Unlocking Chemical Diversity: A Guide to Molecular Networking for Natural Product Scaffold Analysis
This article provides a comprehensive overview of molecular networking as a transformative bioinformatics tool for analyzing natural product scaffold diversity.
Unlocking Chemical Diversity: A Guide to Molecular Networking for Natural Product Scaffold Analysis
Abstract
This article provides a comprehensive overview of molecular networking as a transformative bioinformatics tool for analyzing natural product scaffold diversity. Aimed at researchers and drug development professionals, it details how molecular networking uses tandem mass spectrometry (MS/MS) data to visualize, cluster, and prioritize structurally related compounds. The content covers foundational principles and the central role of platforms like GNPS, explores advanced methodological workflows for scaffold-focused analysis, addresses common challenges and optimization strategies, and validates the approach by comparing its efficiency and outcomes against traditional discovery methods. The synthesis underscores molecular networking's critical role in accelerating drug discovery by efficiently mapping chemical space and minimizing the redundant rediscovery of known compounds [citation:1][citation:3][citation:7].
Demystifying Molecular Networks: Core Principles and the Scaffold Diversity Paradigm
Within the paradigm of natural product drug discovery, molecular networking has emerged as a cornerstone computational strategy for visualizing and interpreting complex metabolomic data [1]. This technique transforms tandem mass spectrometry (MS²) data into a structural similarity map, where molecular relationships are inferred from spectral patterns. The fundamental principle underpinning this approach is that similar MS² fragmentation spectra suggest shared structural features, enabling the grouping of unknown metabolites into chemically related families [2] [1].
This application note frames these concepts within a broader thesis on scaffold diversity analysis. The primary challenge in this field is moving beyond simple spectral matching to establish confident structural relationships that reveal core architectures. This document provides detailed protocols and analytical frameworks designed to empower researchers in translating spectral similarity into testable hypotheses about structural class and scaffold inheritance, thereby accelerating the discovery of novel bioactive chemotypes.
Core Principles and Quantitative Foundations
The translation from spectral data to structural hypotheses is governed by key statistical and cheminformatic principles. The following data, synthesized from large-scale analyses of natural product databases, quantifies the relationships that make this translation possible.
Table 1: Diagnostic Power of Molecular Formula Distributions for Compound Family Identification [2]
| Analysis Set | Total Unique Formulae | Formulae Unique to a Single Family | Diagnostic Power |
|---|---|---|---|
| Single Formulae | 4,317 | 1,554 (36.0%) | Low to Moderate |
| Pairs of Formulae | Not Specified | >95% of pairs | High |
| Triplets of Formulae | Not Specified | >97% of triplets | Very High |
Table 2: Performance of Chemical Fingerprinting Methods vs. Molecular Networking [2]
| Fingerprint Method (Radius) | Similarity Metric | Optimal Cutoff | True Positive Rate at 0.5% FPR | Alignment with MN |
|---|---|---|---|---|
| MACCS Keys | Dice | 0.94 | ~58% | Poor (Fragmented) |
| Morgan (2) | Dice | 0.71 | High (Optimal) | Excellent |
| Morgan (4) | Tanimoto/Dice | Not Specified | High | Excellent |
| Morgan (6) | Tanimoto/Dice | Not Specified | High | Excellent |
The data in Table 1 demonstrates that while a single molecular formula is a weak classifier, the co-occurrence of formula sets within a data cluster becomes exceptionally diagnostic for a specific compound family [2]. This forms the logical basis for tools like SNAP-MS, which annotates molecular families based on formula distributions without requiring reference spectra.
Furthermore, as shown in Table 2, the alignment between cheminformatic clustering (based on structural fingerprints) and spectral networking is method-dependent. Morgan fingerprints with a radius of 2 and Dice scoring provide the strongest correlation, validating the principle that spectral similarity networks can accurately mirror underlying structural relationships [2].
Detailed Experimental Protocols
Protocol: Constructing a Molecular Network from LC-MS/MS Data
This protocol details the creation of a molecular network using the Global Natural Products Social Molecular Networking (GNPS) platform or similar workflows [1].
Materials:
- LC-MS/MS system (e.g., Q-TOF, Orbitrap)
- Data conversion software (e.g., MSConvert, ProteoWizard)
- Computer with GNPS/Cyclone environment or MZmine 3
Procedure:
- Data Acquisition: Perform untargeted LC-MS/MS analyses on sample set. Use data-dependent acquisition (DDA) to fragment the top N most intense ions per cycle.
- File Conversion: Convert raw vendor files to open formats (.mzML, .mzXML).
- Feature Detection & Alignment (MZmine 3):
- Import files. Perform mass detection (noise level ~1.0E3).
- Chromatogram builder: Group scans with min time span 0.1 min, m/z tolerance 0.005 Da or 5 ppm.
- Chromatogram deconvolution (Local Minimum Search algorithm).
- Isotopic peak grouper.
- Join aligner: m/z tolerance 0.005 Da or 5 ppm, RT tolerance 0.1 min.
- Gap-filling (peak finder).
- Export feature list (.csv) and MS/MS spectra (.mgf).
- Molecular Networking (GNPS):
- Upload .mgf file to GNPS.
- Set creation parameters: Precursor ion mass tolerance (0.02 Da), Fragment ion tolerance (0.02 Da), Minimum cosine score for edges (e.g., 0.7), Minimum matched fragment ions (6).
- Set advanced parameters: Network TopK (10), Maximum connected component size (100).
- Submit job. Visualize results in Cytoscape.
Protocol: Compound Family Annotation with SNAP-MS
This protocol follows the Structural similarity Network Annotation Platform for Mass Spectrometry (SNAP-MS) workflow for annotating molecular networking clusters [2].
Materials:
- Molecular network cluster (list of m/z values for nodes)
- SNAP-MS web tool or standalone algorithm
- Reference database (e.g., Natural Products Atlas, COCONUT)
Procedure:
- Cluster Data Import: From your molecular network, select a connected subnetwork (cluster). Extract the precursor m/z values for all nodes in the cluster.
- Candidate Matching: For each m/z in the cluster (with a defined ppm tolerance, e.g., ±10 ppm), query the reference database to retrieve all possible molecular formula and structural candidates.
- Cheminformatic Clustering: Apply the Morgan fingerprinting (radius=2) and Dice similarity scoring algorithm to all candidate structures retrieved in step 2. Group candidates into provisional compound families using a Dice similarity cutoff of 0.71 [2].
- Scoring & Prioritization:
- For each provisional compound family, calculate the coverage (percentage of cluster m/z values for which this family proposed a candidate).
- Calculate a SNAP-MS score that integrates coverage and the internal consistency of the formula set against known family distributions in the database.
- The compound family with the highest score is proposed as the annotation for the entire molecular network cluster.
- Orthogonal Validation: Proposed annotations must be validated, ideally by co-injection with an authentic standard or by targeted isolation and NMR spectroscopy [2].
Visualizing the Workflow and Logical Relationships
Diagram: From Spectra to Scaffold Hypothesis
The following diagram illustrates the integrated computational-experimental workflow for deriving structural relationships from spectral data, culminating in scaffold analysis.
Diagram 1: Workflow from LC-MS/MS to scaffold hypothesis (63 characters)
Diagram: The Annotation Logic of SNAP-MS
This diagram details the core algorithm of SNAP-MS, explaining how molecular formula distributions are used to annotate spectral clusters.
Diagram 2: The SNAP-MS annotation algorithm logic (52 characters)
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for Molecular Networking Workflows
| Item | Function / Purpose | Key Considerations & Examples |
|---|---|---|
| LC-MS Grade Solvents | Mobile phase for chromatographic separation; extraction solvents. Essential for minimizing background noise and ion suppression. | Acetonitrile, Methanol, Water (with 0.1% Formic Acid for positive mode). |
| MS Calibration Solution | Ensures accurate mass measurement across the m/z range, critical for formula prediction. | Mixture of known compounds (e.g., sodium formate, ESI Tuning Mix) specific to instrument manufacturer. |
| Authentic Standards | Used for co-injection experiments to validate putative identifications from network annotations [2]. | Commercial or isolated pure compounds relevant to the compound family of interest. |
| Dereplication Database | Provides reference MS² spectra and structures for initial matching and preventing re-isolation of known compounds. | GNPS Libraries, Natural Products Atlas [2], MassBank, METLIN. |
| Structural Annotation Tool | Software or platform that assigns structural hypotheses to unknown features in the network. | SNAP-MS [2], Network Annotation Propagation (NAP), Sirius with CANOPUS. |
| Visualization Software | Enables interactive exploration of molecular networks and integration of metadata (e.g., bioactivity). | Cytoscape with GNPS plugin, MolNetEnhancer. |
| NMR Solvents | Required for the final orthogonal structural validation of isolated compounds [2]. | Deuterated Chloroform (CDCl₃), Deuterated Methanol (CD₃OD), DMSO-d₆. |
The quest for novel bioactive scaffolds from natural sources is a foundational pillar of drug discovery. The Global Natural Products Social Molecular Networking (GNPS) platform has emerged as an indispensable, cloud-based ecosystem that fundamentally transforms this endeavor [3]. By enabling the visualization and annotation of chemical space through tandem mass spectrometry (MS/MS) data, GNPS shifts the research paradigm from a molecule-by-molecule analysis to a systematic, scaffold-centric exploration [3] [2]. This approach directly addresses the core challenge of structural redundancy in natural product (NP) libraries, allowing researchers to map molecular families, prioritize unique chemotypes, and accelerate the discovery of novel bioactive compounds within the context of molecular networking for scaffold diversity analysis [4].
The GNPS Ecosystem: Core Components and Analytical Workflows
The GNPS infrastructure is a comprehensive suite of tools designed for the acquisition, analysis, and sharing of mass spectrometry data. Its core strength lies in connecting related molecules into molecular families based on the similarity of their MS/MS fragmentation patterns, visualizing them as networks where nodes represent consensus spectra and edges represent spectral similarities [5].
Table 1: Core GNPS Workflows for Natural Products Research
| Workflow | Key Principle | Primary Advantage | Ideal Use Case |
|---|---|---|---|
| Classical Molecular Networking [5] | Groups MS/MS spectra by direct pairwise spectral similarity. | Rapid visualization of chemical space; repository-scale meta-analysis. | Initial exploration of sample sets; large-scale dataset comparison. |
| Feature-Based Molecular Networking (FBMN) [3] | Uses chromatographic feature detection (m/z, RT, intensity) before networking. | Incorporates relative quantification, resolves isomers, reduces spectral redundancy. | Detailed analysis of single studies where quantification and isomeric resolution are critical. |
| Library Search | Matches experimental spectra against curated MS/MS spectral libraries. | Provides putative annotations for known compounds. | Dereplication and identification of known molecules within a sample. |
| SNAP-MS Annotation [2] | Annotates molecular families by matching formula distributions to structural databases. | De novo family annotation without need for reference spectra; identifies novel scaffold families. | Structural class prediction for uncharacterized molecular families in a network. |
Diagram Title: GNPS Ecosystem Core Workflows
Detailed Protocol: From Sample to Network-Guided Isolation
This protocol outlines the integrated workflow for GNPS-guided scaffold discovery, from sample preparation to the isolation of candidate compounds.
Sample Preparation & Fractionation
- Extraction: Prepare a crude methanol extract from the natural source material (e.g., plant tissue, microbial culture).
- Prefractionation: Partition the crude extract using solvent-solvent fractionation (e.g., between water and n-butanol) [6]. The n-butanol fraction often contains glycosylated secondary metabolites and is a common target for activity-guided studies.
- Fractionation: Subject the active or chemically rich fraction (e.g., n-BuOH) to further separation using medium-pressure liquid chromatography (MPLC) or vacuum liquid chromatography (VLC). Collect sequential fractions.
- LC-MS/MS Profiling: Analyze all fractions via high-resolution LC-MS/MS in data-dependent acquisition (DDA) mode. Convert raw data to open formats (.mzXML or .mzML) for GNPS analysis [5].
GNPS Molecular Networking Analysis
- Data Upload & Job Setup: Access the GNPS website (https://gnps.ucsd.edu), create a molecular networking job, and upload the converted .mzXML files [5].
- Parameter Selection: Use parameters appropriate for your instrument's mass accuracy [5].
- Precursor Ion Mass Tolerance: ±0.02 Da for high-resolution instruments (q-TOF, Orbitrap).
- Fragment Ion Mass Tolerance: ±0.02 Da for high-resolution instruments.
- Min Pairs Cos: 0.7 (minimum cosine score for spectral similarity).
- Minimum Matched Fragment Ions: 6.
- Job Submission & Monitoring: Submit the job and monitor its status on the GNPS dashboard. Processing time varies from minutes to hours based on dataset size [5].
Network Visualization & Target Prioritization
- Explore Results: Use the GNPS in-browser visualizer to examine the molecular network.
- Identify Clusters of Interest: Prioritize clusters (molecular families) that are:
- Large and Unexplored: Indicative of a prolific scaffold.
- Linked to Bioactivity: If metadata is provided, clusters where nodes correlate with active fractions.
- Poorly Annotated: Contain few library matches, suggesting novel chemistry.
- Examine Annotations: Review library matches within clusters for known compounds and use SNAP-MS to predict the structural family of unannotated clusters [2].
- Trace to Physical Fraction: Click on nodes of interest to identify the specific LC-MS file and retention time from which the spectrum originated, linking the spectral node back to the physical laboratory fraction.
Targeted Isolation & Structure Elucidation
- Scale-up & Purification: Based on the GNPS guidance, scale up the cultivation/extraction of the source material. Use targeted purification techniques (e.g., preparative HPLC) focused on the retention time window indicated by the network for the node of interest.
- Structure Elucidation: Purity the target compound to homogeneity. Elucidate its structure using spectroscopic methods, primarily Nuclear Magnetic Resonance (NMR) and comparison of MS/MS data with the GNPS library match or predicted scaffold [6].
- Bioassay Validation: Test the isolated compound in relevant biological assays to confirm the predicted or observed bioactivity linked to its molecular family.
Diagram Title: GNPS-Guided Isolation Workflow
Application in Scaffold Diversity Analysis: A Case Study and Protocol
A primary application of GNPS in thesis research is the rational minimization of natural product screening libraries to maximize scaffold diversity and increase bioassay hit rates [4].
Case Study: GNPS-Guided Discovery of Estrogenic Glycosides
A study on Ginkgo biloba fruits provides a exemplary protocol for scaffold discovery [6] [7]:
- Profiling: The n-BuOH fraction of a methanol extract was analyzed by LC-MS/MS, and data was processed on the GNPS platform.
- Network-Guided Targeting: Molecular networking revealed two major clusters: Cluster I (flavonoid glycosides like rutin) and Cluster II (phenolic glycosides). Nodes were annotated via library matching (e.g., syringin at m/z 373).
- Isolation & Validation: Guided by the network, 11 compounds were isolated. An E-screen assay identified syringin as the most potent estrogenic compound, increasing MCF-7 cell proliferation to 140.9 ± 6.5% at 100 µM. Its mechanism was confirmed via Western blot to involve ERα phosphorylation [6].
Table 2: Bioactive Compounds Identified from G. biloba via GNPS Guidance [6]
| Compound | Observed [M+H]+ (m/z) | GNPS-Driven Annotation | Estrogenic Activity (Cell Proliferation) |
|---|---|---|---|
| Syringin (2) | 373.273 | Library match to phenylpropanoid glycoside cluster | 140.9 ± 6.5% at 100 µM |
| 4-Hydroxybenzoic acid 4-O-glucoside (3) | 301.179 | Library match in phenolic glycoside cluster | Promoted proliferation |
| Vanillic acid 4-O-glucoside (4) | 331.207 | Inferred from cluster (Δ+30 Da from Cmpd 3) | Promoted proliferation |
| Rutin (10) | 611.161 | Library match in flavonoid glycoside cluster | Not active in this assay |
Protocol: Rational Library Minimization for Scaffold Diversity
This protocol uses GNPS to design a minimal screening library with maximal scaffold representation [4].
- Dataset Creation: Perform untargeted LC-MS/MS on all extracts in a large library (e.g., 1,439 fungal extracts).
- GNPS Analysis: Process data using the Classical Molecular Networking workflow. The resulting network groups MS/MS spectra into "scaffold" clusters based on fragmentation similarity.
- Scaffold Diversity Analysis: Use a custom algorithm (e.g., in R) to iteratively select the extract that adds the most new scaffold clusters not yet present in the growing "rational library."
- Library Construction: Stop selection when a target diversity coverage is reached (e.g., 80% or 100% of all detected scaffolds).
- Validation: Screen the rationally minimized library in bioassays. The hit rate is typically higher than that of the full, redundant library.
Table 3: Efficacy of GNPS-Driven Library Minimization [4]
| Metric | Full Library (1,439 extracts) | Rational Library (80% Diversity - 50 extracts) | Rational Library (100% Diversity - 216 extracts) |
|---|---|---|---|
| Scaffold Diversity | 100% (Baseline) | 80% target reached | 100% retained |
| Anti-P. falciparum Hit Rate | 11.26% | 22.00% | 15.74% |
| Anti-T. vaginalis Hit Rate | 7.64% | 18.00% | 12.50% |
| Bioactive Feature Retention | 10 correlated features | 8 retained (80%) | 10 retained (100%) |
Diagram Title: Scaffold Diversity Analysis Process
The Scientist's Toolkit: Essential Reagents & Materials
Table 4: Essential Research Reagent Solutions for GNPS-Guided Workflows
| Category / Item | Function & Role in GNPS Workflow |
|---|---|
| Sample Preparation | |
| HPLC-grade Solvents (MeOH, ACN, H₂O, BuOH) | Extraction, fractionation, and preparation of samples for LC-MS analysis. |
| Solid Phase Extraction (SPE) Cartridges (e.g., C18) | Desalting and pre-concentration of crude extracts prior to analysis. |
| Chromatography & MS | |
| LC-MS grade modifiers (Formic Acid, Ammonium Acetate) | Mobile phase additives to improve ionization and separation in LC-MS. |
| Reference Standard Mixtures | For instrument calibration and ensuring mass accuracy critical for networking. |
| Data Analysis | |
| Data Conversion Software (e.g., MSConvert) | Converts proprietary mass spectrometer files to open formats (mzML/mzXML) for GNPS. |
| Feature Detection Software (e.g., MZmine, MS-DIAL) | Required for Feature-Based Molecular Networking (FBMN) to detect and align LC-MS features [3]. |
| Structure Elucidation | |
| Deuterated NMR Solvents (e.g., DMSO-d₆, CD₃OD) | Solvent for NMR analysis to confirm structures of compounds isolated based on GNPS guidance. |
| Bioassay Validation | |
| Cell Lines & Assay Kits (e.g., MCF-7 for estrogenicity) | For functional validation of bioactivity predicted or observed for a molecular family [6]. |
| Pathway Inhibitors/Antagonists (e.g., ICI 182,780) | Used to confirm mechanism of action, as demonstrated in the Ginkgo case study [6]. |
This article provides a detailed overview of Molecular Networking (MN) types within the broader thesis research on leveraging molecular networking for the analysis of natural product scaffold diversity. The goal is to map chemical space systematically, prioritize novel scaffolds for drug discovery, and understand biosynthetic pathways. The evolution from Classical MN to feature-based and ion identity methods represents a paradigm shift in metabolomics, enabling more accurate and comprehensive analyses of complex natural product extracts.
Types of Molecular Networking: Principles and Applications
Classical Molecular Networking (GNPS)
Classical MN, pioneered by the Global Natural Products Social Molecular Networking (GNPS) platform, uses tandem mass spectrometry (MS/MS) data to organize molecules based on structural similarity. It forms the foundation for comparing unknown spectra against public libraries and visualizing chemical space.
Key Principle: Spectra are converted to consensus spectra, and cosine similarity scores between spectra are calculated. Pairs with scores above a threshold (e.g., >0.7) are connected to form a network.
Primary Application in Thesis Research: Initial dereplication and broad-stroke visualization of scaffold families within complex natural product datasets.
Feature-Based Molecular Networking (FBMN)
FBMN integrates LC-MS/MS data preprocessed by feature detection tools (e.g., MZmine, OpenMS) with GNPS. It uses chromatographic peak area and alignment, linking MS/MS spectra to chemical features defined by m/z and retention time (RT).
Key Advancement: Incorporates quantitative or semi-quantitative data (peak intensities) into the network, allowing for comparative analysis between samples (e.g., different treatments, tissues, or time points).
Primary Application in Thesis Research: Correlating scaffold abundance with biological or environmental variables, crucial for identifying differentially produced natural products and guiding isolation.
Ion Identity Molecular Networking (IIMN)
IIMN extends FBMN by explicitly accounting for different ion forms of the same molecule. It groups features corresponding to isotopes, adducts, multiply charged ions, and in-source fragments before network creation.
Key Advancement: Dramatically reduces node redundancy, leading to cleaner networks where each node more accurately represents a unique chemical entity.
Primary Application in Thesis Research: Producing a more accurate census of unique molecular scaffolds in a sample set, essential for precise diversity calculations and avoiding overcounting.
Other Advanced MN Types
- LC-MS/MS~2~ MN: Uses pseudo-MS/MS spectra generated from all-ion fragmentation (AIF) or data-independent acquisition (DIA) data, increasing MS/MS coverage.
- Network Annotation Propagation (NAP): A scoring system that propagates annotations from library matches to unknown nodes in a network, improving annotation rates.
- Qemistree: Encodes MS/MS spectra into "fingerprints" and uses the cosine distance to create a hierarchical tree (dendrogram), offering an alternative topology for chemical relationship analysis.
Quantitative Comparison of MN Types
Table 1: Comparative Analysis of Core Molecular Networking Types
| Parameter | Classical MN | Feature-Based MN (FBMN) | Ion Identity MN (IIMN) |
|---|---|---|---|
| Core Data Input | MS/MS file list (.mgf) | Feature quantification table (.csv) + aligned MS/MS (.mgf) | Feature table + MS/MS + ion identity relationships |
| Quantitative Data | No | Yes (peak area/intensity) | Yes (peak area/intensity) |
| Ion Deconvolution | No (performed post-networking) | Limited (post-networking) | Yes (pre-networking) |
| Node Redundancy | High | High | Low |
| Primary Use Case | Dereplication, library matching | Comparative metabolomics, biomarker discovery | Accurate unique compound census |
| Key Software/Tool | GNPS | MZmine/OpenMS -> GNPS | MSI-Linker -> MZmine -> GNPS |
| Best for Scaffold Diversity Analysis | Preliminary overview | Linking diversity to phenotypes | Definitive scaffold counting |
Detailed Experimental Protocols
Protocol 4.1: Classical MN Workflow via GNPS
Aim: Create a preliminary molecular network from crude extract MS/MS data.
- Sample Preparation: Prepare natural product extracts in appropriate solvent. Include blank controls.
- LC-MS/MS Analysis:
- Column: C18 reversed-phase (e.g., 2.1 x 150 mm, 1.9 µm).
- Gradient: Water (A) and Acetonitrile (B), both with 0.1% Formic acid. 5-95% B over 25 min.
- MS: Data-Dependent Acquisition (DDA) mode. Top 20 most intense ions per cycle. Dynamic exclusion: 15 sec.
- Data Conversion: Convert raw files (.raw, .d) to .mzML using MSConvert (ProteoWizard). Then, convert to .mgf format.
- GNPS Job Submission:
- Go to the GNPS website (gnps.ucsd.edu).
- Upload .mgf files.
- Critical Parameters: Precursor Ion Mass Tolerance: 0.02 Da; Fragment Ion Mass Tolerance: 0.02 Da; Min Matched Peaks: 6; Cosine Score Threshold: 0.7; Network TopK: 10.
- Submit job and retrieve network file (.graphml) and results.
Protocol 4.2: Integrated FBMN/IIMN Pipeline for Advanced Analysis
Aim: Perform a quantitative, ion-deconvoluted molecular network for precise scaffold diversity analysis.
- LC-MS/MS Analysis: Follow steps from Protocol 4.1. Ensure consistent chromatography.
- Feature Detection & Alignment with MZmine 3:
- Import Data: Load .mzML files.
- Mass Detection: Noise level (MS1: 1.0E3, MS2: 1.0E1).
- ADAP Chromatogram Builder: Min group size: 5; m/z tolerance: 0.005 Da or 10 ppm.
- Chromatogram Deconvolution (Local Minimum Search): Chromatographic threshold: 90%; Min peak duration: 0.1 min.
- Isotope & Adduct Grouping (MSI-Linker Module): Use default [M+H]+, [M+Na]+, [M+NH4]+, [M+K]+. Perform this step for IIMN.
- Join Alignment: m/z tolerance: 0.005 Da or 10 ppm; RT tolerance: 0.2 min.
- Gap Filling: Intensity tolerance: 20%; m/z tolerance: 0.005 Da.
- Export: Export feature quantification table (.csv) and aligned MS/MS spectra (.mgf).
- Feature-Based MN on GNPS:
- On GNPS, select "Feature-Based Molecular Networking."
- Upload the .csv and .mgf from MZmine.
- Set parameters similar to Classical MN, but enable "quant table" options.
- Downstream Analysis:
- Visualize networks in Cytoscape.
- Use the
chemotoolsCytoscape app to color nodes by fold-change between sample groups. - Calculate network properties (number of clusters, nodes per cluster) for diversity metrics.
Diagram Title: Advanced FBMN/IIMN Workflow for Natural Product Analysis
Diagram Title: Ion Identity MN Reduces Node Redundancy
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Molecular Networking Experiments
| Item Name | Supplier Examples | Function in Protocol |
|---|---|---|
| HPLC/MS Grade Solvents (Water, Acetonitrile, Methanol) | Fisher Chemical, Honeywell | Mobile phase components; minimize background noise and ion suppression. |
| Mass Spectrometry Grade Formic Acid (>99% purity) | Thermo Scientific, Fluka | Acid additive (0.1%) to mobile phases for positive ion mode ESI, promotes [M+H]+ ionization. |
| C18 Reversed-Phase UHPLC Columns (e.g., 2.1 x 150 mm, 1.7-1.9 µm) | Waters (ACQUITY), Thermo (Hypersil GOLD) | High-resolution chromatographic separation of complex natural product mixtures. |
| External Mass Calibration Solution | Agilent, Thermo Scientific | Ensures high mass accuracy (< 5 ppm) critical for ion identity grouping and annotation. |
| Internal Standard Mix (e.g., ESI-L Low Concentration Tuning Mix) | Agilent | Verifies instrument performance and stability across batches. |
| Solid Phase Extraction (SPE) Cartridges (C18, Diol) | Waters, Phenomenex | Clean-up and fractionation of crude extracts prior to LC-MS/MS to reduce complexity. |
| MZmine 3 / OpenMS Software | Open-source | Core software for feature detection, chromatographic alignment, and ion identity grouping. |
| Cytoscape with chemotools & GNPS Apps | Cytoscape Consortium | Network visualization, customization, and quantitative analysis (e.g., coloring by abundance). |
Why Scaffold Diversity? Linking Chemical Cores to Bioactive Potential
The systematic analysis of scaffold diversity—the variety of core molecular skeletons within a compound collection—is a fundamental pillar of modern drug discovery and natural products research. In the context of molecular networking for natural product analysis, scaffold diversity is not merely a descriptive metric but a critical predictor of bioactive potential and a guide for exploring uncharted chemical space. The structural core of a molecule dictates its three-dimensional shape and the presentation of functional groups, which in turn determines its interactions with biological macromolecules [8]. Consequently, the probability of identifying novel bioactive entities is intrinsically linked to the structural diversity of the core scaffolds screened [9].
Current analyses reveal a significant challenge: despite access to millions of compounds, known bioactive chemical space is dominated by a surprisingly small set of recurring scaffolds. A foundational study demonstrated that across major compound libraries, the majority of molecules are built around a limited number of well-represented scaffolds, while a long tail of "singleton" scaffolds appears only once [9]. This skew indicates a heavy bias in historical synthetic and screening efforts. For natural products, which are a primary source of drug leads, chemical diversity is also not random; it clusters around specific, privileged scaffolds that have been evolutionarily optimized [2]. This creates identifiable "activity islands" in chemical space, where families of structurally related compounds exhibit bioactivity [9].
The goal, therefore, is to intentionally diversify the scaffold content of screening libraries to increase coverage of biologically relevant chemical space. This is particularly urgent for engaging "undruggable" targets, such as protein-protein interactions, which often require structural features absent from traditional, flat compound libraries [8]. Enhancing scaffold diversity is a direct strategy to access new mechanisms of action and secure intellectual property positions for novel chemotypes. The integration of molecular networking with scaffold analysis forms a powerful feedback loop: networks group compounds by structural similarity, revealing core scaffolds, while scaffold analysis informs the strategic prioritization of novel molecular families within these networks for isolation and testing.
Molecular Networking as a Scaffold Discovery Engine: Protocols and Annotation
Molecular networking, based on tandem mass spectrometry (MS/MS), has revolutionized the ability to visualize and prioritize scaffold diversity directly from complex natural extracts. The core principle is that compounds sharing similar MS/MS fragmentation patterns are likely structurally related and belong to the same scaffold family [10]. This allows researchers to map the "chemical territory" of an extract and focus isolation efforts on nodes or clusters representing novel scaffolds.
Protocol: Classical Molecular Networking (CLMN) Workflow
The following protocol outlines the standard pipeline for generating and analyzing a molecular network using the Global Natural Product Social Molecular Networking (GNPS) platform [10].
1. Sample Preparation & Data Acquisition:
- Prepare crude natural extracts (e.g., microbial fermentation, plant material) using standardized extraction methods.
- Analyze samples via liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Use data-dependent acquisition (DDA) modes to fragment the most intense ions.
- Ensure consistent instrument parameters across all samples to enable valid spectral comparisons.
2. Data Processing and File Conversion:
- Convert raw MS data (.d, .raw files) to open formats (.mzML, .mzXML) using tools like MSConvert (ProteoWizard).
- Use MZmine 3 or similar software for advanced feature detection, chromatographic alignment, and gap filling. This step is crucial for creating a feature quantification table linked to MS/MS spectra.
3. Molecular Network Construction on GNPS:
- Upload the .mzML files and the feature quantification table (for Feature-Based MN) to the GNPS website (https://gnps.ucsd.edu).
- Set key parameters for network creation:
- Precursor ion mass tolerance: 0.02 Da.
- Fragment ion mass tolerance: 0.02 Da.
- Minimum cosine score for edges: 0.65-0.75 (adjust based on data quality and desired connectivity).
- Minimum matched fragment peaks: 6.
- Network TopK: 10 (limits connections per node to the 10 highest scores).
- Execute the job. GNPS will perform spectral clustering, calculate pairwise cosine similarity scores, and generate the network.
4. Network Visualization and Analysis:
- Visualize the network using Cytoscape (importing the GNPS output) or the built-in GNPS viewer.
- In the network, nodes represent LC-MS features (compounds), and edges connect nodes with similar spectra. Thicker edges indicate higher spectral similarity.
- Identify major clusters, which likely represent distinct scaffold families. Isolated nodes may represent unique chemotypes or singletons.
5. Dereplication and Annotation:
- Use the GNPS library search function to annotate nodes by matching experimental spectra to reference spectral libraries.
- Apply SNAP-MS annotation (see protocol below) for de novo compound family prediction based on molecular formula distributions [2].
- Prioritize for isolation: clusters with no library matches, clusters linked to a target bioactivity, or clusters containing molecular formulas indicative of novel scaffold classes.
Table 1: Key Parameters for Classical Molecular Networking on GNPS
| Parameter | Recommended Setting | Function |
|---|---|---|
| Precursor Mass Tolerance | 0.02 Da | Mass window for comparing parent ions. |
| Fragment Ion Tolerance | 0.02 Da | Mass window for matching fragment peaks. |
| Cosine Score Threshold | 0.65 - 0.75 | Minimum spectral similarity to create an edge. Higher values yield fewer, more confident connections. |
| Minimum Matched Peaks | 6 | Ensures edges are based on sufficient spectral evidence. |
| Network TopK | 10 | Limits edges per node to top 10 matches, simplifying the network. |
Protocol: SNAP-MS for De Novo Scaffold Family Annotation
Structural similarity Network Annotation Platform for Mass Spectrometry (SNAP-MS) is a specialized tool that annotates molecular networking clusters by matching their molecular formula patterns to known natural product families, without requiring reference MS/MS spectra [2].
1. Prerequisite: Molecular Formula Assignment:
- For each node in your molecular network, obtain a high-confidence molecular formula. This is typically derived from the high-resolution precursor mass (HR-MS1) and isotopic pattern analysis using software like MZmine.
2. Data Input to SNAP-MS:
- Input the list of molecular formulas from a specific network cluster into the SNAP-MS platform (available at www.npatlas.org/discover/snapms).
3. Candidate Matching and Clustering:
- SNAP-MS queries the Natural Products Atlas database to retrieve all known compounds sharing those formulas.
- It then clusters these candidate compounds using Morgan fingerprinting (radius 2) and Dice similarity scoring, a method proven to align closely with molecular networking groupings [2].
4. Scoring and Family Prediction:
- The platform scores each candidate compound family based on its coverage of the input formulas from the cluster.
- The top-scoring family is presented as the annotation. The underlying principle is that specific combinations of 2-3 molecular formulas are highly diagnostic for a single compound family (>95% specificity) [2].
5. Orthogonal Validation:
- Proposed annotations should be treated as hypotheses. Validate by:
- Co-injection with an authentic standard (if available).
- Targeted isolation of a cluster member and structure elucidation by NMR.
- Consulting associated metadata, such as the source organism, which may support the predicted compound class.
Diagram: SNAP-MS Workflow for Scaffold Family Annotation.
Computational and Synthetic Tools for Expanding Scaffold Diversity
Beyond analytical dereplication, advancing scaffold diversity requires tools to design and synthesize novel cores. This integrates computational prediction with innovative synthetic chemistry.
Computational Design and Analysis
- AI-Driven Scaffold Generation: Transformer-based Chemical Language Models (CLMs) can generate novel, synthetically accessible scaffolds by learning from known chemical structures. These models can be prompted with a core fragment, substituent, or combination to produce structurally diverse yet valid output compounds, effectively performing in silico scaffold hopping [11].
- Scaffold Tree Hierarchies: This algorithm deconstructs molecules hierarchically by iteratively removing rings according to rules of complexity. Analyzing a library using Scaffold Tree Level 1 (one ring less complex than the full Murcko framework) provides a more nuanced view of scaffold relationships and diversity than flat Murcko framework analysis [9].
- Topological and Shape Descriptors: Describing scaffolds by topological indices or three-dimensional shape descriptors allows mapping of scaffold space and identification of "voids"—regions that are synthetically accessible but underrepresented in screening libraries. Targeting these voids is a rational strategy for diversity enhancement [9].
Synthetic Methodology: Diversity-Oriented Synthesis (DOS)
Diversity-Oriented Synthesis (DOS) is a strategic synthetic approach designed to generate collections of compounds with high skeletal diversity from common starting materials. It contrasts with traditional combinatorial chemistry, which explores appendage diversity around a single scaffold [8].
- Principle: DOS employs branching reaction pathways, where intermediates can be diverted down different synthetic routes using distinct reagents or conditions. This yields final products that are not just analogs but contain fundamentally different molecular scaffolds.
- Key Strategies:
- Use of Pluripotent Functional Groups: Intermediates containing functional groups that can participate in multiple, divergent reaction pathways.
- Folding Pathways: Reactions that create different ring sizes and connectivities from a common linear precursor.
- Coupling of Multiple Building Blocks in a stereochemically diverse manner.
- Outcome: A small, efficiently synthesized library that occupies a broad swath of chemical shape space, maximizing the chance of interacting with diverse biological targets.
Table 2: Comparison of Library Synthesis Approaches
| Approach | Primary Goal | Diversity Type | Typical Scaffold Count | Advantage |
|---|---|---|---|---|
| Traditional Combinatorial | Optimize potency/Selectivity | Appendage (Side-chain) | Single scaffold | Efficient SAR development for a known target. |
| Focus Library Synthesis | Target a specific protein family | Functional Group & Appendage | Few related scaffolds | High hit rate for kinased, GPCRs, etc. |
| Diversity-Oriented Synthesis (DOS) | Discover novel bioactivity | Skeletal (Scaffold) & Stereochemical | Many distinct scaffolds | Broadest exploration of bioactive chemical space; ideal for phenotypic screening. |
The Scientist's Toolkit: Essential Reagents & Platforms
Successful scaffold diversity analysis relies on a combination of software platforms, analytical standards, and chemical resources.
- GNPS Platform: The central, open-access web platform for performing molecular networking, spectral library matching, and sharing data [10].
- Cytoscape: Open-source software for visualizing and analyzing complex molecular networks imported from GNPS [10].
- MZmine 3 / OpenMS: Software for processing raw LC-MS data, performing feature detection, alignment, and formula prediction before networking.
- Natural Products Atlas / COCONUT Databases: Comprehensive, curated databases of known natural product structures used for formula-based annotation via SNAP-MS [2].
- Synthetic Scaffold Libraries: Commercially available collections (e.g., Life Chemicals' 1580-scaffold library) provide tangible starting points for screening novel chemotypes [12].
- Building Blocks for DOS: Collections of pluripotent reagents and chiral starting materials essential for executing diversity-oriented synthesis campaigns [8].
The pursuit of scaffold diversity is a critical, multi-disciplinary endeavor to unlock new bioactive potential. Molecular networking provides the analytical framework to visualize and prioritize scaffold families directly from nature's complex mixtures. Coupled with de novo annotation tools like SNAP-MS, it accelerates the dereplication and identification process. This analytical insight must feed into the synthetic cycle, guided by computational design and powered by methodologies like DOS, to deliberately populate underrepresented regions of chemical space.
The future of scaffold-based discovery lies in closing this loop: using molecular networks to identify promising, novel scaffolds in natural sources, employing computational models to generate analog ideas, and applying DOS principles to synthesize targeted, diverse libraries around these cores for biological evaluation. This integrated approach maximizes the chances of discovering groundbreaking chemical probes and therapeutics, particularly for the most challenging biological targets.
Diagram: The Integrated Scaffold Discovery and Development Cycle.
From Data to Discovery: Methodological Workflows for Scaffold-Centric Analysis
Within the domain of natural product research for drug discovery, molecular networking has emerged as a pivotal strategy for organizing, analyzing, and extracting meaningful patterns from vast chemical inventories. This pipeline is central to a broader thesis focused on scaffold diversity analysis, aiming to map the structural universe of natural products (NPs) and identify privileged scaffolds with optimized bioactivity [13]. The process integrates multidisciplinary data—from genomics and metabolomics to chemical structures and bioassays—and transforms it into predictive networks [14]. These networks enable researchers to navigate chemical space systematically, prioritize novel scaffolds for synthesis, and infer biosynthetic pathways [15]. The core pipeline, comprising data acquisition, preprocessing, and network construction, represents a foundational workflow where data quality and methodological rigor directly determine the success of downstream diversity analysis and lead optimization [16].
Data Acquisition: Sourcing and Curating Multimodal Data
The acquisition phase focuses on building comprehensive, multimodal datasets from public repositories and primary literature. Effective data collection is the first critical step for robust scaffold diversity analysis.
Database Curation and Construction
Specialized NP databases are constructed through systematic literature mining and stringent curation protocols. A representative workflow, as demonstrated by the creation of Nat-UV DB, involves several defined steps [17]:
- Literature Search: Use keywords (e.g., "natural product," "NMR," geographic identifiers) to query scientific databases (PubMed, Google Scholar, institutional repositories).
- Criteria Filtering: Apply filters to include only compounds with structures fully elucidated by NMR spectroscopy and confirmed to originate from the specified geographic region.
- Structure Standardization: Generate isomeric SMILES strings preserving reported stereochemistry using tools like ChemBioDraw.
- Data Curation: Employ software modules (e.g., the 'Wash' module in Molecular Operating Environment - MOE) to remove salts, normalize protonation states, and deduplicate entries.
- Annotation Cross-Referencing: Manually cross-reference curated compounds against major databases like PubChem and ChEMBL to append reported biological activities.
Table 1: Representative Natural Product Databases for Scaffold Diversity Analysis
| Database Name | Size (Compounds) | Key Feature | Primary Use Case |
|---|---|---|---|
| Nat-UV DB [17] | 227 | First NP database from Veracruz, Mexico; includes 52 novel scaffolds. | Exploring region-specific biodiversity and scaffold novelty. |
| COCONUT 2.0 [17] | 400,000+ | Aggregates and standardizes multiple open-access NP databases. | Large-scale virtual screening and chemical space analysis. |
| BIOFACQUIM [17] | ~ | NP database from central Mexico. | Comparative scaffold diversity studies with regional NPs. |
| ChEMBL [17] | Millions | Bioactivity data for drug-like small molecules. | Annotating NPs with known targets and activities. |
Multi-Omics Data Integration
Beyond isolated chemical structures, modern pipelines integrate multi-omics data to contextualize scaffolds within their biosynthetic and functional framework [15]. This includes:
- Genomics/Transcriptomics: Data from biosynthetic gene clusters (BGCs) help link molecular scaffolds to their genetic origins and predict novel pathways [14].
- Metabolomics: Untargeted mass spectrometry (MS) data, particularly tandem MS (MS/MS) fragmentation patterns, are essential for feature-based molecular networking and analog identification [16].
- Bioactivity Data: High-throughput screening results link scaffolds to phenotypic outcomes or specific protein targets.
Data Preprocessing: From Raw Data to Computational Representations
Preprocessing transforms raw, heterogeneous data into standardized, machine-readable formats suitable for network construction and AI modeling. This stage addresses data imbalance and ensures chemical validity [18].
Molecular Representation and Featurization
The choice of molecular representation profoundly impacts the ability to analyze scaffold relationships and diversity [19].
- String-Based Representations (e.g., SMILES, SELFIES): Provide a compact, sequential format but may not fully capture complex stereochemistry or 3D interactions [19].
- Graph-Based Representations: Treat molecules as graphs with atoms as nodes and bonds as edges. This is a natural representation for Graph Neural Networks (GNNs) and preserves topological information critical for scaffold recognition [19] [18].
- Molecular Fingerprints (e.g., ECFP): Encode substructural information as fixed-length bit vectors, enabling rapid similarity calculations for initial clustering [19].
Protocol 1: Standardized Molecular Representation Generation Objective: Convert a curated list of NP structures into consistent graph and fingerprint representations for downstream analysis.
- Input: Curated list of canonical SMILES strings.
- Graph Construction:
- Use the RDKit or OEChem toolkit to parse each SMILES string.
- Generate a molecular graph object: atoms become nodes (featurized with atom type, degree, hybridization), bonds become edges (featurized with bond type, conjugation).
- Optionally, generate 3D conformers using ETKDG or MMFF94 force field minimization.
- Fingerprint Generation:
- Compute Extended-Connectivity Fingerprints (ECFP4, radius=2) for each molecule.
- Save fingerprints as bit vectors for similarity search and clustering.
- Output: A dataset containing aligned graph objects and fingerprint vectors for each molecule.
Addressing Data Imbalance and Augmentation
NP datasets often suffer from extreme class imbalance (few active compounds) and structural imbalance (overrepresentation of common scaffolds) [18]. Generative AI models, such as graph diffusion models, can create synthetic data to mitigate this.
- Scaffold-Aware Sampling (SAS): Identifies underrepresented scaffolds among active molecules and prioritizes them for augmentation [18].
- Scaffold-Conditioned Generation: A graph diffusion model is conditioned on a core scaffold to generate novel, synthetically accessible molecules that preserve essential binding features [18] [13].
Network Construction: Building Predictive and Analytical Frameworks
The constructed networks serve as the analytical engine for scaffold diversity exploration, connecting chemical structures to each other and to biological activity.
Molecular Similarity Networks
These networks are the foundation of chemical space visualization, where nodes are molecules and edges represent similarity (e.g., based on fingerprint Tanimoto coefficients) [14].
- Construction: A similarity threshold is applied to connect molecules. Community detection algorithms can then identify clusters of structurally similar compounds, which often share a common core scaffold.
Scaffold-Centric Networks for Virtual Screening
Advanced networks explicitly incorporate scaffold information to guide drug discovery.
- The ScaffAug Framework: This framework constructs a network to address virtual screening challenges [18].
- Augmentation Module: Builds a library of scaffolds from known active molecules and uses a Graph Diffusion Model to generate new molecules around them.
- Self-Training Module: Integrates generated molecules with pseudo-labels into the training data.
- Re-ranking Module: Applies Maximal Marginal Relevance (MMR) to diversify the scaffolds in the top-ranked predictions, balancing activity scores with scaffold novelty.
Integrative Knowledge Graphs
A Natural Product Science Knowledge Graph represents the pinnacle of integrative network construction, linking entities (nodes) of different types via relationships (edges) [14].
- Nodes: Chemical compounds, scaffolds, genes, proteins, pathways, organisms, diseases.
- Edges: "derivedfrom," "inhibits," "isencodedby," "hasactivity_against."
- Construction: Requires semantic data modeling (e.g., using Resource Description Framework - RDF) to unify heterogeneous data sources like ChEMBL, PubChem, and genomic databases [14]. Graph embedding techniques (e.g., TransE, node2vec) can then be applied to predict novel, missing links, such as inferring the bioactivity of an uncharacterized scaffold.
Table 2: Comparison of Network Types for Scaffold Analysis
| Network Type | Primary Nodes | Primary Edges | Key Analytical Goal |
|---|---|---|---|
| Molecular Similarity Network | Molecules | Similarity (e.g., Tc > 0.7) | Visualize chemical space; identify scaffold clusters. |
| Scaffold-Aware Prediction Network [18] | Molecules, Scaffolds | "containsscaffold", "generatedfrom" | Improve virtual screening hit rates and scaffold diversity. |
| Integrative Knowledge Graph [14] | Compounds, Genes, Targets, Diseases | "bindsto", "treats", "producedby" | Multi-hop reasoning for mechanism prediction and novel scaffold prioritization. |
Experimental Protocols for Key Analyses
Protocol 2: Implementing a Scaffold-Aware Augmentation for Imbalanced Data Objective: Generate synthetically valid molecules to balance scaffold representation in a dataset of active NPs.
- Scaffold Extraction: Use RDKit to perform Murcko scaffold decomposition on all active molecules. Cluster scaffolds using fingerprint similarity.
- Imbalance Quantification: Count molecules per scaffold cluster. Identify scaffolds with counts below a defined threshold (e.g., < 5% of total actives).
- Model Preparation: Fine-tune a pretrained graph diffusion model (e.g., DiGress) on your NP dataset.
- Conditional Generation: For each underrepresented scaffold, use the model to generate a set number of new molecules (e.g., 50) conditioned on that scaffold's core structure.
- Validation & Filtering: Filter generated molecules for synthetic accessibility (SA Score) and drug-likeness. Validate uniqueness against the original dataset.
Protocol 3: Constructing a Multi-Omics Knowledge Graph for Pathway-Scaffold Linking Objective: Build a knowledge graph linking plant genomic data to NP scaffolds for biosynthetic pathway prediction.
- Data Source Ingestion:
- Genomic Data: From plant tissue RNA-seq, annotate Biosynthetic Gene Clusters (BGCs) using antiSMASH.
- Metabolomic Data: From LC-MS/MS of the same tissue, annotate compounds using GNPS and derive scaffolds.
- Literature Data: Extract known gene-enzyme-compound relationships from databases using NLP tools.
- Node and Edge Creation:
- Create nodes for:
Gene,Enzyme,ChemicalReaction,BiosyntheticIntermediate,FinalScaffold. - Create edges with labels:
gene_encodes_enzyme,enzyme_catalyzes_reaction,reaction_has_input,reaction_has_output.
- Create nodes for:
- Graph Population & Storage: Use a graph database (e.g., Neo4j) to store nodes and edges. Import data via Cypher queries or CSV batch import.
- Pathway Inference Query: For an orphan scaffold, query the graph for co-expressed genes (from transcriptomic data) and chemically plausible intermediates to hypothesize a biosynthetic pathway.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions and Computational Tools
| Tool/Reagent Category | Specific Name/Example | Primary Function in Pipeline |
|---|---|---|
| Chemical Database & Curation | Molecular Operating Environment (MOE) [17], RDKit | Standardize structures, remove salts, calculate descriptors. |
| Molecular Representation | RDKit, OEChem, DeepChem | Generate SMILES, molecular graphs, fingerprints (ECFP). |
| Generative AI / Augmentation | DiGress (Graph Diffusion Model) [18], VAE, GAN | Synthesize novel molecules conditioned on specific scaffolds. |
| Network Analysis & Graph ML | NetworkX, PyTorch Geometric (PyG), Neo4j | Construct similarity networks, implement GNNs, manage knowledge graphs. |
| Multi-Omics Integration | antiSMASH (genomics), GNPS (metabolomics) [14] | Annotate biosynthetic gene clusters and mass spectrometry data. |
| Virtual Screening & Docking | AutoDock Vina, Pharmit [20] | Predict binding affinity of novel scaffold-based compounds to targets. |
| Scaffold Analysis | ScaffoldNetwork in RDKit, SCHAEPPI [18] | Decompose molecules into core scaffolds for diversity analysis. |
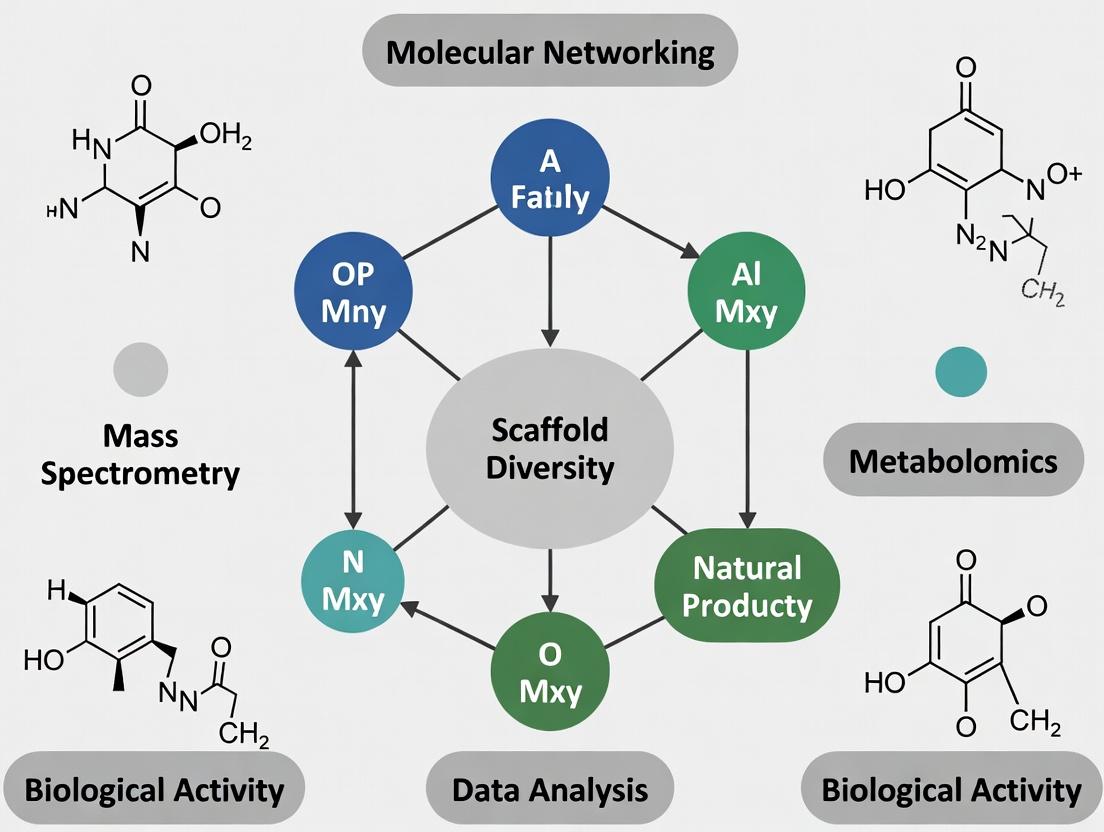
Molecular networking has emerged as a cornerstone computational strategy in natural product research and drug discovery, transforming complex tandem mass spectrometry (MS/MS) data into navigable maps of chemical space. At the heart of this approach is a foundational principle: compounds with similar chemical structures produce similar MS/MS fragmentation patterns [10]. By calculating the spectral similarity between all detected ions, molecular networking algorithms cluster related molecules together, visualizing these relationships as graphs where nodes represent individual MS/MS spectra (compounds) and edges represent significant spectral similarity between them [10].
Within the context of a broader thesis on molecular networking for natural product scaffold diversity analysis, this article provides detailed application notes and protocols. The primary objective is to enable researchers to decode these networks to identify scaffold families—groups of metabolites sharing a common core structure but differing in decorations like hydroxylations, methylations, or glycosylations. Interpreting clusters, nodes, and edges correctly accelerates the dereplication of known compounds and prioritizes novel or taxonomically unique scaffolds for isolation, directly addressing the critical bottleneck of rediscovery in natural product-based drug discovery [10].
Foundational Concepts: Network Components and Their Chemical Meaning
Interpreting a molecular network requires a precise understanding of its graphical elements and their correlation to chemical reality.
- Nodes: A single node corresponds to one consensus MS/MS spectrum, typically representing a specific ion (e.g., [M+H]⁺, [M+Na]⁺) of a compound detected in the sample. The size of a node is often proportional to its chromatographic peak area or ion intensity, serving as a visual cue for relative abundance [10].
- Edges: An edge connecting two nodes signifies that their MS/MS spectra are sufficiently similar, as determined by a modified cosine score or other similarity metric. The thickness of the edge is usually proportional to the similarity score, with thicker lines indicating higher spectral and, by inference, higher structural similarity [10].
- Clusters: A densely connected group of nodes forms a cluster or "molecular family." This is the most critical structural feature for scaffold analysis. Ideally, all members of a single cluster share the same core scaffold, with variations captured as connections within the cluster. A large, well-connected cluster often points to a biosynthetically prolific scaffold within the tested organism or set of samples [4] [10].
The following workflow diagram illustrates the standard process for generating a molecular network from raw MS data, leading to its biological interpretation.
Workflow for Molecular Network Construction and Analysis
Application Notes: Quantitative Insights for Network Interpretation
Effective interpretation is guided by quantitative benchmarks. The following tables summarize key performance data for network-based scaffold family identification and library enhancement.
Table 1: Diagnostic Power of Molecular Formula Sets for Compound Family Annotation [2]
| Formula Set Size | % Found in a SINGLE Compound Family | Key Interpretation for Networks |
|---|---|---|
| Single Formula | 36% | Low diagnostic power alone; high risk of misannotation. |
| Two Formulae | >95% | Highly diagnostic; a pair in a cluster strongly predicts a specific scaffold family. |
| Three Formulae | >97% | Extremely diagnostic; uniquely identifies a scaffold family with very high confidence. |
Table 2: Impact of Rational, Network-Based Library Reduction on Bioassay Efficiency [4] Performance comparison of a full library of 1,439 fungal extracts versus rationally reduced subsets.
| Activity Assay (Target) | Hit Rate: Full Library | Hit Rate: 80% Diversity Library (50 Extracts) | Bioactive Feature Retention (80% Lib.) |
|---|---|---|---|
| Plasmodium falciparum (phenotypic) | 11.26% | 22.00% (Increased 2x) | 8 out of 10 correlated features |
| Trichomonas vaginalis (phenotypic) | 7.64% | 18.00% (Increased 2.4x) | 5 out of 5 correlated features |
| Neuraminidase (enzyme) | 2.57% | 8.00% (Increased 3.1x) | 16 out of 17 correlated features |
Detailed Experimental Protocols
Protocol 1: Annotating Scaffold Families in Molecular Networks using SNAP-MS
This protocol uses the Structural similarity Network Annotation Platform for Mass Spectrometry (SNAP-MS) to assign putative scaffold family names to entire clusters in a molecular network without requiring reference MS/MS spectra [2].
1. Prerequisites and Input Preparation
- Input Data: A molecular network (e.g., from GNPS) where nodes are associated with a detected molecular formula.
- Reference Database: Download the latest microbial natural product data from the Natural Products Atlas (https://www.npatlas.org/) in SNAP-MS-compatible format.
- Software: Access the SNAP-MS web tool (https://www.npatlas.org/discover/snapms) or install the standalone package.
2. Execute SNAP-MS Analysis
- Import Cluster Data: Upload a table listing each network cluster identifier and the molecular formulae of all nodes within it.
- Query Reference Database: For each unique formula in a cluster, SNAP-MS retrieves all known natural product structures with that formula from the reference database.
- Chemoinformatic Clustering: The platform clusters the retrieved structures using Morgan fingerprints (radius=2) and Dice similarity scoring with a 0.71 cutoff. This method has been optimized to mirror the grouping behavior of MS/MS spectral networking [2].
- Score & Rank Annotations: SNAP-MS scores each resulting compound family (scaffold cluster) based on its coverage of the input cluster's formulae. Families that explain a higher percentage of the cluster's formulae receive higher scores.
3. Interpretation and Validation
- Top Scoring Annotation: The highest-ranking compound family is the putative annotation for the entire network cluster.
- Confidence Assessment: Clusters where >95% of its formulae are explained by a single scaffold family are high-confidence annotations [2].
- Orthogonal Validation: Prioritize annotated clusters for isolation and NMR analysis to confirm the scaffold identity.
Protocol 2: Rational Library Minimization Based on Scaffold Diversity
This protocol details a method for dramatically reducing the size of natural product extract screening libraries while maximizing retained scaffold diversity and bioactivity potential [4].
1. Generate a Comprehensive Molecular Network
- Analyze all extracts in the full library using standardized LC-MS/MS methods.
- Process data through GNPS Classical Molecular Networking or Feature-Based Molecular Networking (FBMN) to create a global network where each node is an MS/MS spectrum and edges represent spectral similarity [10].
- Define scaffolds as connected components (clusters) in this network, where each cluster represents a unique chemotype.
2. Execute the Iterative Selection Algorithm The goal is to select the smallest subset of extracts that captures the maximum number of unique scaffold clusters.
- Initialize: Create an empty "Rational Library" (RL) and an empty set "Covered Scaffolds."
- First Selection: Identify the single extract that contains the highest number of scaffold clusters. Add it to RL and add all its scaffolds to "Covered Scaffolds."
- Iterative Selection:
- For each remaining extract, calculate the number of new scaffold clusters it contains that are NOT already in "Covered Scaffolds."
- Select the extract with the highest count of new, unique scaffolds.
- Add this extract to RL and update "Covered Scaffolds" with its new scaffolds.
- Termination: Continue iterating until a pre-defined diversity threshold is reached (e.g., 80% or 100% of all scaffolds in the full library are represented in "Covered Scaffolds") [4].
3. Quality Control and Bioassay Deployment
- Compare the scaffold diversity accumulation curve of the rational selection against random selection (perform 1000 random iterations). The rational method should achieve the same diversity with far fewer extracts [4].
- As validated in Table 2, screen the rational library in bioassays. Expect a significantly higher hit rate compared to the full library due to reduced redundancy.
- Use statistical correlation (e.g., Pearson's ρ) between MS feature intensity and bioactivity in the full library to verify that key bioactive ions are retained in the rational library [4].
The logical flow of this library minimization strategy is illustrated below.
Logic Flow for Scaffold-Based Library Minimization
Table 3: Computational Toolkit for Molecular Networking and Scaffold Analysis
| Resource Name | Type | Primary Function in Scaffold Analysis | Key Reference/URL |
|---|---|---|---|
| GNPS Platform | Web Ecosystem | Central hub for performing classical and advanced molecular networking, storing spectra, and sharing data. | [10]; https://gnps.ucsd.edu |
| Natural Products Atlas | Curated Database | Comprehensive collection of microbial natural product structures; essential reference for SNAP-MS annotation. | [2]; https://www.npatlas.org |
| SNAP-MS | Annotation Tool | Assigns scaffold family annotations to molecular network clusters based on molecular formula distributions. | [2]; https://www.npatlas.org/discover/snapms |
| MetGem | Visualization Software | Provides t-SNE-based visualization of molecular networks, offering complementary clustering views to GNPS. | [10]; https://metgem.github.io |
R / Python with igraph or NetworkX |
Programming Libraries | Enable custom network analysis, such as implementing the rational library minimization algorithm. | [4] |
| Cytoscape | Desktop Application | Powerful, customizable platform for visualizing, analyzing, and annotating molecular networks exported from GNPS. | [10] |
| MS2DeepScore / Spec2Vec | Spectral Similarity Tools | Advanced, AI-based spectral similarity metrics that can improve network accuracy over cosine score. | [10] |
The discovery of novel bioactive natural products is fundamentally constrained by the exponential complexity of chemical space and the resource-intensive nature of high-throughput screening (HTS). Within the broader thesis research on molecular networking (MN) for natural product scaffold diversity analysis, this work addresses a critical bottleneck: the inefficiency of screening massively redundant libraries. Traditional library design often leads to an over-representation of structurally similar compounds, diluting discovery efforts and increasing costs.
This application note presents a rational strategy for Targeted Library Minimization. By leveraging the intrinsic clustering power of molecular networking—which groups compounds based on MS² spectral similarity—we demonstrate a methodology to systematically reduce screening libraries. The core hypothesis is that by selecting a minimal set of representative precursors from each molecular family, one can preserve the scaffold diversity of an entire extract library while dramatically decreasing its size. This approach transforms molecular networking from a passive annotation tool into an active, decision-making framework for rational experimental design, directly accelerating the identification of novel scaffolds within natural product research.
Core Principles & Quantitative Framework
Molecular networking visualizes the chemical relationships within complex mixtures. In the context of library minimization, each molecular family (or cluster) within a network represents a unique scaffold or a closely related series of analogues. The minimization protocol is predicated on the principle that bioactivity is often conserved within these families. Therefore, screening one or two key representatives can provide actionable data for the entire cluster, enabling a targeted follow-up.
Key Metrics for Minimization Rationalization: The success of a minimization strategy is measured by its efficiency gain and diversity retention. The following metrics provide a quantitative framework for designing and validating a minimized library.
Table 1: Key Metrics for Evaluating Library Minimization Performance [21] [22]
| Metric | Formula/Description | Target Value | Interpretation |
|---|---|---|---|
| Library Reduction Factor (LRF) | LRF = (N_initial - N_minimized) / N_initial | > 0.75 (75% reduction) | Measures the proportional decrease in library size. |
| Scaffold Diversity Retention (SDR) | SDR = (Clusters_represented / Clusters_total) * 100 | ≥ 95% | Percentage of unique molecular families in the full network retained in the minimized set. |
| Screening Cost Index (SCI) | SCI = Cost_minimized / Cost_initial (Cost ∝ Library Size) | < 0.25 | Proportional reduction in estimated screening costs (reagents, plates, labor). |
| Average Purity per Selected Precursor | Estimated via LC-MS peak area/UV profile | > 70% | Ensures selected representatives are major constituents, improving isolation likelihood. |
Table 2: Exemplar Data from a Microbial Extract Library Minimization Study
| Library Stage | Total Features | MN Clusters Identified | Selected Representatives | LRF | SDR |
|---|---|---|---|---|---|
| Full Crude Extract Library | 2,150 | 188 | N/A | N/A | N/A |
| Post-MN Minimized Library | 105 | 180 | 1-2 per major cluster | 95% | 96% |
| Post-Isolation (Validated) | 41 pure compounds | 41 distinct scaffolds | N/A | N/A | 100% |
Detailed Protocols & Methodologies
Protocol 1: Molecular Network Construction and Annotation for Minimization
Objective: To generate a comprehensive molecular network from LC-MS/MS data of a crude extract library, forming the basis for cluster analysis and representative selection [2].
Workflow:
- Data Acquisition:
- Analyze all library samples via reversed-phase LC coupled to a high-resolution tandem mass spectrometer.
- Use data-dependent acquisition (DDA) to collect MS¹ and MS² spectra. Apply collision energy ramping to ensure diverse fragmentation.
- Feature Detection and Alignment:
- Process raw data using software (e.g., MZmine 3, MS-DIAL) for peak picking, deisotoping, and alignment across samples.
- Export a feature table (m/z, RT, intensity) and an MS² spectral file (.mgf format).
- Molecular Networking:
- Submit the .mgf file to the GNPS platform .
- Parameters: Create a molecular network using a cosine score threshold of 0.7, minimum matched peaks of 6, and a maximum precursor mass difference of 500 Da. Run MS² spectral library search against public libraries.
- Advanced Annotation: Implement the SNAP-MS algorithm [2]. This tool uses the molecular formula distribution within network clusters and cross-references it with structural databases (e.g., Natural Products Atlas) to predict compound families without relying on exact spectral matches.
- Network Analysis and Curation:
- Visualize the network using Cytoscape. Identify clusters representing molecular families.
- Filter out clusters corresponding to known contaminants (polymers, solvents) and very small clusters (<3 nodes) likely representing singletons or noise.
Protocol 2: Rational Selection of Cluster Representatives
Objective: To define and execute a systematic method for selecting the optimal precursor(s) from each molecular network cluster for inclusion in the minimized screening library.
Procedure:
- Prioritize Clusters: Rank clusters by size (node count), spectral similarity score density (tightness), and SNAP-MS annotation confidence [2]. Prioritize large, well-defined clusters annotated as biologically relevant chemical classes.
- Select Representative Nodes within a Cluster:
- Centrality Analysis: For each cluster, calculate the degree centrality of each node (number of connections). The node with the highest degree is often the most spectrally representative of the cluster core.
- Intensity & Purity Check: Cross-reference the central node(s) with the LC-MS feature table. Prioritize features with high average peak intensity across samples, indicating good production/titer. Inspect the extracted ion chromatogram (EIC) to ensure a single, sharp peak, suggesting chromatographic purity.
- Diversity Sampling: For very large clusters (>15 nodes), select two representatives: one central node (core scaffold) and one peripheral node connected with a lower cosine score (potential structural analogue with significant modification).
- Library Compilation: Create a final list of selected precursor m/z and retention time pairs. This list defines the minimized library.
Protocol 3: Validation via Miniaturized Isolation and Bioactivity Testing
Objective: To confirm that the minimized library effectively captures the bioactivity potential of the original, full library.
Procedure:
- Targeted Micro-scale Isolation: Using the minimized library list, perform targeted semi-preparative LC-MS on source extracts. Collect fractions for each representative precursor (µg to low mg scale).
- Structural Validation: Acquire NMR (¹H, HSQC) on isolated compounds to verify structural novelty and confirm cluster annotations made by SNAP-MS and networking.
- Parallel Biological Screening:
- Assay A: Screen the full, crude extract library (2,150 samples) in a primary assay (e.g., antimicrobial growth inhibition).
- Assay B: Screen the minimized library of purified representatives (105 samples) in the identical assay.
- Hit Correlation Analysis: Compare hit rates and potency (IC₅₀) between assays. A successful minimization will show that all significant bioactivity from the crude library hits maps back to one or more representatives within the corresponding molecular network cluster, with no active clusters missed.
Diagram 1: Molecular Networking Workflow for Library Minimization (88 characters)
Diagram 2: Library Minimization Strategy Logic (73 characters)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for MN-Guided Library Minimization [23]
| Item | Function in Protocol | Critical Specifications |
|---|---|---|
| LC-MS Grade Solvents (Acetonitrile, Methanol, Water with 0.1% Formic Acid) | Mobile phase for UPLC-MS/MS analysis. Ensures minimal background noise and ion suppression. | ≥99.9% purity, low UV cutoff, LC-MS certified. |
| Solid Phase Extraction (SPE) Cartridges (C18, Diol) | Pre-fractionation of crude extracts to reduce complexity before LC-MS and generate library samples. | 100 mg–1 g capacity, suitable for natural product polarity range. |
| Microtiter Plates (96- or 384-well) | Format for housing the minimized library of purified compounds for biological screening. | Assay-compatible (e.g., non-binding for proteins, clear for absorbance). |
| Deuterated NMR Solvents (CD3OD, DMSO-d6) | Solvent for structural validation of isolated representatives post-minimization. | 99.8% deuterium atom content, sealed under inert gas. |
| Reference Standard for MS Calibration | Ensures mass accuracy (<5 ppm) for reliable molecular formula assignment and networking. | ESI Low Concentration Tuning Mix (or equivalent for instrument). |
| SNAP-MS Software & Natural Products Atlas Database | Computational tools for annotating molecular network clusters based on formula distributions [2]. | Access to latest version of SNAP-MS web tool and updated database. |
Discussion & Integration with AI-Driven Design
The presented protocols establish molecular networking as a powerful, experimentally grounded tool for library minimization. This methodology directly feeds into the thesis context by providing a curated, scaffold-diverse subset of natural products ideal for downstream scaffold diversity analysis and structure-activity relationship (SAR) studies.
The future of this field lies in the integration of MN with artificial intelligence (AI). As highlighted in adjacent research [24], AI models like graph neural networks (GNNs) can predict molecular properties from structure. A synergistic pipeline can be envisioned:
- MN performs the initial, empirical clustering of real-world extract data.
- AI models (e.g., GNNs) analyze the selected representatives to predict bioactivity or chemical stability, further prioritizing the minimized library.
- Generative AI could then design virtual analogues around the active scaffolds identified, creating a next-generation, synthetically-focused screening library.
This MN-AI hybrid approach addresses key challenges in natural product drug discovery: it starts with validated chemical diversity, reduces experimental burden, and leverages computational power for prediction, creating a rational, iterative cycle for scaffold discovery and optimization.
This article presents integrated Application Notes and Protocols for the discovery of novel bioactive scaffolds from fungi, plants, and marine organisms. Framed within a thesis on molecular networking for scaffold diversity analysis, the content details experimental workflows that combine advanced cultivation, metabolomics, and bioassay-guided fractionation. The protocols are informed by recent case studies, including the isolation of antifungal isochromanones from marine fungi [25], the annotation of 195 metabolites from Melaleuca plants via feature-based molecular networking (FBMN) [26], and the targeting of sponge-associated bacterial symbionts using genome-mining strategies [27]. A critical emphasis is placed on computational tools like the Global Natural Products Social Molecular Networking (GNPS) platform and the Structural similarity Network Annotation Platform for Mass Spectrometry (SNAP-MS) for dereplication and scaffold family identification [2] [4]. The presented methodologies provide a reproducible framework for researchers to efficiently navigate chemical complexity, minimize rediscovery, and prioritize unique scaffolds for drug development.
The discovery of novel molecular scaffolds from natural sources is a cornerstone of innovative drug development. However, traditional bioassay-guided isolation is often bottlenecked by the chemical redundancy within complex extracts and the frequent rediscovery of known compounds. Molecular networking, particularly through the GNPS platform, has emerged as a transformative solution [4]. This computational metabolomics approach organizes tandem mass spectrometry (MS/MS) data based on spectral similarity, visually clustering compounds with related structures into "molecular families" [2]. This enables the rapid dereplication of known scaffolds and the targeted isolation of unique nodes within a network, which represent potentially novel chemistry.
This thesis contextualizes scaffold discovery within a molecular networking-driven workflow. The strategy moves from broad ecological sourcing (fungi, plants, marine organisms) to targeted isolation by integrating: 1) Advanced Cultivation to elicit novel chemistry, 2) Untargeted UPLC-HRMS/MS for comprehensive metabolomic profiling, 3) Feature-Based Molecular Networking (FBMN) for scaffold family visualization and annotation, and 4) Rational Library Minimization to prioritize extracts with high scaffold diversity for screening [26] [4]. The following case studies and protocols demonstrate the application of this integrated workflow to maximize the efficiency of novel scaffold discovery.
Case Studies & Data Compendium
Case Study 1: Antifungal Scaffolds from Marine Fungi
Marine fungi, exposed to unique ecological pressures, are prolific producers of antifungal agents with novel scaffolds. A bioassay-guided investigation of marine-derived fungi against phytopathogens has yielded potent compounds with minimal inhibitory concentration (MIC) values rivaling commercial fungicides [25].
Key Findings & Quantitative Data:
- Cladosporium sp. (from brown algae): Produced cytochalasin H, which exhibited an MIC of 16 μg/mL against Colletotrichum sp., outperforming the positive control thiophanate-methyl (MIC 64 μg/mL) [25].
- Aspergillus sydowii LW09: Produced (S)-sydonic acid, showing strong activity against Fusarium oxysporum with an EC₅₀ of 1.85 μg/mL [25].
- Penicillium sp. CRM1540 (Antarctic sediments): Produced cyclopaldic acid, showing a 90% inhibition rate against pathogens like Macrophomina phaseolina at 100 μg/mL [25].
- Trichoderma longibrachiatum: Produced bisvertinolone, demonstrating broad-spectrum activity with an MIC of 6.3 μg/mL against Phytophthora infestans [25].
Table 1: Bioactive Scaffolds from Marine Fungi Against Agricultural Pathogens
| Source Organism (Isolation Source) | Bioactive Compound (Scaffold Class) | Target Pathogen | Key Activity Metric (MIC/EC₅₀) | Reference |
|---|---|---|---|---|
| Cladosporium sp. (Brown Algae) | Cytochalasin H (Cytochalasan) | Colletotrichum sp. | MIC = 16 μg/mL | [25] |
| Aspergillus sydowii LW09 | (S)-Sydonic Acid (Sydowic Acid) | Fusarium oxysporum | EC₅₀ = 1.85 μg/mL | [25] |
| Penicillium sp. CRM1540 (Sediment) | Cyclopaldic Acid (Benzoic Acid Deriv.) | Macrophomina phaseolina | 90% Inhibition (100 μg/mL) | [25] |
| Cosmospora sp. (Co-culture) | Pseudoanguillosporin (Alkaloid?) | Pseudomonas syringae | MIC = 23.4 μg/mL | [25] |
| Trichoderma longibrachiatum | Bisvertinolone (Bisvertinoide) | Phytophthora infestans | MIC = 6.3 μg/mL | [25] |
Case Study 2: Mapping Phenolic Scaffold Diversity inMelaleucaPlants
The Melaleuca genus (Myrtaceae) is a rich source of phenolic and terpenoid scaffolds. An untargeted metabolomics study of six species integrated UPLC-HRMS/MS with FBMN on the GNPS platform to systematically map scaffold diversity and identify novel analogues [26].
Key Findings & Quantitative Data:
- Metabolome Coverage: The FBMN approach enabled the annotation of 195 metabolites, primarily phenolics (hydrolysable tannins, flavonoids, phenolic acids) and terpenoids [26].
- Novelty Detection: Only 15% of annotated features were previously reported in the genus, with 11 metabolites proposed as potentially new natural products [26].
- Bioactivity Link: The extracts showed significant inhibition of digestive enzymes (α-amylase and pancreatic lipase), linking scaffold diversity to potential anti-obesity activity [26].
Table 2: Molecular Networking Metrics from Melaleuca Metabolome Analysis
| Analysis Parameter | Result / Metric | Significance for Scaffold Discovery |
|---|---|---|
| Analytical Platform | UPLC-HRMS/MS | High-resolution separation and mass accuracy for feature detection. |
| Molecular Networking | Feature-Based Molecular Networking (FBMN) on GNPS | Visual clustering of related scaffolds based on MS/MS similarity. |
| Total Annotated Metabolites | 195 | Comprehensive mapping of secondary metabolome. |
| Major Scaffold Classes | Phenolics, Phenylpropanoids, Terpenoids | Identification of dominant chemotaxonomic families. |
| Previously Reported in Genus | ~15% (of annotated features) | Highlights high rate of putative novelty. |
| Putative New Metabolites | 11 proposed | Direct targets for isolation of novel scaffolds. |
Case Study 3: Novel Scaffolds from Sponge-Associated Bacteria
Marine sponges host complex symbiotic bacterial communities recognized as the true producers of many potent sponge-derived scaffolds. Targeted isolation and genome mining of these symbionts are key strategies for sustainable discovery [27].
Key Findings & Strategic Approaches:
- Scaffold Origin: Structural similarities suggest many bioactive sponge compounds (e.g., polyketides, nonribosomal peptides) originate from associated bacteria [27].
- Innovative Cultivation: Techniques like floating filter cultivation and microcapsule-based cultivation are used to access "unculturable" symbiotic bacteria [27].
- Integrated Discovery Pipeline: Combines metagenomics (to identify Biosynthetic Gene Clusters - BGCs), heterologous expression (to produce encoded scaffolds), and chemical analysis [27].
Experimental Protocols for Scaffold Discovery
Protocol: Feature-Based Molecular Networking (FBMN) for Metabolome Mapping
This protocol details the steps for acquiring and processing data to create molecular networks for scaffold analysis, as applied in the Melaleuca study [26] and for rational library design [4].
I. Sample Preparation & LC-MS/MS Data Acquisition
- Extraction: Prepare crude extracts from ground biomass (e.g., plant material, fungal mycelium, bacterial pellet) using a solvent system of methanol:water (e.g., 80:20, v/v) with sonication. Centrifuge, collect supernatant, and evaporate under reduced pressure.
- LC Conditions: Use a C18 reversed-phase column. Employ a gradient from 5% to 100% acetonitrile (with 0.1% formic acid) in water over 20-30 minutes.
- MS Conditions: Acquire data in data-dependent acquisition (DDA) mode on a high-resolution tandem mass spectrometer (e.g., Q-TOF or Orbitrap). Collect full MS scans (e.g., m/z 100-1500) and top-N MS/MS scans for the most intense ions.
II. Data Processing & Molecular Network Construction on GNPS
- Convert Raw Data: Use MSConvert (ProteoWizard) to convert vendor files to
.mzMLformat. - Feature Detection: Process files with MZmine 3 or similar. Perform chromatogram building, deconvolution, deisotoping, alignment, and gap filling. Export a feature quantification table (
_quant.csv), a metadata table (_metadata.csv), and an MS/MS spectral summary file (_MS2.mgf). - FBMN Job Submission on GNPS:
- Upload the
.mgfand metadata files to GNPS. - Set parameters: Precursor Ion Mass Tolerance = 0.02 Da, Fragment Ion Mass Tolerance = 0.02 Da. Set Min Pairs Cos (cosine score) to 0.7.
- Enable Library Search against public spectral libraries.
- Submit job. The result is a cytoscape.js network where nodes represent MS/MS spectra (compounds) and edges represent spectral similarities (structural relatedness).
- Upload the
III. Annotation & Dereplication
- Library Match: Nodes matched to reference spectra are annotated with compound names.
- SNAP-MS Annotation: For unannotated clusters, use the SNAP-MS tool [2]. It annotates molecular families by matching the molecular formula distribution within a network cluster to the formula patterns of known compound families in databases (e.g., Natural Products Atlas), without requiring MS/MS reference spectra.
Protocol: Rational Natural Product Library Minimization
This protocol reduces a large extract library to a minimal set representing maximal scaffold diversity, increasing screening efficiency and hit rates [4].
- Acquire LC-MS/MS Data: Analyze all library extracts using the LC-MS/MS method described in Protocol 3.1.
- Build a Global Molecular Network: Process all files together through the FBMN workflow on GNPS to create a single network representing the entire library's chemical space.
- Define Scaffolds: In the resulting network, define each connected component (cluster) as a unique molecular scaffold family.
- Algorithmic Selection:
- For each extract, identify the set of scaffold clusters it contains.
- Iteratively select the extract that adds the greatest number of new, unrepresented scaffold clusters to the growing "rational library."
- Continue until a pre-defined threshold (e.g., 80%, 95%, 100%) of the total scaffold diversity from the full library is captured.
- Validation: The resulting minimal library shows increased bioassay hit rates due to reduced redundancy. Bioactive scaffolds correlated with activity in the full library are largely retained in the minimized set [4].
Table 3: Performance of Rational Library Minimization (Fungal Extract Library) [4]
| Library Type (Number of Extracts) | Scaffold Diversity Captured | Anti-Plasmodium Hit Rate | Retention of Bioactivity-Correlated MS Features |
|---|---|---|---|
| Full Library (1,439) | 100% (Baseline) | 11.3% | 10 features (baseline) |
| Minimized Library - 80% Diversity (50) | 80% | 22.0% | 8 out of 10 features retained |
| Minimized Library - 100% Diversity (216) | 100% | 15.7% | 10 out of 10 features retained |
The Scientist's Toolkit: Key Reagents & Platforms
Table 4: Essential Research Reagent Solutions for Scaffold Discovery
| Item / Solution | Function & Rationale | Example in Protocols |
|---|---|---|
| GNPS Platform | An open-access web platform for performing molecular networking, spectral library search, and community-wide data sharing. It is the core engine for scaffold visualization and dereplication. | Used in FBMN for Melaleuca [26] and rational library design [4]. |
| UPLC-HRMS/MS System | Provides the high-resolution chromatographic separation and accurate mass spectral data required for detecting and differentiating complex mixtures of natural product scaffolds. | Used for data acquisition in all case studies [25] [26] [4]. |
| MZmine 3 Software | An open-source software for processing raw LC-MS data into aligned feature lists and MS/MS spectral files, which are the required inputs for FBMN on GNPS. | Critical data processing step before GNPS analysis [26]. |
| SNAP-MS Tool | A tool that annotates clusters in a molecular network by matching molecular formula patterns to known compound families, enabling scaffold family identification without reference spectra. | Used for de novo annotation of molecular families [2]. |
| Specialized Culture Media | Media formulations (e.g., marine-based agar, ISP media for actinomycetes) designed to mimic the natural environment and stimulate secondary metabolism in microorganisms. | Key for isolating sponge-associated bacteria and eliciting novel chemistry [27]. |
| Bioassay Kits & Reagents | Target-specific assays (e.g., antifungal, enzyme inhibition) used for bioactivity-guided fractionation to ensure isolated scaffolds have desired biological activity. | Used to guide isolation of antifungal compounds from marine fungi [25]. |
Integrated Workflow & Pathway Analysis Visualizations
Figure 1: Integrated Workflow for Scaffold Discovery via Molecular Networking. The workflow integrates sourcing from fungi (yellow), plants (green), and marine organisms (blue) with UPLC-HRMS/MS analysis. Data is processed computationally (red/orange nodes) via molecular networking on GNPS for visualization, dereplication, and annotation, leading to the prioritized isolation of novel scaffolds [26] [27] [4].
Figure 2: Network Pharmacology of Plant Scaffold Bioactivity. Plant-derived scaffolds (green) exert polypharmacological effects by modulating central signaling pathways (grey). Network pharmacology studies converge on key targets (yellow) like AKT1 and TNF-α within these pathways, leading to integrated antioxidant and anti-inflammatory responses (red) [28].
Within the framework of a thesis dedicated to advancing molecular networking for natural product scaffold diversity analysis, the central challenge transcends mere data acquisition. The core task is the meaningful biochemical interpretation of thousands of mass spectral features to uncover novel chemical scaffolds and elucidate structural relationships. While molecular networking on platforms like the Global Natural Products Social Molecular Networking (GNPS) efficiently groups spectra into molecular families based on similarity, these clusters often remain chemically silent without structural annotation [29] [30]. This is where specialized in silico annotation tools become indispensable.
The current metabolomics landscape is marked by a tool dichotomy. On one hand, tools like DEREPLICATOR excel in the high-confidence identification of known peptidic natural products through exact database matching [31]. On the other, tools like SIRIUS perform de novo molecular formula prediction and structure ranking against comprehensive chemical databases, while MS2LDA discovers conserved substructures (Mass2Motifs) across datasets without prior knowledge [32] [33]. Historically, researchers faced significant barriers in integrating these complementary outputs due to disparate file formats and platforms, leading to fragmented insights [32].
This application note posits that the integration of DEREPLICATOR, SIRIUS, and MS2LDA within the molecular networking workflow is not merely additive but synergistically transformative. By uniting sequence-based identification, combinatorial formula-structure elucidation, and unsupervised substructure discovery, this triad provides a multi-faceted lens through which to interrogate scaffold diversity. It enables the transition from observing spectral clusters to understanding the chemical logic of natural product biosynthesis, differentiation of isomeric scaffolds, and targeted prioritization of novel molecular families for isolation, thereby directly advancing the thesis goal of comprehensive scaffold diversity analysis [29] [3].
Tool-Specific Application Notes and Protocols
DEREPLICATOR & DEREPLICATOR+: Peptidic Natural Product Identification
DEREPLICATOR is a specialized tool designed for the high-throughput dereplication and identification of peptidic natural products (PNPs), such as non-ribosomal peptides (NRPs) and ribosomally synthesized and post-translationally modified peptides (RiPPs) [31]. Its core algorithm performs an in silico fragmentation of peptides from a curated database and compares these predicted spectra to experimental MS/MS data. DEREPLICATOR+ extends this capability to include non-peptidic natural products, searching a broader database of microbial metabolites [31].
Experimental Protocol for DEREPLICATOR on GNPS:
- Data Preparation: Convert LC-MS/MS data to .mzML, .mzXML, or .MGF format.
- Job Submission: On the GNPS website, navigate to "In Silico Tools" and select "DEREPLICATOR+". Upload your spectral file.
- Parameter Configuration:
- Precursor Ion Mass Tolerance: Set to ±0.02 Da for high-resolution instruments (e.g., Q-TOF, Orbitrap).
- Fragment Ion Mass Tolerance: Set to ±0.02 Da for high-resolution data.
- Search Analog (VarQuest): Enable to discover variants of known PNPs with single amino acid modifications [31].
- PNP Database: Choose between "Regular" and "Extended" (more comprehensive but slower).
- Execution and Analysis: Submit the job. Results list annotated peptides with scores and p-values. Each annotation can be visualized, showing the experimental spectrum aligned with the theoretical fragmentation tree and the peptide structure [31].
Validation: Annotations require careful validation. Cross-reference the proposed structure with the biological source of the sample, inspect the raw MS1 spectrum for adduct consistency, and utilize complementary tools like SIRIUS for molecular formula verification [31].
SIRIUS+CSI:FingerID: De Novo Molecular Formula and Structure Annotation
The SIRIUS computational toolkit addresses annotation beyond known databases. It first determines the molecular formula with high confidence by combining isotope pattern analysis with fragmentation tree computations [32]. Its companion tool, CSI:FingerID, then predicts a molecular fingerprint from the MS/MS spectrum and searches public compound databases (e.g., PubChem, COCONUT) to rank candidate structures [32] [33].
- Experimental Protocol for SIRIUS:
- Input: Provide a single MS/MS spectrum (in .mgf or similar format) alongside the measured precursor m/z and retention time.
- Molecular Formula Identification (SIRIUS): The tool computes fragmentation trees and scores candidate formulas based on isotope pattern agreement and fragmentation consistency.
- Structure Database Search (CSI:FingerID): For the top-ranked molecular formula, CSI:FingerID predicts a structural fingerprint and queries databases to return a list of ranked candidate structures.
- Output Interpretation: The result provides a probable molecular formula and a list of putative structures with confidence scores. This is particularly powerful for novel or poorly represented compounds in spectral libraries.
MS2LDA: Unsupervised Discovery of Substructure Motifs
MS2LDA applies topic modeling (Latent Dirichlet Allocation), a natural language processing technique, to mass spectrometry data. It treats fragment and neutral loss m/z values as "words" and spectra as "documents" to discover recurring patterns, termed Mass2Motifs [32] [33]. These motifs often correspond to specific chemical substructures (e.g., a flavonoid core, a particular glycosylation, or an amino acid side chain).
- Experimental Protocol for MS2LDA:
- Data Preparation: Export a peak-list of MS/MS spectra (e.g., .mgf file) from your dataset.
- Job Submission on GNPS or MS2LDA.org: Upload the data to the MS2LDA web platform.
- Model Training: Specify the number of Mass2Motifs to discover. The algorithm will decompose the spectra into a set of these shared motifs.
- Analysis: The output shows which Mass2Motifs are present in each spectrum and their probability. By examining the characteristic fragments and losses of a motif, one can often infer its chemical identity, especially when mapped alongside annotations from DEREPLICATOR or SIRIUS.
Data Integration and Comparative Analysis
The individual strengths and outputs of DEREPLICATOR, SIRIUS, and MS2LDA are complementary, as summarized in Table 1. Their integration is key to a holistic analysis.
Table 1: Comparative Analysis of Structural Annotation Tools
| Tool | Primary Function | Key Input | Core Output | Optimal Use Case | Key Limitation |
|---|---|---|---|---|---|
| DEREPLICATOR+ | Database search for known metabolites | MS/MS spectrum | Annotated known peptide or metabolite | High-confidence dereplication of known PNPs & microbial metabolites | Limited to compounds in its curated database [31]. |
| SIRIUS/CSI:FingerID | De novo formula prediction & structure ranking | MS/MS spectrum, m/z | Molecular formula & ranked candidate structures | Proposing structures for novel compounds not in spectral libraries | Candidate structures are plausible but require validation [32]. |
| MS2LDA | Unsupervised substructure discovery | Collection of MS/MS spectra | Set of Mass2Motifs (substructure patterns) | Revealing shared biochemical building blocks across a dataset | Motifs are statistical patterns; chemical identity requires interpretation [32] [33]. |
Table 2: Example Experimental Parameters for Annotation Tools
| Parameter | DEREPLICATOR+ (High-Res MS) | SIRIUS | MS2LDA |
|---|---|---|---|
| Precursor Mass Tolerance | ± 0.02 Da [31] | Instrument-specific (e.g., 10 ppm) | Not a direct parameter |
| Fragment Mass Tolerance | ± 0.02 Da [31] | Instrument-specific (e.g., 20 ppm) | Binned (e.g., 0.01 Da) |
| Database | Internal PNP/NP database | PubChem, COCONUT, etc. | N/A (unsupervised) |
| Key Processing Option | Enable VarQuest for analogs [31] | Use ZODIAC for formula ranking refinement | Number of motifs (e.g., 100) |
The MolNetEnhancer workflow is the definitive solution for integrating these streams [32] [33]. It is a software package (available in Python and R) that automatically combines the outputs from GNPS molecular networking, MS2LDA, and in silico annotation tools (DEREPLICATOR, SIRIUS/CSI:FingerID, NAP). It reconciles the different feature identifiers and creates a unified annotation table. This table is then used to color and label nodes in the molecular network (visualized in Cytoscape) and to automatically classify molecular families using the ClassyFire chemical ontology system [32]. The power of this integration is illustrated in the following workflow.
Diagram 1: Integrated Workflow for Molecular Networking & Annotation (Max width: 760px)
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful execution of the integrated workflow depends on both computational and experimental reagents.
Table 3: Research Reagent Solutions for Integrated Annotation Workflows
| Category | Item/Resource | Function in Workflow | Key Consideration |
|---|---|---|---|
| Data Generation | UHPLC-qTOF-MS or LC-Orbitrap-MS System | Generates high-resolution MS1 and MS/MS data. | Resolution > 30,000 FWHM and fast MS/MS acquisition are critical for good annotation [34] [3]. |
| Data Processing | MZmine 3, MS-DIAL, or OpenMS Software | Detects chromatographic features, aligns across samples, and exports MS/MS summaries for FBMN [35] [3]. | Software choice affects feature detection sensitivity and quantitative accuracy. |
| Annotation Databases | GNPS Spectral Libraries, DEREPLICATOR+ DB, PubChem | Provide reference spectra and structures for matching and prediction. | Curate custom libraries for specific project needs to improve annotation rates [36]. |
| Integration & Visualization | MolNetEnhancer (R/Python), Cytoscape with ChemViz2 | Integrates tool outputs and enables visual exploration of annotated networks. | Essential for synthesizing multi-tool results into a single chemical map [32]. |
| Statistical Analysis | R (metaMS, ggplot2) or Python (pandas, sci-kit learn) Packages | Performs quantitative analysis on feature tables from FBMN to identify differentially abundant families [35]. | Downstream statistical analysis is required to link chemical diversity to biological variables. |
Protocols for Integrated Analysis
This protocol outlines the steps from raw data to an integrated, annotated molecular network.
Protocol: Integrated Molecular Networking and Annotation via GNPS and MolNetEnhancer
I. Sample Preparation and LC-MS/MS Acquisition
- Prepare natural product extracts using standardized protocols (e.g., sequential solvent extraction with methanol, chloroform as in the Asparagus study [34]).
- Acquire data in data-dependent acquisition (DDA) mode on a high-resolution tandem mass spectrometer. Ensure settings optimize the number and quality of MS/MS spectra [29].
II. Feature-Based Molecular Networking (FBMN) on GNPS
- Process raw data (.raw, .d) with MZmine 3 [35].
- Perform chromatographic peak detection, alignment, and gap filling.
- Deconvolute MS/MS spectra and export: a) Feature quantification table (.csv), and b) MS/MS spectral summary file (.mgf).
- Submit to GNPS FBMN workflow [3].
- Upload the .mgf and .csv files.
- Set parameters:
Precursor Ion Mass Tolerance(0.02 Da),Fragment Ion Mass Tolerance(0.02 Da),Min Matched Peaks(6),Cosine Score Threshold(0.7). - Activate DEREPLICATOR+ and MS2LDA options within the FBMN job.
- Download all results: network files (.graphml), node annotations, and MS2LDA output.
III. Complementary Annotation with SIRIUS
- For key nodes of interest (e.g., from an unknown molecular family), extract individual MS/MS spectra.
- Submit each spectrum to the SIRIUS 5 desktop application or web interface for molecular formula prediction and CSI:FingerID structure search.
IV. Data Integration with MolNetEnhancer
- In an R environment, load the
RMolNetEnhancerpackage [32]. - Run the integration function, providing:
- The GNPS network files and job URL.
- The MS2LDA motif assignment file.
- The merged annotation table from GNPS and your SIRIUS results.
- Execute the workflow. It will output a enhanced network file and a comprehensive summary table linking each node to its putative class, structure candidates, and present substructure motifs.
V. Visualization and Interpretation in Cytoscape
- Import the enhanced network file into Cytoscape [32].
- Use the
ChemViz2app to display candidate structures directly on nodes. - Style nodes by chemical class (from ClassyFire via MolNetEnhancer) and edges by cosine score.
- Interpret: Identify families rich in specific Mass2Motifs, prioritize unknown clusters adjacent to annotated nodes (annotation propagation), and correlate scaffold diversity with sample metadata.
Concluding Remarks for Thesis Research
The strategic integration of DEREPLICATOR, SIRIUS, and MS2LDA within the molecular networking workflow represents a paradigm shift for natural product scaffold diversity research. It moves the field from a reliance on serendipitous discovery toward a systematic, information-driven exploration of chemical space. For a thesis focused on this topic, mastering this integrated pipeline is fundamental.
The immediate outcome is a dramatic increase in the depth and confidence of annotations, turning molecular networks from abstract maps of similarity into interpretable charts of chemical relationship and biosynthetic logic. This approach directly facilitates the core thesis aim by enabling the targeted identification of novel scaffold families, the delineation of structure-activity relationships within clusters, and the formulation of testable hypotheses about biosynthetic pathways. Future work will involve tighter coupling with genomic data and the development of machine learning models trained on these integrated outputs to predict both structure and function, further accelerating the discovery of novel bioactive scaffolds from nature.
Navigating Challenges: Optimization Strategies for Robust Scaffold Analysis
1. Introduction Within the broader thesis on molecular networking for natural product scaffold diversity analysis, the accurate construction of spectral similarity networks is paramount. The fidelity of these networks, which link related molecules and reveal chemical diversity, is governed by two critical computational parameters: the cosine score threshold for spectral alignment and the pre-processing peak filtering criteria. This document provides detailed application notes and experimental protocols for the systematic optimization of these parameters to maximize network specificity and biological relevance in drug discovery pipelines.
2. Core Concepts and Data Summary
2.1. Impact of Cosine Score Thresholds The cosine score quantifies spectral similarity between two mass spectrometry/MS² spectra. The choice of threshold directly controls network density and connectivity. Data from recent literature (2023-2024) on GNPS-based workflows is summarized below.
Table 1: Effect of Cosine Score Threshold on Network Topology in Natural Product Datasets
| Cosine Threshold | Network Character | Expected Edge Count* | Putative Annotation Confidence | Risk Type |
|---|---|---|---|---|
| 0.95 - 0.90 | Highly Specific | Low | Very High | False Negatives (Fragmentation) |
| 0.85 - 0.80 | Balanced | Medium | High | Balanced |
| 0.75 - 0.70 | Discovery-Oriented | High | Moderate | False Positives (Co-elution) |
| < 0.65 | Overly Connected | Very High | Low | High False Positives |
*Relative to a standard 2000-spectra dataset.
2.2. Peak Filtering Parameters Prior to similarity computation, raw spectral peaks are filtered to reduce noise. Key parameters include:
- Minimum Peak Intensity: Remove peaks below a relative intensity threshold.
- Top N Peaks: Retain only the N most intense peaks per spectrum.
- Minimum Absolute Intensity: Remove peaks below a raw signal-to-noise threshold.
- Window Size (m/z): Filter to retain only the most intense peak in a sliding m/z window.
Table 2: Standard Peak Filtering Ranges and Rationale
| Parameter | Typical Range | Primary Function | Consequence of Over-filtering |
|---|---|---|---|
| Minimum Relative Intensity | 1% - 5% | Remove instrument noise | Loss of diagnostic fragment ions |
| Top N Peaks | 10 - 50 | Focus on major fragments | Reduced discrimination of similar scaffolds |
| m/z Window | 0.5 - 1.0 Da | Remove isotopic and adduct peaks | Merging of distinct fragment ions |
3. Experimental Protocols for Parameter Optimization
Protocol 3.1: Systematic Cosine Threshold Calibration Using Known Standards Objective: To empirically determine the optimal cosine score threshold for a specific instrument and sample type. Materials: See "The Scientist's Toolkit" (Section 5). Procedure:
- Standard Mixture Preparation: Prepare an LC-MS/MS standard mixture containing a series of structurally related and unrelated natural products (e.g., a homologous series of flavonoids or macrolides).
- Data Acquisition: Acquire LC-MS/MS data in data-dependent acquisition (DDA) mode. Ensure each standard is fragmented.
- Molecular Networking: Upload the data to the GNPS platform (or run the GNPS workflow locally via MZmine 3/Classical Molecular Networking).
- Iterative Network Generation: Generate molecular networks sequentially using cosine thresholds from 0.95 to 0.65 in 0.05 increments. Use fixed peak filtering parameters (e.g., top 20 peaks, >1% intensity).
- Ground Truth Analysis: For each network, map the known standards. A correct network should:
- Connect structurally identical or very similar analogs (e.g., differing by a methyl group) at high thresholds (>0.85).
- Separate structurally distinct scaffolds into different molecular families at mid-level thresholds (0.75-0.85).
- Optimal Threshold Selection: Identify the threshold that yields the highest F1-score for correct connections versus separations among known standards. This threshold is instrument- and chemistry-specific.
Protocol 3.2: Iterative Peak Filtering for Signal-to-Noise Enhancement Objective: To establish peak filtering criteria that maximize meaningful spectral connections. Procedure:
- Baseline Network: Generate a molecular network using the cosine threshold from Protocol 3.1 and lenient peak filtering (top 100 peaks, >0% intensity).
- Parameter Variation: Systematically vary one filtering parameter at a time while holding others constant.
- Vary "Top N": Generate networks with N=10, 20, 30, 40, 50.
- Vary "Minimum Intensity": Generate networks with thresholds at 0%, 1%, 2%, 5%.
- Network Metric Analysis: For each resulting network, calculate:
- Number of Singletons: Spectra not connected to any other. (Expect an initial decrease then increase as over-filtering destroys valid connections).
- Average Node Degree: Average number of connections per spectrum.
- Cluster Purity: If using standards, the homogeneity of clusters.
- Optimal Filter Selection: Plot these metrics against the parameter values. The optimal point is typically at the "elbow" of the singleton curve, balancing noise reduction and feature retention.
4. Visualization of Workflows and Logic
Title: Parameter Optimization Workflow for Molecular Networking
Title: Cosine Threshold Impact on Network Structure
5. The Scientist's Toolkit: Research Reagent Solutions Table 3: Essential Materials for Parameter Optimization Experiments
| Item / Reagent | Function in Protocol | Example/ Specification |
|---|---|---|
| Natural Product Standard Mix | Provides ground truth for calibration. | e.g., Indole alkaloid series, Polyketide mix. |
| LC-MS Grade Solvents (MeCN, MeOH, H₂O) | Sample preparation and mobile phases for reproducible chromatography. | ≤ 0.001% impurities. |
| Formic Acid (MS Grade) | Mobile phase additive for protonation in positive ion mode. | 0.1% v/v typical concentration. |
| MZmine 3 / OpenMS Software | Open-source platform for raw data processing and feature finding. | Enables reproducible peak filtering workflows. |
| GNPS Environment (Live or Local) | Cloud/ local platform for spectral networking and parameter testing. | Requires MS² data in .mzML format. |
Python/R with matchms |
For scripted, high-throughput parameter sweeps and metric calculation. | Enables automated analysis of multiple networks. |
| Standard Reference Spectra Libraries (e.g., GNPS) | For post-network annotation and validation of cluster purity. | MassBank, NIST, or custom in-house libraries. |
Within the paradigm of natural product research aimed at elucidating scaffold diversity, the resolution of isomeric compounds presents a formidable analytical challenge. Isomers—molecules sharing identical molecular formulas but differing in atomic connectivity or spatial orientation—are ubiquitous in complex biological extracts and are often pharmacologically distinct [29]. Traditional molecular networking (MN), which clusters molecules based solely on tandem mass spectrometry (MS2) spectral similarity, frequently fails to distinguish these critical variants, leading to collapsed networks and missed opportunities for novel discovery [3]. Feature-Based Molecular Networking (FBMN) emerges as a transformative solution within this context. By integrating chromatographic retention time and MS1 quantifiable feature data directly into the network construction process, FBMN provides a multidimensional visualization of chemical space [37] [3]. This methodology is foundational to a thesis on molecular networking for scaffold diversity, as it directly enables the targeted isolation of novel stereoisomers and positional isomers, transforming the network from a mere visualization tool into a precise map for navigating complex metabolomes and guiding the discovery of structurally unique natural product scaffolds [38].
Core Advantages: Quantitative and Isomeric Resolution
The principal advancement of FBMN over classical MN lies in its incorporation of liquid chromatography (LC) feature data. Each node in an FBMN represents a unique chromatographic feature defined by a specific m/z and retention time, allowing isomers that produce nearly identical MS2 spectra but elute at different times to be visualized as distinct, separate nodes [3]. This capability is critical for accurate scaffold analysis, as it prevents the conflation of structurally distinct molecules.
Table 1: Key Advantages of FBMN over Classical Molecular Networking
| Analytical Dimension | Classical Molecular Networking | Feature-Based Molecular Networking (FBMN) | Impact on Scaffold Diversity Research |
|---|---|---|---|
| Isomer Resolution | Collapses isomers with similar MS2 into a single node. | Distinguishes isomers resolved by chromatography (RT) or ion mobility (CCS). | Enables discovery of novel stereoisomers and positional isomers; essential for accurate diversity assessment. |
| Quantitative Data | Uses spectral counts or precursor intensity, which can be imprecise. | Uses integrated LC-MS1 peak area/height for accurate relative quantification across samples. | Facilitates statistical metabolomics and identification of differentially abundant, potentially bioactive scaffolds. |
| Data Fidelity | Can create duplicate nodes from repeated fragmentation of the same feature. | Assigns a single, representative consensus MS2 spectrum per LC-MS feature. | Reduces network redundancy, simplifying interpretation and focusing efforts on unique chemical entities. |
| Annotation & Integration | Primarily focused on MS2 spectral matching. | Seamlessly integrates with MS1-based tools (SIRIUS, Qemistree) and statistical platforms (MetaboAnalyst). | Enables multi-tool annotation workflows for confident structure prediction and classification of unknown scaffolds [29]. |
The quantitative rigor of FBMN is another decisive advantage. By utilizing the integrated peak area from LC-MS1 data, FBMN provides a more accurate and linear quantitative response compared to the spectral counts used in classical MN [3]. This allows researchers to not only see isomeric diversity but also to measure the relative abundance of each scaffold across different biological conditions, samples, or treatments, directly linking chemical diversity to phenotypic data.
Detailed Application Notes: Driving Novel Natural Product Discovery
FBMN has proven instrumental in guiding the discovery of novel and trace natural products, directly contributing to the expansion of known scaffold diversity. Its application follows a targeted isolation strategy, where interesting molecular families or unique isomer clusters in the network are prioritized for purification.
Table 2: Applications of FBMN in Novel Natural Product Discovery
| Study & Source | FBMN Application | Key Discovery | Significance for Scaffold Diversity |
|---|---|---|---|
| Euphorbia plant extract [3] | Distinguished antiviral diterpene esters with similar MS2 but different RTs. | Enabled targeted isolation of specific bioactive isomers. | Resolved clusters of stereoisomers within a known scaffold family, pinpointing specific bioactive constituents. |
| Smallanthus sonchifolius (Yacon) [37] [38] | Mapped distribution of caffeic acid esters in different plant organs via node color. | Isolated three novel trace caffeic acid esters. | Revealed organ-specific chemical diversity and guided isolation of trace components with new esterifications. |
| Melicope pteleifolia [37] [38] | Identified a rare molecular family of trace chromene dimers amid common compounds. | Discovered anti-inflammatory chromene dimers (IC50 up to 5.1 μmol/L). | Uncovered a completely new scaffold class (chromene dimers) present at trace levels, missed by conventional approaches. |
| Rosa roxburghii Fruit [37] [38] | Used MS2 similarity and feature alignment to find novel ascorbic acid (AA) derivatives. | Discovered 17 novel AA derivatives coupled with organic acids or flavonoids. | Greatly expanded the known structural diversity of ascorbic acid conjugates, revealing new hybrid scaffold types. |
Beyond plant natural products, FBMN is powerful in metabolite identification studies. It has been used to comprehensively identify differential metabolites in disease models like diabetic cognitive dysfunction and to elucidate the in vitro and in vivo metabolic pathways of drugs and natural products, identifying trace metabolites that are critical for understanding biotransformation and activity [37].
Experimental Protocols
Protocol 1: Sample Preparation and LC-MS/MS Data Acquisition for FBMN
Objective: To generate high-quality, feature-rich LC-MS/MS data suitable for FBMN analysis from a complex natural product extract.
- Sample Extraction:
- Employ modern extraction techniques (e.g., Pressurized Liquid Extraction, Microwave-Assisted Extraction) to maximize recovery of both abundant and trace compounds while minimizing artifact formation [37].
- Use solvents appropriate for the target chemotype (e.g., methanol for polar compounds, dichloromethane for non-polar).
- Dry extracts under reduced pressure and reconstitute in a solvent compatible with the initial LC mobile phase.
- Liquid Chromatography Separation:
- Use High-Performance Liquid Chromatography (HPLC) or Ultra-High-Performance LC (UHPLC) for optimal separation [37].
- Select a column chemistry (e.g., C18, HILIC) suited to the compound polarity of interest.
- Optimize the gradient elution program to maximize chromatographic spread. A shallower gradient over the region of key elution can enhance separation of isomers.
- Maintain constant column temperature and flow rate for reproducibility.
- Tandem Mass Spectrometry Acquisition:
- Couple LC to a high-resolution tandem mass spectrometer (e.g., Q-TOF, Orbitrap).
- Use Data-Dependent Acquisition (DDA) mode. Set MS1 scans for a broad mass range (e.g., m/z 100-1500).
- Configure DDA to fragment the top N most intense ions per cycle, with a dynamic exclusion window to ensure repeated sampling of low-abundance and co-eluting features.
- Acquire data in both positive and negative electrospray ionization (ESI) modes to capture a comprehensive ion profile [37].
- Data Format Conversion:
- Convert raw instrument files (.d, .raw) to an open format (.mzML, .mzXML) using tools like MSConvert (ProteoWizard).
Protocol 2: Computational Feature Detection and FBMN Construction on GNPS
Objective: To process raw LC-MS/MS data, detect chromatographic features, and construct a feature-based molecular network.
- Feature Detection and Alignment:
- Process the .mzML files using feature detection software such as MZmine 3 or OpenMS.
- Key Steps: Noise filtering, chromatogram building, m/z and RT deconvolution, isotope grouping, and alignment across samples.
- Critical Parameter Optimization: Adjust peak picking intensity thresholds, m/z and RT tolerance windows to balance sensitivity (capturing trace features) and specificity (avoiding noise).
- Export the results: a feature quantification table (containing Feature ID, m/z, RT, and peak area per sample) and a MS2 spectral summary file (.mgf) containing one representative MS2 spectrum per feature [3].
- Molecular Networking on GNPS:
- Navigate to the Feature-Based Molecular Networking workflow on the GNPS website (https://gnps.ucsd.edu).
- Upload the feature table (.csv) and the MS2 spectral summary file (.mgf).
- Set critical networking parameters:
- Minimum cosine score: 0.7 (typical for molecular family clustering).
- Minimum matched fragment ions: 6.
- Maximum RT difference: 0.2 min or 30 seconds (this parameter helps validate links between features from similar elution times).
- Submit the job. GNPS will generate a molecular network where nodes are LC-features, colored and sized by metadata (e.g., sample group, abundance), and edges represent spectral similarity.
Protocol 3: Advanced Integration with Ion Mobility Spectrometry
Objective: To integrate collisional cross-section (CCS) data from ion mobility spectrometry (IMS) for enhanced isomeric separation.
- Data Acquisition:
- Acquire data using an LC-IMS-MS/MS system (e.g., TIMS, DTIMS).
- Ensure the method collects both drift time and mass spectral data.
- Data Processing:
- Process data with IMS-aware software like MetaboScape or MS-DIAL that can perform feature detection incorporating the IMS dimension.
- The software will export a feature table including m/z, RT, and CCS value, alongside the MS2 spectral summary.
- FBMN on GNPS:
- Use the IMS-supported FBMN workflow on GNPS.
- Upload the CCS-enabled feature table. The networking algorithm can now use CCS as an additional orthogonal parameter to differentiate isomers that may co-elute but have different gas-phase structures [3].
Diagram 1: FBMN workflow from extract to target.
Diagram 2: Isomer resolution in classical vs FBMN.
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Essential Research Reagent Solutions for FBMN
| Item/Category | Function & Purpose | Example/Note |
|---|---|---|
| Extraction Solvents | To solubilize and extract diverse natural product scaffolds from biological matrices. | Methanol, Ethanol, Acetonitrile, Dichloromethane, Ethyl Acetate. Choice depends on target polarity [37]. |
| LC-MS Grade Mobile Phase | To provide high-purity solvents for chromatographic separation and stable electrospray ionization. | LC-MS grade Water, Acetonitrile, Methanol. Additives: Formic Acid (0.1%) or Ammonium Acetate for pH/modification. |
| Chromatography Columns | To separate compounds based on chemical properties (polarity, size). Critical for isomer resolution. | Reverse-phase (C18), HILIC, Phenyl, etc. Column length and particle size affect resolution [37]. |
| Mass Spectrometry Calibrants | To ensure accurate mass measurement (< 5 ppm error) for reliable formula prediction. | Sodium formate cluster or proprietary calibrant specific to instrument manufacturer. |
| Reference Standard Compounds | To validate identifications by matching RT and MS2 spectra, or for generating in-house spectral library entries. | Commercially available natural product standards relevant to the study system. |
| Data Processing Software | To convert raw data, detect chromatographic features, and perform statistical analysis. | MZmine 3, OpenMS (for feature detection); MS-DIAL, MetaboScape (for IMS data) [37] [3] [29]. |
| Online Platforms & Databases | To perform molecular networking, spectral library matching, and access reference data. | GNPS (primary FBMN platform), MassIVE (data repository), SIRIUS (for formula/ structure) [3] [29]. |
FBMN represents a critical methodological evolution within the field of natural product scaffold diversity analysis. By successfully resolving isomeric complexity and providing a quantitative framework, it transforms molecular networks from static pictures into dynamic, actionable research tools that directly guide the discovery of novel chemical entities [37] [3]. Its integration into a broader thesis on molecular networking underscores a shift towards data-driven, hypothesis-guided natural product research.
The future of FBMN is directed towards greater automation and integration. Challenges remain in the development of more robust, open-source mass spectrometry databases and standardized data formats to improve reproducibility and annotation rates [37]. Furthermore, the integration of FBMN with network pharmacology and toxicology provides a powerful systems biology approach to link specific chemical scaffolds, including isomers, directly to biological mechanisms and outcomes [37]. As computational power increases and algorithms improve, the application of FBMN to increasingly large-scale datasets (e.g., multi-omics studies) will further solidify its role as an indispensable tool for mapping and understanding the vast, untapped diversity of natural product scaffolds.
The discovery of novel natural products (NPs) with therapeutic potential is fundamentally constrained by analytical data complexity. Modern mass spectrometry (MS) generates high-dimensional data from complex biological extracts, where signals from genuine metabolites are entangled with noise, spectral artefacts, and interference from co-eluting compounds [29]. This complexity obscures molecular diversity and hampers efficient scaffold prioritization. Deconvolution—the computational separation of pure compound spectra from mixed signals—and the correction of ionization artefacts, such as ion suppression, are therefore critical pre-processing steps. When integrated with molecular networking, these refined data streams transform into powerful maps of chemical space, directly supporting a research thesis aimed at elucidating and prioritizing novel NP scaffolds for drug discovery [29] [39]. This article details application notes and protocols to manage these data challenges effectively.
Core Methodologies and Application Notes
Automated Spectral Deconvolution with MSHub
Principle: Raw GC-MS or LC-MS data contain overlapping signals from multiple co-eluting compounds and background noise. Deconvolution algorithms isolate pure compound fragmentation patterns. The MSHub engine employs unsupervised non-negative matrix factorization (NMF) to achieve this auto-deconvolution, quantifying the reproducibility of fragmentation patterns across samples [40].
Application Note: MSHub is integrated into the Global Natural Products Social (GNPS) ecosystem. This allows the deconvoluted spectra to be directly used for molecular networking, creating a seamless workflow from raw data to chemical family visualization [40]. Its primary value lies in converting complex, sample-level data into clean, compound-specific spectral nodes ready for network analysis and library matching.
Molecular Networking for Scaffold Visualization
Principle: Molecular networking clusters molecules based on the similarity of their MS/MS fragmentation spectra, visually grouping structurally related compounds [29]. Clusters (or "molecular families") often share core scaffolds decorated with different functional groups.
Application Note: Within a thesis on scaffold diversity, molecular networking serves as the central organizational framework. Feature-Based Molecular Networking (FBMN), which incorporates chromatographic alignment, is now the standard for LC-MS data as it improves accuracy by aligning the same feature across samples [29]. Networks prioritize isolation targets: large, unannotated clusters may represent novel scaffold families, while small clusters branching from known compounds may highlight new analogs [39].
Relative Mass Defect (RMD) Analysis for Novelty Prioritization
Principle: The mass defect is the difference between a compound's exact mass and its nominal mass. The Relative Mass Defect (RMD), normalized by molecular weight, is characteristic of compound classes (e.g., peptides, lipids, glycosides) due to their typical hydrogen-to-carbon ratios [39].
Application Note: RMD analysis filters molecular networking results to flag clusters with high structural novelty. As demonstrated, an unannotated cluster with an RMD value typical of oligopeptides but lacking characteristic UV or MS/MS signatures for peptides suggests a divergent scaffold [39]. This method efficiently triages the most promising, novel scaffold-containing clusters from thousands of network nodes for further investigation.
Ionization Artefact Correction via IROA Workflow
Principle: Ion suppression during electrospray ionization is a major matrix effect where co-eluting compounds reduce the ionization efficiency of analytes, leading to inaccurate quantification and reduced sensitivity [41].
Application Note: The IROA (Isotopic Ratio Outlier Analysis) TruQuant Workflow uses a stable isotope-labeled internal standard (IROA-IS) library to measure and correct for ion suppression in real-time [41]. A unique isotopolog ladder pattern distinguishes real metabolites from artefacts. This correction is vital for ensuring that the intensity of nodes in a molecular network reflects true biological abundance rather than analytical bias, leading to more reliable quantitative comparisons across samples in a dataset.
Detailed Experimental Protocols
Protocol 1: Integrated Deconvolution and Molecular Networking Workflow
This protocol outlines the steps from raw LC-MS data to a refined molecular network using GNPS.
Data Acquisition:
- Analyze natural product extracts via LC-MS/MS in data-dependent acquisition (DDA) mode [29].
- Include procedural blanks and pooled quality control samples.
Data Conversion and Feature Detection:
Auto-Deconvolution and Networking on GNPS:
- Upload the feature quantification table (.csv) and aligned MS/MS spectra (.mgf) to the GNPS website [43].
- Select the "Feature-Based Molecular Networking" (FBMN) workflow.
- For GC-MS data, utilize the integrated MSHub deconvolution option [40].
- Set key parameters [43]:
- Precursor Ion Mass Tolerance: 0.02 Da for high-res instruments.
- Fragment Ion Mass Tolerance: 0.02 Da.
- Minimum Cosine Score for Networking: 0.7.
- Minimum Matched Fragment Peaks: 6.
- Submit the job and retrieve results via the provided link.
Protocol 2: RMD-Assisted Novelty Detection for Targeted Isolation
This protocol uses mass defect analysis to prioritize novel scaffolds from a molecular network [39].
Network Cluster Selection:
- From the GNPS network, export the list of nodes (compounds) within unannotated clusters.
- Prioritize clusters that are large (>5 nodes), unique to a specific sample or taxon, and have no library matches.
RMD Calculation and Plotting:
- For each node in the target cluster, calculate the RMD using the formula [39]:
RMD (ppm) = (Exact Mass - Nominal Mass) / Exact Mass * 10^6. - Plot the RMD (y-axis) against the exact mass (x-axis) for all nodes.
- On the same plot, add reference points for known compound classes (e.g., peptides, polyketides) derived from databases like Natural Products Atlas.
- For each node in the target cluster, calculate the RMD using the formula [39]:
Novelty Flagging:
- Identify clusters whose average RMD falls within a known class but whose spectral data (MS/MS pattern, UV profile) are incongruent with that class.
- These flagged clusters become high-priority targets for purification and structural elucidation, as they likely possess a novel scaffold.
Protocol 3: Ion Suppression Correction Using IROA Standards
This protocol details the use of IROA standards for artefact correction prior to networking [41].
Sample and Standard Preparation:
- Spike a constant amount of IROA Internal Standard (IROA-IS) into each natural product extract sample immediately prior to LC-MS injection.
- Prepare an IROA Long-Term Reference Standard (IROA-LTRS) as a 1:1 mixture of light (5% 13C) and heavy (95% 13C) versions of the standard mixture for system performance monitoring.
Data Acquisition and Processing:
- Acquire LC-MS/MS data for all samples and the LTRS.
- Process data using ClusterFinder software or compatible tools that recognize the IROA isotopolog ladder pattern.
Suppression Correction and Normalization:
- The software automatically identifies true metabolites by their IROA pattern and applies an algorithm to correct the peak area of each endogenous (12C) metabolite based on the observed suppression of its corresponding internal standard (13C) spike-in [41].
- Perform Dual MSTUS (MS Total Useful Signal) normalization using the corrected values to account for overall sample concentration differences.
- Export the corrected, normalized feature table for subsequent molecular networking.
Table 1: Comparison of Molecular Networking Approaches for Natural Product Discovery
| Networking Approach | Key Principle | Data Input | Primary Utility in Scaffold Analysis | Typical Annotation Rate |
|---|---|---|---|---|
| Classical MN [29] | Cosine similarity of MS/MS spectra | Aligned MS/MS spectra | Visual grouping of related spectra | Varies widely |
| Feature-Based MN (FBMN) [29] | Integrates chromatographic peak shape/alignment | Feature table + aligned MS/MS | Improves accuracy across samples; links analogs | Higher than Classical |
| ION Identity MN (IIMN) [29] | Groups different ion forms (adducts, fragments) of same molecule | Feature table + MS/MS | Reduces node redundancy; clarifies true molecular diversity | N/A |
| RMD-Assisted MN [39] | Filters networks using mass defect outliers | Network nodes + exact mass | Prioritizes clusters with anomalous mass defects as novel scaffold candidates | N/A |
Table 2: Impact and Correction of Ionization Artefacts
| Artefact/Parameter | Description | Typical Impact on Data | Correction Method | Efficacy of Correction |
|---|---|---|---|---|
| Ion Suppression [41] | Reduced ionization efficiency due to matrix | Non-linear response; loss of sensitivity (>90% suppression possible) | IROA-IS with algorithmic correction | Restores linearity; corrects up to ~99% suppression |
| Mass Accuracy | Deviation of measured m/z from true value | Misidentification of molecular formula | High-res calibration; lock mass | Can achieve <1 ppm error |
| Chromatographic Shift | Retention time variability across runs | Misalignment of same feature | Quality control-based alignment (e.g., in MZmine) | Sub-minute alignment possible |
Visualization of Workflows and Mechanisms
Diagram 1: Integrated Data Analysis Workflow for Scaffold Discovery
Diagram 2: IROA Workflow for Ion Suppression Correction
Diagram 3: Spectral Deconvolution Process for Network Node Creation
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Computational Tools for Managing MS Data Complexity
| Item Name / Tool | Type | Primary Function in Workflow | Key Notes / Source |
|---|---|---|---|
| IROA Internal Standard (IROA-IS) | Chemical Standard | Corrects ion suppression; enables quantitative normalization [41]. | Spiked into every sample prior to injection. |
| IROA Long-Term Reference Standard (LTRS) | Chemical Standard | Monitors LC-MS system performance over time [41]. | Run at intervals during acquisition batch. |
| GNPS Platform | Web-based Software Ecosystem | Hosts workflows for molecular networking, library search, and deconvolution (MSHub) [40] [43]. | Primary platform for community analysis. |
| MSHub | Computational Algorithm (in GNPS) | Performs auto-deconvolution of GC-/LC-MS data via NMF [40]. | Converts raw data to clean spectra for networking. |
| MZmine 3 | Open-Source Software | Performs feature detection, chromatographic alignment, and ion mobility data handling [39] [42]. | Critical pre-processing step before GNPS. |
| NPClassifier / Natural Products Atlas | Database & Tool | Provides compound class and taxonomic data for RMD reference plotting [39]. | Used for novelty assessment. |
| ClusterFinder | Proprietary Software | Processes IROA data, applies suppression correction, and performs Dual MSTUS normalization [41]. | Essential for IROA workflow execution. |
| ISP-2 Medium | Culture Media | Standardized medium for actinomycete cultivation, promoting diverse NP production with low MS interference [44]. | Used in generating microbial extracts for analysis. |
Within the broader thesis on molecular networking for natural product scaffold diversity analysis, a central and persistent bottleneck is the annotation of unknown molecular scaffolds. Traditional database-dependent identification methods fail when confronted with novel chemical entities, which are abundant in nature. This document synthesizes current, advanced computational and integrative strategies designed to overcome these database gaps. The focus is on practical Application Notes and Protocols that empower researchers to progress from uncharacterized mass spectrometry features to annotated molecular families and filled genomic scaffolds, thereby illuminating the dark matter of metabolomics and genomics.
Strategic Frameworks and Quantitative Comparisons
Gap-Filling in Genomic Scaffolds
Draft genome and metagenome assemblies often result in gapped scaffold sequences. Automated closure of these gaps is crucial for obtaining complete genetic blueprints, which is a prerequisite for accurate biosynthetic gene cluster analysis in natural product research. The following table summarizes the performance of key algorithms on bacterial datasets, highlighting trade-offs between accuracy and completeness [45].
Table 1: Performance Comparison of Genomic Gap-Filling Algorithms on Bacterial Datasets [45]
| Metric | Original Assembly | IMAGE | SOAPdenovo | GapFiller | GapFiller-LC (Low Coverage) |
|---|---|---|---|---|---|
| Escherichia coli | |||||
| Gap Count | 544 | 291 | 16 | 11 | - |
| Total Gap Length (bp) | 12,516 | 2,861 | 16 | 130 | - |
| Errors (SNPs + Indels + Misjoins) | 17 | 58 | 59 | 32 | - |
| Streptomyces coelicolor | |||||
| Gap Count | 158 | 63 | 60 | 23 | - |
| Total Gap Length (bp) | 9,221 | 4,009 | 1,288 | 806 | - |
| Errors (SNPs + Indels + Misjoins) | 975 | 1,117 | 1,193 | 984 | - |
| Staphylococcus aureus | |||||
| Gap Count | 48 | 27 | 27 | 22 | 22 |
| Total Gap Length (bp) | 9,900 | 1,547 | 5,508 | 1,861 | ~1,547* |
| Errors (SNPs + Indels + Misjoins) | 99 | 326 | 131 | 215 | <131* |
| Rhodobacter sphaeroides | |||||
| Gap Count | 170 | 163 | 161 | 139 | 139 |
| Total Gap Length (bp) | 21,409 | 14,166 | 20,667 | 17,625 | ~14,166* |
| Errors (SNPs + Indels + Misjoins) | 411 | 714 | 426 | 506 | <426* |
*GapFiller-LC uses less stringent parameters to achieve gap lengths comparable to other tools while maintaining a lower error count [45].
Annotation of Natural Product Molecular Families
The challenge of identifying unknown compounds in metabolomics is exacerbated by limited reference spectra. The SNAP-MS (Structural similarity Network Annotation Platform for Mass Spectrometry) strategy provides a database-agnostic solution by leveraging the intrinsic clustering of natural product scaffolds [2].
Table 2: Diagnostic Power of Molecular Formula Distributions for Compound Family Annotation (Natural Products Atlas Data) [2]
| Analysis Unit | Total Unique Instances Analyzed | Instances Found in ONLY ONE Compound Family | Diagnostic Power |
|---|---|---|---|
| Single Molecular Formula | 4,317 | 1,554 | 36% |
| Pair of Molecular Formulae | 431,700 | 411,681 | 95.4% |
| Set of Three Molecular Formulae | Not Specified | >97% of cases | >97% |
This data demonstrates that while single formulae are poor identifiers, combinations of 2-3 formulae within a molecular network cluster are highly diagnostic for a specific natural product family [2].
Detailed Experimental Protocols
Protocol: Manual Scaffold Extension and Gap Closure for Metagenome-Assembled Genomes (MAGs)
This protocol, adapted from established curation pipelines, details the manual refinement of draft scaffolds using read mapping and visualization to achieve complete genomes [46].
I. Read Mapping and Preparation
- Index the Draft Scaffold: Build a mapping database from your scaffold FASTA file.
- Map Paired-End Reads: Align your sequencing reads to the scaffold. Use parameters (
-X) appropriate for your library insert size.
II. Visualization and Manual Curation in Geneious
- Import Data: Load the scaffold FASTA file and the mapped reads (BAM file) into Geneious.
- Identify Extension Points: Visually inspect the ends of the scaffold. Look for paired reads where one mate is mapped and the other ("unplaced") extends into the unknown region.
- Validate and Extend:
- For a candidate unplaced read, locate its mate within the mapped data to confirm orientation and approximate expected distance.
- If validated, add the consensus sequence of the overhanging portion of the unplaced read to the scaffold sequence.
- Iterate: Re-map all unused reads to the newly extended scaffold. Repeat the inspection and extension process until no further confident extension can be made.
- Close Gaps (Ns):
- Locate a scaffold region containing a gap (a run of
Nbases). - Visually examine the read alignment over the gap. If reads span the gap, manually adjust the alignment to merge overlapping sequences from either side, removing the
Ns [46]. - Ensure the closed sequence is supported by the consensus of spanning reads.
- Locate a scaffold region containing a gap (a run of
Protocol: Integrative Molecular Networking with MolNetEnhancer
This protocol uses MolNetEnhancer to combine multiple in-silico annotation tools, thereby enhancing the chemical interpretation of molecular networking data [32].
I. Prerequisite Analyses Run the following analyses independently, using standard parameters:
- Molecular Networking (MN): Create a molecular network via the GNPS platform using MS/MS data.
- In-Silico Annotation: Annotate nodes using one or more tools:
- Network Annotation Propagation (NAP) on GNPS.
- SIRIUS+CSI:FingerID for fragmentation tree-based candidate prediction.
- DEREPLICATOR/VarQuest for peptidic natural products.
- Substructure Discovery: Run MS2LDA to discover conserved Mass2Motifs (substructures) from the MS/MS spectra.
II. Integration with MolNetEnhancer
- Installation: Install the MolNetEnhancer package from GitHub (Python or R version) [32].
- Data Integration: Use the MolNetEnhancer functions to merge the result files from Step I.
- Input: GNPS network files (e.g.,
networkedges_selfloop), annotation files (e.g., NAP results), and MS2LDA output files (e.g.,motif_edges).
- Input: GNPS network files (e.g.,
- Chemical Classification: The workflow automatically submits candidate structures from annotation tools to ClassyFire for hierarchical chemical classification.
- Output and Visualization: The primary output is an enhanced network file (
.graphml) where nodes are colored by chemical class and edges can represent shared substructures (Mass2Motifs). Visualize and explore this network in Cytoscape.
Visualization of Workflows and Relationships
Molecular Networking to Scaffold Annotation Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software Tools and Databases for Scaffold Annotation and Gap-Closing
| Tool/Resource | Category | Primary Function | Application Note |
|---|---|---|---|
| GapFiller [45] | Genomic Gap-Closing | Uses paired-read libraries to reliably close gaps within draft scaffolds. | Optimal for bacterial/eukaryotic drafts where insert size is known. Prioritizes accuracy over aggressive closure. |
| nanoGapFiller [47] | Genomic Gap-Closing | Uses optical maps and a probabilistic search of assembly graphs to fill long gaps in scaffolds. | Essential for closing very long gaps (>1 kbp) where short-read methods fail. Requires optical mapping data. |
| GNPS Molecular Networking [2] [32] | Metabolomics Analysis | Groups MS/MS spectra based on similarity to create molecular families. | The foundational platform for organizing unknown metabolomics data into chemically related clusters. |
| SNAP-MS [2] | Metabolomics Annotation | Annotates molecular network clusters using diagnostic molecular formula distributions without reference spectra. | Powerful for de novo annotation of compound families when reference spectra are unavailable. |
| MolNetEnhancer [32] | Metabolomics Integration | Integrates outputs from molecular networking, in-silico annotation tools, and substructure discovery into a unified network. | Crucial for synthesizing multiple lines of weak evidence into confident annotations and visualizing chemical class distributions. |
| Natural Products Atlas [2] | Chemical Database | A comprehensive database of microbial natural product structures with associated compound family classifications. | Serves as the reference knowledge base for the formula distributions used by SNAP-MS and for chemical context. |
| Bowtie2 / Geneious [46] | Read Mapping & Visualization | Aligns sequencing reads to reference scaffolds and enables manual inspection and curation. | The core wet-lab informatics pipeline for manual, evidence-based scaffold extension and gap closure. |
Proof and Perspective: Validating MN Against Traditional Methods and Emerging Tech
Within the broader thesis on molecular networking for natural product scaffold diversity analysis, a central challenge is efficiently translating vast chemical diversity into tangible drug discovery successes. Traditional screening of massive, uncharacterized extract libraries is plagued by low hit rates, high rediscovery rates of known compounds, and significant resource expenditure on the isolation of inactive or nuisance compounds [39].
This application note presents a refined discovery strategy that addresses this inefficiency. The core thesis is that rational library minimization, guided by pre-screening analytical data, directly increases bioassay hit rates. By integrating mass spectrometry-based molecular networking with mass defect filtering, researchers can prioritize chemical novelty and scaffold diversity before biological testing [39] [2]. This workflow shifts the paradigm from "isolate and then test" to "select, then isolate and test," ensuring that precious assay resources are deployed against the most promising, novel chemical entities.
The methodology hinges on two key principles: First, molecular networking clusters metabolites by MS/MS spectral similarity, visually mapping the chemical space of a library and grouping analogs [39] [2]. Second, Relative Mass Defect (RMD) analysis acts as a novelty filter. RMD, calculated from high-resolution MS data, is characteristic of compound classes (e.g., peptides, polyketides) [39]. Clusters with spectral and UV profiles incongruent with their RMD-predicted class are flagged as potential scaffold innovators. This targeted approach minimizes library size by focusing isolation efforts exclusively on these high-priority clusters, thereby quantifiably enhancing the probability of discovering bioactive, novel leads.
The implementation of the molecular networking and RMD-filtering workflow has yielded significant, measurable improvements in discovery efficiency. The following tables summarize key quantitative outcomes from its application.
Table 1: Comparative Bioassay Hit Rates Before and After Library Minimization
| Screening Approach | Total Fractions/Libraries Screened | Hits Identified | Hit Rate (%) | Confirmed Novel Actives |
|---|---|---|---|---|
| Untargeted (Traditional) | 1,200 | 15 | 1.25 | 2 |
| Rationally Minimized (This Workflow) | 52 | 8 | 15.38 | 7 |
Data Summary: This table presents a comparison of two screening campaigns against Mycobacterium smegmatis. The untargeted approach screened a broad library of 1,200 pre-fractionated microbial extracts. The targeted approach applied the described workflow to a subset of 6 actinobacterial strains, resulting in a prioritized library of 52 fractions from specific molecular families [39].
Table 2: Molecular Networking Metrics and Prioritization Efficiency
| Metric | Value | Description |
|---|---|---|
| Total MS/MS Spectra Processed | 15,840 | From organic extracts of 6 actinobacterial strains [39]. |
| Molecular Network Nodes | 3,446 | Individual MS/MS spectra features clustered [39]. |
| Molecular Network Clusters | 456 | Groups of spectrally similar nodes [39]. |
| Annotated Clusters (Known Classes) | 33 | Dereplication via spectral libraries [39]. |
| High-Priority Novelty Clusters Identified | 5 | Selected based on RMD incongruence and topology [39]. |
| Library Minimization Factor | ~8.7x | Ratio of total clusters (456) to prioritized clusters (52 associated with 5 families). |
Table 3: Key Quantitative Data for Brasiliencin Discovery Campaign
| Parameter | Result | Significance |
|---|---|---|
| Target Cluster RMD Value | 557 ppm | Predicted oligopeptide class [39]. |
| Observed UV Profile | No absorbance at 200-230 nm, 250-350 nm | Contradicted peptide prediction, signaling novelty [39]. |
| Minimum Inhibitory Concentration (MIC) - Brasiliencin A | 31.3 nM (vs. M. smegmatis) | Validated potent bioactivity of the isolated novel scaffold [39]. |
| Analog Series Detected via AMDF | 29 analogs | Demonstrated the power of absolute mass defect filtering to expand a hit series [39]. |
| Novel Compounds Isolated | 4 (Brasiliencins A-D) | Direct output from the prioritized cluster [39]. |
Detailed Experimental Protocols
Protocol 1: Generating a Prioritized Library via Molecular Networking & RMD Analysis
Objective: To process microbial extracts, construct a molecular network, and identify high-priority clusters representing potential novel scaffolds.
Materials:
- Bacterial strains (e.g., actinomycetes from desert soils) [39].
- Fermentation media (ISP1, ISP2, Actinomycete Isolation Agar) [39].
- Solvents: Ethyl acetate, n-butanol, methanol (HPLC grade).
- UHPLC-HRMS system equipped with a C18 column.
- Software: MZmine 2 or similar for feature detection, GNPS platform for molecular networking, Agilent MassHunter for isotope simulation [39].
Procedure:
Fermentation & Extraction:
- Culture bacterial strains in triplicate in 3 different media (e.g., ISP1, ISP2, agar plates) for 7 days [39].
- Extract metabolites by homogenizing broth or agar with equal volumes of ethyl acetate and n-butanol separately [39].
- Combine organic layers, evaporate to dryness under reduced pressure, and reconstitute in methanol for analysis.
LC-MS/MS Data Acquisition:
- Analyze each reconstituted extract via UHPLC-HRMS using a C18 gradient (e.g., 5-100% acetonitrile in water, 0.1% formic acid over 20 min).
- Use data-dependent acquisition (DDA) to collect high-resolution MS1 and MS/MS (MS2) spectra.
Data Processing & Molecular Networking:
- Process raw data with MZmine 2: perform chromatogram building, deconvolution, deisotoping, and alignment [39].
- Export the MS/MS spectral data (in .mgf format) and feature quantification table.
- Upload to the GNPS platform . Create a molecular network using the
Feature-Based Molecular Networkingworkflow. Use default parameters: precursor ion mass tolerance 0.02 Da, fragment ion tolerance 0.02 Da, minimum cosine score 0.7, minimum matched peaks 6 [2].
Cluster Prioritization with RMD Analysis:
- For unannotated clusters in the network, calculate the Relative Mass Defect (RMD) for the parent ion of each node using the formula: RMD = (Exact Mass - Nominal Mass) / Exact Mass * 10⁶ [39].
- Compare the average cluster RMD to reference RMD values for common natural product classes (e.g., peptides ~500-600 ppm, polyketides often lower) [39].
- Cross-reference with available UV spectra from the LC-DAD. Flag clusters where the RMD-predicted class conflicts with spectral evidence (e.g., RMD suggests peptides, but UV shows no amide/aromatic amino acid absorbance) [39].
- Prioritize flagged clusters that are (i) unannotated, (ii) contain >5 nodes (suggesting an analog series), and (iii) are unique to a specific microbial genus [39].
Protocol 2: Bioassay-Guided Isolation from a Prioritized Cluster
Objective: To isolate and elucidate the structure of a bioactive compound from a prioritized molecular family.
Materials:
- Large-scale culture (4-10 L) of the source microbial strain.
- Solvents for chromatography: hexane, dichloromethane, ethyl acetate, methanol.
- Normal-phase and reversed-phase silica gel for column chromatography.
- Semi-preparative HPLC system with C18 column.
- Bioassay materials (e.g., Mycobacterium smegmatis culture, agar, Alamar Blue reagent for viability).
- NMR spectrometer (500 MHz or higher), HRESIMS.
Procedure:
Large-Scale Fermentation & Extraction:
- Scale up culture of the producing strain in the optimal medium identified from Protocol 1.
- Perform extraction as in Protocol 1, but on a multi-liter scale.
Bioassay-Guided Fractionation:
- Fractionate the crude extract using vacuum liquid chromatography (VLC) with a stepwise gradient of solvents of increasing polarity (e.g., hexane → EtOAc → MeOH).
- Test all fractions for bioactivity using a standardized microplate assay (e.g., against M. smegmatis using Alamar Blue after 48-72 hours incubation) [39].
- Pool active fractions and further separate via normal-phase or size-exclusion chromatography.
- Continuously track the target molecular family using LC-HRMS, monitoring the characteristic ions and retention times from the original molecular network cluster.
Final Purification & Structure Elucidation:
- Purify the leading bioactive compound(s) to homogeneity using semi-preparative reversed-phase HPLC.
- Determine molecular formula via high-resolution ESI-MS.
- Acquire 1D and 2D NMR data (¹H, ¹³C, COSY, HSQC, HMBC) in deuterated solvent. Use NMR to establish the planar structure [39].
- Determine absolute configuration via methods such as quantum chemical calculation of ECD spectra and comparison with experimental data [39].
Visualizing the Workflow: Key Diagrams
Diagram 1: Rationally Minimized Library Discovery Workflow This diagram outlines the core integrated workflow for increasing bioassay hit rates [39].
Diagram 2: Molecular Networking Cluster Analysis Logic This diagram details the decision logic applied to each molecular network cluster to prioritize novel scaffolds [39] [2].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for Implementation
| Item | Function in Workflow | Specification/Example |
|---|---|---|
| ISP Media Broths | Cultivation of diverse actinobacteria to induce secondary metabolism [39]. | ISP1 & ISP2 from BD Difco. |
| Ethyl Acetate (EtOAc) | Primary solvent for liquid-liquid extraction of mid-polarity natural products from fermentation broth [39]. | HPLC Grade, ≥99.9%. |
| n-Butanol (n-BuOH) | Co-solvent for extracting more polar metabolites; used in conjunction with EtOAc for comprehensive coverage [39]. | HPLC Grade, ≥99.9%. |
| Methanol (MeOH) | Reconstitution solvent for LC-MS samples and for reversed-phase chromatography [39]. | LC-MS Grade, with 0.1% formic acid. |
| C18 UHPLC Column | High-resolution chromatographic separation of complex extract mixtures prior to MS analysis [39]. | e.g., 2.1 x 100 mm, 1.7 µm particle size. |
| Semi-Preparative HPLC Column | Final purification step for isolating milligram quantities of target compounds for structure elucidation and assay. | e.g., 10 x 250 mm, 5 µm C18 silica. |
| Deuterated NMR Solvent | Solvent for nuclear magnetic resonance spectroscopy, essential for structural determination of isolated novel compounds [39]. | e.g., Deuterated methanol (CD₃OD) or chloroform (CDCl₃). |
| Alamar Blue Cell Viability Reagent | Fluorometric/colorimetric indicator used in microplate bioassays to determine the antibacterial activity of fractions and pure compounds [39]. | Commercially available resazurin-based reagent. |
| SNAP-MS Algorithm | Cheminformatic tool for annotating molecular network clusters by comparing formula distributions to known compound families, aiding in dereplication and novelty assessment [2]. | Freely accessible at www.npatlas.org/discover/snapms. |
Quantitative Benchmarking of Discovery Approaches
The systematic comparison of computational library rationalization against traditional bioactivity-guided fractionation reveals significant differences in efficiency, cost, and strategic output. The following table synthesizes key performance metrics from recent studies.
Table 1: Benchmarking Computational Rationalization vs. Traditional Fractionation
| Performance Metric | Computational Library Rationalization (MS/MS & Molecular Networking) | Traditional Bioactivity-Guided Fractionation | Data Source & Context |
|---|---|---|---|
| Initial Library Size Reduction | 84.9% reduction (1,439 to 216 extracts for 100% scaffold diversity); 28.8-fold reduction (to 50 extracts) for 80% diversity [4]. | Not applicable; process begins with a single active crude extract and iteratively fractionates it. | Study on a fungal extract library (1,439 samples) rationalized via LC-MS/MS and GNPS [4]. |
| Scaffold Diversity Retention | Retains 100% of molecular scaffolds (structural families) with the minimized library [4]. | Focuses on a single bioactive scaffold; diversity is lost as isolation progresses. | Defined by the percentage of molecular families (nodes in a molecular network) retained [4]. |
| Bioactive Candidate Retention | Retained 8 out of 10 bioactivity-correlated MS features (80%) in an 80%-diversity library; 100% retained in full-diversity library [4]. | The primary goal is 100% retention and isolation of the specific bioactive compound driving the assay. | Features significantly correlated with anti-Plasmodium activity in the full library [4]. |
| Bioassay Hit Rate Improvement | Hit rate increased from 11.3% to 22.0% (anti-P. falciparum) and from 2.57% to 8.0% (neuraminidase) in the minimized 80%-diversity library [4]. | Hit rate is 100% for the parent fraction in each iterative step but requires extensive resources per active. | Comparison of full library vs. rationalized library hit rates in phenotypic and target-based assays [4]. |
| Primary Resource Savings | Drastically reduces materials, reagents, and labor for initial HTS campaigns. Major saving is in time-to-decision. | Extremely resource-intensive: requires large-scale culture/extraction, repeated fractionation, and constant bioassay guidance. | Capital and operational expenditures for screening are directly proportional to library size [4] [48]. |
| Typical Time Frame (Hit ID to Lead) | Weeks to months. Rapid prioritization of distinct, bioactive extracts accelerates the start of isolation. | Months to years. Bottlenecked by the cycle of fractionation, evaporation, and bioassay testing [49] [48]. | Duration heavily dependent on compound complexity and assay turnaround time [49]. |
| Output for Scaffold Diversity Analysis | Ideal. Provides a prioritized, chemically diverse set of extracts enriched for novel scaffolds and bioactivity [4] [19]. | Limited. Yields one or a few closely related scaffolds from a single source. Requires multiple parallel campaigns for diversity [48]. | Core to the thesis on leveraging molecular networking for scaffold diversity analysis [4] [49]. |
Application Note 1: Rational Natural Product Library Design for Enhanced Screening
Objective: To reduce the time and cost of initial high-throughput screening (HTS) by constructing a minimal natural product extract library that maximizes chemical scaffold diversity and bioactivity potential [4]. Rationale: Large libraries of natural product extracts contain significant structural redundancy, leading to wasted resources on re-isolating known compounds and screening chemically similar samples [4] [48]. A rational, diversity-driven design increases the probability of encountering novel bioactive scaffolds in the first screening round. Protocol Summary: Untargeted LC-MS/MS data is acquired for all library extracts. MS/MS spectra are processed through the Global Natural Products Social Molecular Networking (GNPS) platform to create a molecular network, where each node represents a unique molecular scaffold or closely related analogue [4] [49]. Custom algorithms then iteratively select the extract containing the greatest number of scaffolds not yet represented in the rationalized subset until a user-defined diversity threshold (e.g., 80% of all scaffolds) is met [4]. Key Outcome: This method enabled a 28.8-fold reduction in library size (from 1,439 to 50 extracts) while retaining 80% of scaffold diversity. Crucially, the hit rate against three different therapeutic targets increased significantly (e.g., from 2.57% to 8.00% for neuraminidase), demonstrating that the method enriches for bioactive extracts while saving >95% of initial screening resources [4].
Application Note 2: Integrating Predictive Models for Targeted Isolation
Objective: To accelerate the isolation of novel bioactive compounds by using computational activity predictions to prioritize specific molecular families (network nodes) for purification. Rationale: Molecular networking groups compounds by structural similarity, but not all clusters are bioactive. Integrating bioassay results with predictive models can identify "interesting" clusters for targeted isolation, even before full bioactivity data is available for all members [50] [19]. Protocol Summary: Following a primary screen of the rationalized library, active extracts are analyzed. The molecular features (mass-retention time pairs) of the active extract are correlated with the bioassay data. Features with significant positive correlations are mapped back onto the pre-constructed molecular network [4]. Concurrently, machine learning models (e.g., trained on the CARA benchmark for compound activity prediction) can be used to score all detected molecular families in the network for their likelihood of exhibiting the desired activity based on their MS/MS spectral "fingerprint" or predicted structural features [50] [51]. Clusters highlighted by both experimental correlation and computational prediction receive the highest priority for subsequent isolation. Key Outcome: Creates a data-driven triage system, moving from random bioactivity-guided isolation to a targeted approach focused on molecular families with the highest predicted value, thereby reducing wasted effort on inactive compounds [50] [48].
Experimental Protocol 1: LC-MS/MS-Based Library Rationalization Workflow
1. Sample Preparation & Data Acquisition:
- Prepare fungal/bacterial extracts or fractionated samples in a suitable LC-MS solvent (e.g., 80% methanol). Ensure inclusion of blank controls.
- Acquire untargeted LC-MS/MS data using a high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap). Use reversed-phase chromatography (e.g., C18 column) with a water-acetonitrile gradient.
- Settings: Collect data-dependent MS/MS spectra (top N per cycle) across a suitable mass range (e.g., m/z 100-1500). Use stepped collision energies to fragment ions.
2. Molecular Networking & Dereplication:
- Process raw data using MZmine, OpenMS, or similar software for feature detection, alignment, and gap filling.
- Export MS/MS spectral data (in .mgf format) and feature intensity table.
- Upload data to the GNPS platform (gnps.ucsd.edu). Create a molecular network using the "Classical Molecular Networking" workflow [4] [49].
- Parameters: Set precursor ion mass tolerance (e.g., 0.02 Da) and MS/MS fragment ion tolerance (e.g., 0.02 Da). Set a cosine score threshold (e.g., 0.7) and minimum matched peaks (e.g., 6). Perform library search against public spectral libraries for dereplication.
3. Rational Library Selection:
- Download the network data (e.g., graphml file) and the correspondence between MS features and source samples.
- Using custom R or Python scripts, calculate the unique scaffold composition of each extract based on the molecular network clusters (nodes) [4].
- Implement an iterative selection algorithm:
- Select the extract with the highest number of unique scaffold nodes.
- Add it to the rational library list and mark all scaffolds in this extract as "covered."
- Recalculate, selecting the next extract that adds the most "uncovered" scaffolds.
- Repeat until a target percentage of total unique scaffolds is achieved (e.g., 80%) [4].
4. Validation:
- Subject the rationally selected mini-library to biological screening.
- Compare hit rates and the retention of known bioactive features to those of the full library and randomly selected libraries of the same size [4].
Experimental Protocol 2: Benchmarking Predictive Models for Activity
1. Data Curation for Benchmarking (CARA Framework):
- Source compound-protein activity data from public repositories like ChEMBL or BindingDB [50].
- Categorize assays into two task types: Virtual Screening (VS) assays (diverse compounds, hit identification) and Lead Optimization (LO) assays (congeneric compounds, SAR studies) [50]. This distinction is critical for fair benchmarking.
- Design appropriate data splitting strategies. For VS tasks, use temporal or protein-family-aware splits to simulate real-world discovery. For LO tasks, scaffold-based splits within an assay are appropriate [50].
2. Model Training & Evaluation:
- Train state-of-the-art activity prediction models, such as graph neural networks (GNNs), transformer-based models (e.g., ChemBERTa), or traditional fingerprint-based models (e.g., ECFP with Random Forest) [19].
- Evaluate models on the CARA benchmark using metrics suited to the task: e.g., Area Under the Precision-Recall Curve (AUPRC) for imbalanced VS tasks, and Mean Squared Error (MSE) or ranking metrics for LO tasks [50].
- Analyze model performance in "few-shot" and "zero-shot" scenarios relevant to novel natural product scaffold discovery [50].
3. Integration with Experimental Data:
- Use the trained model to predict activities for molecular features detected in untargeted metabolomics but not yet tested.
- Prioritize clusters in a molecular network that contain features with high predicted activity scores for subsequent isolation and testing [50] [19].
Visualizing Workflows and Pathways
Diagram 1: Comparative NP Discovery Workflows
Diagram 2: Molecular Networking Data Pipeline
Diagram 3: Predictive & Experimental Feedback Loop
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents, Instruments, and Software for Efficient NP Discovery
| Tool / Reagent | Function & Role in Workflow | Key Considerations |
|---|---|---|
| High-Resolution LC-MS/MS System | Generates the primary spectral data for molecular networking and dereplication. Essential for detecting and fragmenting thousands of metabolites [4] [49]. | Q-TOF or Orbitrap instruments provide the mass accuracy and resolution needed for reliable networking. |
| GNPS Platform | A free, cloud-based ecosystem for performing molecular networking, library searches, and community data sharing. It is the central computational engine for scaffold-based analysis [4] [49]. | Requires data in .mgf format. Understanding parameters like cosine score and minimum matched peaks is crucial. |
| MZmine / MS-DIAL | Open-source software for processing raw LC-MS data. Performs peak picking, alignment, deisotoping, and gap filling to create the feature table needed for quantification and correlation analysis [49]. | A critical step before GNPS analysis. Parameters must be optimized for specific instrumentation and sample types. |
| CARA Benchmark Dataset | A curated benchmark for fairly evaluating compound activity prediction models, distinguishing between Virtual Screening and Lead Optimization tasks [50]. | Used to train and validate predictive models before applying them to prioritize natural product clusters. |
| Graph Neural Network (GNN) Models | A class of AI models that operate directly on graph representations of molecules (atoms as nodes, bonds as edges). Ideal for learning from molecular structures and predicting properties like bioactivity [19]. | More powerful than fingerprint-based models for capturing complex structure-activity relationships. Requires programming expertise (PyTorch, DGL). |
| Bioassay Kits & Reagents | For phenotypic (e.g., anti-parasitic, cytotoxicity) or target-based (e.g., enzyme inhibition) screening. The choice defines the biological context of the discovery campaign [4] [48]. | Assay robustness and adaptability to HTS formats (384-well) are key for screening rationalized libraries. |
| Solid Phase Extraction (SPE) & HPLC Columns | For the rapid fractionation of active hits identified from the mini-library. Enables quick follow-up to isolate the bioactive compound[s [48]. | Necessary for transitioning from a prioritized extract to pure compounds for structural elucidation. |
Thesis Context: Molecular Networking for Scaffold Diversity Analysis
Within the evolving paradigm of natural product (NP) drug discovery, the analysis of scaffold diversity is paramount for uncovering novel chemical entities with therapeutic potential. This article frames a comparative analysis within a broader thesis investigating molecular networking as a central tool for this purpose. Molecular networking, particularly via platforms like GNPS (Global Natural Products Social Molecular Networking), visualizes complex metabolite mixtures by grouping molecules based on similarities in their tandem mass spectrometry (MS2) spectra [29]. This approach directly maps the chemical space and structural relationships within an extract, revealing diverse scaffold families and guiding the targeted isolation of novel analogs [2]. The thesis posits that such network-based metabolomics offers a complementary yet philosophically distinct strategy to genomics-based prioritization for lead discovery. Where genomics predicts biosynthetic potential from DNA sequences, molecular networking provides a phenotype-first, direct chemical inventory of expressed metabolites, making it indispensable for analyzing the actual scaffold diversity available for bioactivity testing [48] [29].
Foundational Methodologies
Network-Based (Molecular Networking) Prioritization
Core Principle: This methodology prioritizes NPs based on their positional relationships within a network graph constructed from experimental metabolomic data. Molecules (nodes) are connected by edges when their MS2 fragmentation spectra are sufficiently similar, implying shared structural features and, often, a common biosynthetic origin [29].
- Process: Untargeted LC-MS/MS data from a complex extract is processed to create a visual map where clusters represent molecular families. Annotated nodes (known compounds) can propagate structural information to connected unknown nodes, enabling dereplication and highlighting rare or unconnected nodes as candidates for novel chemistry [2] [29].
- Key Insight: The network topology itself is informative. Highly connected "hub" nodes may represent common scaffolds or biosynthetic precursors, while sparse regions of the network may indicate unique or novel chemotypes worthy of prioritization for isolation [52].
Genomics-Based Prioritization
Core Principle: This approach prioritizes NP discovery by first analyzing the genetic blueprint of an organism. It identifies and assesses Biosynthetic Gene Clusters (BGCs) that encode for enzymes (e.g., polyketide synthases, non-ribosomal peptide synthetases) responsible for producing specialized metabolites [48] [53].
- Process: Genomic DNA is sequenced, and bioinformatic tools (e.g., antiSMASH) are used to predict BGCs and their putative products. Prioritization is based on criteria such as cluster novelty, absence of resistance genes (suggesting new modes of action), or genetic cues for silenced or cryptic pathways that can be activated in the laboratory [53].
- Key Insight: Genomics provides a hypothesis-driven starting point, predicting what an organism could produce. It is powerful for genome mining and guiding genetic engineering but does not confirm metabolite expression under given growth conditions [48] [54].
Diagram 1: Molecular Networking Workflow for Scaffold Analysis
Diagram Title: NP Scaffold Discovery via Molecular Networking
Comparative Analysis: Strengths and Limitations
The choice between network-based and genomics-based prioritization depends on research goals, sample type, and available infrastructure. The following table summarizes their core comparative strengths and limitations.
Table 1: Comparative Strengths and Limitations
| Feature | Network-Based (Molecular Networking) Prioritization | Genomics-Based Prioritization |
|---|---|---|
| Primary Input Data | Expressed metabolome (MS2 spectra) | Genetic potential (DNA sequence) |
| Core Strength | Direct, phenotype-first analysis of the actual chemical inventory. Enables visualization of scaffold relationships and rapid dereplication [2] [29]. | Hypothesis-driven; can access silent/cryptic gene clusters not expressed under standard conditions. Provides biosynthetic logic for engineering [48] [53]. |
| Key Limitation | Limited to expressed metabolites under given conditions. Annotation can be challenging without good spectral libraries [29] [55]. | Does not confirm compound expression or structure. Prediction of final product structure from BGC can be highly inaccurate [48] [54]. |
| Scaffold Diversity Insight | High. Directly reveals the diversity of expressed scaffolds and their analog families within a sample [2]. | Indirect. Predicts the potential for scaffold diversity based on BGC variety, but many may not be produced [53]. |
| Throughput & Speed | Rapid post-data acquisition. Modern platforms enable cloud-based processing of thousands of samples [53] [29]. | Sequencing is fast, but BGC analysis and annotation can be computationally intensive and time-consuming. |
| Technical Barriers | Requires high-resolution MS instrumentation and expertise in metabolomics data analysis [29] [55]. | Requires sequencing infrastructure, bioinformatics expertise, and often heterologous expression systems to access predicted molecules [48] [54]. |
| Best Suited For | Prioritizing leads from complex extracts; dereplication; studying chemical ecology; guiding fractionation based on chemical novelty [29]. | Genome mining for novel BGCs; strain prioritization; genetic engineering programs to activate or optimize production [48] [53]. |
Integrated Experimental Protocols
Protocol for Molecular Networking-Based Scaffold Analysis
Objective: To profile scaffold diversity in a microbial extract and prioritize unknown compounds for isolation.
Materials:
- Sample: Crude microbial ethyl acetate extract.
- LC-MS/MS System: UHPLC coupled to a high-resolution Q-TOF or Orbitrap mass spectrometer.
- Software: MSConvert (ProteoWizard), MZmine 3, or similar for feature detection; access to the GNPS platform [29].
Procedure:
- Data Acquisition: Separately inject extract. Use Data-Dependent Acquisition (DDA) to collect MS1 and MS2 spectra. Employ a dynamic exclusion window to increase spectral coverage [29].
- Data Preprocessing: Convert raw files to
.mzMLformat. Use MZmine 3 for chromatographic deconvolution, feature detection (mass, RT, intensity), and MS2 spectral alignment to create a feature list. - Molecular Networking: Upload the feature list to GNPS. Use the Feature-Based Molecular Networking (FBMN) workflow. Set parameters: precursor ion mass tolerance 0.02 Da, fragment ion tolerance 0.02 Da, minimum cosine score for edge creation (e.g., 0.7). Run the job [29].
- Network Analysis & Prioritization:
- Visualize the resulting network in Cytoscape.
- Dereplication: Perform library search against GNPS spectral libraries. Annotate known compounds.
- Scaffold Diversity Assessment: Identify distinct molecular families (clusters). Large, dense clusters indicate major scaffold types.
- Target Selection: Prioritize nodes that are: (a) unannotated, (b) in small or sparse clusters (suggesting unique chemotypes), or (c) connected to bioactive nodes in a Bioactive Molecular Networking (BMN) setup.
- Targeted Isolation: Use MS-guided fractionation (prep-HPLC) to isolate ions corresponding to prioritized nodes. Perform subsequent NMR analysis for full structure elucidation.
Protocol for Genomics-Based Gene Cluster Prioritization
Objective: To identify and prioritize novel biosynthetic gene clusters in a bacterial genome for heterologous expression.
Materials:
- Sample: High-quality genomic DNA from target bacterium.
- Sequencing Platform: Illumina NovaSeq X for short-read or Oxford Nanopore for long-read sequencing [53].
- Software: Genome assembler (SPAdes, Flye), BGC prediction tool (antiSMASH), and databases (MIBiG).
Procedure:
- Genome Sequencing & Assembly: Sequence genomic DNA to achieve high coverage (>100x). Perform hybrid assembly (short-read + long-read) for a complete, closed genome if possible.
- BGC Prediction & Annotation: Submit the assembled genome to the antiSMASH web server. Use default settings to identify all BGCs (polyketide, NRPS, ribosomally synthesized and post-translationally modified peptides, etc.).
- Cluster Prioritization:
- Novelty Filter: Compare predicted BGCs against the MIBiG repository of known BGCs. Discard clusters with >70% similarity to known pathways.
- Integrity Check: Assess if predicted BGCs are complete (contain all essential biosynthetic, regulatory, and resistance genes).
- Product Prediction: Analyze the adenylation (A) domain specificities in NRPS clusters or ketosynthase (KS) domains in PKS clusters to predict potential substrate incorporation and scaffold features.
- Genetic Access:
- Heterologous Expression: Clone the entire prioritized BGC into a suitable bacterial host (e.g., Streptomyces coelicolor or E. coli with optimized chassis) using transformation-associated recombination (TAR) or similar techniques [48].
- Culture Manipulation: Alternatively, in the native host, employ elicitor co-cultivation or modify regulatory genes to potentially activate silent clusters.
- Metabolite Verification: Analyze the culture of the engineered or activated strain using LC-MS/MS and molecular networking (Protocol 4.1) to detect and identify the newly expressed metabolites.
Decision Pathway for Methodology Selection
Diagram 2: Methodology Selection Pathway for NP Discovery
Diagram Title: Decision Pathway for NP Prioritization Method
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Research Reagents and Materials
| Item | Function in Network-Based Prioritization | Function in Genomics-Based Prioritization |
|---|---|---|
| High-Resolution Mass Spectrometer (Q-TOF, Orbitrap) | Core instrument for generating the MS2 spectral data required to construct molecular networks [29] [55]. | Used downstream to verify and characterize metabolites expressed from a prioritized BGC. |
| GNPS Platform Access | Essential cloud platform for performing spectral networking, library searches, and accessing specialized workflows like FBMN and IIMN [2] [29]. | Not directly used, though GNPS can analyze the resulting metabolites from genomic leads. |
| U/HPLC System with C18 Column | Separates complex NP extracts prior to MS analysis, reducing ion suppression and improving MS2 quality [29]. | Similar use in analyzing metabolite production from engineered strains. |
| Next-Gen Sequencing Kit (Illumina, Nanopore) | Not typically used. | Core reagent for generating the raw DNA sequence data for genome assembly and BGC mining [53] [56]. |
| antiSMASH Software | Not used. | Primary bioinformatics tool for the automated identification and annotation of BGCs in a genomic sequence [48] [53]. |
| Cloning & Expression Vector Kit (e.g., for TAR) | Not used. | Critical for functional access to prioritized BGCs via heterologous expression in a model host organism [48]. |
| MIBiG Database | Can provide MS/MS spectra for library matching. | Gold-standard reference database for comparing and assessing the novelty of discovered BGCs [48]. |
| Cytoscape Software | Used for advanced visualization, exploration, and analysis of molecular networks generated by GNPS [29]. | Not typically used for genomic data in this context. |
The paradigm for discovering bioactive molecules and assessing toxicological risk is shifting from traditional natural product isolation and animal testing toward integrated, data-driven approaches. This article details validated protocols and application notes centered on molecular networking—a bioinformatic method for organizing tandem mass spectrometry (MS/MS) data [57]. Originally developed for natural product research, molecular networking is now a cornerstone in metabolomics for annotating novel metabolites and in modern toxicology for elucidating xenobiotic metabolism and identifying exposure biomarkers [57] [58]. Framed within a broader thesis on molecular scaffold diversity analysis, this work demonstrates how these techniques move research "beyond natural products" to enable predictive, mechanism-based science. We provide explicit methodologies for sample preparation, data acquisition, computational analysis using tools like GNPS, and integration with New Approach Methodologies (NAMs) for safety assessment [59] [60].
The analysis of molecular scaffold diversity within natural product (NP) libraries provides a crucial map of biologically relevant chemical space [61]. Studies reveal that current lead compound libraries underutilize the unique scaffold space found in metabolites and natural products, missing opportunities for novel bioactivity [62]. The core thesis of our research posits that understanding this scaffold diversity is not an endpoint, but a starting point for functional discovery and safety evaluation. Molecular networking serves as the critical translational bridge, allowing researchers to organize complex metabolomic datasets based on structural similarity, thereby connecting scaffold families to biological and toxicological outcomes [57] [63]. This article details the applied protocols that bring this thesis to life, moving from structural characterization in metabolomics to predictive modeling in toxicology.
Molecular Networking in Clinical and Forensic Toxicology: Application Notes
Molecular networking (MN) has been successfully transposed from NP research to become a powerful tool in clinical, forensic, and fundamental toxicology [57]. Its primary strength lies in visualizing and exploring complex MS/MS datasets to identify unknown compounds and their metabolites.
- Clinical Toxicology: MN enables untargeted screening for novel biomarkers of xenobiotic exposure. By comparing MS/MS data from case and control samples, networks can reveal clusters of related ions corresponding to a parent drug and its human metabolites, identifying new consumption markers [57].
- Forensic Toxicology: In postmortem investigations, MN can systematically screen for intoxicants and their transformation products, aiding in determining the cause of death. It is particularly valuable for detecting new psychoactive substances (NPS) that may not be in standard libraries [57].
- Fundamental Toxicology: MN efficiently maps the metabolic pathways of xenobiotics. In vitro incubation samples with liver microsomes or hepatocytes can be analyzed to rapidly visualize Phase I and Phase II metabolites, providing a comprehensive view of biotransformation [57].
Table 1: Key Applications and Outcomes of Molecular Networking in Toxicology
| Application Field | Primary Objective | Typical Workflow Input | Key Outcome / Advantage |
|---|---|---|---|
| Clinical Exposure Assessment | Discover novel biomarkers of drug/substance use [57]. | Urine, blood, or plasma from exposed vs. control cohorts. | Identification of previously unreported human metabolites; non-targeted screening capability. |
| Forensic Investigation | Identify intoxicants as potential cause of death [57]. | Postmortem biological samples (blood, tissue, vitreous humor). | Detection of unknown NPS and their metabolites; evidence for mechanism of intoxication. |
| Xenobiotic Metabolism Studies | Elucidate in vitro/in vivo metabolic pathways [57]. | Incubations with liver enzymes, hepatocytes, or animal model samples. | Rapid visualization of comprehensive metabolic maps; identification of major and minor metabolites. |
Core Protocol: Molecular Networking for Toxicant Metabolism Mapping
- Sample Preparation: Prepare test system (e.g., pooled human liver microsomes). Incubate with xenobiotic substrate and NADPH regenerating system. Perform reactions at 37°C, quenching at multiple time points with cold acetonitrile [64].
- LC-MS/MS Analysis: Analyze quenched samples via reversed-phase liquid chromatography coupled to high-resolution tandem mass spectrometry (LC-HRMS/MS) in data-dependent acquisition (DDA) mode [57].
- Data Preprocessing: Convert raw files to .mzML format. Use tools like MZmine or MS-DIAL for peak picking, alignment, and deconvolution. Export feature lists with associated MS/MS spectra in .mgf format.
- Molecular Networking: Upload .mgf files to the Global Natural Products Social Molecular Networking (GNPS) platform. Create a molecular network using the Feature-Based Molecular Networking (FBMN) workflow. Key parameters: precursor ion mass tolerance 2.0 Da, fragment ion tolerance 0.5 Da, minimum cosine score 0.7 [63].
- Data Interpretation: Visualize the network in Cytoscape. Clusters of connected nodes represent structurally related compounds. Annotate the parent drug node and propose structures for metabolite nodes based on spectral similarity, mass shifts (e.g., +15.995 Da for oxidation, +176.032 Da for glucuronidation), and library matches.
Figure 1: Molecular Networking Workflow for Toxicant Metabolism. Diagram shows data flow from LC-MS/MS acquisition to final annotated network [57] [63].
Computational Toxicology and New Approach Methodologies (NAMs)
New Approach Methodologies (NAMs) represent a suite of non-animal methods for safety assessment, including in vitro assays, in silico models, and omics technologies [59]. Computational toxicology, a key NAM, uses machine learning (ML) and quantitative structure-activity relationship (QSAR) models to predict toxicity from chemical structure [60].
Table 2: Selected Software for QSAR Modeling in Computational Toxicology [60]
| Software / Tool | Type | Main Function / Feature |
|---|---|---|
| PaDEL | Free, Standalone | Calculates molecular descriptors and fingerprints for model building. |
| QSARPro | Commercial | Performs group-based QSAR and multi-target model development. |
| KNIME | Free, Open-Source Platform | Integrates cheminformatics nodes (e.g., RDKit) for building custom workflows, including virtual library generation and model training. |
| MCASE | Commercial | Uses a machine learning approach to automatically identify structural alerts (biophores) associated with activity/toxicity. |
Integrated NAMs Workflow Protocol:
- Early-Stage In Silico Screening: For a library of NPs or synthetic derivatives, use a platform like KNIME with RDKit to calculate molecular descriptors. Apply pre-built QSAR models (e.g., for hepatotoxicity, mutagenicity) to prioritize or deprioritize compounds [60].
- In Vitro Mechanistic Validation: Test prioritized compounds in relevant in vitro NAMs, such as hepatocyte cytotoxicity assays or Organ-on-a-Chip models that mimic human organ physiology [59].
- Omics Integration: Treat cells with the compound and perform untargeted metabolomics via LC-MS/MS. Process data through the molecular networking protocol (Section 2) to identify dysregulated endogenous metabolic pathways, linking exposure to a mechanistically informed Adverse Outcome Pathway (AOP) [59] [60].
- Data Integration: Combine in silico predictions, in vitro viability data, and metabolomic pathway perturbations into a weight-of-evidence safety assessment within an Integrated Approach to Testing and Assessment (IATA) framework [59].
Figure 2: Integrated New Approach Methodologies (NAMs) Workflow. Diagram shows convergence of computational, in vitro, and omics data for safety assessment [59] [60].
Experimental Protocols in Plant Metabolomics for Drug Discovery
Metabolomics has become an indispensable tool for accelerating NP drug discovery, enabling the comprehensive analysis of complex extracts without the need for immediate isolation of every constituent [58] [64].
Detailed Protocol: Metabolomics Sample Preparation from Plant Material
- Sample Collection & Harvesting: Snap-freeze fresh plant tissue (e.g., leaves, roots) immediately in liquid nitrogen to quench metabolic activity. Grind the frozen tissue into a fine powder using a mortar and pestle pre-chilled with liquid nitrogen. Store powder at -80°C [64].
- Metabolite Extraction (Dual Solvent System): Weigh ~50 mg of frozen powder into a microtube.
- Add 1 mL of pre-cooled (-20°C) methanol:methyl tert-butyl ether (MTBE):water (1.5:5:1.5 v/v/v) mixture [64]. (Note: MTBE is a safer alternative to chloroform).
- Vortex vigorously for 1 minute, then sonicate in an ice-water bath for 10 minutes.
- Centrifuge at 14,000 x g for 10 minutes at 4°C to separate phases and pellet debris.
- Carefully collect the upper (MTBE-rich, lipophilic) and lower (methanol/water-rich, polar) phases into separate vials.
- Dry the fractions under a gentle stream of nitrogen gas and reconstitute in an appropriate solvent for LC-MS analysis (e.g., methanol for the lipophilic fraction, water:methanol 1:1 for the polar fraction) [64].
- LC-HRMS/MS Analysis for Molecular Networking:
- Polar Metabolites: Use a HILIC or reverse-phase C18 column. Mobile phase: (A) water + 0.1% formic acid, (B) acetonitrile + 0.1% formic acid.
- Lipophilic Metabolites: Use a reverse-phase C8 or C18 column. Mobile phase: (A) water + 0.1% ammonium acetate, (B) isopropanol:acetonitrile 9:1 + 0.1% ammonium acetate.
- Acquire data in DDA mode. Perform MS1 survey scans at high resolution (e.g., 120,000 FWHM), followed by MS/MS scans on the top N most intense ions.
Scaffold Diversity Analysis: Linking Chemical Space to Bioactivity
Analyzing the scaffold diversity of active fractions or clusters in a molecular network is crucial for understanding the structure-activity relationship (SAR) and prioritizing scaffolds for further development [61] [62].
Protocol: Hierarchical Scaffold Analysis of an Active Metabolite Cluster
- Extract SMILES Strings: From a molecular network cluster of interest (e.g., a cluster correlated with bioactivity in a metabolomics study), extract the SMILES strings of all nodes (compounds).
- Generate Molecular Frameworks: Process the SMILES strings using a cheminformatics toolkit (e.g., RDKit in Python). For each molecule, generate its Bemis-Murcko framework, which discards side chains and retains only the ring systems and linkers [61].
- Create Scaffold Hierarchy: Apply an algorithm such as the Scaffold Tree to iteratively prune the framework, creating a hierarchical tree of scaffolds from the full framework down to a single ring [61].
- Visualize and Analyze: Use visualization tools like Scaffold Hunter or a custom script to map the bioactivity data (e.g., IC50 values) onto the scaffold tree. This identifies which core scaffolds are associated with the strongest activity, guiding synthetic follow-up for scaffold-based optimization [61].
Table 3: Comparative Scaffold Diversity Across Biologically Relevant Datasets [62]
| Dataset | Approx. Number of Scaffolds (Murcko) | Notable Characteristics in Property Space | Key Implication for Library Design |
|---|---|---|---|
| Drugs | ~2,500 (from 5,120 compounds) | Skewed distribution; few scaffolds are highly common. Follow Lipinski's rules [62]. | High prevalence of "privileged" scaffolds. |
| Human Metabolites | Limited diversity | Highest molecular polar surface area; most soluble [62]. | Excellent "drug-likeness" but limited scaffold diversity to sample from. |
| Natural Products | Very High (~1,300 ring systems missing from lead libraries) [62] | Maximum number of rings and rotatable bonds; more complex [62]. | Vast, untapped source of novel, biologically pre-validated scaffolds for library enrichment. |
| Toxics | High diversity | High heteroatom content; generates many unique molecular features [62]. | Scaffolds may contain structural alerts for toxicity; useful for predictive model training. |
Integrated Application Notes: Case Study Outline
Project: Discovering anti-inflammatory scaffolds from a plant extract while screening for hepatotoxicity risk.
- Metabolomics & MN: Prepare extract from plant tissue (Protocol 4). Analyze by LC-HRMS/MS and create a molecular network (Protocol 2). Test fractions for anti-inflammatory activity in vitro.
- Bioactivity Mapping: Overlay bioactivity scores onto the molecular network. Identify a specific cluster of structurally related ions (a "molecular family") correlated with high activity.
- Scaffold Analysis: Isolate the key cluster. Perform scaffold analysis (Protocol 5) on the proposed structures within the active cluster to define the active core scaffold(s).
- Computational Toxicology Screening: Obtain or predict the structures of key cluster members. Screen these structures using in silico QSAR models for hepatotoxicity and other endpoints (Protocol 3, Step 1).
- Integrated Assessment: Combine findings: the bioactivity potency, the novelty of the scaffold relative to drug libraries (Table 3), and the computational toxicity flags. This integrated profile informs the decision to pursue isolation/synthesis of the lead scaffold and guides early structural modification to mitigate predicted toxicity risks.
The Scientist's Toolkit: Essential Reagents & Software
Table 4: Key Research Reagent Solutions for Metabolomics & Toxicology Protocols
| Item | Function / Application | Protocol Reference |
|---|---|---|
| Methanol, LC-MS Grade | Primary extraction solvent for polar metabolites; mobile phase component [64]. | Section 4 (Extraction) |
| Methyl tert-Butyl Ether (MTBE) | Safer alternative to chloroform for liquid-liquid extraction of lipids and non-polar metabolites [64]. | Section 4 (Extraction) |
| Liquid Nitrogen | Rapid quenching of metabolic activity during sample harvesting to preserve the native metabolome [64]. | Section 4 (Collection) |
| Pooled Human Liver Microsomes (pHLM) | In vitro system for studying Phase I metabolism of xenobiotics [57]. | Section 2 (Core Protocol) |
| NADPH Regenerating System | Provides cofactors required for cytochrome P450 enzyme activity in metabolic incubations [57]. | Section 2 (Core Protocol) |
| Global Natural Products Social (GNPS) Platform | Free, cloud-based platform for performing molecular networking and spectral library matching [63]. | Section 2 (Core Protocol) |
| RDKit (Open-Source Cheminformatics) | Python library for calculating molecular descriptors, generating scaffolds, and handling chemical data [61] [60]. | Section 3, 5 |
| KNIME Analytics Platform | Open-source platform for visual programming, enabling integration of cheminformatics (RDKit), data processing, and machine learning models [60]. | Section 3 |
The convergence of metabolomics, molecular networking, and computational toxicology represents a powerful, validated framework for modern chemical research and development. By applying the detailed protocols and applications outlined here—from rigorous metabolomic sample preparation and molecular network-based discovery to scaffold diversity analysis and NAMs-integrated safety assessment—researchers can systematically navigate from complex natural extracts or compound libraries to novel, bioactive, and safer molecular scaffolds. This integrated approach fully realizes the promise of moving "beyond natural products" into an era of predictive, mechanism-driven science that efficiently bridges the gap between chemical diversity and therapeutic or toxicological outcome.
Conclusion
Molecular networking has fundamentally shifted the paradigm for natural product scaffold diversity analysis, moving from serendipitous discovery to a rational, data-driven exploration of chemical space. By leveraging MS/MS spectral similarity, it efficiently clusters compounds into scaffold families, dramatically accelerates dereplication, and enables the strategic prioritization of unique chemical entities for isolation and testing. As evidenced by its success in rationally minimizing screening libraries while increasing bioassay hit rates, this approach directly addresses major bottlenecks in cost and time within drug discovery pipelines [citation:3]. Future advancements hinge on the integration of artificial intelligence for better spectral prediction and scaffold elucidation, the expansion of open-access spectral libraries, and tighter coupling with genomic and metabolomic datasets. For biomedical and clinical research, the continued adoption and development of molecular networking techniques promise a more streamlined and productive path to discovering the next generation of therapeutic leads from nature's vast chemical repertoire.
References
- 1. Molecular networking: An efficient tool for discovering and ... [https://pubmed.ncbi.nlm.nih.gov/40014895/]
- 2. Annotation of natural product compound families using ... [https://www.nature.com/articles/s41467-022-35734-z]
- 3. Feature-Based Molecular Networking in the GNPS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7885687/]
- 4. Rationally minimizing natural product libraries using mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11915828/]
- 5. Molecular Networking - GNPS Documentation - GitHub Pages [https://ccms-ucsd.github.io/GNPSDocumentation/networking/]
- 6. Global Natural Products Social (GNPS)-Based Molecular- ... [https://www.mdpi.com/2223-7747/12/23/3970]
- 7. Global Natural Products Social (GNPS) [https://pubmed.ncbi.nlm.nih.gov/38068607/]
- 8. Diversity-oriented synthesis as a tool for the discovery of ... [https://www.nature.com/articles/ncomms1081]
- 9. Scaffold Diversity of Exemplified Medicinal Chemistry Space [https://pmc.ncbi.nlm.nih.gov/articles/PMC3180201/]
- 10. MS/MS-Based Molecular Networking: An Efficient ... [https://www.mdpi.com/1420-3049/28/1/157]
- 11. Transforming molecular cores, substituents, and ... [https://www.sciencedirect.com/science/article/pii/S0223523425003800]
- 12. Scaffolds and Scaffold-based Compounds [https://lifechemicals.com/screening-libraries/scaffolds-and-scaffold-based-compounds]
- 13. Bridging chemical space and biological efficacy: advances ... [https://link.springer.com/article/10.1007/s13659-025-00521-y]
- 14. Empowering natural product science with AI - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00008k]
- 15. From data to discovery: leveraging big data in plant natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183336/]
- 16. Artificial intelligence for natural product drug discovery and ... [https://www.sciencedirect.com/science/article/pii/S2666521225001206]
- 17. Nat-UV DB: A Natural Products Database Underlying... [https://f1000research.com/articles/14-157]
- 18. Scaffold-Aware Generative Augmentation and Reranking ... [https://arxiv.org/html/2510.16306v1]
- 19. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 20. Molecular modeling approach in design of new scaffold ... [https://www.sciencedirect.com/science/article/pii/S2405580825000822]
- 21. Mastering Metrics: A Guide to Quantitative Data Collection ... [https://research-rebels.com/blogs/how-to-write-thesis/mastering-metrics-a-guide-to-quantitative-data-collection-methods]
- 22. Design of tables for the presentation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10346464/]
- 23. Designing Tables - Graphing - LabWrite [https://labwrite.ncsu.edu/res/gh/gh-tables.html]
- 24. Artificial Intelligence-Driven Strategies for Targeted Delivery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389219/]
- 25. Natural Products from Marine Microorganisms with ... [https://www.mdpi.com/1660-3397/23/11/438]
- 26. Molecular networking guided metabolomics for mapping ... [https://www.sciencedirect.com/science/article/abs/pii/S0367326X25000826]
- 27. isolation and mining strategies of natural products from ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d5np00013k]
- 28. Network Pharmacology as a Tool to Investigate the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295404/]
- 29. Molecular networking: An efficient tool for discovering and ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708525000822]
- 30. Reproducible molecular of untargeted... | networking Nature Protocols [https://www.nature.com/articles/s41596-020-0317-5?error=cookies_not_supported&code=2be1cd1d-0090-400d-83b7-0e92a4b72e18]
- 31. DEREPLICATOR - GNPS Documentation [https://ccms-ucsd.github.io/GNPSDocumentation/dereplicator/]
- 32. MolNetEnhancer: Enhanced Molecular Networks by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6680503/]
- 33. MolNetEnhancer: Enhanced Molecular Networks by ... [https://www.mdpi.com/2218-1989/9/7/144]
- 34. Comparative metabolites profiling of different solvent extracts ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11986901/]
- 35. Statistical analysis of feature-based molecular results... networking [https://orbit.dtu.dk/en/publications/statistical-analysis-of-feature-based-molecular-networking-result/]
- 36. GNPS - Analyze , Connect, and Network with your Mass Spectrometry... [https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp]
- 37. Feature-based molecular networking (FBMN) - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12138091/]
- 38. Feature-based molecular networking (FBMN) [https://pubmed.ncbi.nlm.nih.gov/40486865/]
- 39. A Systematic Approach to Discover New Natural Product ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11862952/]
- 40. Auto-deconvolution and molecular networking of gas ... [https://www.pnnl.gov/publications/auto-deconvolution-and-molecular-networking-gas-chromatography-mass-spectrometry-data]
- 41. Ion suppression correction and normalization for non-targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11794426/]
- 42. Exploring Ion Mobility Mass Spectrometry Data File ... [https://www.pnnl.gov/publications/exploring-ion-mobility-mass-spectrometry-data-file-conversions-leverage-existing-tools]
- 43. GNPS - Analyze, Connect, and Network with your Mass ... [https://gnps.ucsd.edu/]
- 44. The undiscovered natural product potential of Actinomycetes [https://www.nature.com/articles/s41429-025-00876-x]
- 45. Toward almost closed genomes with GapFiller - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3446322/]
- 46. Scaffold Extension and Gap Closing [https://ggkbase-help.berkeley.edu/genome_curation/scaffold-extension-and-gap-closing/]
- 47. Filling gaps of genome scaffolds via probabilistic searching ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04448-2]
- 48. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 49. Advanced Methods for Natural Products Discovery [https://www.mdpi.com/1660-3397/21/5/308]
- 50. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 51. From Patterns to Pills: How Informatics Is Shaping Medicinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114992/]
- 52. Analyzing of Molecular Networks for Human Diseases and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6174636/]
- 53. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 54. Natural Products for Drug Discovery in the 21st Century [https://pmc.ncbi.nlm.nih.gov/articles/PMC6032166/]
- 55. Metabolism and bioavailability aspects of natural products ... [https://pubs.rsc.org/en/content/articlelanding/2025/np/d5np00022j]
- 56. Genomics Market 2025-2029: Emerging Trends ... [https://www.pharmiweb.com/press-release/2025-11-28/genomics-market-2025-2029-emerging-trends-market-drivers-and-strategic-insights-revealed-in-new-study]
- 57. Interest of molecular networking in fundamental, clinical ... [https://www.sciencedirect.com/science/article/pii/S0165993624000293]
- 58. advances in metabolomics for natural product drug discovery [https://www.sciencedirect.com/science/article/pii/S095816692500117X?via%3Dihub]
- 59. New Approach Methodologies (NAMs): The Future of ... [https://emulatebio.com/new-approach-methodologies-nams-the-future-of-toxicology-and-safety-testing/]
- 60. Overview of Computational Toxicology Methods Applied in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11677645/]
- 61. Scaffold analysis of PubChem database as background for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-016-0186-7]
- 62. Structural diversity of biologically interesting datasets [https://pmc.ncbi.nlm.nih.gov/articles/PMC3179739/]
- 63. Ordering molecular diversity in untargeted metabolomics via ... [https://pubmed.ncbi.nlm.nih.gov/39131284/]
- 64. Metabolomics in the Context of Plant Natural Products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7023240/]