retrosynthetic analysis to plan a feasible route.
- Iterative Synthesis & Testing: Synthesize the core scaffold. Test binding affinity (e.g., surface plasmon resonance) or inhibitory activity in a biochemical assay. Iteratively decorate the scaffold with substituents to optimize interactions, guided by structural biology.
Table 3: The Scientist's Toolkit: Key Reagents & Platforms for Ring System Exploration
| Tool / Reagent Category | Specific Examples / Platforms | Primary Function in Novel Ring Discovery |
|---|---|---|
| Chemical Space Libraries | Enamine REAL Space, GalaXi, GDB-17 [62]; "Dark chemical matter" libraries [65] | Provide physical or virtual sources of billions of molecules containing novel ring systems for screening. |
| AI/ML Modeling Platforms | D-MPNN models [65]; Graph convolutional networks (GCNs); Biological foundation models (e.g., Bioptimus, Evo) [66] | Predict bioactivity of novel structures; identify patterns in vast biological datasets to propose novel target-ring system pairs. |
| Synthetic Building Blocks | Commercially available rare heterocycles; DNA-encoded library (DEL) building blocks | Enable the practical synthesis of novel ring systems and the construction of diverse libraries around a novel core. |
| High-Throughput Screening Assays | Phenotypic whole-cell screens (e.g., for antibiotics); Target-based enzymatic assays | Experimentally validate the bioactivity of novel ring systems identified in silico or from natural product inspiration. |
| Structural Biology Tools | X-ray crystallography; Cryo-EM; AlphaFold protein structure prediction | Enable target-driven design by revealing the precise binding site geometry a novel ring must engage. |
The novelty paradox in drug discovery is a rational equilibrium, not an intellectual failure. The overwhelming reliance on known ring systems is a risk-averse strategy calibrated against the staggering costs of clinical failure and the profound complexities of human biology [62] [67]. However, this equilibrium must be strategically disrupted in the face of existential challenges like antimicrobial resistance and undruggable targets.
The future lies in precision exploration. By coupling causal, driver-based target validation [67] with advanced computational methods like foundation models and D-MPNNs [65] [66], researchers can identify the precise scenarios where novel ring systems are not just chemically interesting but biologically and therapeutically necessary. The vast chemical space of rings, much of it inspired by natural product frameworks, thus transforms from a daunting wilderness into a mapped territory where calculated expeditions can yield the next generation of transformative medicines.
1. Introduction: The Centrality of Ring Systems in Molecular Frameworks Within the broader thesis of natural products research, complex ring systems are not merely structural motifs; they are the architectural keystones defining biological activity, conformational rigidity, and metabolic stability. The synthesis of polycyclic frameworks—bridged, fused, or spiro—represents a persistent frontier in accessing drug candidates and probing chemical space. This guide provides a technical framework for assessing synthetic accessibility (SA) and navigating the unique challenges posed by complex ring synthesis.
2. Predictive Models for Synthetic Accessibility Quantitative SA scores combine algorithmic analysis of structural complexity with empirical reaction data. Key metrics are summarized below.
Table 1: Comparative Analysis of Synthetic Accessibility Prediction Tools
| Tool/Method | Core Algorithm | Key Output | Strengths | Limitations |
|---|---|---|---|---|
| SCScore | Neural network trained on reaction data | Score (1-5, simple-complex) | Correlates with expert intuition | Less interpretable; trained on historical data |
| SAscore (RDKit) | Fragment contribution & complexity penalty | Score (1-10, easy-hard) | Fast, computable from structure | Can over-penalize novel scaffolds |
| SYBA | Bayesian classifier | Probability (0-1, easy-hard) | Identifies synthetically suspicious fragments | Requires predefined fragment library |
| DELS | Deep learning from ELN data | Score & route suggestions | Incorporates actual lab success data | Proprietary; requires large dataset |
3. Key Hurdles in Complex Ring Synthesis and Strategic Solutions The primary hurdles are topological strain, stereochemical control, and strategic bond disconnection.
Table 2: Major Hurdles and Corresponding Synthetic Strategies
| Synthesis Hurdle | Underlying Challenge | Strategic Solution | Exemplar Reaction/Technique |
|---|---|---|---|
| High-Ring Strain | Unfavorable thermodynamics & kinetics | Strain-release driven cyclization | Vinylcyclopropane rearrangements, photochemical [2+2] |
| Concurrent Stereocenters | Diastereoselective control in constrained systems | Tandem asymmetric catalysis & substrate control | Organocascade reactions, directed hydrogenation |
| Bridgehead Bonds | Geometric inaccessibility | Fragment coupling via radical or metal insertion | Late-stage C-H functionalization, cycloaddition |
| Spiro Junctures | Quaternary center formation | Cyclization onto pre-formed quaternary centers | Dieckmann condensation, radical cyclization |
4. Experimental Protocol: A Representative Methodology for Bridged System Construction Protocol: Gold(I)-Catalyzed Endo-Selective Cycloisomerization to Access Bicyclo[3.2.1] Scaffolds This protocol exemplifies the use of late-stage, complexity-generating transformations to overcome entropic barriers.
Materials:
- Dien-yne substrate (1.0 equiv, 0.1 mmol)
- (JohnPhos)Au(MeCN)SbF₆ (2 mol%)
- Anhydrous 1,2-Dichloroethane (DCE)
- Molecular sieves (4 Å)
- Argon atmosphere & standard Schlenk line equipment
Procedure:
- In a flame-dried Schlenk tube under argon, add molecular sieves (50 mg).
- Charge the tube with the substrate and dissolve in anhydrous DCE (0.01 M concentration).
- In a separate vial, dissolve the gold(I) catalyst in 0.5 mL anhydrous DCE.
- Add the catalyst solution to the reaction mixture via syringe at room temperature (RT).
- Stir the reaction at RT, monitoring by TLC/LC-MS until complete (typically 2-6 h).
- Quench by filtering through a short plug of silica gel, eluting with ethyl acetate.
- Concentrate under reduced pressure and purify the residue by flash chromatography.
Mechanistic Workflow:
Diagram Title: Au(I)-Catalyzed Cycloisomerization Mechanism
5. The Scientist's Toolkit: Key Reagent Solutions Table 3: Essential Research Reagents for Complex Ring Synthesis
| Reagent/Material | Primary Function in Complex Ring Synthesis | Technical Note |
|---|---|---|
| Gold(I) Catalysts (e.g., JohnPhosAu(MeCN)SbF₆) | Activates alkynes/allenes towards diverse cyclizations; soft Lewis acid. | Air-stable but moisture-sensitive. Use with polar, non-nucleophilic solvents (DCE, MeCN). |
| Chiral Organocatalysts (e.g., MacMillan's imidazolidinone) | Enables asymmetric induction in pericyclic and cascade reactions via enamine/iminium. | Often require acidic co-catalysts. Performance is highly substrate-specific. |
| Grubbs II / Hoveyda-Grubbs Metathesis Catalysts | Enables ring-closing metathesis (RCM) to form medium/large rings and macrocycles. | Strict exclusion of air and water is critical. Purify substrates to remove catalyst poisons. |
| Photoredox Catalysts (e.g., Ir(ppy)₃, Ru(bpy)₃²⁺) | Generates radical intermediates under mild conditions via single-electron transfer (SET). | Requires compatible light source (LEDs). Optimal solvent is typically DMF or MeCN. |
| DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene) | Non-nucleophilic, strong base for promoting eliminations or isomerizations in sensitive systems. | Can cause side reactions (e.g., nucleophilic attack) on electrophilic centers. |
| Molecular Sieves (3Å, 4Å) | Scavenges trace water from reactions, critical for moisture-sensitive metal catalysis. | Activate by heating (200-300°C) under vacuum before use. |
6. Strategic Workflow for Route Design and Optimization A systematic approach integrating prediction, disconnection, and experimental validation is crucial.
Diagram Title: Workflow for Complex Ring Synthesis Design
7. Conclusion: Integrating Prediction and Execution The synthesis of complex ring frameworks remains a defining challenge in natural products research. Success hinges on the iterative integration of predictive SA tools, which highlight topological vulnerabilities, with modern catalytic methods that convert these vulnerabilities into strategic opportunities. This synergy between in silico assessment and innovative experimental execution is essential for advancing the frontiers of synthesizable molecular space.
The exploration of chemical space, estimated to contain between 10²³ to 10⁶⁰ molecules, represents the primary challenge in molecular design and drug discovery [68] [69]. Within this vast expanse, the molecular scaffold serves as the indispensable core framework, guiding critical processes such as diversity assessment and scaffold hopping in medicinal chemistry [69]. While approximately 70% of approved drugs are based on known scaffolds, a staggering 98.6% of ring-based scaffolds in virtual libraries remain unvalidated [68] [69]. Traditional metrics like the Ring Complexity Index (RCI) have provided initial insights but are limited by their reliance on a single parameter—the count of ring atoms. To address this, we introduce the Quantitative Ring Complexity Index (QRCI), a novel metric that integrates ring diversity, topological complexity, and macrocyclic properties into a unified, computable score [68]. This whitpaper details the formulation, computational methodology, and application of QRCI, framing it as an essential tool within the broader thesis of advancing natural products research and the design of bioactive molecular frameworks.
Natural products (NPs) are recognized as privileged structures with an inherent capacity to interact with therapeutically relevant protein targets [70]. Their unique and diverse chemical architectures, often dominated by intricate ring systems, have made them a cornerstone of modern medicine. Historically, a significant proportion of approved drugs originate from NPs, their analogues, or contain NP-derived pharmacophores [70]. Despite this proven value, NP research faces significant hurdles, including complex syntheses, challenging dereplication processes, and molecular frameworks that often possess high complexity in the form of fused ring systems and stereocenters [70].
The molecular scaffold, particularly its ring system, is the central pillar defining a compound's shape, physicochemical properties, and biological interactions. In the era of artificial intelligence (AI) and big data, the need for quantitative, computable descriptors of molecular complexity has never been greater [71]. Advanced cheminformatic frameworks are being developed to categorize and predict the activity of NPs, but these methods fundamentally depend on robust numerical representations of molecular structure [8]. The proposed QRCI responds directly to this need, offering a sophisticated yet easily computed metric to quantify the complexity of the core ring framework. This enables more informed decisions in virtual screening, library design, and the prioritization of NP-inspired compounds for synthesis and testing, thereby accelerating the drug discovery pipeline [68].
Beyond Atom Count: The Need for a Quantitative Ring Complexity Index
The traditional Ring Complexity Index (RCI) is defined simply as the number of atoms that are part of a ring system. While straightforward, this one-dimensional metric fails to capture the qualitative nuances that define true molecular complexity. A linear six-membered ring and a complex, bridged polycyclic system with the same number of ring atoms would receive an identical RCI score, despite vast differences in their topological intricacy, synthetic challenge, and potential for unique bioactivity.
The Quantitative Ring Complexity Index (QRCI) is engineered to overcome this critical limitation. It moves beyond mere atom counting to integrate multiple dimensions of ring system sophistication into a single, comprehensive value [68]. The index is designed to be calculable directly from a molecule's connection table or SMILES string, requiring no 3D conformational data, which ensures computational efficiency and broad applicability across large chemical databases [69].
Table 1: Core Limitations of RCI and How QRCI Addresses Them
| Aspect of Complexity | RCI Limitation | QRCI Solution |
|---|---|---|
| Topological Diversity | Only counts atoms; cannot distinguish between a single ring and a fused/spiro system. | Incorporates metrics for ring fusion, bridgeheads, and system interconnectedness. |
| Structural Saturation | Does not consider the saturation/unsaturation of the ring system. | Accounts for pi-bond density and aromaticity within rings. |
| Macrocyclic Presence | Treats all ring sizes equally. | Includes a specific term to weight the presence and size of macrocycles (typically rings >12 atoms). |
| Synthetic Accessibility | Poor correlation with the actual synthetic difficulty of a ring system. | Designed to correlate strongly with synthetic accessibility scores and topological complexity metrics. |
Computational Methodology and Calculation of QRCI
The QRCI is formulated as a weighted sum of contributing factors that collectively define ring system complexity. The following is a detailed protocol for its calculation.
Input Data and Preprocessing
- Molecular Representation: Input a molecule using a standard chemical representation (e.g., SMILES string, Mol file, InChI).
- Ring Perception: Perform a canonical ring perception algorithm (e.g., SSSR - Smallest Set of Smallest Rings, or relevant cycles) to identify all unique rings and ring systems within the molecule.
- System Definition: Group individual rings that share atoms into distinct ring systems (e.g., fused, bridged, or spiro systems).
Core QRCI Calculation Formula
The QRCI for a single molecule is calculated using the following foundational equation, which aggregates scores from its constituent ring systems [68]:
QRCI_molecule = Σ (Base_Score(System_i) * Diversity_Modifier(System_i) * Macrocyclic_Modifier(System_i))
Table 2: Breakdown of QRCI Formula Components and Calculation Protocol
| Component | Description | Experimental/Computational Protocol |
|---|---|---|
| Base_Score(System) | Foundation score for a ring system. | Base_Score = log(N_ring_atoms + 1) + (N_rings_in_system * 0.5) + (N_bridgehead_atoms * 0.3) Protocol: 1. Count total ring atoms (N_ring_atoms) in the system. 2. Count number of individual rings (N_rings_in_system) in the fused/bridged system. 3. Identify and count bridgehead atoms (N_bridgehead_atoms). |
| Diversity_Modifier(System) | Adjusts for heteroatom and bond-type diversity. | Diversity_Modifier = 1 + (Heteroatom_Ratio * 0.2) + (Pi_Bond_Ratio * 0.15) Protocol: 1. Calculate Heteroatom_Ratio = (NO + NN + NS + NP) / Nringatoms. 2. Calculate Pi_Bond_Ratio = (Number of ring pi-bonds) / Nringatoms. |
| Macrocyclic_Modifier(System) | Accounts for the complexity of large rings. | Macrocyclic_Modifier = 1.0 if largest_ring_size < 12 Macrocyclic_Modifier = 1.0 + (0.05 * (largest_ring_size - 12)) if largest_ring_size >= 12 Protocol: Determine the size (number of atoms) of the largest ring in the system. |
| Final QRCI | Total complexity score for the molecule. | Sum the weighted scores from all independent ring systems in the molecule. |
Workflow for QRCI Implementation
The following diagram illustrates the logical workflow for computing the QRCI from a molecular structure.
Applications in Natural Products Research and Drug Discovery
The QRCI is not merely a theoretical metric; it is a practical tool designed to inform key decisions in the research pipeline.
1. Virtual Screening and Library Prioritization: In large-scale virtual screens of NP databases or synthetic libraries, QRCI can be used as a filter or sorting criterion. Researchers can prioritize compounds within a desired complexity range—avoiding overly simple scaffolds with low potential for novel interactions or prohibitively complex scaffolds with high synthetic burden [68] [69].
2. Scaffold Hopping and Bioisostere Design: When seeking to replace a core ring system with a novel scaffold (scaffold hopping), QRCI provides a quantitative measure to ensure the replacement maintains a similar level of topological complexity, which is often linked to maintaining target binding and overall molecular properties.
3. Guiding Synthetic Campaigns: The strong correlation between QRCI and synthetic accessibility allows medicinal chemists to anticipate the synthetic challenge of a target molecule early in the design phase. This can guide the selection of lead compounds or inspire the design of simplified analogues with retained activity [68].
4. Chemical Space Analysis: Mapping compounds based on QRCI and other descriptors (e.g., polarity, molecular weight) provides a powerful visual representation of chemical space. This can reveal clustering of complex NPs from specific biological sources and identify underexplored regions of complexity-bioactivity space [70].
Table 3: Illustrative QRCI Scores for Representative Molecular Frameworks
| Molecule Class | Example Scaffold | Approx. QRCI | Interpretation & Utility |
|---|---|---|---|
| Simple Drug-like | Benzene, Pyridine | 0.8 - 1.5 | Low complexity. High prevalence. Useful for QSAR studies of simple substituent effects. |
| Fused Bicyclic | Naphthalene, Quinoline | 2.5 - 3.5 | Moderate complexity. Common in drugs. Good balance of stability and interaction potential. |
| Complex NP Core | Steroid backbone, Pentacyclic triterpene | 5.0 - 8.0 | High complexity. High potential for unique, potent bioactivity. High synthetic challenge. |
| Macrocyclic NP | Cyclosporin-like core, Macrocyclic lactone | 7.0 - 12.0+ | Very high complexity. Often associated with specific target engagement (e.g., protein-protein inhibition). Major synthetic endeavor. |
Integration with AI and Modern Cheminformatics
The true power of QRCI is unlocked when integrated with contemporary AI-driven drug discovery paradigms. As a numerical descriptor, QRCI is perfectly suited for machine learning (ML) workflows [71].
Feature in ML Models: QRCI can be incorporated as an essential feature in models predicting bioactivity, toxicity, or physicochemical properties. Its inclusion provides the model with direct, quantifiable information about the core structural complexity that other fingerprints may only implicitly capture [8].
Guiding Generative AI: In de novo molecular design using generative models (e.g., Generative Adversarial Networks, Variational Autoencoders), QRCI can be used as a constraint or optimization target. This allows for the direct generation of novel molecular structures within a user-defined complexity range, enabling the exploration of "optimally complex" chemical space [71] [70].
Enhancing Explainability: The components of QRCI (ring count, heteroatom ratio, macrocyclic presence) offer a chemically intuitive breakdown of why a molecule receives a certain score. This enhances the explainability of "black-box" AI models by linking model decisions to understandable chemical concepts.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Implementing QRCI analysis requires a combination of software tools and databases. The following toolkit is essential for researchers in this field.
Table 4: Research Reagent Solutions for QRCI Analysis
| Item / Resource | Function / Description | Application in QRCI Workflow |
|---|---|---|
| RDKit (Open-source) | A comprehensive cheminformatics toolkit for Python/C++. | Core library for reading molecules, performing ring perception, and calculating component metrics (heteroatoms, bond types, etc.) required for QRCI. |
| Python/NumPy/SciPy | Programming language and scientific computing libraries. | Environment for implementing the QRCI calculation formula, data manipulation, and statistical analysis of results. |
| NP Databases (e.g., LOTUS, NPASS, COCONUT) | Public databases cataloging natural products and their sources. | Source of natural product structures for analysis, benchmarking, and exploring the distribution of QRCI in nature [8]. |
| DrugBank | Database of approved and investigational drugs. | Source of drug molecules for comparative analysis, establishing complexity trends in successful therapeutics [69]. |
| Jupyter Notebooks | Interactive computing environment. | Platform for developing, documenting, and sharing reproducible QRCI analysis pipelines and visualizations. |
| Cheminformatics Platform (e.g., Knime, Pipeline Pilot) | Visual workflow tools with chemistry extensions. | Alternative GUI-based environment for building and executing QRCI calculation workflows without extensive programming. |
The Quantitative Ring Complexity Index (QRCI) represents a significant advance in our ability to quantify, analyze, and ultimately design sophisticated molecular frameworks. By moving beyond the simplistic atom-counting of the RCI to integrate topology, diversity, and macrocyclic character, QRCI provides a robust, computable metric that strongly aligns with the synthetic and topological realities of molecular complexity.
Framed within the broader thesis of natural products research, QRCI serves as a critical bridge between the intricate architectures found in nature and the pragmatic demands of modern drug discovery. It enables the intelligent navigation of chemical space, from prioritizing novel NP scaffolds to designing synthetically tractable, NP-inspired lead compounds.
Future development will focus on refining the weighting schemes of the QRCI components using larger-scale validation against synthetic feasibility databases and bioactivity data. Furthermore, integration with real-time synthesis planning algorithms and advanced generative AI models promises to create a closed-loop design system where desired complexity is a fundamental parameter. As the field continues to embrace data-driven strategies, tools like QRCI will be indispensable for the informed design of the next generation of therapeutics.
Appendix: Diagram of QRCI's Role in the NP Drug Discovery Pipeline
The intricate molecular frameworks of natural products represent a cornerstone of drug discovery, offering unparalleled structural diversity and validated bioactivity against human targets [4]. However, these complex molecules—characterized by high sp³ carbon content, numerous chiral centers, aliphatic ring systems, and low nitrogen/halogen content—often present significant challenges for development as oral therapeutics [72]. Their inherent structural complexity frequently translates to suboptimal physicochemical properties, including poor aqueous solubility, limited membrane permeability, and rapid metabolic clearance [73] [72].
This technical guide addresses the critical optimization of these three interdependent properties—solubility, permeability, and metabolic stability—within the specific context of natural product-derived scaffolds. Optimization is not merely an exercise in empirical modification but a rational process guided by an understanding of molecular conformation, ring system dynamics, and their interaction with biological environments [73]. The ultimate goal is to transform a bioactive natural lead compound into a drug-like molecule capable of efficient oral absorption, adequate tissue distribution, and sustained exposure at the target site, thereby fulfilling its therapeutic promise [74].
Strategic Framework for Physicochemical Optimization
The optimization of natural products requires a holistic and iterative strategy that acknowledges the interconnectedness of physicochemical properties. The following workflow outlines a rational approach, integrating computational prediction, strategic molecular design, and rigorous experimental validation.
Diagram 1: A strategic workflow for optimizing natural product leads
Optimization of Aqueous Solubility
3.1. The Solubility Challenge in Natural Products Poor aqueous solubility is a predominant issue for natural products, stemming from their high molecular rigidity, extensive lipophilic regions, and low aromatic content [72]. Low solubility directly compromises oral bioavailability by limiting the dissolved fraction available for absorption across the gastrointestinal membrane.
3.2. Key Strategic Approaches
- Molecular Disruption of Crystal Packing: Introducing ionizable groups (e.g., amines, carboxylic acids) or polar, hydrogen-bonding motifs (e.g., alcohols, amides) disrupts efficient crystal lattice packing, reducing the enthalpy of fusion and increasing solubility.
- Pro-drug Strategies: Temporary conjugation of polar, ionizable, or enzymatically cleavable moieties (e.g., phosphate esters, amino acid conjugates) can dramatically enhance apparent solubility, which is regenerated in vivo.
- Strategic Use of Halogens: While rare in nature, introducing fluorine or chlorine can be a powerful tool. Fluorine's high electronegativity can modulate pKa and dipole moments, while its small size minimizes steric perturbation, potentially improving solubility without compromising target binding [72].
- Ring System Manipulation: Altering ring systems can significantly impact solubility. This includes ring contraction or expansion to alter molecular planarity, or the introduction of heteroatoms (N, O, S) into aliphatic rings to create hydrogen-bond acceptors/donors [75] [4].
3.3. Experimental Protocols for Solubility Assessment
- Shake-Flask Method (Gold Standard): An excess of the solid compound is added to a buffered aqueous solution (e.g., phosphate buffer pH 7.4) and agitated at constant temperature (e.g., 37°C) for 24-72 hours to achieve equilibrium. The suspension is then filtered or centrifuged, and the concentration of the compound in the supernatant is quantified using a validated analytical method, typically HPLC-UV or LC-MS/MS.
- High-Throughput Thermodynamic Solubility Assay: Utilizes 96-well or 384-well plates. A concentrated DMSO stock of the compound is dispensed into buffer, inducing precipitation. After equilibration, plates are centrifuged, and the supernatant is analyzed via UV-plate readers coupled with cheminformatic correction or directly by LC-MS.
- In Silico Prediction: Tools like the General Solubility Equation (GSE), which uses melting point and logP estimates, or more advanced machine learning models trained on large chemical datasets, provide early-stage rankings of synthetic analogues [76].
Table 1: Impact of Structural Modifications on Solubility and Related Properties [72] [76]
| Modification Strategy | Example Structural Change | Expected Impact on Solubility | Potential Trade-off/Effect on Other Properties |
|---|---|---|---|
| Introduction of Ionizable Group | Addition of a basic aliphatic amine or acidic carboxylic acid. | High increase (salt formation possible). | May increase susceptibility to efflux transporters (if charged at physiological pH); can affect permeability. |
| Reduction of Lipophilicity (LogP/D) | Replacement of a -CH2- with a polar group (e.g., -O-), removal of alkyl chain. | Moderate to high increase. | May decrease passive membrane permeability; could reduce target affinity if group is critical for hydrophobic interactions. |
| Pro-drug Formation | Esterification of an alcohol, phosphorylation. | Very high increase for the pro-drug itself. | Adds a synthetic step; requires predictable in vivo activation; intrinsic solubility of active moiety unchanged. |
| Introduction of Fluorine | Strategic replacement of -H or -CH3 with -F. | Variable, often moderate increase due to altered electronic properties and crystal packing. | Can improve metabolic stability and membrane permeability; minimal steric impact. |
| Ring Distortion/Scaffold Hop | Changing a 6-membered cyclohexane to a piperidine. | Moderate increase (introduction of H-bond acceptor). | Can dramatically alter conformation and thus target binding affinity; requires careful SAR study. |
Optimization of Membrane Permeability
4.1. Permeability and the Natural Product Conformation Permeability, particularly passive transcellular permeability, is governed by a molecule's ability to partition into and diffuse across lipid bilayers. For flexible natural products, permeability is not a property of a single structure but of an ensemble of conformations. The "functional structure" that permeates a membrane may differ from the bioactive conformation bound to a protein target [73]. Natural products' high sp³ character often provides the flexibility to adopt membrane-permeable conformations.
4.2. Key Strategic Approaches
- Modulating Lipophilicity (Log D): Optimizing the distribution coefficient at physiological pH (Log D₇.₄) is crucial. A balance is needed: sufficient lipophilicity for membrane partitioning but not so high that solubility is devastated. This is often achieved by fine-tuning the number and nature of alkyl groups and aromatic systems [72].
- Minimizing Hydrogen Bond Donors (HBDs): The total number of HBDs (e.g., OH, NH) is a strong negative predictor of permeability due to the desolvation penalty required for membrane partitioning. Masking HBDs as esters or ethers, or their intramolecular engagement in hydrogen bonds, can improve permeability.
- Controlling Molecular Flexibility and Size: While flexibility can be beneficial, excessive molecular weight (>500 Da) and polar surface area (>140 Ų) generally hinder permeability. "Pruning" redundant atoms or peripheral groups that do not contribute to target binding can streamline the molecule for better permeation [72] [4].
- Managing Transporter Interactions: Many natural products are substrates for efflux transporters like P-glycoprotein (P-gp). Structural modifications to reduce P-gp recognition—such as reducing the number of HBDs, masking amines, or altering planar aromatic domains—can enhance net cellular uptake [77].
4.3. Experimental Protocols for Permeability Assessment
- Parallel Artificial Membrane Permeability Assay (PAMPA): A high-throughput model for passive transcellular permeability. A filter coated with a lipid/oil mixture (e.g., lecithin in dodecane) separates donor and acceptor compartments. Compound movement from donor to acceptor over time is quantified by HPLC-UV or LC-MS, and an effective permeability (Pe) is calculated.
- Caco-2 Cell Monolayer Assay: The gold standard for predicting human intestinal absorption. Differentiated human colon adenocarcinoma cells form confluent, polarized monolayers with tight junctions. Test compound is added to the apical (A) or basolateral (B) side, and appearance on the opposite side is measured. Apparent permeability (Papp) is calculated, and asymmetry in A→B vs B→A flux indicates active transport/efflux.
- PBPK Modeling for Prediction: Bottom-up physiologically-based pharmacokinetic (PBPK) modeling integrates in vitro permeability data (e.g., from Caco-2) with physiological parameters to predict in vivo absorption. Recent optimizations, such as using updated gastrointestinal physiology models and refined transporter parameters (e.g., P-gp Relative Expression Factor), have significantly improved prediction accuracy for challenging compounds [77] [78].
Table 2: Permeability Optimization Strategies and Their In Vitro/In Vivo Correlates [77] [73] [74]
| Optimization Goal | Structural Tactic | Primary In Vitro Assay | Key Model Parameter | Link to In Vivo Outcome |
|---|---|---|---|---|
| Increase Passive Transcellular Permeability | Reduce polar surface area; optimize Log D (~2-4); mask H-bond donors. | PAMPA | Effective Permeability (Pe) | Predicts fraction absorbed (fa) in humans when combined with solubility data. |
| Assess Active Transport/Efflux | Modify structure to reduce recognition by efflux pump pharmacophores. | Caco-2 Monolayer | Apparent Permeability (Papp) and Efflux Ratio (Papp B→A / Papp A→B) | High efflux ratio predicts potential for low/dariable oral bioavailability and drug-drug interactions. |
| Predict Integrated Oral Absorption | Combine optimized solubility, permeability, and stability data. | PBPK Model Simulation (e.g., Simcyp) | Predicted vs. Observed AUC and Cmax | Retrospective clinical validation shows optimized models can predict human exposure within 3-fold for >80% of compounds [77]. |
Optimization of Metabolic Stability
5.1. Metabolic Vulnerability of Natural Product Scaffolds Natural products are evolutionarily optimized for ecological function, not human pharmacokinetics. They often contain motifs susceptible to Phase I metabolism (e.g., oxidation by Cytochrome P450 enzymes at unsubstituted aromatic rings, allylic or benzylic positions) and Phase II conjugation (e.g., glucuronidation of phenols, alcohols) [72].
5.2. Key Strategic Approaches
- Blocking Metabolic Soft Spots: Identifying and modifying the site of fastest metabolism is the most direct strategy. This can involve replacing a metabolically labile hydrogen with a deuterium (deuterium isotope effect), substituting a susceptible aromatic C-H with a CF group, or introducing a blocking methyl group.
- Bioisosteric Replacement: Replacing a metabolically vulnerable group with a bioisostere that retains target affinity but resists metabolism. For example, replacing an ester (susceptible to hydrolysis) with an amide or heterocycle.
- Conformational Rigidification: Reducing molecular flexibility by introducing constraints (e.g., ring fusion, macrocyclization) can shield vulnerable sites from accessing the active pocket of metabolic enzymes [73] [4].
- Strategic Use of Three-Membered Rings: Incorporating rings like cyclopropane, aziridine, or epoxide can serve dual purposes. They can act as rigid, polarity-modifying bioisosteres for functional groups and, due to their inherent ring strain and unique geometry, often exhibit unusual metabolic stability profiles [75].
5.3. Experimental Protocols for Metabolic Stability Assessment
- Human Liver Microsome (HLM) Stability Assay: Compound is incubated with pooled HLMs in the presence of NADPH cofactor for Phase I metabolism. Aliquots are taken over time (e.g., 0, 5, 15, 30, 60 min), reactions quenched with acetonitrile, and parent compound loss is quantified by LC-MS/MS. Intrinsic clearance (CLint) is calculated from the depletion half-life.
- Recombinant CYP Enzyme (rCYP) Assay: To identify specific CYP isoforms responsible for metabolism, the compound is incubated with individual rCYPs (e.g., CYP3A4, 2D6, 2C9). Analysis of metabolite formation or parent loss pinpoints the involved enzymes, guiding targeted structural blocking.
- Hepatocyte Stability Assay: Provides the most physiologically relevant in vitro system, containing full complement of Phase I and Phase II enzymes. Similar incubation and analysis as HLM assay yields a more comprehensive intrinsic clearance value.
- In Vitro-In Vivo Extrapolation (IVIVE) for PBPK: Clearance data from HLMs or hepatocytes, scaled using physiological scaling factors (e.g., microsomal protein per gram of liver) and system-specific factors (e.g., Intersystem Extrapolation Factor - ISEF for rCYPs), is integrated into PBPK models to predict human hepatic clearance and plasma half-life [77].
Diagram 2: A decision-based workflow for metabolic stability optimization
The Scientist's Toolkit: Essential Reagents and Platforms
Table 3: Key Research Reagent Solutions for Physicochemical Optimization [77] [78] [74]
| Tool/Reagent Category | Specific Example/Platform | Primary Function in Optimization | Key Application & Relevance |
|---|---|---|---|
| In Vitro ADME Assay Systems | Pooled Human Liver Microsomes (HLMs); Cryopreserved Human Hepatocytes; Caco-2 Cells. | Provide experimental measurement of metabolic stability, metabolite ID, and permeability/transport. | Foundation for generating drug-specific parameters (CLint, Papp) for IVIVE and PBPK modeling. |
| Recombinant Metabolic Enzymes | Gentest Supersomes (rCYPs, UGTs). | Identify specific enzyme isoforms responsible for metabolite formation. | Enables targeted structural blocking and accurate scaling using Intersystem Extrapolation Factors (ISEF) [77]. |
| PBPK Modeling Software | Simcyp Simulator, GastroPlus, PK-Sim. | Mechanistic, bottom-up prediction of human pharmacokinetics by integrating in vitro ADME and physicochemical data. | Critical for predicting exposure (AUC, Cmax) and DDIs before clinical studies; allows virtual screening of analogue profiles [77] [78]. |
| Computational Prediction Tools | Quantum Mechanics/Molecular Dynamics (QM/MD); Quantitative Structure-Property Relationship (QSPR) models. | Predict conformation, pKa, logP, intrinsic solubility, and metabolic sites from molecular structure. | Guides initial analogue design and prioritization, especially using graph-based topological indices [76]. |
| Analytical Core Technology | High-Resolution LC-MS/MS Systems. | Quantify parent compound and identify metabolites in complex biological matrices (e.g., from stability assays). | Essential for generating accurate, high-quality data for all experimental assays and model parameterization [74]. |
The optimization of solubility, permeability, and metabolic stability in natural product-derived leads is a multidisciplinary endeavor that blends classical medicinal chemistry with modern computational and systems pharmacology approaches. The strategies outlined—from strategic halogenation and ring distortion to conformational analysis and PBPK-led design—provide a roadmap for navigating this complex space.
The future of this field lies in the deeper integration of advanced computational methods. Machine learning and graph-based QSPR models will enhance our ability to predict the properties of complex, natural product-like scaffolds [79] [76]. Furthermore, the increasing sophistication and regulatory acceptance of PBPK modeling transforms optimization from a sequential process into a holistic, predictive exercise, where the in vivo consequences of structural changes can be forecast with greater confidence [77] [78]. By leveraging these tools within a framework that respects the unique structural and conformational biology of natural products, researchers can more efficiently translate these privileged scaffolds into the next generation of effective therapeutics.
The structural core of most bioactive small molecules, including drugs and natural products, is formed by ring systems. These cyclic frameworks dictate the three-dimensional shape, electronic distribution, and ultimate biological activity of a compound [58]. In the context of natural products research, ring systems are not merely passive scaffolds but the primary architects of bioactivity. Their immense structural diversity, evolved over millennia, offers an unparalleled source of novel molecular frameworks for drug discovery. A comprehensive analysis of 38,662 natural product ring systems reveals a universe of chemical space dominated by a few common scaffolds, with a long tail of rare, structurally unique systems [58]. Notably, only about 2% of these naturally occurring ring systems are found in approved drugs, indicating a vast reservoir of untapped potential [58].
However, this potential is counterbalanced by intrinsic risk. The same structural features that confer potent, desirable biological activity can also be responsible for mechanism-based toxicity. Novel ring systems, particularly those heteroatom-rich or with unusual steric strain, may introduce unforeseen toxicophores—structural alerts that can trigger adverse biological responses such as DNA damage, enzyme inhibition, or pathological cellular stress signaling [80] [81]. The challenge for modern research is to harness the inspirational power of natural product ring systems while developing robust strategies to identify and mitigate their associated toxicological risks early in the development pipeline.
Structural Alerts and Toxicity Endpoints in Novel Ring Systems
A structural alert is a molecular substructure or fragment known to be associated with a specific toxicological outcome. For novel ring systems derived from or inspired by natural products, these alerts are often embedded within the core framework itself.
Common Toxicity Endpoints and Associated Alerts: Certain ring system features are empirically linked to major toxicity endpoints. Predictive models for reproductive and developmental toxicity, for instance, must account for complex molecular pathways, including hormone receptor interactions and cellular apoptosis signaling [82]. Hepatotoxicity (drug-induced liver injury) is a critical endpoint where natural products pose a significant concern; compounds like pyrrolizidine alkaloids contain a specific heterocyclic ring system that, upon metabolic activation, becomes a reactive electrophile causing liver damage [80]. Table 1 summarizes key toxicity endpoints and the ring system features often implicated in them.
Table 1: Key Toxicity Endpoints and Associated Ring System Alerts
| Toxicity Endpoint | Description & Impact | Common Ring System Alerts/Features | Relevant Signaling Pathways |
|---|---|---|---|
| Reproductive & Developmental Toxicity [82] | Adverse effects on fertility, embryonic development, and offspring health; a major regulatory hurdle. | Michael acceptors, epoxides, specific aromatic amine motifs, certain heterocycles (e.g., imidazoles). | Androgen/Estrogen receptor signaling, apoptosis pathways, DNA damage response. |
| Hepatotoxicity [80] | Drug-induced liver injury (DILI); leading cause of drug attrition and post-market withdrawal. | Furan rings (via metabolic activation to reactive cis-enedials), pyrroles, unsaturated lactones. | Nrf2 (oxidative stress), NF-κB (inflammation), P53 (DNA damage/apoptosis). |
| Mutagenicity/Carcinogenicity | Potential to cause DNA damage leading to mutations and cancer. | Aromatic nitro groups, polycyclic aromatic hydrocarbons, aziridines, N-nitrosamines. | DNA damage response, cell cycle arrest, P53 pathway. |
| Cardiotoxicity | Interference with cardiac ion channels (e.g., hERG), leading to arrhythmia. | Broad structural feature, often linked to lipophilic bases separated by a rigid spacer from aromatic rings. | hERG potassium channel blockade. |
| Endocrine Disruption [83] | Interference with hormone synthesis, transport, or receptor binding. | Phenolic rings (mimicking estrogens), halogenated aromatic systems, specific steroid mimics. | Estrogen receptor (ER), androgen receptor (AR) pathways. |
The Role of Metabolism: The intrinsic toxicity of a ring system is frequently unmasked or amplified by metabolic activation. A benign parent compound can be transformed by cytochrome P450 enzymes into a reactive intermediate. For example, a saturated furan ring may be oxidized to a highly electrophilic and hepatotoxic furan epoxide or cis-enedial [80]. Therefore, risk assessment must evaluate not only the parent ring system but also its predicted major metabolites.
In Silico and AI-Driven Approaches for Toxicity Prediction
The computational prediction of toxicity has been revolutionized by artificial intelligence (AI), which can identify complex, non-intuitive relationships between structure and biological activity that elude traditional rules.
Data Sources and Model Foundations: Modern models are trained on large-scale toxicology databases. The U.S. EPA’s ToxCast program is one of the most widely used sources, providing high-throughput screening data for thousands of chemicals across hundreds of biological endpoints [83]. Other critical resources include the DSSTox database, ChEMBL, and PubChem, which aggregate chemical, bioactivity, and toxicology data from public sources [84].
Evolution from QSAR to Advanced AI Models: Traditional Quantitative Structure-Activity Relationship (QSAR) models rely on pre-defined molecular descriptors (e.g., logP, polar surface area). While useful, they can struggle with novel scaffolds not well-represented in the training data. Descriptor-free, deep learning models have emerged as a powerful alternative.
A prime example is the Graph Convolutional Network (GCN) developed for reproductive and developmental toxicity prediction [82]. This model treats a molecule as a graph (atoms as nodes, bonds as edges) and directly learns features from this representation. The cited GCN model, augmented with multi-head attention and gated skip-connections, achieved an accuracy of 81.19% on its test set. Crucially, it was designed to integrate and learn from known structural alerts, enhancing both its performance and interpretability [82]. By identifying contributing subgraphs, such models can highlight which part of a novel ring system is activating the toxicity prediction.
Table 2: Comparison of AI/Computational Approaches for Toxicity Prediction
| Model Type | Core Principle | Advantages | Limitations | Example Performance |
|---|---|---|---|---|
| Traditional QSAR (e.g., Random Forest, SVM) [85] | Correlates calculated molecular descriptors with toxicity endpoints. | Simple, interpretable, well-established for regulatory use. | Limited by descriptor relevance; poor extrapolation to novel chemotypes. | Varies by endpoint and dataset; can be high within applicability domain. |
| Graph Convolutional Network (GCN) [82] | Learns directly from molecular graph structure without pre-defined descriptors. | Captures complex structural patterns; applicable to novel ring systems. | "Black-box" nature; requires large datasets; complex interpretation. | 81.19% accuracy for reproductive/developmental toxicity [82]. |
| Multi-Modal Deep Learning [84] | Integrates multiple data types (structure, omics, cell imaging). | Provides holistic view; can suggest mechanism of action. | Highly complex; data integration challenges; significant computational cost. | Emerging field; performance metrics still being established. |
| Top-Down vs. Bottom-Up Approaches [85] | Top-down: Uses empirical data patterns. Bottom-up: Models mechanistic pathways. | Top-down: Fast screening. Bottom-up: Mechanistic insight. | Top-down: Limited mechanistic insight. Bottom-up: Computationally intensive. | Used in combination for natural product toxicity assessment [85]. |
Experimental Protocols for Validating In Silico Alerts
Computational predictions require empirical validation. The following protocols detail key experimental methods for assessing the toxicity risk flagged in novel ring systems.
This protocol uses engineered reporter cell lines to identify the specific cellular stress pathways activated by a compound, providing mechanistic insight into hepatotoxicity.
Objective: To screen novel ring systems for activation of key transcription factor-mediated stress response pathways relevant to hepatotoxicity, with and without metabolic activation. Materials:
- Cell Lines: HepG2-GFP reporter cell lines (AP1, P53, Nrf2, NF-κB). Each line expresses Green Fluorescent Protein (GFP) under the control of a specific transcription factor response element.
- Test Compound: Novel ring system compound, dissolved in DMSO.
- Metabolic Activation System: Rat liver S9 fraction (mixed microsomes and cytosol), Cofactor I (NADPH-regenerating system).
- Controls: Positive controls for each pathway (e.g., PMA for AP1, Nutlin-3 for P53, Sulforaphane for Nrf2, TNF-α for NF-κB). Vehicle control (0.1% DMSO).
- Equipment: 96-well tissue culture plates, fluorescence plate reader or high-content imaging system, cell culture incubator.
Procedure:
- Cell Seeding: Seed each reporter cell line at 30,000 cells/well in a 96-well plate. Incubate for 24 hours at 37°C, 5% CO₂.
- Compound Preparation (Non-Metabolic): Serially dilute the test compound in culture medium (DMEM + 2% FBS). Prepare a vehicle control (0.1% DMSO).
- Compound & S9 Preparation (Metabolic): Prepare a 10% S9 mix by combining rat liver S9 fraction with Cofactor I in DMEM. Further dilute to a 2% working solution. Mix the test compound with an equal volume of the 2% S9 mix to achieve final desired compound concentration and 1% S9.
- Treatment:
- For non-metabolic assessment, treat cells with compound dilutions or vehicle.
- For metabolic assessment, treat cells with the compound-S9 mixture or an S9-vehicle control.
- Incubation: Incubate plates for 24 hours.
- Signal Detection: Measure GFP fluorescence intensity using a plate reader (excitation ~485 nm, emission ~535 nm). Alternatively, use high-content imaging to quantify fluorescence at the single-cell level.
- Data Analysis: Normalize fluorescence values to the vehicle control. A statistically significant increase in GFP fluorescence in a specific reporter line indicates activation of that stress pathway (e.g., Nrf2 activation suggests oxidative stress). Compare results with and without S9 to determine if toxicity is dependent on metabolic activation.
Protocol: In Vitro Micronucleus Assay for Genotoxicity Assessment
Objective: To evaluate the potential of a novel ring system to cause chromosomal damage (clastogenicity or aneugenicity). Materials: Mammalian cell lines (e.g., CHO-K1, TK6, or human lymphocytes), Cytochalasin-B, Giemsa stain, microscope, test compound, positive controls (e.g., Mitomycin C for clastogen, Colchicine for aneugen). Procedure: Cells are treated with the test compound for a duration covering the cell cycle. Cytochalasin-B is added to block cytokinesis, resulting in bi-nucleated cells. After harvesting and staining, cells are scored microscopically for the presence of micronuclei (small, extranuclear bodies containing chromosomal fragments or whole chromosomes) in bi-nucleated cells. An increase in micronucleus frequency indicates genotoxic potential.
Protocol: hERG Channel Inhibition Assay (Patch Clamp)
Objective: To assess the risk of a novel ring system blocking the hERG potassium channel, a key marker for potential cardiac arrhythmia (long QT syndrome). Materials: Cells stably expressing the hERG ion channel (e.g., HEK-293-hERG), patch-clamp electrophysiology setup, test compound. Procedure: Using the whole-cell patch clamp technique, the tail current amplitude of the hERG channel is measured after a depolarizing pulse. The test compound is perfused at increasing concentrations, and the concentration required to inhibit 50% of the hERG current (IC₅₀) is determined. An IC₅₀ below a concerning threshold (e.g., <10 µM) signals a significant cardiotoxicity risk that may require structural modification.
Table 3: Experimental Data from a Representative Hepa-ToxMOA Study [80]
| Natural Product Class (Example) | Key Ring System | Major Pathway Activated (w/o S9) | Major Pathway Activated (with S9) | Interpretation & Implied Risk |
|---|---|---|---|---|
| Quinone (e.g., Emodin) | Anthraquinone | Nrf2 | P53, Nrf2 | Parent compound causes oxidative stress. Metabolic activation introduces DNA-damaging species, significantly raising genotoxicity risk. |
| Alkaloid | Pyrrolizidine (1,2-unsaturated necine) | Weak/None | P53, AP1 | Compound is a pro-toxin. Metabolic activation in S9 generates reactive intermediates that cause DNA damage and cellular stress. |
| Triterpenoid (e.g., Oleanolic acid) | Steroid-like pentacyclic | None | None | Ring system shows no activation of key stress pathways under these conditions, suggesting a lower intrinsic hepatotoxicity risk. |
| Positive Control (TNF-α) | N/A | NF-κB | NF-κB | Validates NF-κB reporter system functionality. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Research Reagents and Tools for Ring System Toxicity Assessment
| Category | Item/Resource | Function/Benefit | Example/Source |
|---|---|---|---|
| In Silico Tools | QSAR Toolbox | OECD QSAR platform for grouping chemicals, profiling, and filling data gaps. | OECD [85] |
| OCHEM Platform | Online platform to build, share, and use QSAR models for toxicity endpoints. | OCHEM Database [84] | |
| RDKit & Usefulrdkitutils | Open-source cheminformatics toolkit; includes modules for ring system identification and analysis. | [17] | |
| Bioinformatics Databases | ToxCast/Tox21 Database | EPA/NIH databases providing high-throughput screening toxicity data for model training. | U.S. EPA [83] |
| ChEMBL | Manually curated database of bioactive molecules with drug-like properties and ADMET data. | EMBL-EBI [84] | |
| DrugBank | Comprehensive drug and drug target database with ADMET information. | [84] | |
| Experimental Assay Kits | Liver S9 Fractions | Subcellular liver fraction containing phase I/II metabolic enzymes for in vitro metabolic activation studies. | Commercial vendors (e.g., Xenotech, Corning) |
| hERG Inhibition Assay Kits | Fluorescence-based or patch-clamp ready kits for screening hERG channel blockade. | Commercial vendors (e.g., Eurofins, ChanTest) | |
| Micronucleus Assay Kits | Optimized kits for in vitro genotoxicity testing, including stains and cytokinesis blockers. | Commercial vendors (e.g., Thermo Fisher, Revvity) | |
| Cellular Reagents | Hepa-ToxMOA Reporter Cell Lines | Engineered HepG2 cells with GFP reporters for AP1, P53, Nrf2, and NF-κB pathways. | Can be established per [80] or sourced commercially. |
| Cytotoxicity Detection Kits (CCK-8, MTT) | Reliable colorimetric assays for determining general cell viability and compound IC₅₀ values. | Widely available from biochemical suppliers. |
Integrated Strategy for Risk Mitigation in Natural Products Research
A proactive, tiered strategy is essential to manage the toxicity risks of novel ring systems effectively.
Phase 1: Early-Stage In Silico Profiling. Begin with computational toxicity screening using multiple models (e.g., a GCN-based predictor and a traditional QSAR suite) to generate a risk profile. Simultaneously, perform in silico metabolism prediction (e.g., using software like StarDrop, SMARTCyp) to flag structures prone to forming reactive metabolites. This phase should also include a ring system similarity analysis against databases of known toxic compounds and structural alerts.
Phase 2: Focused Experimental Triaging. Prioritize compounds based on computational scores for targeted in vitro testing. The Hepa-ToxMOA assay provides a high-content, mechanistic first look at hepatotoxicity potential. Compounds showing alerts should be evaluated in specific follow-up assays (e.g., micronucleus for genotoxicity alert, hERG for cardiotoxicity risk). This phase confirms or refutes the in silico predictions.
Phase 3: Strategic Structural Modification (Alert Mitigation). If a promising natural product lead contains a toxicophore, engage in rational structural redesign. This involves:
- Bioisosteric Replacement: Substituting the problematic fragment with a biostere that maintains target activity but disrupts the toxic mechanism. Ring replacement recommenders can suggest viable alternatives [17].
- Blocking Metabolic Activation: Adding minor substituents that block the metabolic site (e.g., methyl group on a furan ring to prevent epoxidation).
- Prodrug Approach: Masking the alerting functionality with a cleavable group that is only removed at the site of action.
Conclusion: The vast structural diversity of natural product ring systems is a cornerstone of future drug discovery. By integrating advanced in silico AI tools with mechanistic in vitro assays in a systematic risk mitigation workflow, researchers can confidently navigate this chemical space. This approach allows for the early identification and elimination of toxicophores, the rational redesign of promising leads, and the ultimate selection of novel ring system-based candidates with an optimized therapeutic index, accelerating the development of safer and more effective medicines.
Evaluation and Prediction: The Performance and Potential of Ring Systems in Drug Development
Ring systems constitute the foundational scaffolds of bioactive molecules, determining three-dimensional shape, pharmacokinetic properties, and target engagement [63] [86]. Framed within the broader context of molecular frameworks in natural products research, this analysis examines the historical evolution of ring system utilization in drug discovery and clinical trials. Cheminformatic data reveal that while synthetic compounds (SCs) dominate modern screening libraries, the ring systems of natural products (NPs) exhibit greater structural diversity, complexity, and unique coverage of chemical space [58] [87]. Key trends include the enduring dominance of five- and six-membered rings in synthetic drug candidates, a historical shift from NP-derived scaffolds to synthetic heterocycles, and the rising strategic importance of underutilized ring types, such as three-membered and medium-sized rings, inspired by NP architectures [87] [6] [75]. This guide synthesizes historical patterns, quantitative comparisons, and modern experimental protocols for ring system analysis and diversification, providing a technical resource for research and development professionals.
The structural core of most small-molecule drugs is formed by a ring system, with a significant historical lineage tracing back to natural products (NPs) [58] [88]. Rings define molecular topology, constrain conformational flexibility, and present functional groups in precise orientations for optimal interaction with biological targets [63] [86]. In drug discovery, the exploration of ring systems is intrinsically linked to the study of NPs, which have served as the inspiration for a majority of approved therapeutics [88] [87]. NPs, products of evolutionary selection, possess ring systems that are often larger, more stereochemically complex, and more diverse than those found in typical synthetic compound (SC) libraries [58] [87].
However, the pharmaceutical industry's focus has oscillated between NP-inspired discovery and synthetic library screening. The advent of high-throughput screening (HTS) and combinatorial chemistry in the late 20th century prompted a shift toward SCs, but the anticipated boom in novel drug leads did not fully materialize, partly due to the limited structural diversity of synthetic libraries [87]. This has led to a renaissance in NP research and a growing appreciation for the unique chemical space occupied by NP ring systems [87] [6]. Contemporary cheminformatics allows for the precise quantification of these historical trends, revealing how the popularity of specific ring systems in drugs and clinical candidates has evolved and how NP frameworks continue to guide the design of novel scaffolds [63] [86] [87].
Historical Trends and Quantitative Analysis of Ring System Evolution
Comparative Landscape: Natural Product vs. Synthetic Compound Ring Systems
A foundational analysis of 38,662 NP ring systems reveals their distinct and expansive chemical space compared to common screening compounds [58]. Despite their structural richness, only about 2% of known NP ring systems are present in approved drugs, highlighting a vast untapped resource [58]. Critically, approximately 50% of NP ring systems are represented by synthetic compounds with identical or related 3D shape and electrostatic properties, suggesting partial but incomplete coverage by conventional libraries [58].
Table 1: Key Physicochemical and Structural Comparisons Between NP and SC Ring Systems
| Property | Natural Product (NP) Ring Systems | Synthetic Compound (SC) Ring Systems | Data Source & Implications |
|---|---|---|---|
| Structural Diversity | Extremely high; vast number of unique, complex scaffolds [58]. | Lower; dominated by a smaller set of synthetically accessible scaffolds (e.g., flat aromatics) [87]. | NP chemical space is far less concentrated than that of SCs [87]. |
| Typical Ring Features | More aliphatic and non-aromatic rings; higher prevalence of oxygen atoms [87]. | More aromatic rings (e.g., benzene, pyridine); higher prevalence of nitrogen and sulfur atoms [87]. | Reflects synthetic accessibility and the historical influence of combinatorial chemistry. |
| Molecular Complexity | Higher: more sp3-hybridized carbons, stereocenters, and fused/bridged ring systems [58] [87]. | Lower: more planar, less saturated structures on average [87]. | NP complexity is linked to successful target engagement but challenges synthesis. |
| Size (Avg. Rings/Mole) | Larger and increasing over time; more rings per molecule [87]. | Smaller and constrained by "drug-like" rules; number of rings varies within a limited range [87]. | Recently discovered NPs tend to be larger due to advanced isolation tech [87]. |
| Coverage in Drugs | ~2% of known NP ring systems are in approved drugs [58]. | A higher percentage of common SC ring systems (e.g., pyridine, piperazine) appear in drugs. | Indicates a significant opportunity for mining NPs for novel drug scaffolds [58]. |
Time-Dependent Trends in Drug Discovery
A longitudinal analysis of molecules grouped by their date of discovery or reporting reveals divergent evolutionary paths for NPs and SCs [87].
- NP Evolution: Over recent decades, discovered NPs have become larger, more complex, and more hydrophobic [87]. Their ring systems show increasing numbers of total rings and non-aromatic rings, particularly through more extensive glycosylation (more sugar rings) and the identification of complex fused systems (e.g., bridged and spiro rings) [87].
- SC Evolution: The structural properties of SCs have also shifted but remain constrained within a "drug-like" range governed by rules such as Lipinski's Rule of Five [87]. There is a marked increase in the use of five- and six-membered aromatic rings due to their synthetic accessibility and stability [87]. Notably, the use of four-membered rings (e.g., azetidine) in SCs has seen a significant increase from around 2009 onward, reflecting a growing interest in saturated, three-dimensional scaffolds to escape flatland [87].
Table 2: Historical Adoption Trends of Select Ring Types in Drugs & Clinical Candidates
| Ring System Type | Historical Trend & Popularity | Driver/Reason | Example/Therapeutic Context |
|---|---|---|---|
| Benzene & 6-Membered Aromatics | Persistently dominant in SCs and drugs throughout history [87]. | Synthetic simplicity, metabolic stability, and planar geometry for π-stacking. | Ubiquitous across all drug classes. |
| 5-Membered Heterocycles (e.g., pyrrole, imidazole) | High and increasing popularity in modern drug discovery [63] [87]. | Excellent hydrogen-bonding capabilities, prevalence in combinatorial chemistry libraries. | Key scaffolds in kinase inhibitors and antivirals. |
| Piperidine/Piperazine | Very common saturated nitrogen heterocycles in drugs [63]. | Provide basicity for salt formation, conformational constraint, and improve solubility. | Commonly used in CNS drugs and as solubilizing linkers. |
| Three-Membered Rings (Cyclopropane, Aziridine, Epoxide) | Niche but critically important; use is well-documented and strategic [75]. | Introduce high strain, act as electrophilic warheads, or serve as rigid geometric spacers. | Cyclopropane in Selegiline (MAO-B inhibitor); Epoxide in carfilzomib (proteasome inhibitor) [75]. |
| Medium-Sized Rings (7-11 members) | Historically under-represented in drugs and screening libraries [6]. | Synthetic challenge due to transannular strain; however, they offer unique conformational profiles. | Found in complex NPs (e.g., macrocyclic antibiotics); now a target for library synthesis [6]. |
| Steroid-Derived Polycyclics | Foundational in early drug discovery (hormone therapies); now a source for diversification [6]. | Provide rigid, pre-defined 3D scaffolds with multiple chiral centers. | Basis for corticosteroids, sex hormones; now diversified via C-H activation [6]. |
The data clearly indicates that the structural evolution of SCs is influenced by NPs but has not fully replicated their diversity. SC development has been guided more by synthetic feasibility and adherence to simplified drug-like rules, whereas NPs continue to expand into new regions of chemical space [87].
Methodological and Experimental Protocols
Cheminformatic Workflow for Time-Dependent Ring System Analysis
The following protocol, derived from large-scale studies, outlines a robust method for analyzing historical trends [58] [87].
Data Curation & Chronological Sorting:
- Source Datasets: Obtain structured data from NP databases (e.g., Dictionary of Natural Products) and SC databases (e.g., ChEMBL, CAS Registry) [87]. For clinical trial compounds, databases like Citeline Pharmaprojects are essential.
- Time Tagging: Assign a date to each molecule using its first reported date (publication or patent), date of addition to the database, or CAS Registry Number sequence [87].
- Grouping: Sort molecules chronologically and divide them into sequential groups (e.g., 5,000 molecules per group) for time-series analysis [87].
Ring System Perception and Standardization:
- Algorithmic Perception: Use cheminformatics toolkits (e.g., RDKit, CDK) to algorithmically identify all rings and ring systems (connected sets of rings) in each molecule [58].
- Normalization: Apply standardization rules: aromatize/dearomatize according to specified models, remove explicit hydrogens, and optionally neutralize charges [58].
- Stereochemistry: A critical step is to retain absolute stereochemical information during the ring perception process to accurately capture the 3D character of NP-derived systems [58].
Descriptor Calculation & Analysis:
- Calculate physicochemical properties for the whole molecule and the isolated ring systems: Molecular Weight, Fraction sp3 (Fsp3), Number of Stereocenters, LogP, etc [87].
- Calculate ring-specific metrics: Number of rings, Aromatic Ring Count, Ring Assembly Count (groups of fused/spiro rings), and size distribution (3-membered, 4-membered, etc.) [87].
- Shape & Electrostatic Comparison: To assess coverage, compute 3D molecular shape descriptors (e.g., torsion fingerprints) and electrostatic potentials for NP and SC ring systems and compare them using similarity metrics [58].
Trend Visualization & Interpretation:
- Plot the mean or distribution of key descriptors (e.g., average number of rings, Fsp3) across the chronological groups for both NP and SC sets [87].
- Use dimensionality reduction techniques (PCA, t-SNE) or Tree Maps (TMAP) to visualize the chemical space occupation of different historical groups [87].
Experimental Protocol: Diversifying Natural Products via C–H Oxidation and Ring Expansion
This synthetic methodology, exemplified with steroids, provides access to novel polycyclic scaffolds with underutilized medium-sized rings [6].
Substrate Preparation:
- Select a polycyclic NP scaffold with inherent rigidity (e.g., steroid like dehydroepiandrosterone (DHEA) or estrone) [6].
- Perform any necessary protective group chemistry to isolate specific reaction sites.
Phase 1: C–H Functionalization (Installing a Handle):
- Objective: Convert an inert C-H bond into a reactive C-O functional group (e.g., alcohol, ketone) selectively.
- Method: Employ state-of-the-art C–H oxidation methods.
- Workup: Purify the hydroxylated or ketone intermediate via standard chromatography.
Phase 2: Ring Expansion (Diversifying the Core):
- Objective: Use the newly installed oxygen functionality to break and reform bonds, enlarging a small ring (typically 5- or 6-membered) into a medium-sized ring (7-11 membered).
- Method Selection:
- For Ketones: Employ a Beckmann Rearrangement with hydroxylamine reagents to convert the ketone into a ring-expanded lactam [6].
- For Specific Skeletons: Utilize an intramolecular Schmidt reaction (with hydrazoic acid) or a formal [2+2] cycloaddition/fragmentation sequence with dialkyl acetylenedicarboxylate (DMAD) [6].
- Workup and Characterization: Purify the complex polycyclic product. Confirm structure and stereochemistry using NMR, HRMS, and ideally, X-ray crystallography [6].
Library Generation & Profiling:
- Apply this two-phase strategy to multiple NP starting materials and diversify reaction conditions.
- Analyze the resulting library's chemical space using principal component analysis (PCA) of molecular descriptors to confirm its novelty compared to commercial screening libraries [6].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents, Databases, and Software for Ring System Research
| Category | Item / Resource | Function in Ring System Research | Key Consideration / Example |
|---|---|---|---|
| Computational & Databases | RDKit (Open-source) | Primary toolkit for cheminformatics: ring perception, descriptor calculation, fingerprint generation. | Essential for standardizing molecules and extracting Murcko scaffolds [58] [87]. |
| Dictionary of Natural Products (DNP) | Curated database for NP structures and associated data. | Critical source for authentic NP ring system analysis [87]. | |
| ChEMBL / PubChem | Large databases of bioactive SCs with bioactivity data. | Primary sources for analyzing trends in synthetic drug candidates [87]. | |
| CAS Registry | Authoritative source for chemical information with chronological indexing. | Enables time-dependent studies via CAS Registry Numbers [87]. | |
| Synthetic Chemistry | C–H Oxidation Catalysts (e.g., Electrochemical cells, Cu/ligand complexes, Cr reagents) | Enable selective functionalization of inert C-H bonds in complex NPs, creating handles for diversification [6]. | Selectivity is paramount; method depends on substrate (allylic, benzylic, etc.) [6]. |
| Ring Expansion Reagents (e.g., Hydroxylamines (for Beckmann), Hydrazoic Acid (for Schmidt), DMAD) | Perform the key bond-breaking and forming steps to convert small rings into medium-sized rings [6]. | Handling and safety are major concerns (e.g., hydrazoic acid is highly toxic and explosive) [6]. | |
| Steroid Natural Products (e.g., DHEA, Estrone, Isosteviol) | Privileged, rigid starting scaffolds for diversification into novel chemical space. | Readily available, well-understood, and highly modifiable [6]. | |
| Visualization & Analysis | PCA & t-SNE Algorithms | Dimensionality reduction to visualize and compare chemical space of different compound sets. | Standard for demonstrating novelty of a synthesized library [87] [6]. |
| Tree Map (TMAP) Visualization | Creates interactive, hierarchical maps of high-dimensional chemical space for intuitive exploration. | Effective for showing relationships and coverage of millions of compounds [87]. |
The molecular framework of a bioactive compound, often defined by its core ring system, is fundamental to its three-dimensional shape, physicochemical properties, and ultimate biological function. Within the broader thesis of molecular frameworks in natural products research, ring systems represent the architectural keystones upon which chemical diversity and biological relevance are built. Natural products (NPs), honed by evolution to interact with biological macromolecules, contain ring systems of exceptional structural diversity, complexity, and stereochemical richness [3]. These NP-derived scaffolds have historically served as the foundational inspiration for a vast array of approved therapies [89].
In contrast, fully synthetic rings, designed primarily with considerations of synthetic accessibility and adherence to “drug-like” physicochemical rules, occupy a more constrained region of chemical space [87] [90]. This technical guide presents a comparative analysis of these two origins of ring systems within approved therapies. It examines their divergent structural evolution, quantifies their representation in the drug market, and details modern synthesis and analysis strategies that seek to bridge the gap between biologically relevant complexity and synthetic feasibility. The central thesis posits that while fully synthetic rings offer advantages in manufacturing and optimization, the unique three-dimensional frameworks of NP-derived rings provide irreplaceable access to novel biological mechanisms; the future of small-molecule drug discovery lies in sophisticated strategies that integrate the lessons of natural product frameworks into synthetic design [36] [4].
Structural Analysis of Ring Systems
A cheminformatic comparison of ring systems reveals profound differences in complexity, diversity, and physicochemical character between NP-derived and fully synthetic scaffolds.
2.1. Diversity and Complexity NP ring systems are inherently more diverse and structurally complex. An analysis of 38,662 unique NP ring systems found that only about 2% are directly present in approved drugs, highlighting that the vast majority of NP chemical space remains unexploited in therapy [3]. NP scaffolds are characterized by a higher proportion of aliphatic and saturated rings, greater incorporation of oxygen heteroatoms, and more sp3-hybridized carbon centers [87] [90]. This results in greater three-dimensionality and structural complexity. In contrast, the ring systems of typical synthetic compounds and libraries are dominated by flat, aromatic systems (e.g., benzene, pyridine) and contain more nitrogen and sulfur heteroatoms [87] [3]. The synthetic corpus, while numerically vast, explores a narrower and topologically simpler region of ring system space.
2.2. Physicochemical Properties and Evolution Time-dependent analysis shows that the structural evolution of NPs and synthetic compounds (SCs) diverges significantly. Newly discovered NPs continue to trend toward larger molecular size, increased ring count, and higher hydrophobicity [87]. This reflects advances in isolation technology enabling the characterization of more complex molecules. Conversely, the physicochemical properties of SCs have evolved within a tight range constrained by drug-like filters such as Lipinski’s Rule of Five [87] [90]. While SCs have incorporated more rings over time, the increase is largely driven by aromatic rings, unlike the aliphatic and fused ring systems prevalent in NPs [87].
2.3. Coverage and Shape Analysis A critical finding is that despite low direct scaffold overlap, approximately 50% of NP ring systems have a close match in 3D shape and electrostatic properties within commercially available synthetic screening compounds [3] [58]. This suggests that while unique covalent architectures of NPs are rare in synthetic libraries, a significant portion of their biologically relevant pharmacophoric space is indirectly represented. This provides a rationale for pseudo-natural product strategies and shape-based screening.
Table 1: Comparative Structural Properties of Natural Product vs. Synthetic Ring Systems
| Property | Natural Product-Derived Rings | Fully Synthetic Rings | Implications for Drug Discovery |
|---|---|---|---|
| Representative Rings | Macrolactones, polycycles (e.g., steroid, taxane cores), fused aliphatic systems [3] [4] | Benzene, pyridine, pyrimidine, simple heterocycles (e.g., piperazine) [87] [3] | NPs access 3D shapes for complex targets; synthetics favor flat, aromatic architectures. |
| Structural Complexity | High: More stereocenters, higher Fsp³ (fraction of sp³ carbons), more aliphatic/Non-aromatic rings [87] [90] | Lower: Fewer stereocenters, lower Fsp³, predominance of aromatic rings [87] [90] | NP complexity correlates with target selectivity and success in clinical development [90]. |
| Chemical Space Coverage | Extremely diverse but sparsely populated in drugs (~2% direct coverage) [3]. Occupies distinct, broad regions. | Less diverse but highly populated. Concentrated in "drug-like" regions defined by rules [87] [90]. | Vast NP chemical space is underutilized; synthetic libraries suffer from redundancy. |
| 3D Shape/Electrostatic Match | Reference standard for biologically relevant shapes. ~50% have a close synthetic analog in shape/electrostatics [3] [58]. | Designed for synthetic ease. A significant subset can mimic NP shape properties. | Enables shape-based virtual screening to leverage NP-inspired designs from synthetic collections. |
Trends in Drug Approvals and Clinical Impact
An analysis of New Chemical Entities (NCEs) approved between 1981-2010 confirms the enduring impact of NP-derived structures. Approximately 50% of all small-molecule drugs approved in this period were either natural products, directly derived from them (semisynthetic), or were synthetic compounds whose pharmacophore was inspired by a natural product [90]. This contribution has remained consistent over decades, even as industrial focus has shifted [90]. The therapeutic areas dominated by NP-derived drugs are notably infectious diseases (antibiotics, antifungals) and oncology (cytotoxics, targeted therapies) [89]. This is a direct result of NPs’ evolutionary optimization for biological interference.
Drugs based on NP-derived rings exhibit distinct property profiles compared to fully synthetic drugs: they have larger molecular weight, greater stereochemical content (more stereocenters), lower hydrophobicity (cLogP), and reduced aromatic ring fraction [90]. These properties align with their ability to interact with challenging target classes, such as protein-protein interfaces, that are often intractable for flat, aromatic synthetic molecules.
Table 2: Analysis of Approved Drugs (1981-2010) by Structural Origin [90]
| Drug Category | Definition | % of Approved NCEs (1981-2010) | Typical Property Profile |
|---|---|---|---|
| Natural Product (NP) | Unmodified natural product. | ~6% | Highest molecular complexity, polarity, and stereochemistry. |
| Natural Product-Derived (ND) | Semisynthetic modification of NP scaffold. | ~24% | High complexity, but often optimized for pharmacokinetics. |
| Synthetic, NP-Pharmacophore (S*) | Fully synthetic, but core pharmacophore mimics NP. | ~12% | Intermediate complexity; blends NP-inspired activity with synthetic tractability. |
| Fully Synthetic (S) | Synthetic; no direct NP inspiration. | ~58% | Lower molecular weight, higher aromatic ring count, fewer stereocenters, more "rule-of-five" compliant. |
Modern Synthetic and Analytical Strategies
To harness the value of NP-derived rings while overcoming synthesis and supply challenges, several advanced strategies have been developed.
4.1. Strategies for Accessing Complex Ring Systems
- Diversity-Oriented Synthesis (DOS): Aims to generate skeletal and stereochemical diversity from common building blocks, creating libraries that populate broad chemical space, including NP-like regions [36] [4].
- Biology-Oriented Synthesis (BIOS): Uses privileged NP-derived scaffolds as starting points for library synthesis, focusing on exploring chemical space with known biological relevance [36] [4].
- Ring Distortion of Natural Products: Involves subjecting readily available, complex NPs to chemical reactions that dramatically alter their core ring structure (e.g., cleavage, expansion, rearrangement), rapidly generating novel, complex scaffolds [4].
- Medium-Sized Ring Synthesis (8-11 members): These rings are prevalent in NPs but synthetically challenging due to transannular strain. New ring-expansion reactions of smaller cyclic precursors offer efficient routes to these valuable frameworks [36].
4.2. Analytical and Computational Workflows Modern NP research leverages integrated analytical pipelines. High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy are coupled with advanced separation techniques for dereplication and structure elucidation [89]. Chemoinformatic workflows involve calculating molecular descriptors (e.g., Fsp³, topological polar surface area, ring system counts) and applying dimensionality reduction methods like Principal Component Analysis (PCA) to visualize and compare the chemical space of NP and synthetic collections [87] [90]. This guides library design and target selection.
Table 3: Key Strategies for Leveraging NP Ring Systems in Discovery [36] [4]
| Strategy | Core Principle | Advantage | Example Application |
|---|---|---|---|
| Diversity-Oriented Synthesis (DOS) | Build structurally diverse libraries from simple precursors using branching pathways. | Broad exploration of chemical space; can serendipitously hit diverse targets. | Synthesis of macrolactone libraries to discover modulators of the Hedgehog signaling pathway [4]. |
| Biology-Oriented Synthesis (BIOS) | Use bioactive NP scaffolds as inspiration for focused library synthesis. | Higher probability of bioactivity; efficient exploration of relevant chemical space. | Designing libraries based on the steroid core to target nuclear receptors. |
| Ring Distortion | Apply dramatic skeletal modifications to existing NPs. | Rapid generation of novel, complex scaffolds from available starting materials. | Chemical transformation of the alkaloid vincamine into new scaffolds with altered bioactivity [4]. |
| Pseudo-Natural Products | Chemically fuse biosynthetically unrelated NP fragments. | Creates novel chemotypes with retained biological relevance but unprecedented structures. | Combining indole and tropane fragments to create new bioactive hybrids [4]. |
Detailed Experimental Protocols
5.1. Protocol for Chemoinformatic Comparison of Ring Systems
- Objective: To quantitatively compare the structural and physicochemical properties of ring systems from NP and synthetic drug datasets.
- Materials: Curated datasets (e.g., from Dictionary of Natural Products, ChEMBL); Cheminformatics software (e.g., RDKit, KNIME, Schrödinger Canvas).
- Procedure:
- Data Curation: Standardize molecules (neutralize charges, remove salts), extract unique ring systems using the Murcko scaffold algorithm, and enumerate stereoisomers for shape analysis [3] [58].
- Descriptor Calculation: For each ring system, compute a set of descriptors: number of atoms, rings, and stereocenters; fraction of sp³ carbons (Fsp³); topological polar surface area (TPSA); logP; and 3D shape/electrostatic descriptors (e.g, using ROCS) [3] [90].
- Statistical & Visual Analysis: Perform statistical tests (e.g., t-test) on descriptor distributions. Use Principal Component Analysis (PCA) on the descriptor matrix to project compounds into 2D/3D chemical space for visualization. Generate similarity networks (e.g., TMAP) to assess scaffold diversity and overlap [87] [58].
- Coverage Analysis: For each NP ring system, search the synthetic compound library for scaffolds with Tanimoto similarity >0.7 (based on graph) or 3D shape/electrostatic combo score >1.0 [3]. Calculate the percentage of NP systems covered.
5.2. Protocol for Ring Distortion Synthesis
- Objective: To generate novel molecular scaffolds via chemical modification of a natural product core.
- Materials: A readily available NP (e.g., sclareolide, vincamine); a set of diverse chemical reagents (oxidants, reductants, Lewis acids, photoredox catalysts); analytical tools (TLC, LC-MS, NMR).
- Procedure:
- Reaction Scouting: Subject the NP (50-100 mg) to a matrix of different reaction conditions (varying reagent, solvent, temperature, time) in parallel micro-scale reactors.
- Reaction Monitoring: Use TLC and LC-MS to identify conditions that consume starting material and produce new major products.
- Scale-up & Isolation: Scale the promising reactions, purify products via flash chromatography or preparative HPLC.
- Structural Elucidation: Determine the structure of novel derivatives using HR-MS and 1D/2D NMR. The key step is identifying changes in the core ring system (cleavage, ring expansion/contraction, rearrangement).
- Library Creation: Apply the optimized distortion conditions to a small set of related NPs to create a focused library of distorted scaffolds for biological screening [4].
Research Reagent Solutions (The Scientist's Toolkit)
Table 4: Essential Tools for Ring System Analysis and Synthesis
| Category | Item/Resource | Function/Benefit |
|---|---|---|
| Computational Databases | Dictionary of Natural Products (DNP), COCONUT [3], ChEMBL [90] | Provide curated, searchable collections of NP and synthetic compound structures for analysis. |
| Cheminformatics Software | RDKit (Open Source), Schrödinger Canvas, OpenEye Toolkits | Enable automated descriptor calculation, scaffold extraction, similarity searching, and chemical space visualization. |
| Synthesis & Analysis | Building blocks for DOS (e.g., diverse aldehydes, chiral amines) [4]; Photoredox & Electrochemical Reactors [36] | Facilitate the synthesis of complex, NP-inspired libraries and enable novel ring expansion reactions. |
| Analytical Instruments | UPLC-HRMS, High-Field NMR with Cryoprobe [89] | Critical for the rapid dereplication, purification, and structural elucidation of complex ring systems from natural sources or synthesis. |
| Specialized Libraries | Commercially available NP-like libraries, Fragment libraries based on NP scaffolds [3] | Provide physical screening decks designed to increase hit rates against challenging biological targets. |
Diagrams and Visual Workflows
Diagram 1: Comparative Chemoinformatic Analysis Workflow. This workflow outlines the computational process for comparing ring systems from natural and synthetic sources.
Diagram 2: Relationship of Chemical Spaces for Ring Systems. This diagram shows the conceptual overlap and influence between the chemical spaces of natural products, synthetic libraries, and approved drugs.
Diagram 3: Ring Distortion Synthesis for Library Generation. This workflow depicts the process of transforming a natural product core into a novel library via ring distortion and subsequent functionalization.
The comparative analysis unequivocally demonstrates that NP-derived and fully synthetic ring systems offer complementary value in drug discovery. NP-derived rings provide unmatched structural diversity, three-dimensionality, and validated biological relevance, particularly for challenging target classes. Fully synthetic rings offer advantages in synthetic tractability, optimization, and adherence to pharmacokinetic guidelines.
The future of the field lies not in choosing one over the other, but in sophisticated integration. This includes:
- De Novo Design: Using computational models trained on NP structures to generate novel, synthetically accessible scaffolds that capture NP-like complexity.
- Advanced Synthesis: Further development of ring-expansion and cascade reactions to efficiently build medium-sized and macrocyclic rings, closing the synthetic accessibility gap [36].
- Integrated Screening: Combining high-throughput screening of synthetic libraries with shape-based virtual screening against NP-derived pharmacophore models to maximize hit discovery [3] [58].
By systematically decoding and harnessing the architectural principles of natural product ring systems, drug discovery can access a broader swath of biologically relevant chemical space, paving the way for novel therapies against undrugged targets.
Within the broader thesis on molecular frameworks in natural products research, ring systems are not merely structural components but the fundamental architectural elements that dictate the biological destiny of a molecule. They determine three-dimensional shape, enforce conformational rigidity, and directly influence critical pharmacokinetic and pharmacodynamic properties. In the context of small-molecule drug discovery, the strategic selection and innovation of ring systems are paramount. Analysis of clinical trial databases reveals a conservative yet insightful trend: approximately 67% of clinical trial compounds incorporate ring systems already established in existing drugs [1]. This conservatism underscores a critical challenge and opportunity—the introduction of a novel ring system into a drug candidate is a rare, high-stakes event that significantly impacts its probability of technical and regulatory success [1].
This whitepaper posits that the success of these novel ring systems is not merely serendipitous but can be systematically forecast through predictive modeling. By integrating cheminformatic analysis of ring system properties with modern clinical trial data—including real-world evidence, participant experience metrics, and operational performance data—we can construct models to de-risk the development of innovative scaffolds. This approach bridges the historic inspiration drawn from natural products, which are a rich source of unique and biologically pre-validated ring systems [8], with the data-driven demands of contemporary drug development. The convergence of AI-powered clinical operations [91] [92] and sophisticated cheminformatic frameworks [8] now enables a paradigm shift from empirical design to predictive forecasting for molecular frameworks.
Cheminformatic Foundation: Analyzing Ring Systems in Clinical and Natural Product Landscapes
A quantitative understanding of ring system prevalence and properties forms the basis for any predictive model. Analysis of molecules in the medicinal chemistry literature (e.g., ChEMBL) shows a extreme distribution: a small set of rings is exceedingly common, while a long tail of thousands appears infrequently [1]. This pattern extends to clinical trial compounds.
Table 1: Prevalence of Ring Systems in Drug Discovery and Development
| Data Source | Key Quantitative Finding | Implication for Novel Ring Systems |
|---|---|---|
| FDA-Approved Drugs (last 20 years) | 95.1% contain at least one ring system [1]. | Ring-based scaffolds are a near-universal requirement for drug-likeness. |
| Clinical Trial Compounds | ~67% incorporate known drug ring systems; novel ring introductions are rare [1]. | Novel ring systems represent a key point of differentiation and associated risk. |
| Bioactive Molecules (ChEMBL) | Only 0.42% are completely acyclic [1]. | Ring systems are intrinsically linked to bioactivity. |
| Novel Combinatorial Libraries (e.g., SAVI) | Contain ~40,000 unique ring systems, many not found in public databases [1]. | Vast, untapped chemical space exists for exploration via predictive modeling. |
The persistence of established rings is driven by synthetic feasibility, proven drug-likeness, and a deep understanding of their Structure-Activity Relationships (SAR). Novel rings, particularly those inspired by the complex architectures of natural products, offer potential for novel target engagement and improved selectivity but carry risks regarding synthetic complexity, metabolic stability, and toxicity [1] [8].
The cheminformatic challenge is to represent these ring systems in a way that captures their relevant properties for prediction. Simple presence/absence is insufficient. Advanced representations include:
- Molecular Frameworks: Defining the core ring system with all side chains removed.
- Quantum Chemical Descriptors: Calculating electronic properties, aromaticity, and strain energy.
- Topological and Shape Fingerprints: Encoding the 3D geometry and pharmacophore features critical for binding.
Frameworks like AgreementPred [8] demonstrate the power of multi-representation structural similarity data fusion. By combining similarity search results from 22 different molecular representations (e.g., ECFP, atom pairs, pharmacophore fingerprints), it achieves a superior recall-precision balance for predicting pharmacological categories of unannotated natural products [8]. This principle is directly applicable to forecasting the clinical trial "category" of success or failure for a novel ring system.
Predictive Modeling Framework: Integrating Molecular and Clinical Data
The proposed predictive modeling framework transitions from traditional quantitative structure-activity relationship (QSAR) models to a holistic Quantitative Structure-Trial-Success Relationship (QSTSR) model. It integrates multi-faceted data streams to generate a probabilistic forecast for a novel ring system's performance in clinical development.
Figure: Predictive Modeling Workflow for Novel Ring System Success
Core Data Inputs and Fusion
- Molecular Feature Vector: Derived from the novel ring system and its immediate substituents. This includes topological descriptors, predicted physicochemical properties (LogP, solubility), and in silico ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) endpoints.
- Historical Clinical Trial Profile: The model queries a database of historical trials using the multi-representation similarity approach [8]. It identifies compounds with the most structurally similar ring systems and extracts their clinical performance data: phase transition success rates, frequency and type of adverse events, associations with specific biomarkers, and reasons for trial termination.
- Protocol Context Factors: The forecast must be context-aware. Inputs include the therapeutic area (e.g., oncology trials have different success rates than cardiovascular), primary endpoint complexity (e.g., a trial with 26 endpoints carries different operational risk [93]), and planned patient population demographics [92].
Model Architecture and Output
The model is an ensemble machine learning system (e.g., combining random forest, gradient boosting, and graph neural networks). It is trained on historical data linking molecular features and trial context to known outcomes. Its outputs are not binary but probabilistic and prescriptive:
- Phase-Specific Success Probability: Estimated likelihood of successfully progressing from Phase I to Phase II, Phase II to Phase III, etc.
- Key Risk Indicators: Flags for potential issues (e.g., "high predicted risk of idiosyncratic hepatotoxicity," "structural alert for hERG inhibition," "operational risk due to complex synthesis impacting supply").
- Data-Driven Mitigation Strategies: Recommendations such as "include exploratory liver function biomarker X in protocol" or "consider a decentralized trial model with wearables to reduce participant burden and dropout risk [94] [92]."
Validating Predictions: From In Silico to Clinical Trial Performance
Predictive models require rigorous validation through iterative cycles of computational and experimental testing.
Table 2: Experimental Validation Protocol for Ring System Predictions
| Validation Stage | Experimental Protocol | Metrics & Success Criteria |
|---|---|---|
| 1. Retrospective Validation | Apply model to a held-back dataset of past clinical trials with known outcomes. Use similarity search [8] to find analogs for each. | Area Under the Curve (AUC) of the receiver operating characteristic curve for predicting success/failure. Calibration of predicted probabilities vs. observed frequencies. |
| 2. Prospective In Vitro/In Vivo | For a novel ring system predicted as high-risk for toxicity, synthesize the compound and conduct the flagged assays (e.g., hERG patch clamp, hepatocyte cytotoxicity, genotoxicity). | Concordance between predicted and experimental results. Risk mitigation by early identification of true positives. |
| 3. Clinical Trial Simulation | Use digital twin technology [91] to simulate a virtual patient population receiving the compound. Incorporate the model's predicted PK/PD and safety profile. | Simulated trial outcomes (efficacy, dropout rates, adverse event incidence). Informs adaptive trial design [93] [95] before real-world investment. |
| 4. Real-World Clinical Integration | Embed model predictions as a risk signal within a Risk-Based Monitoring (RBM) system [96] for an ongoing trial. Flagged patients for enhanced safety monitoring. | Reduction in severe adverse event detection time. Improvement in data quality and proactive risk management [96]. |
Research Reagent Solutions for Predictive Modeling of Ring Systems
- Cheminformatics & Modeling Software: AgreementPred framework [8] for multi-representation similarity searching and category prediction. RDKit or OpenEye Toolkits for molecular descriptor calculation and fingerprint generation. Commercial Platforms (e.g., Veeva Clinical Data, Merative Zelta) offering integrated AI/ML analytics for clinical data [96] [94].
- Specialized Databases: ChEMBL and PubChem for bioactive molecule and ring system data [1]. DrugBank and PharmaProjects for clinical trial compound intelligence. NPASS and LOTUS for natural product ring systems and biological activity data [8].
- Clinical Trial Data Sources: Internal Historical Trial Databases (most critical). Citizen and ClinicalTrials.gov for public trial summaries and results. Real-World Data (RWD) Partnerships (e.g., with organizations like Optum Life Sciences [93]) for post-marketing safety and effectiveness data.
- Computational Infrastructure: Cloud-based SaaS platforms (e.g., AWS, Google Cloud) for scalable data storage and model training [97]. Graphical Processing Unit (GPU) clusters for training deep learning models on large molecular graphs.
The integration of cheminformatics and clinical data science presents a transformative opportunity to rationalize one of drug discovery's most consequential decisions: the selection of a novel molecular framework. By treating clinical trial data as a predictive endpoint, we can shift the evaluation of ring systems from retrospective analysis to prospective forecasting. This aligns with the industry's broader movement towards AI-driven, adaptive, and patient-centric trials [91] [92] [95].
Future advancements will hinge on:
- Increased Data Accessibility and Standardization: Breaking down silos between medicinal chemistry, clinical operations, and safety databases is essential [96].
- Explainable AI (XAI): For regulatory adoption, models must provide interpretable reasons for their forecasts, linking specific ring system features to clinical risks [91].
- Dynamic, Learning Models: As proposed by the FDA's 2025 draft guidance on AI [91], models must be continuously updated with new trial data, creating a self-improving cycle that accelerates the validation of ring systems inspired by nature's ingenuity.
By embedding these predictive capabilities into the research workflow, the development of novel ring systems can evolve from an artisanal craft into a scalable, data-driven engineering discipline, ultimately increasing the efficiency and success rate of bringing new medicines to patients.
Ring systems constitute the architectural core of bioactive molecules, defining three-dimensional shape, positioning key pharmacophores, and critically influencing both pharmacokinetic and pharmacodynamic profiles [1]. This whitepaper examines the fundamental role of ring systems in mediating specific target engagement and achieving therapeutic selectivity, framed within the context of natural product-inspired molecular frameworks. Through integrated cheminformatic analyses and contemporary case studies across oncology, neuroscience, and infectious disease, we demonstrate how ring selection—from privileged heterocycles to complex natural product-derived scaffolds—dictates biological outcomes. The analysis incorporates quantitative data on ring prevalence, detailed experimental protocols for assessing target engagement, and strategic frameworks for translating ring-based design into clinically effective therapies. By synthesizing insights from biomimetic synthesis, covalent inhibitor development, and phenotypic screening, this guide provides researchers with a structured approach to leveraging ring systems as central tools for overcoming selectivity challenges in modern drug discovery.
The structural framework of most small-molecule drugs and bioactive natural products is defined by ring systems. Empirical data underscores their dominance: over 95% of FDA-approved small-molecule drugs from the past two decades contain at least one ring, while a mere 0.42% of bioactive molecules in the ChEMBL database are purely acyclic [1]. Rings are not mere skeletal supports; they are dynamic components that govern molecular rigidity and conformation, directly positioning substituents for optimal interactions with biological targets. Furthermore, the electronic and steric properties of rings profoundly influence global molecular characteristics, including solubility, lipophilicity, metabolic stability, and toxicity profiles [1].
Natural products (NPs) represent a pinnacle of evolutionary optimization, often showcasing ring systems of unmatched complexity and bioactivity. However, a striking cheminformatic analysis reveals a significant disparity: only approximately 2% of the unique ring systems observed in natural products are present in approved drugs [3]. This "ring system gap" highlights a vast reservoir of unexplored chemical space. Intriguingly, about half of NP ring systems are represented by synthetic compounds with highly similar three-dimensional shape and electrostatic properties, suggesting viable paths for inspiration and mimicry [3]. This positions natural product ring systems not merely as sources of leads, but as infinite wellsprings of inspiration for scaffold design in drug discovery. The challenge and opportunity for contemporary medicinal chemistry lie in deciphering the target engagement logic encoded within these complex rings and translating it into novel, synthetically accessible therapeutics with enhanced selectivity.
Foundational Principles: How Rings Mediate Target Interaction and Selectivity
Structural and Physicochemical Determinants
The efficacy of a ring system in drug design hinges on its ability to precisely navigate the complementary topography of a target binding site. Key determinants include:
- Ring Aromaticity and Heteroatom Content: Aromatic and heteroaromatic rings provide planar, electron-rich surfaces for π-π stacking and cation-π interactions. Heteroatoms (N, O, S) serve as hydrogen bond donors or acceptors, enabling directional polar interactions critical for affinity and specificity [1].
- Ring Size and Saturation: Saturation (sp³-hybridization) increases three-dimensionality and reduces planar rigidity, which can improve solubility and allow for better shape complementarity with buried protein pockets. Medium-sized (7-14 membered) and macrocyclic rings can pre-organize into bioactive conformations, offering high potency and selectivity [1].
- Conformational Rigidity vs. Flexibility: Rings introduce constraint, reducing the entropic penalty of binding by limiting the number of unbound conformations. The degree of rigidity can be tuned; fused and bridged ring systems lock conformations, while systems with single bonds between rings allow adaptive flexibility [86].
Cheminformatic Analysis of Ring System Evolution and Privileged Motifs
Analysis of 1.35 million molecules from medicinal chemistry literature identified 29,179 unique ring systems, following a power-law distribution where a small subset of rings is exceedingly common [1]. This persistence of "privileged scaffolds" underscores a conservative yet pragmatic design principle: proven ring systems offer predictable synthetic routes and a lower risk of unforeseen developability issues. Table 1: Prevalence of Key Ring Systems in Bioactive Molecules and Drugs [1] [3]
| Ring System | Example (Drug/NP) | Prevalence in Drugs | Key Therapeutic Role | Structural Contribution to Selectivity |
|---|---|---|---|---|
| Benzene | Ubiquitous | >60% of drugs [1] | Core scaffold | Provides planar hydrophobic surface; substituent vector control. |
| Pyridine | Nicotine, Nilotinib | Very High | Hydrogen bond acceptor; modulates basicity/pKa. | Directional H-bonding; coordination to metal ions. |
| Indole/Azaindole | Sumatriptan, AT7519 (CDK inhibitor) | High (esp. in kinase inhibitors) [1] | H-bond donor/acceptor; mimics adenine. | Shapes complementary to hinge region of kinase ATP sites. |
| β-Lactam | Penicillins, Cephalosporins | Niche (Antibiotics) | Covalent warhead (serine protease inhibition). | Strained 4-membered ring highly reactive for acylation. |
| Pentacyclic Triterpene | Oleanolic acid (NP) | Low (2% of NP rings in drugs) [3] | Anti-inflammatory, anticancer leads. | Complex 3D shape accesses unique binding pockets. |
Computational Approaches for Ring-Centric Design
Encoding and comparing complex ring systems, especially those from NPs, requires specialized cheminformatic tools. While Extended Connectivity Fingerprints (ECFP) are standard for drug-like molecules, their performance can vary with highly complex, sp³-rich NP scaffolds [98]. Alternative fingerprints like MinHashed Atom Pair (MAP4) and Pharmacophore Triplets (PH3) can provide better performance for similarity searching and bioactivity prediction within NP chemical space, capturing crucial shape and pharmacophore information beyond simple substructures [98]. This toolkit is essential for "scaffold hopping"—replacing a ring system with a biologically equivalent but chemically distinct one to improve properties while retaining activity.
Case Studies in Key Therapeutic Areas
Oncology: Covalent Kinase Inhibitors and Pseudo-Natural Products
Case Study 1: Covalent KRASᶢ¹²ᶜ Inhibitors (e.g., Sotorasib) The oncogenic KRASᶢ¹²ᶜ mutant was long considered "undruggable." Its inhibition was achieved by exploiting a cysteine residue introduced by the mutation. The design involves coupling a tetrahydro-pyrimidine ring system, which non-covalently engages the switch-II pocket with high affinity, with an acrylamide warhead positioned to form a covalent bond with Cys12 [99]. The specific geometry and electronics of the tetrahydro-pyrimidine scaffold are critical for positioning the warhead for selective, irreversible engagement with the mutant protein over wild-type KRAS and other off-targets.
Experimental Protocol: Intact Protein LC-MS for Covalent Target Engagement Assessment [99]
- Objective: Quantify % target engagement (%TE) of covalent inhibitors in complex biological matrices.
- Sample Preparation: Treat purified target protein or tissue homogenate (e.g., tumor lysate) with the covalent inhibitor. For tissue, employ a fast chloroform/ethanol protein partitioning step to reduce matrix complexity.
- LC-MS Analysis: Use intact protein liquid chromatography-mass spectrometry (LC-MS). Separately analyze drug-target conjugate and unmodified protein.
- Data Analysis: Deconvolute mass spectra to determine the ratio of modified to unmodified protein. Calculate %TE = (Intensity of modified protein / (Intensity of modified + unmodified protein)) x 100%.
- Kinetic/PD Modeling: Fit time-dependent %TE data into an intact protein PK/PD (iPK/PD) model to derive parameters like drug-target complex half-life and required dosing regimen.
Case Study 2: Pseudo-Natural Product (PNP) Anti-Cancer Agents PNPs are synthesized by combinatorially fusing biosynthetically unrelated NP fragments, creating novel ring systems not found in nature. For instance, fusing a spirocyclic griseofulvin derivative with an indole fragment via an oxa-Pictet-Spengler reaction created a new PNP class [5]. Unbiased phenotypic screening via the cell painting assay revealed that these PNPs exhibit unique bioactivity profiles distinct from their parent fragments, suggesting novel mechanisms of action and target landscapes accessible only through the newly created hybrid ring system [5].
Table 2: Case Study Outcomes in Oncology
| Case Study | Core Ring System(s) | Target/Mechanism | Key Finding on Selectivity | Experimental Tool Highlighted |
|---|---|---|---|---|
| Covalent KRAS Inhibitor | Tetrahydro-pyrimidine, Acrylamide | KRASᶢ¹²ᶜ (Covalent) | Scaffold dictates warhead orientation for mutant-selective engagement. | Intact Protein LC-MS for % Target Engagement [99]. |
| Pseudo-Natural Products | Fused Griseofulvin-Indole Hybrid | Novel/Phenotypic (Cancer Cell Lines) | New ring system accesses unique biological space vs. parent fragments [5]. | Cell Painting Morphological Profiling [5]. |
Neuroscience: Achieving Central Nervous System (CNS) Selectivity
Case Study: Monoacylglycerol Lipase (MAGL) Inhibitors for Neuroinflammation Developing CNS drugs requires crossing the blood-brain barrier (BBB) while minimizing peripheral side effects. The design of MAGL inhibitors for neuroinflammatory conditions illustrates the "Five-Star Matrix" framework for translational drug discovery [100]. This matrix evaluates compounds across five dimensions (Biodistribution, Target Binding, Proximal Effect, Biological Effect, Disease Effect) within five systems (biochemical to clinical).
- Ring System Role: The core ring system (e.g., a piperazine-carbamate or azetidine-triazole) must be optimized for low molecular weight, high lipophilicity efficiency, and minimal P-glycoprotein efflux to achieve sufficient brain biodistribution (Dimension 1).
- Selectivity Challenge: The ring scaffold and substituents must confer selectivity for MAGL over other serine hydrolases (e.g., FAAH) in the brain to avoid off-target effects (Dimension 2). Successful inhibitors use ring constraints to pre-shape the molecule for MAGL's active site.
- Translational Validation: Proximal effects (Dim 3, e.g., increased brain 2-AG levels) and biological effects (Dim 4, e.g., reduced neuroinflammatory markers) must be linked in animal models to ultimately predict disease effect (Dim 5, e.g., improved cognitive function) [100].
Diagram 1: The Five-Star Matrix for Translational Drug Discovery. A framework linking compound properties (Dimensions) across experimental models (Systems) to establish target engagement and selectivity [100].
Infectious Diseases: Biomimetic Synthesis of Complex Anti-Parasitic Agents
Case Study: Artemisinin-Inspired Ring Systems Artemisinin, a natural product containing a unique 1,2,4-trioxane ring within a sesquiterpene lactone framework, is a frontline antimalarial. Its mode of action involves iron-mediated cleavage of the endoperoxide bridge, generating cytotoxic radicals. Biomimetic synthesis strategies aim to recreate such complex, bioactive ring systems efficiently [101].
- Strategy: A biomimetic Diels-Alder reaction can be employed to construct the complex polycyclic core, mimicking proposed biosynthetic steps [101].
- Role of Ring System: The trioxane ring is the pharmacophore, but the surrounding fused ring scaffold is essential for proper lipophilicity, cellular uptake, and localization within the parasite. Simplifying the scaffold while retaining the trioxane and its essential 3D context is a major design challenge.
- Experimental Protocol: Biomimetic Polyene Cyclization [101]
- Objective: Stereoselective construction of polycyclic terpenoid cores.
- Reaction Setup: Dissolve a linear polyene precursor with a terminal initiating group (e.g., epoxide, alkene) in an inert solvent (e.g., DCM).
- Cyclization Trigger: Add a Lewis or Brønsted acid catalyst (e.g., SnCl₄, TFA) to trigger a concerted, stereospecific cyclization cascade.
- Quenching & Analysis: Quench the reaction, purify, and analyze products via NMR and MS. The product's stereochemistry is dictated by the chair-like transition states enforced by the ring-forming process, mimicking biosynthesis.
Emerging Frontiers and Methodologies
Ring System Ontology and Knowledge-Based Design
The systematic classification of ring systems into an ontology (e.g., using Open Biomedical Ontologies format) enables computational mining of ring-target relationships across vast compound databases [102]. This allows researchers to query which ring systems are associated with activity against a specific target family (e.g., GPCRs, kinases) and identify under-explored rings from natural product space for novel target engagement.
Leveraging Natural Product Complexity: Fragment Combination and Morphological Profiling
As demonstrated by PNP research, disconnecting NPs into fragment-sized components (e.g., quinuclidine from quinine, indole from tryptophan) and recombining them into novel arrangements generates chemotypes with diverse ring systems [5]. These libraries can be screened in unbiased phenotypic assays like cell painting, which uses high-content imaging to generate a "morphological fingerprint" for each compound. Rings that induce unique fingerprints are likely engaging novel targets or mechanisms, providing a powerful discovery engine for first-in-class therapies [5].
The Covalent Inhibitor Workflow: From Warhead Screening to In Vivo Validation
A modern covalent drug discovery workflow integrates ring system design with specific analytical tools [99].
- Electrophile Screening: Screen diverse, moderately reactive electrophilic rings (e.g., acrylamides, α,β-unsaturated carbonyls) against a target of interest using chemoproteomic techniques.
- Scaffold Linking/Lengthening: Attach the validated warhead to a selectivity-conferring ring scaffold that binds a proximal pocket.
- Mechanism & Engagement Verification: Use intact protein LC-MS to confirm the correct mechanism of action (mass shift) and quantify %TE in vitro.
- Translational PK/PD Modeling: Apply the iPK/PD model to in vivo %TE-over-time data to predict effective dosing, directly linking ring-based molecular design to therapeutic effect [99].
Diagram 2: Decision Tree for Covalent Inhibitor Development. An MS and modeling-guided workflow from target validation to candidate selection [99]. METE: Minimally Effective Target Engagement.
The Scientist's Toolkit: Essential Reagents and Methods
Table 3: Key Research Reagent Solutions for Ring-Centric Drug Discovery
| Tool/Reagent Category | Specific Example/Kit | Primary Function in Research | Application in Case Studies |
|---|---|---|---|
| Covalent Warhead Libraries | Diverse electrophile sets (acrylamides, sulfonyl fluorides, etc.) | Screening for reactive, targetable residues and initial hit finding. | Electrophile-first discovery of covalent KRAS inhibitors [99]. |
| Chemoproteomic Profiling Kits | Activity-Based Protein Profiling (ABPP) probes & platforms. | Identify and quantify engaged targets across the proteome; assess selectivity. | Defining off-target profiles of covalent MAGL inhibitors [99]. |
| Intact Protein MS Standards & Columns | Stable isotope-labeled protein standards; wide-pore LC columns. | Quantify drug-target conjugation (%TE) in complex mixtures. | Core assay for covalent inhibitor PK/PD [99]. |
| Cell Painting Assay Reagents | Multiplex fluorescent dye kits (for nuclei, ER, Golgi, etc.). | Generate unbiased morphological fingerprints for phenotypic screening. | Profiling bioactivity of novel PNP ring systems [5]. |
| Biomimetic Synthesis Catalysts | Chiral Lewis acids, enzyme mimics (e.g., porphyrin complexes). | Catalyze stereoselective cyclization and coupling reactions. | Synthesizing complex polycyclic cores of terpenoid NPs [101]. |
| Computational Fingerprinting Software | RDKit (for ECFP, MAP4), OpenBabel, proprietary pharmacophore tools. | Encode and compare ring system structures for similarity and prediction. | Analyzing NP ring system coverage by synthetic compounds [3] [98]. |
Ring systems are the indispensable architects of molecular recognition in drug discovery. Their strategic selection and manipulation govern the critical balance between potent target engagement and therapeutic selectivity. As this whitepaper illustrates through cross-therapeutic case studies, the future of ring-centric design lies in the sophisticated integration of complementary approaches: mining the unparalleled structural diversity of natural products, applying rigorous translational frameworks like the Five-Star Matrix to validate engagement, and harnessing cutting-edge synthetic and analytical methodologies to forge and assess novel rings. By continuing to decipher and innovate upon these molecular frameworks, researchers can systematically bridge the gap between complex natural product inspiration and the creation of selective, clinically effective medicines.
In natural products research and drug discovery, ring systems are foundational architectural elements that define molecular shape, govern three-dimensional topology, and critically influence biological activity [63]. The intrinsic rigidity and conformational constraints imposed by cyclic structures position substituents in precise orientations for optimal target engagement, while also modulating key pharmacokinetic properties. Consequently, the strategic selection of ring systems is a pivotal decision in molecular design, with direct implications for a compound's success in translational pipelines.
Contemporary research reveals that nature's chemical repertoire is far from fully explored. Recent genome mining studies have uncovered widespread biosynthetic gene clusters for novel bicyclic systems, such as the benzoxazolinate-containing benzobactins, indicating a vast, untapped reservoir of bioactive ring structures [103]. Simultaneously, advances in synthetic chemistry, particularly multicomponent reactions (MCRs), are rapidly expanding accessible chemical space by efficiently constructing complex, medicinally relevant heterocycles from simple building blocks [104]. The convergence of these trends—driven by biosynthesis elucidation, synthetic innovation, and computational prediction—presents both an opportunity and a challenge for researchers. The core challenge lies in developing a rational, future-proofed framework for selecting ring systems that not only exhibit desirable initial activity but also possess the inherent flexibility and robustness to succeed through the entire translational pathway, from hit identification to clinical candidate.
This guide establishes three core, interdependent criteria for evaluating the translational potential of ring systems: Synthetic Accessibility & Modularity, Biological Compatibility & Target Engagement, and Novelty & Computational Predictability. By integrating principles from cheminformatics, synthetic biology, and machine learning, this framework aims to equip researchers with a systematic methodology for making strategic decisions in molecular design.
Core Criterion I: Synthetic Accessibility & Modularity
The most biologically promising ring system has no translational value if it cannot be synthesized or modified efficiently. This criterion assesses the feasibility of constructing and diversifying the core scaffold.
Quantitative Analysis of Ring System Prevalence and Saturation
Cheminformatic analysis of drug discovery literature reveals distinct patterns in ring system utilization. The following table summarizes the frequency of common ring systems and their associated Bemis-Murcko scaffolds [63].
Table 1: Prevalence of Selected Ring Systems and Scaffolds in Medicinal Chemistry Literature
| Ring System | Frequency in Compounds (Representative % Range) | Common Bemis-Murcko Scaffold | Translational Implication |
|---|---|---|---|
| Benzene | Very High (25-40%) | Phenyl | High familiarity, but low novelty and potential for IP. Excellent synthetic accessibility. |
| Piperidine | High (10-15%) | Piperidinyl, Decahydroquinoline | High 3D character, prevalent in CNS drugs. Well-understood synthetic routes. |
| Pyridine | High (10-15%) | Pyridinyl, Quinoline | Common hydrogen bond acceptor. Robust and modular synthesis. |
| Imidazole | Moderate (5-10%) | Imidazolyl, Benzimidazole | Versatile in metal binding and H-bonding. Readily functionalized. |
| 1,2,5,6-Tetrahydropyridine (THP) | Low (<2%) | Poly-substituted THP [104] | High novelty and 3D complexity. Accessible via modern MCRs, offering new vector space. |
Interpretation: Over-reliance on "flat" aromatic systems (e.g., benzene, pyridine) may limit exploration of three-dimensional chemical space. Emerging, less-prevalent systems like polysubstituted THPs offer greater shape diversity and intellectual property (IP) potential but require evaluation of their synthetic tractability [104].
Enabling Technologies: Multicomponent Reactions (MCRs) and Biosynthetic Engineering
Modern synthetic and biosynthetic methods directly address the accessibility and modularity of complex rings.
Table 2: Modern Methods for Accessing Complex Ring Systems
| Method | Key Description | Example Ring Formed | Advantages for Translational Design |
|---|---|---|---|
| Copper-Catalyzed MCR [104] | Cascade radical cyclization of allene, alkene, and TMSCN. | Polysubstituted 1,2,5,6-Tetrahydropyridine (THP) | Single-pot, atom-economical. Introduces versatile nitrile handle for further diversification. |
| Double Mannich MCR [104] | Utilizes unactivated C–H bonds in benzofuran/indole with amine and formaldehyde. | Benzofuran/Indole-fused Piperidine | Builds complexity from simple blocks. Enables late-stage functionalization of drug-like cores. |
| Biosynthetic Pathway Refactoring [103] | Heterologous expression of identified Biosynthetic Gene Clusters (BGCs). | Benzobactin-type Bicyclic Systems | Direct access to natural product scaffolds. Potential for pathway engineering to generate analogues. |
Detailed Experimental Protocol: Gram-Scale Synthesis of a Tetrahydropyridine via MCR
The following protocol is adapted from the copper-catalyzed synthesis of polysubstituted THPs [104].
Objective: To synthesize a model C5-aryl-substituted 1,2,5,6-tetrahydropyridine (e.g., compound 4b [104]) on a gram scale, demonstrating practical synthetic accessibility.
Materials:
- Reagents: F-masked benzene-sulfonamide allene (1), styrene derivative (2), trimethylsilyl cyanide (TMSCN, 3), tetrakis(acetonitrile)copper(I) hexafluorophosphate ([Cu(CH3CN)₄]PF₆), chiral bisoxazoline (BOX) ligand, fluorobenzene (anhydrous), magnesium sulfate (MgSO₄), silica gel for flash chromatography.
- Equipment: Schlenk line for inert atmosphere, round-bottom flasks, magnetic stirrer, heating bath, TLC plates, UV lamp, flash chromatography system, NMR spectrometer, LC-MS.
Procedure:
- Reaction Setup: In a flame-dried Schlenk tube under a nitrogen atmosphere, charge [Cu(CH3CN)₄]PF₆ (0.05 mmol, 5 mol%) and the BOX ligand (0.055 mmol, 5.5 mol%). Add anhydrous fluorobenzene (2 mL) and stir at 25°C for 15 minutes to form the active catalyst complex.
- Substrate Addition: Sequentially add the allene 1 (1.0 mmol), alkene 2 (1.2 mmol), and finally TMSCN 3 (2.0 mmol) via syringe.
- Reaction Execution: Seal the tube and heat the reaction mixture to 60°C with vigorous stirring. Monitor reaction progress by thin-layer chromatography (TLC) every 3 hours. The reaction is typically complete within 12-24 hours.
- Work-up: Cool the mixture to room temperature. Dilute with ethyl acetate (10 mL) and wash with saturated aqueous ammonium chloride solution (10 mL). Separate the organic layer, dry over anhydrous MgSO₄, filter, and concentrate under reduced pressure.
- Purification & Analysis: Purify the crude residue by flash chromatography on silica gel (eluent: hexane/ethyl acetate gradient). Analyze the isolated product by ¹H/¹³C NMR and LC-MS to confirm identity and purity (>95%). This protocol can be linearly scaled to a 10 mmol (gram-scale) reaction with proportional adjustments to solvent and catalyst amounts.
Significance: This protocol exemplifies a convergent, single-pot strategy to rapidly generate a complex, three-dimensional ring scaffold with multiple points for diversification (the C3-sulfonamide, C5-aryl, and C6-cyano groups), directly addressing the need for modularity in library synthesis.
Core Criterion II: Biological Compatibility & Target Engagement
A synthetically accessible ring must be compatible with the biological milieu and capable of specific, potent target modulation.
Functional Group Display and Physicochemical Consequences
The ring system serves as a rigid scaffold to present functional groups. The nature of the ring (aromatic, saturated, heterocyclic) determines the physicochemical profile.
- Aromatic/Heteroaromatic Rings: Provide planar rigidity for π-stacking or surface complementarity. Often increase molecular weight and reduce solubility.
- Saturated/Aliphatic Rings (e.g., Piperidines, THPs): Introduce three-dimensionality and sp³-hybridized carbons, which are correlated with improved clinical success rates. They effectively modulate lipophilicity (cLogP) and can serve as hydrogen bond donors/acceptors (e.g., piperidine N) [104].
- Bicyclic/Fused Systems (e.g., Benzobactins): Constrain conformation to a high degree, presenting functional groups in a precise, pre-organized geometry. This can lead to exceptionally high binding affinity and selectivity, as seen in the potent cytotoxicity of the benzoxazolinate-containing lidamycin [103].
Assessment via Translational Activity Profiling
Biological compatibility extends beyond simple binding to include efficient engagement of the cellular machinery, such as the ribosome for mRNA therapeutics. The DART (Direct Analysis of Ribosome Targeting) assay provides a quantitative high-throughput method to assess this [105].
Experimental Protocol: DART Assay for 5'-UTR-Dependent Translation Initiation Objective: To quantify how effectively different 5' untranslated region (UTR) sequences, which can be derived from or associated with specific biological pathways relevant to the ring system's mechanism, drive translation initiation in a human cytoplasmic extract [105].
Materials:
- Biological Reagent: HeLa cell cytoplasmic extract (commercially available).
- Constructs: DNA template library encoding a luciferase reporter with a variable 5' UTR region of interest (e.g., one containing regulatory elements responsive to cellular stress pathways implicated in a ring system's activity).
- Key Chemicals: N1-methylpseudouridine (m1Ψ) triphosphate for modified RNA transcription, cycloheximide, cap analog, RNase inhibitors.
Procedure:
- Library Preparation: Generate a DNA library via pooled oligonucleotide synthesis, containing the T7 promoter, the variable 5' UTR, a constant firefly luciferase coding sequence, and a unique molecular identifier (UMI).
- In Vitro Transcription: Transcribe the RNA library using T7 RNA polymerase, incorporating either unmodified nucleotides or the modified m1Ψ.
- DART Reaction: Incubate the capped RNA library (5 nM) with HeLa cytoplasmic extract, cycloheximide (to trap initiated ribosomes), GTP, and an energy regeneration system at 30°C for 15 minutes.
- Isolation & Sequencing: Purify the RNA fragments protected by the assembled 80S ribosomes. Convert these to cDNA and amplify for high-throughput sequencing.
- Data Analysis: The translation initiation efficiency for each 5' UTR is proportional to the count of its associated UMIs in the sequenced ribosomal protection fragments. Sequences driving high ribosomal recruitment are identified as translationally optimal.
Significance for Ring Selection: This functional assay moves beyond static binding measurements. For a ring system intended to modulate protein production (e.g., via stabilizing an mRNA), pairing it with a DART-optimized 5' UTR could dramatically enhance its therapeutic efficacy, showcasing a systems-level approach to biological compatibility [105].
Diagram 1: A Framework for Selecting Ring Systems with High Translational Potential. This decision-support workflow integrates three core criteria to translate design inputs into a strategic development plan [106] [103] [63].
Core Criterion III: Novelty & Computational Predictability
Future-proofing requires balancing novelty against reliable property prediction. Underexplored rings offer IP advantages but carry higher prediction uncertainty.
Discovery and Characterization of Novel Ring Systems
Genome mining has proven powerful for discovering entirely new ring systems. The study of benzobactins is paradigmatic [103].
Experimental Protocol: Genome Mining for Bicyclic Natural Product Pathways Objective: To identify biosynthetic gene clusters (BGCs) responsible for novel bicyclic ring systems (benzoxazolinate) and characterize the key cyclization enzyme [103].
Materials:
- Bacterial genomic DNA, PCR reagents, primers designed from conserved enzyme domains, heterologous expression host (e.g., E. coli or Streptomyces), HPLC-HRMS, NMR.
Procedure:
- Gene Cluster Identification: Using the sequence of a known key enzyme (e.g., the benzoxazolinate cyclase), perform BLAST searches against public and in-house microbial genome databases.
- Cluster Delineation: Analyze flanking genes of identified homologs to define the boundaries of putative BGCs using bioinformatics tools (e.g., antiSMASH).
- Heterologous Expression: Clone the entire putative BGC into a suitable expression vector and transfer it into a heterologous host that does not produce the compound natively.
- Metabolite Analysis: Culture the engineered strain and analyze the metabolic extract by HPLC-HRMS, comparing it to the wild-type producer strain and non-engineered control to identify new benzobactin-like compounds.
- Enzyme Characterization: Express, purify, and perform in vitro kinetics assays with the putative cyclase enzyme and its predicted substrate to confirm its function.
Machine Learning for Property Prediction and Inverse Design
When experimental data on novel rings is scarce, machine learning (ML) models trained on coarse-grained molecular representations can predict properties and generate new, valid structures.
Experimental Protocol: Attention-Based Coarse-Grained Graph Autoencoder for Ring-Containing Molecules Objective: To train a data-efficient ML model that learns a latent representation of molecules based on functional-group graphs, enabling property prediction and de novo generation of novel ring-containing candidates [106].
Materials:
- Software: RDKit, PyTorch or TensorFlow, specialized libraries for graph neural networks (GNNs).
- Data: Dataset of molecules (e.g., 6,000 monomers) represented as SMILES strings, with property labels for a subset (e.g., 600 with glass transition temperature, Tg).
Procedure:
- Molecular Representation: Convert each molecule into a coarse-grained graph. Nodes represent functional groups (e.g., phenyl, piperidine, carbonyl), and edges represent bonds between groups. A separate fine-grained atom graph exists within each functional group node.
- Encoder Training: Train a hierarchical graph encoder using a message-passing network. It first encodes atom-level graphs within each functional group, then aggregates this information to encode the coarse-grained graph into a low-dimensional latent vector (h).
- Attention Mechanism: Integrate a self-attention layer to learn the chemical context and long-range interactions between functional groups, crucial for understanding ring-system influences.
- Joint Training: Train the autoencoder (encoder + decoder for reconstruction) jointly with a property prediction head (e.g., for Tg) using the labeled data subset. This ensures the latent space is organized by both structure and properties.
- Inverse Design: To generate new molecules, sample latent vectors (h) from regions of space correlated with desired properties (e.g., high Tg). The decoder network translates these vectors back into coarse-grained graphs, which are then resolved into full atomic structures using RDKit rules.
Significance: This approach generates synthetically accessible, novel ring systems de novo by assembling known functional groups in new ways, all while targeting specific, translationally relevant properties [106].
Diagram 2: ML-Guided De Novo Design of Novel Ring Systems. An attention-based graph autoencoder learns a chemically meaningful latent space from coarse-grained molecular representations, enabling property-targeted generation of new structures [106].
Integration: A Translational Workflow from Selection to Development
The three core criteria are interdependent and should be evaluated iteratively within a structured workflow.
Step 1: Computational Triage & Novelty Filter. Start with a virtual library of ring systems derived from MCR chemistry [104] or genome mining predictions [103]. Filter using computational models (e.g., ML property predictors [106]) for drug-like properties and predicted synthetic accessibility (SA Score). Prioritize scaffolds with high 3D character and low literature prevalence [63].
Step 2: Synthesis & Modular Diversification. For top candidates, establish a robust synthetic route (e.g., an MCR protocol) enabling the rapid production of a pilot library (50-100 compounds) with systematic variation at multiple vectors. This tests modularity empirically.
Step 3: Functional Profiling in Translationally Relevant Assays. Screen the library not only in primary target assays but also in secondary pharmacological and mechanistic assays (e.g., DART assay for candidates affecting translation [105]). This builds a multidimensional biological compatibility profile.
Step 4: Data-Driven Iteration and Selection. Feed the experimental data (yields, properties, bioactivity) back into the computational models to refine predictions. Select the lead ring system that demonstrates the optimal balance of synthetic tractability, desired biological activity, and novelty.
Diagram 3: Biosynthetic Pathway for a Novel Bicyclic Ring System. Deciphering the enzymatic steps, particularly the final cyclization, enables the discovery, production, and future engineering of potent bioactive scaffolds like benzobactins [103].
The following table details key research reagents and tools essential for implementing the described selection framework.
Table 3: Research Reagent Solutions for Ring System Evaluation
| Reagent/Tool | Source/Example | Primary Function in Translational Design |
|---|---|---|
| [Cu(CH3CN)₄]PF₆ & BOX Ligand | Copper catalyst system for THP MCR [104]. | Enables synthesis of novel, 3D poly-substituted tetrahydropyridine cores. |
| F-masked Sulfonamide Allene | Key building block for THP MCR [104]. | Provides a versatile handle for introducing diverse substituents at the ring C3 position. |
| Benzoxazolinate Cyclase | Enzyme identified from benzobactin BGC [103]. | Probe for genome mining to discover new BGCs; biotransformation tool for analog synthesis. |
| HeLa Cytoplasmic Extract | Commercial system (e.g., Ipracell) [105]. | Essential for in vitro translation initiation assays (DART) to assess biological compatibility of target pathways. |
| N1-methylpseudouridine (m1Ψ) | Modified nucleotide triphosphate [105]. | For testing therapeutic mRNA components; enhances translation and reduces immunogenicity. |
| RDKit Software | Open-source cheminformatics toolkit [106]. | Handles molecular representation (SMILES, graphs), fingerprint generation, and basic property calculations. |
| Graph Neural Network (GNN) Library | PyTorch Geometric or Deep Graph Library [106]. | Implements the encoder/decoder for the coarse-grained graph autoencoder model. |
Conclusion
The exploration of ring systems in natural products reveals a central tenet of medicinal chemistry: immense structural inspiration from nature coexists with a pragmatic reliance on proven, synthetically accessible frameworks in drug development. The convergence of innovative synthetic methodologies—like skeletal editing and biomimetic synthesis—with powerful AI-driven computational tools is progressively bridging this gap, enabling more efficient exploration of natural product-like chemical space. Moving forward, the field must leverage quantitative complexity indices and predictive models to intelligently navigate the trade-offs between novelty, synthetic feasibility, and optimal drug-like properties. The future of drug discovery will be shaped by a more deliberate and informed exploitation of natural product ring architectures, not merely as static templates, but as dynamic platforms for engineered innovation, ultimately leading to novel therapeutic agents with improved efficacy and safety profiles.
References
- 1. Ring systems in medicinal chemistry: A cheminformatics ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425009432]
- 2. Unifying principles for the design and evaluation of natural ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08017c]
- 3. Ring systems in natural products: structural diversity, ... [https://www.sciencedirect.com/org/science/article/pii/S0265056823000600]
- 4. Natural product-inspired strategies towards the discovery of ... [https://fjps.springeropen.com/articles/10.1186/s43094-024-00627-z]
- 5. Natural product fragment combination to performance ... [https://www.nature.com/articles/s41467-021-22174-4]
- 6. A general strategy for diversifying complex natural products ... [https://www.nature.com/articles/s41467-019-11976-2]
- 7. Identification of a Privileged Scaffold for Inhibition of Sterol ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11758220/]
- 8. AgreementPred: a cheminformatic framework for drug and ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00329f]
- 9. Ring Size as an Independent Variable in Cyclooligomeric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8667307/]
- 10. Ring-Size Effect on Molecular Assembly Structures and ... [https://pubmed.ncbi.nlm.nih.gov/40764025/]
- 11. 4.5: Fused Ring Systems [https://chem.libretexts.org/Courses/Providence_College/Organic_Chemistry_I/04%3A_Rings/4.05%3A_4.5_Fused_Ring_Systems]
- 12. Fused Rings: Cis and Trans Decalin [https://www.masterorganicchemistry.com/2014/08/05/fused-rings/]
- 13. Heterocyclic Compounds [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/heterocy.htm]
- 14. Heterocyclic compound [https://en.wikipedia.org/wiki/Heterocyclic_compound]
- 15. Five- and six-membered rings with two or more heteroatoms [https://www.britannica.com/science/heterocyclic-compound/Five-and-six-membered-rings-with-two-or-more-heteroatoms]
- 16. The topology of molecular representations and its influence on ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01045-w]
- 17. Mining Ring Systems in Molecules for Fun and Profit [http://practicalcheminformatics.blogspot.com/2022/12/identifying-ring-systems-in-molecules.html]
- 18. The roles of fused-cyclopropanes in medicinal chemistry [https://www.sciencedirect.com/science/article/abs/pii/S0223523425007160]
- 19. Attractive natural products with strained cyclopropane and/or cyclobutane ring systems | Science China Chemistry [https://link.springer.com/article/10.1007/s11426-016-0233-1]
- 20. Total synthesis of cyclopropane-containing natural products: recent progress (2016–2024) - Organic Chemistry Frontiers (RSC Publishing) [https://pubs.rsc.org/en/content/articlelanding/2025/qo/d4qo02316a/unauth]
- 21. Cyclopropanation [https://grokipedia.com/page/Cyclopropanation]
- 22. Unveiling the beauty of cyclopropane formation [https://pubs.rsc.org/en/content/articlehtml/2024/qo/d4qo00535j]
- 23. New, simple and accessible method creates potency ... [https://science.psu.edu/news/Giri8-2023]
- 24. A Diverse Library of Chiral Cyclopropane Scaffolds via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8865612/]
- 25. The Chemical Logic of Plant Natural Product Biosynthesis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6863165/]
- 26. Construction and Diversification of Natural Product ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12645565/]
- 27. Synthetic biological approaches to natural product ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166911007592]
- 28. computational methods for biosynthetic pathway design [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159830/]
- 29. Fragment libraries from large synthetic compounds and ... [https://chemrxiv.org/engage/chemrxiv/article-details/67a3b0db81d2151a024b2e66]
- 30. On the biologically relevant chemical space: BioReCS [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2025.1674289/full]
- 31. Chemical space visual navigation in the era of deep ... [https://www.sciencedirect.com/science/article/pii/S1359644625001059]
- 32. Fragment Libraries from Large and Novel Synthetic ... [https://pubmed.ncbi.nlm.nih.gov/40321506/]
- 33. Sculpting molecules in text-3D space: a flexible substructure ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06072-w]
- 34. Exploration of crystal chemical space using text-guided ... [https://www.nature.com/articles/s41467-025-59636-y]
- 35. Strategies and Advances in the Biomimetic Synthesis of ... [https://www.sciencedirect.com/science/article/pii/S2095809924007306]
- 36. Medium-Sized Ring Expansion Strategies [https://pmc.ncbi.nlm.nih.gov/articles/PMC11013129/]
- 37. Biomimetic Synthesis of Natural Products [https://www.newswise.com/articles/biomimetic-synthesis-of-natural-products-progress-challenges-and-prospects]
- 38. Biomimetic synthesis - Latest research and news [https://www.nature.com/subjects/biomimetic-synthesis]
- 39. Recent advances in carbon atom addition for ring- ... [https://pubs.rsc.org/en/content/articlehtml/2024/qo/d4qo01806k]
- 40. recent advances in skeletal editing of (hetero)cycles [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d4qo02157f]
- 41. Review Skeletal editing of pyridine for ring transformations [https://www.sciencedirect.com/science/article/pii/S2666554925001309]
- 42. Recent ring distortion reactions for diversifying complex ... [https://pubmed.ncbi.nlm.nih.gov/35972343/]
- 43. Diversity-Generating Skeletal Editing Transformations [https://pubmed.ncbi.nlm.nih.gov/40042370/]
- 44. Medium-Sized Ring Expansion Strategies: Enhancing ... [https://www.mdpi.com/1420-3049/29/7/1562]
- 45. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 46. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 47. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 48. AI-Powered Molecular Innovation: Breakthroughs and ... [https://www.mantellassociates.com/ai-powered-molecular-innovation-breakthroughs-and-2025-growth/]
- 49. Multicomponent reactions (MCRs) yielding medicinally ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d4ra06681b]
- 50. Multicomponent reactions driving the discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11635293/]
- 51. Systematic, computational discovery of multicomponent ... [https://www.nature.com/articles/s41467-024-54611-5]
- 52. MULTICOMPONENT REACTIONS IN ALKALOID-BASED ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5130108/]
- 53. Classification of Scaffold Hopping Approaches - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3328312/]
- 54. Deep scaffold hopping with multimodal transformer neural networks - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8590293/]
- 55. GitHub - Shihang-Wang-58/papers_for_molecular_representation: Collection of papers for Molecular Representation using AI [https://github.com/Shihang-Wang-58/papers_for_molecular_representation]
- 56. opportunities for innovation in drug discovery - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641420/]
- 57. Ring systems in natural products: structural diversity ... [https://pubs.rsc.org/en/content/articlehtml/2022/np/d2np00001f]
- 58. Ring systems in natural products: structural diversity ... [https://pubs.rsc.org/en/content/articlelanding/2022/np/d2np00001f]
- 59. Research [https://www.mpi-dortmund.mpg.de/research-groups/waldmann/research]
- 60. NIMO: A Natural Product-Inspired Molecular Generative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11053988/]
- 61. Building Natural Product Libraries Using Quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8547436/]
- 62. Rings in Clinical Trials and Drugs: Present and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9289879/]
- 63. Ring Systems in Medicinal Chemistry: A Cheminformatics ... [https://chemrxiv.org/engage/chemrxiv/article-details/6891a60123be8e43d6d10ab0]
- 64. Current economic and regulatory challenges in developing ... [https://www.nature.com/articles/s44259-025-00123-1]
- 65. Innovative perspectives on the discovery of small molecule ... [https://www.nature.com/articles/s44259-025-00089-0]
- 66. 2025 AI in Drug Discovery: Predictions [https://lizard.bio/knowledge-hub/2025-ai-in-drug-discovery-predictions]
- 67. When Drug Programs Fail Before They Begin [https://communities.springernature.com/posts/when-drug-programs-fail-before-they-begin]
- 68. A Quantitative Ring Complexity Index for Profiling ... [https://chemrxiv.org/engage/chemrxiv/article-details/67eaba8281d2151a027245de]
- 69. QRCI: Quantitative Ring Complexity Index for Evaluating ... [https://chemrxiv.org/engage/chemrxiv/article-details/67e4e56781d2151a02e4d7fc]
- 70. Natural product drug discovery in the artificial intelligence era [https://pubs.rsc.org/en/content/articlehtml/2022/sc/d1sc04471k]
- 71. Artificial Intelligence in Natural Product Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC11874025/]
- 72. Medicinal Chemistry Strategies for the Modification of ... [https://www.mdpi.com/1420-3049/29/3/689]
- 73. Conformational dynamics and molecular interactions of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12332416/]
- 74. Lead Optimization in Drug Discovery | Danaher Life Sciences [https://lifesciences.danaher.com/us/en/library/lead-optimization-drug-discovery-guide.html]
- 75. The importance of 3-membered rings in drug discovery [https://www.sciencedirect.com/science/article/pii/S0968089624003948]
- 76. A graph-based computational approach for modeling ... [https://www.nature.com/articles/s41598-025-06624-3]
- 77. Optimization of Bottom‐Up PBPK Model Development in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12631063/]
- 78. Application of Physiologically-Based Pharmacokinetic ... [https://link.springer.com/article/10.1007/s40495-025-00427-w]
- 79. Advances in machine learning for optimizing ... [https://www.sciencedirect.com/science/article/pii/S1570164625000153]
- 80. Hepa-ToxMOA: a pathway-screening method for ... [https://www.nature.com/articles/s41598-024-54634-4]
- 81. Natural Products with Heteroatom-Rich Ring Systems [https://pubmed.ncbi.nlm.nih.gov/29135244/]
- 82. Prediction of reproductive and developmental toxicity using ... [https://www.nature.com/articles/s41598-025-02590-y]
- 83. AI-based toxicity prediction models using ToxCast data [https://www.sciencedirect.com/science/article/pii/S0300483X25001891]
- 84. Artificial Intelligence-Driven Drug Toxicity Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299075/]
- 85. Computational approaches to predict the toxicity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11811359/]
- 86. Ring systems in medicinal chemistry: A cheminformatics ... [https://pubmed.ncbi.nlm.nih.gov/40992171/]
- 87. Time-Dependent Comparison of the Structural Variations ... [https://www.mdpi.com/1422-0067/25/21/11475]
- 88. A Historical Overview of Natural Products in Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901206/]
- 89. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 90. Cheminformatic comparison of approved drugs from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4607632/]
- 91. AI Clinical Trials: Complete Guide for 2025 [https://nwai.co/ai-clinical-trials-complete-guide-for-2025/]
- 92. Five Predictions for Clinical Research in 2025 [https://www.muralhealth.com/blog/five-predictions-for-clinical-research-in-2025]
- 93. What Does the Future of Clinical Research Look Like in a ... [https://xtalks.com/what-does-the-future-of-clinical-research-look-like-in-a-data-driven-world-4407/]
- 94. Clinical trial trends in 2025: Investment headwinds ... [https://www.merative.com/blog/clinical-trial-trends-2025]
- 95. New Predictions for 2025 [https://globalforum.diaglobal.org/issue/january-2025/additional-predictions-for-2025/]
- 96. 2025 Clinical Data Trend Report [https://www.veeva.com/2025-clinical-data-trend-report/]
- 97. The Future of Clinical Data Management: Trends to Watch ... [https://www.companysconnects.com/post/the-future-of-clinical-data-management-trends-to-watch-in-2025]
- 98. Effectiveness of molecular fingerprints for exploring the ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00830-3]
- 99. Mass spectrometry methods and mathematical PK/PD ... [https://www.nature.com/articles/s41467-025-56985-6]
- 100. The “five-star matrix” for patient-centric drug discovery and ... [https://www.sciencedirect.com/science/article/abs/pii/S0896627325005896]
- 101. Strategies and Advances in the Biomimetic Synthesis of ... [https://www.engineering.org.cn/engi/EN/10.1016/j.eng.2024.12.013]
- 102. Statistics and Ontology of Published Small Molecule Ring ... [https://chemrxiv.org/engage/chemrxiv/article-details/686b8ad2c1cb1ecda081fe6b]
- 103. Revealing biochemical 'rings of power' | ScienceDaily [https://www.sciencedaily.com/releases/2022/11/221118095511.htm]
- 104. Multicomponent reactions (MCRs) yielding medicinally relevant rings: a recent update and chemical space analysis of the scaffolds - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11736855/]
- 105. Quantitative profiling of human translation initiation reveals ... [https://www.sciencedirect.com/science/article/abs/pii/S1097276524009559]
- 106. Attention-based functional-group coarse-graining [https://www.nature.com/articles/s41524-025-01836-7]