Synergistic Integration of HR-MS and NMR for Comprehensive Metabolite Identification in Biomedical Research
This article provides a comprehensive overview of the integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy for enhanced metabolite identification in metabolomics.
Synergistic Integration of HR-MS and NMR for Comprehensive Metabolite Identification in Biomedical Research
Abstract
This article provides a comprehensive overview of the integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy for enhanced metabolite identification in metabolomics. Tailored for researchers, scientists, and drug development professionals, it explores foundational principles, methodological workflows, troubleshooting strategies, and validation approaches. By combining the high sensitivity and broad coverage of HR-MS with the non-destructive, quantitative, and structural elucidation capabilities of NMR, this integrated approach enables more accurate biomarker discovery, drug development, and clinical diagnostics, offering a holistic view of metabolic profiles in biological systems.
Foundations of HR-MS and NMR Synergy in Metabolomics
Core Principles of HR-MS and NMR Spectroscopy in Metabolite Analysis
Abstract Metabolite identification research is fundamentally enhanced by the strategic integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy. This integration leverages their complementary analytical strengths: HR-MS provides exceptional sensitivity and broad metabolite coverage, while NMR offers unparalleled structural elucidation power, quantitative accuracy, and high reproducibility. Framed within a thesis on advancing metabolic phenotyping, this article details the core principles of each technique and presents standardized protocols for their synergistic application. We outline specific workflows for sample preparation, data acquisition, and multi-level data fusion, supported by application notes in clinical biofluid analysis, plant biology, foodomics, and drug discovery. The presented framework aims to establish robust, reproducible methodologies for comprehensive metabolome characterization.
Metabolomics, the comprehensive study of small-molecule metabolites, provides a direct functional readout of cellular state and phenotype. No single analytical platform can capture the full chemical diversity, concentration range, and structural complexity of the metabolome. Consequently, the integration of HR-MS and NMR has emerged as a cornerstone for rigorous metabolite identification research [1].
HR-MS excels in sensitivity, capable of detecting thousands of features at nano- to picomolar concentrations. When coupled with chromatography (LC or GC), it provides high resolution for complex mixtures. However, its drawbacks include destructiveness, susceptibility to ion suppression effects, and often ambiguous identification without authentic standards [2] [1]. In contrast, NMR spectroscopy is non-destructive, inherently quantitative, and highly reproducible, offering definitive structural insights through phenomena like chemical shift, J-coupling, and nuclear Overhauser effects. Its primary limitation is lower sensitivity, typically detecting metabolites in the micromolar range [3] [2].
The synergistic combination of these techniques overcomes individual limitations. NMR can validate and quantify metabolites tentatively identified by HR-MS, while HR-MS can extend metabolite coverage to low-abundance species missed by NMR. This multi-platform approach maximizes confidence in metabolite annotation, expands metabolome coverage, and strengthens biological interpretation, forming the basis for a robust thesis in metabolic research [4] [2].
Core Analytical Principles
2.1. Principles of NMR Spectroscopy in Metabolomics
NMR detects magnetically active nuclei (e.g., ¹H, ¹³C) in a strong magnetic field. The core principles utilized in metabolomics are:
- Chemical Shift (δ): The resonant frequency of a nucleus, reported in parts per million (ppm), which is influenced by its local electronic environment. This provides primary information on functional groups.
- J-Coupling: The splitting of NMR signals due to spin-spin interactions between neighboring nuclei, revealing atomic connectivity.
- Signal Intensity: For
¹HNMR, the integrated area under a signal is directly proportional to the number of nuclei contributing to it, enabling absolute quantification with a single internal standard [3]. - Reproducibility: NMR offers exceptional analytical precision, with coefficients of variance (CVs) often ≤5%, making it ideal for longitudinal and large-scale studies [5].
Quantitative NMR (qNMR) workflows use an internal reference standard of known concentration (e.g., TSP, DSS) for absolute quantification. Profiling approaches use multivariate statistics on entire spectral datasets to discriminate sample groups [5]. High-Resolution Magic Angle Spinning (HR-MAS) NMR is a critical variant for semi-solid samples, spinning the sample at 54.7° to the magnetic field to average out anisotropic interactions, allowing for the analysis of intact tissues with minimal preparation [3] [6].
2.2. Principles of HR-MS in Metabolomics
HR-MS separates ions based on their mass-to-charge ratio (m/z) with high accuracy (often <5 ppm), enabling the determination of elemental composition.
- Ionization: Soft ionization techniques like Electrospray Ionization (ESI) are predominant, generating molecular ions ([M+H]⁺, [M-H]⁻) with minimal fragmentation.
- Mass Analyzers: Time-of-flight (TOF) and Orbitrap analyzers provide the high resolution and mass accuracy required to distinguish between isobaric metabolites.
- Chromatography Coupling: Liquid Chromatography (LC) or Gas Chromatography (GC) is essential for separating isomers and reducing ion suppression prior to MS analysis. Hydrophilic Interaction Liquid Chromatography (HILIC) is often used alongside Reverse-Phase (RP) chromatography to capture a wider range of metabolite polarities [4].
- Tandem MS (MS/MS): Provides fragmentation patterns that are crucial for structural characterization and confident database matching.
Experimental Protocols for Integrated Analysis
A robust integrated workflow requires meticulous planning from sample preparation through data acquisition.
3.1. Unified Sample Preparation Protocol
- Objective: To generate a single sample extract compatible with both LC-HRMS and
¹HNMR analysis. - Materials: Biofluid (e.g., urine, serum) or tissue homogenate; cold LC-MS grade methanol, acetonitrile, and water; deuterated NMR buffer (e.g., 75 mM Na₂HPO₄ in D₂O, pD 7.4); internal standard for NMR (e.g., 0.5 mM TSP); internal standard for MS (e.g., stable isotope-labeled compounds).
- Procedure:
- For biofluids, mix 300 µL of sample with 600 µL of cold methanol (-20°C) to precipitate proteins. Vortex vigorously for 1 minute.
- Incubate at -20°C for 1 hour.
- Centrifuge at 14,000 × g for 15 minutes at 4°C.
- Split the supernatant into two equal aliquots (~400 µL each) in labeled tubes.
- Aliquot for LC-HRMS: Dry completely under a gentle stream of nitrogen gas. Reconstitute in 100 µL of a solvent mix compatible with your LC method (e.g., 98:2 water:acetonitrile + 0.1% formic acid). Add MS internal standards. Centrifuge and transfer to an LC vial [7].
- Aliquot for
¹HNMR: Dry completely. Reconstitute in 600 µL of deuterated phosphate buffer containing TSP. Vortex, centrifuge, and transfer to a 5 mm NMR tube [4].
3.2. Instrumental Data Acquisition Parameters Table 1: Standardized Acquisition Parameters for HR-MS and NMR.
| Parameter | LC-HRMS (Orbitrap Example) | ¹H NMR (600 MHz Example) |
|---|---|---|
| Chromatography | Column: C18 (2.1 x 100 mm, 1.7 µm). Gradient: 5-95% B over 18 min. Flow: 0.3 mL/min. Temp: 40°C [4]. | Not Applicable |
| Ionization | ESI Positive & Negative Mode. Spray Voltage: ±3.5 kV. Capillary Temp: 320°C [4]. | Not Applicable |
| Mass Analysis | Resolution: 70,000 (at m/z 200). Scan Range: m/z 85-1275. AGC Target: 1e6 [4]. | Not Applicable |
| NMR Pulse Sequence | Not Applicable | 1D NOESY with presaturation (noesygppr1d). Mixing time: 10 ms. Presat frequency set on water peak [3]. |
| Spectral Width | Not Applicable | 20 ppm (typically -1 to 19 ppm) |
| Acquisition Time | ~20 min/sample | ~4-5 min/sample (64 scans) |
| Lock/Reference | Not Applicable | Deuterium lock; TSP referenced to δ 0.0 ppm |
3.3. Protocol for HR-MAS NMR on Intact Plant Tissue
- Objective: To acquire metabolic profiles from intact plant tissue without solvent extraction [3].
- Materials: Fresh or flash-frozen plant tissue (e.g., leaf); HR-MAS probe with 4 mm zirconia rotor and Kel-F cap; D₂O for lock.
- Procedure:
- Harvest material under controlled conditions (consistent time of day) and immediately flash-freeze in liquid N₂.
- Weigh ~20 mg of frozen tissue into a pre-chilled 4 mm HR-MAS rotor.
- Add 10 µL of D₂O containing a reference compound (e.g., TSP) for lock and chemical shift reference.
- Insert a cylindrical insert to minimize sample volume and vortexing. Seal the rotor tightly.
- Insert the rotor into the spectrometer pre-cooled to 4°C.
- Set the magic angle (54.7°) and spin rate to 4 kHz.
- Acquire a 1D
¹Hspectrum using a CPMG (Carr-Purcell-Meiboom-Gill) pulse sequence to suppress broad signals from macromolecules [3] [6].
3.4. SYNHMET Protocol for Assisted Metabolite Quantification
- Objective: To use HR-MS data to guide and improve the accuracy of quantitative NMR spectral deconvolution, as exemplified by the SYNHMET method for urine [4].
- Procedure:
- Acquire
¹HNMR and LC-HRMS (RP and HILIC, positive/negative mode) data from the same sample set. - Perform an initial NMR deconvolution (e.g., using Chenomx software) to obtain a preliminary concentration list for a database of expected metabolites.
- For each metabolite, search the HR-MS dataset for all chromatographic peaks with an accurate mass matching its theoretical mass within 5 ppm.
- For each candidate MS peak, plot its intensity against the preliminary NMR-derived concentration across all samples.
- Identify the MS peak that shows the highest linear correlation (R² > 0.8). This peak is assigned to the metabolite.
- Use the slope of this correlation to convert the MS peak intensity into an MS-informed concentration.
- Use this MS-informed concentration as a fixed, accurate constraint in a subsequent round of NMR spectral deconvolution. This dramatically improves the accuracy of quantifying metabolites with low-concentration or overlapped NMR signals [4].
- Acquire
Diagram 1: Core Workflow for Integrated HR-MS/NMR Metabolite Analysis (89 characters).
Data Integration and Fusion Strategies
Data fusion is the core computational challenge of integrated metabolomics. Strategies are classified by the level of data abstraction [1].
Table 2: Levels of Data Fusion for Integrating NMR and HR-MS Datasets [1].
| Fusion Level | Description | Process | Advantages | Disadvantages |
|---|---|---|---|---|
| Low-Level | Concatenation of raw or pre-processed data matrices. | NMR bins and MS peak intensities are scaled and merged into one matrix. | Simple; retains all raw information. | Very high dimensionality; requires careful scaling to balance technique contributions. |
| Mid-Level | Fusion of extracted features. | Separate PCA is run on each dataset; scores or selected variables are fused. | Reduces dimensionality; focuses on most relevant features. | Risk of losing information during initial feature selection. |
| High-Level | Fusion of model decisions or predictions. | Separate classification models are built for NMR and MS; their outputs (e.g., class probabilities) are combined. | Flexible; allows use of optimal model for each data type. | Complex; requires separate modeling before integration. |
A common mid-level approach is Statistical Heterospectroscopy (SHY), which identifies statistical correlations between NMR chemical shifts and MS m/z features across a sample set, directly linking signals from the same metabolite [4] [8]. Another powerful model is Multiblock PCA/PLS, which analyzes multiple data blocks simultaneously while preserving their individual structures [2] [1].
Diagram 2: Hierarchical Data Fusion Strategies (Low, Mid, High-Level) (73 characters).
Application Notes
5.1. Clinical Biofluid Profiling (SYNHMET Case Study)
- Thesis Context: Developing personalized metabolic profiles for disease stratification.
- Application: Analysis of urine from healthy controls, chronic cystitis, and bladder cancer patients [4].
- Protocol Highlights: Use of the SYNHMET protocol (Section 3.4). Quantification of 165 metabolites with minimal missing values, outperforming either technique alone. HR-MS data resolved ambiguities in NMR deconvolution of crowded spectral regions (e.g., 2.37-2.47 ppm), enabling accurate quantification of 2-oxoglutarate, glutamine, and others [4].
- Outcome: Creation of a detailed, quantitative personalized metabolic profile for monitoring health status and disease progression.
5.2. Plant Metabolomics and HR-MAS NMR
- Thesis Context: Understanding plant phenotype and stress responses in a systems biology framework.
- Application: Metabolic profiling of intact plant leaves or tissues to study abiotic stress, development, or crop improvement [3].
- Protocol Highlights: Use of HR-MAS NMR protocol (Section 3.3) on intact tissue, preserving spatial information and avoiding extraction bias. Complementary LC-HRMS analysis of extracts provides coverage of low-abundance secondary metabolites.
- Outcome: A more holistic view of in vivo plant metabolism, linking metabolic changes directly to phenotype.
5.3. Foodomics & Authentication
- Thesis Context: Ensuring food quality, safety, and authenticity through metabolic fingerprinting.
- Application: Classifying Amarone wines by grape withering time and yeast strain, or authenticating table olive origin and processing method [7] [8].
- Protocol Highlights: Parallel untargeted
¹HNMR and LC-HRMS profiling. Data fusion using Multi-block Consensus PCA or sPLS-DA. SHY analysis used to correlate polyphenol NMR signals with HR-MS features for confident marker identification [7] [8]. - Outcome: Enhanced predictive accuracy for sample classification and identification of robust, cross-validated biomarker compounds.
5.4. Drug Discovery from Natural Products (ELINA Workflow)
- Thesis Context: Accelerating the identification of bioactive lead compounds from complex mixtures.
- Application: Discovering steroid sulfatase (STS) inhibiting lanostane triterpenes from a fungal extract [9].
- Protocol Highlights:
- Microfractionate a bioactive crude extract.
- Acquire
¹HNMR and LC-HRMS data for all fractions. - Test all fractions in a bioassay (e.g., STS inhibition).
- Apply Heterocovariance Analysis (HetCA) to correlate spectral features with bioactivity, generating "hot" (positive correlation) and "cold" spectral traces.
- Use "hot" NMR signals and correlated HR-MS ions to target the isolation of active constituents [9].
Diagram 3: ELINA Workflow for Bioactive Natural Product Discovery (78 characters).
Table 3: Quantitative Comparison of NMR and HR-MS Performance in a Model Study [2].
| Metric | NMR Alone | GC/LC-HRMS Alone | Combined NMR & MS |
|---|---|---|---|
| Total Metabolites Detected | 20 | 82 | 102 |
| Unique Metabolites Identified | 14 | 16 | 47 (Perturbed) |
| Metabolites Identified by Both | - | - | 17 |
| Pathway Coverage (e.g., TCA Cycle) | Partial (e.g., misses fumarate) | Partial (e.g., misses key amino acids) | Most Comprehensive |
| Confidence in Identification | Very High | Moderate to High | Highest (Orthogonal) |
The Scientist's Toolkit: Essential Reagents & Materials
Table 4: Key Research Reagent Solutions for Integrated HR-MS/NMR Metabolomics.
| Item | Function & Specification | Example & Notes |
|---|---|---|
| Deuterated NMR Solvent | Provides a field-frequency lock for stable NMR acquisition; minimizes large solvent proton signals. | D₂O (99.9% D), with phosphate buffer for biofluids; CD₃OD for lipid extracts. |
| NMR Chemical Shift Reference | Provides a precise internal reference point (0 ppm) for all chemical shifts. | TSP-d₄ (sodium 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionate) or DSS-d₆ (4,4-dimethyl-4-silapentane-1-sulfonic acid). |
| MS Internal Standards | Monitors and corrects for instrumental drift and matrix effects during LC-HRMS runs. | Stable isotope-labeled compound mix (e.g., ¹³C, ¹⁵N-labeled amino acids, fatty acids). Added prior to injection. |
| Protein Precipitation Solvent | Removes proteins and macromolecules for clean metabolite analysis of biofluids/tissue homogenates. | Cold methanol, acetonitrile, or methanol:acetonitrile:water mixtures. Maintain at -20°C [7]. |
| LC-MS Grade Solvents & Additives | Ensures minimal background noise and ion suppression in HR-MS analysis. | Water, methanol, acetonitrile, formic acid, ammonium acetate/formate. |
| HILIC & RP UHPLC Columns | Separates the highly polar (HILIC) and mid-to-non-polar (RP) fractions of the metabolome. | e.g., BEH Amide (HILIC) and BEH C18 (RP) columns, 2.1 x 100 mm, 1.7 µm particle size [4]. |
| HR-MAS NMR Consumables | Enables analysis of intact tissues. | 4 mm zirconia rotor, Kel-F caps, cylindrical inserts to reduce sample volume [3]. |
| Data Analysis Software | Processes, aligns, and statistically analyzes complex multi-platform datasets. | NMR: Chenomx, MestReNova, NMRPipe. MS: XCMS, MS-DIAL, Compound Discoverer. Fusion: R packages (mixOmics, omicFusion), SIMCA. |
Diagram 4: SYNHMET Protocol for MS-Assisted NMR Quantification (77 characters).
The comprehensive identification and characterization of metabolites represent a central challenge in modern life sciences and drug development. No single analytical technique can capture the full chemical diversity of the metabolome, which is estimated to contain upwards of 150,000 metabolites in humans [2]. High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy have emerged as the two most powerful techniques for metabolomic analysis, yet they possess fundamentally different and complementary strengths and limitations [10]. This application note frames their synergy within the critical thesis that the integration of HR-MS and NMR data is not merely beneficial but essential for conclusive metabolite identification, transforming ambiguous detection into unambiguous structural elucidation.
HR-MS excels in sensitivity, capable of detecting metabolites at femtomolar to attomolar concentrations, and offers high mass resolution for determining precise molecular formulas [11] [10]. However, its reliance on ionization efficiency and its susceptibility to matrix-induced ion suppression can limit coverage, and it often cannot distinguish between structural isomers [2] [10]. Conversely, NMR spectroscopy is inherently quantitative, non-destructive, and provides unparalleled atomic-level structural information through parameters like chemical shift, J-coupling, and spatial correlations, making it a gold standard for de novo structure elucidation [12] [13]. Its primary limitation is sensitivity, typically requiring metabolites to be present at micromolar (≥1 μM) or higher concentrations for detection [2] [14]. This core dichotomy between the exquisite sensitivity of MS and the definitive structural elucidation power of NMR forms the basis for a synergistic workflow. By strategically combining these techniques, researchers can leverage the broad screening capability of HR-MS to identify targets of interest, which are then subjected to the definitive structural characterization provided by NMR, thereby achieving a more complete and accurate metabolic profile than either technique could provide alone [15] [2].
Quantitative Comparison of HR-MS and NMR Performance
The complementary nature of HR-MS and NMR is quantitatively demonstrated by their differential detection of metabolites in complex biological samples. Studies show that a combined approach significantly expands metabolome coverage.
Table 1: Comparative Metabolite Detection by NMR and GC-MS in a Plant Study
Data from a study on Chlamydomonas reinhardtii treated with lipid modulators [2].
| Detection Category | Number of Metabolites | Key Characteristics of Metabolites Detected |
|---|---|---|
| Detected by GC-MS Only | 82 | Broad range, including many amino acids (Asn, Cys, His, Ser, Trp), fructose-6-phosphate, fumarate, uracil. Relies on volatility and ionization. |
| Detected by NMR Only | 20 | Includes key glycolytic intermediates (fructose, glycerol, pyruvate), amino acids (Gly, Lys, Met, Val), TCA cycle intermediates (acetate, isocitrate), cytosine, uridine. Often highly polar or overlapping isomers. |
| Detected by Both Techniques | 22 | Common central metabolites: e.g., adenosine, glutamate, malate, succinate. Provides cross-validated, high-confidence identifications. |
| Total Unique Metabolites | 102 | Combined coverage is 24% greater than GC-MS alone. |
Table 2: Core Analytical Characteristics of HR-MS and NMR
Summary of fundamental performance parameters [2] [16] [10].
| Parameter | High-Resolution Mass Spectrometry (HR-MS) | Nuclear Magnetic Resonance (NMR) Spectroscopy |
|---|---|---|
| Primary Strength | Ultra-high sensitivity (fmol-amol) | Definitive structural elucidation and isomer discrimination |
| Typical LOD | Femtomolar to picomolar range | Low micromolar range (≥1 μM) |
| Quantitation | Possible but requires internal standards and can be affected by ion suppression | Inherently quantitative; direct proportionality between signal and nucleus count [12] [17] |
| Throughput | High (with fast chromatography) | Moderate to High (especially with automation) |
| Sample Preparation | Often requires extraction, derivatization (for GC-MS), chromatography | Minimal; often none beyond stabilization and buffering in D₂O |
| Key Limitation | Cannot distinguish isomers without prior separation; subject to ion suppression | Lower sensitivity; spectral overlap in complex mixtures (esp. ¹H NMR) |
| Information Gained | Molecular formula (exact mass), fragment ions for substructures | Atomic connectivity, functional groups, stereochemistry, molecular dynamics |
Table 3: qNMR Performance Metrics for Quantitative Analysis
Performance characteristics of quantitative NMR (qNMR) in metabolomic and pharmaceutical applications [12] [17] [14].
| Metric | Typical qNMR Performance | Context & Application |
|---|---|---|
| Accuracy | ± 1% to ± 2% | For purity assessment of pharmaceutical compounds [17] |
| Precision (RSD) | < 5% (often < 2%) | Achievable with optimized protocols and automation [14] |
| Linear Dynamic Range | 4–5 orders of magnitude | From ~10 μM to 1 M [14] |
| Limit of Detection (LOD) | ~1–4 μM (for ¹H) | Dependent on magnet field strength, probe technology, and experiment time [12] [14] |
| Key Advantage | Single internal standard can quantify all detectable components | Unlike MS, does not require compound-specific calibration curves [17] |
Integrated Experimental Protocols for Metabolite Identification
Protocol: Integrated HR-MS/NMR Sample Preparation from Bacterial Cell Culture
This protocol, adapted from the SUMMIT MS/NMR strategy, details the parallel preparation of a single biological sample for both HR-MS and NMR analysis [15].
Objective: To extract hydrophilic metabolites from E. coli cells and prepare aliquots suitable for direct infusion HR-MS and high-field NMR spectroscopy.
Materials:
- E. coli BL21(DE3) cell pellet from 1L culture (OD ~3) in M9 minimal medium.
- Ice-cold phosphate buffer (50 mM, pH 7.0), methanol, chloroform, D₂O, LC-MS grade water, acetonitrile (ACN), formic acid.
- Equipment: Centrifuge, rotary evaporator, lyophilizer, vortex, 3 mm NMR tube.
Procedure:
Cell Lysis & Metabolite Extraction:
- Resuspend cell pellet in 10 mL ice-cold water. Perform three freeze-thaw cycles (liquid nitrogen/ice water) to lyse cells.
- Centrifuge lysate at 20,000 × g for 15 min at 4°C. Retain the supernatant containing hydrophilic metabolites.
- To the supernatant, sequentially add chilled methanol and chloroform under vigorous vortexing to a final ratio of 1:1:1 (sample:methanol:chloroform, v/v/v).
- Incubate the mixture at -20°C overnight for phase separation.
- Centrifuge at 4,000 × g for 20 min at 4°C. Collect the clear top hydrophilic (aqueous methanol) phase.
Sample Concentration and Division:
- Reduce the methanol content in the collected phase using a rotary evaporator.
- Lyophilize the resulting aqueous solution to complete dryness.
- Crucially, divide the dry metabolite extract into two equal portions: one dedicated to MS analysis and one for NMR analysis.
NMR Sample Preparation:
- Dissolve the NMR portion in approximately 200 μL of D₂O containing 0.1-1 mM internal reference standard (e.g., DSS or TSP-d₄).
- Transfer the solution to a 3 mm NMR tube.
HR-MS Sample Preparation:
- Dissolve the MS portion in 200 μL of LC-MS grade water.
- Dilute 10 μL of this stock 10-fold with 50%/50% (v/v) ACN/H₂O containing 0.1% formic acid (to promote positive ionization).
- Centrifuge the diluted sample at 13,000 rpm for 5 min at 4°C to remove any particulates.
- Transfer the supernatant to an appropriate vial for direct infusion or LC-MS analysis.
Protocol: Quantitative ¹H NMR (qNMR) for Metabolite Concentration Determination
This protocol outlines the steps for absolute quantification of metabolites in a complex mixture using ¹H qNMR [12] [17].
Objective: To determine the absolute concentration of target metabolites in a biofluid or extract using an internal reference standard.
Materials:
- NMR sample in D₂O (as prepared in Section 2.1).
- Internal reference standard of known concentration and high purity (e.g., DSS, TSP). The standard must have a singlet resonance in a clear spectral region.
- High-field NMR spectrometer (≥ 500 MHz recommended) with a cryoprobe for optimal sensitivity.
Procedure:
Experiment Setup:
- Insert the sample into the magnet and lock, shim, and tune the probe.
- Use a pulse sequence with sufficient relaxation delay (d1). A standard ¹H pulse-acquire sequence with a d1 ≥ 5 times the longest T1 of the resonances of interest is recommended to ensure full longitudinal relaxation for quantitative accuracy.
- Acquire a 1D ¹H spectrum with sufficient digital resolution (e.g., 64k data points) and a high signal-to-noise ratio (≥ 150:1 for the reference peak), typically achieved with 64-256 scans.
Data Processing:
- Process the Free Induction Decay (FID): Apply exponential line broadening (0.3-1.0 Hz), Fourier transform, phase correction, and baseline correction.
- Reference the spectrum to the internal standard peak (e.g., DSS methyl singlet at 0.00 ppm).
Quantification Calculation:
- Select a well-resolved, characteristic signal for the target metabolite (e.g., a singlet or clean doublet).
- Integrate the area of the target metabolite's signal (Imet) and the area of the internal standard's signal (Istd).
- Calculate the absolute concentration of the metabolite ([Met]) using the formula [17]:
[Met] = (I_met / I_std) × (N_std / N_met) × [Std]where:I_metandI_stdare the integrated peak areas.N_metandN_stdare the number of protons giving rise to each integrated signal.[Std]is the known molar concentration of the internal standard in the sample.
Protocol: HR-MS Analysis for Molecular Formula and Fragmentation Assignment
This protocol describes key steps for obtaining structural information via HR-MS, focusing on accurate mass measurement and tandem MS [11] [18].
Objective: To obtain the accurate mass and diagnostic fragment ions of a metabolite for molecular formula assignment and partial structural characterization.
Materials:
- Prepared MS sample (from Section 2.1).
- Calibrant for the mass spectrometer (e.g., Agilent Tuning Mix).
- HR-MS instrument (e.g., Q-TOF, Orbitrap, or FT-ICR) with electrospray ionization (ESI) source.
Procedure:
Instrument Calibration:
- Perform mass calibration according to the manufacturer's protocol using the appropriate calibrant to ensure mass accuracy within ±5 ppm (or better).
Data Acquisition:
- For direct infusion: Infuse the sample at a low, steady flow rate (e.g., 2-5 μL/min). Acquire spectra in both positive and negative ionization modes to maximize coverage [15].
- For LC-MS/MS: Inject the sample onto a suitable column (e.g., HILIC for polar metabolites). Use a data-dependent acquisition (DDA) or data-independent acquisition (DIA) method.
- The method should include a full MS1 scan at high resolution (R > 30,000) to obtain accurate mass, followed by MS2 scans on selected precursor ions using collision-induced dissociation (CID) to generate fragment ions.
Data Analysis:
- Molecular Formula Assignment: For a detected ion of interest, use the accurate m/z value from the MS1 scan. Input the value into formula calculation software, constraining elements (C, H, N, O, S, P, etc.) and setting a tight mass tolerance (e.g., 3-5 ppm). The software will generate a ranked list of candidate formulas [15] [18].
- Fragment Analysis: Interpret the MS2 spectrum by assigning plausible structures to major fragment ions. This helps identify functional groups and partial substructures (e.g., loss of water, phosphate, or amino acids).
Integrated Workflow for De Novo Metabolite Identification
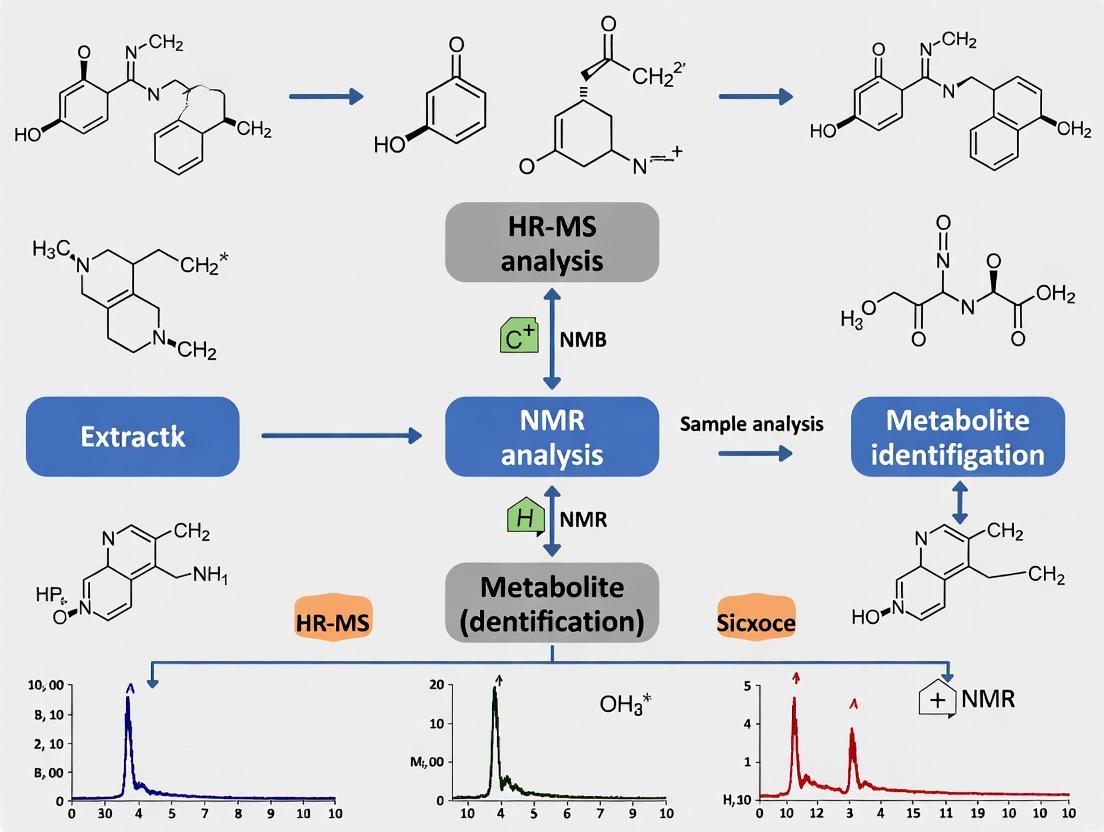
The SUMMIT MS/NMR strategy provides a powerful framework for identifying unknown metabolites without pure standards or database matches [15].
Step 1: HR-MS Analysis and Formula Enumeration.
- Acquire high-resolution mass data to determine the accurate mass of the unknown.
- Calculate all plausible molecular formulas within a defined error tolerance (e.g., ±5 ppm).
Step 2: In Silico Structure Generation.
- For each candidate molecular formula, query databases (e.g., ChemSpider) or use structure generators to create a "structural manifold" – a list of all possible isomers consistent with that formula.
Step 3: In Silico NMR Spectrum Prediction.
- Use computational chemistry tools to predict the NMR spectra (e.g., ¹H and ¹³C chemical shifts) for every structure in the manifold.
Step 4: Experimental NMR Acquisition and Matching.
- Acquire 1D and 2D NMR spectra (e.g., ¹H, HSQC, HMBC, COSY) of the complex mixture or a fraction containing the unknown.
- Deconvolute the experimental NMR signals belonging to the unknown compound.
- Compare the experimental NMR data with the predicted spectra for the entire structural manifold. The correct isomer is identified as the one with the highest degree of match.
Diagram 1: Integrated HR-MS/NMR Workflow for De Novo Identification
Pathway and Data Integration Logic
The power of integrating MS and NMR data extends to mapping metabolic pathway activity, where each technique informs different parts of the network.
Diagram 2: Metabolic Pathway Inferred by Multi-Technique Detection
The Scientist's Toolkit: Essential Reagents and Materials
A successful integrated metabolomics study requires careful selection of reagents and standards.
Table 4: Essential Research Reagent Solutions for HR-MS/NMR Metabolomics
| Item | Function & Importance | Key Considerations |
|---|---|---|
| Deuterated NMR Solvent (D₂O) | Provides the NMR lock signal and minimizes strong solvent proton background in ¹H NMR. | High isotopic purity (99.9% D or higher). May require buffering with deuterated buffers for pH-sensitive samples. |
| NMR Internal Reference Standard (e.g., DSS, TSP) | Provides chemical shift reference (0 ppm) and is essential for quantitative concentration determination in qNMR. | Must be stable, soluble, and give a singlet resonance in a clear region of the spectrum. Concentration must be known precisely [12]. |
| LC-MS Grade Solvents (Water, ACN, MeOH) | Used for sample preparation, dilution, and mobile phases in LC-MS. Minimizes background ions and suppresses ion suppression. | Low volatile organic content and particulate matter. Acid/Base modifiers (formic acid, ammonium acetate) must also be high purity. |
| Mass Calibration Solution | Calibrates the m/z axis of the mass spectrometer to ensure high mass accuracy for formula assignment. | Use manufacturer-recommended solutions (e.g., Agilent Tuning Mix). Calibrate regularly. |
| Chemical Derivatization Reagents (e.g., MSTFA for GC-MS) | Increases volatility and thermal stability of polar metabolites for GC-MS analysis. Can also improve ionization efficiency. | Derivatization must be complete and reproducible. Can introduce artifacts if not carefully controlled [2]. |
| Solid Phase Extraction (SPE) Cartridges | Fractionates complex mixtures or removes interfering salts and macromolecules prior to analysis. | Select sorbent chemistry (C18, HILIC, ion exchange) based on target metabolite polarity. |
The comprehensive identification and characterization of metabolites represent a central challenge in modern bioscience, with direct implications for drug discovery, toxicology, and systems biology. Individual analytical techniques, such as High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy, provide powerful but incomplete windows into the metabolome. HR-MS excels in sensitivity and the ability to detect a vast number of features, offering tentative identifications, while 1H NMR provides highly reproducible, quantitative structural information in a non-destructive manner [19]. The integration of these orthogonal data streams is therefore not merely beneficial but essential for confident metabolite annotation and biological insight.
This article details the strategic framework of data fusion, specifically structured into low, mid, and high-level integration, within the context of a broader thesis on HR-MS/NMR integration for metabolite identification. Moving beyond simple data reporting, advanced fusion strategies enable researchers to transcend the limitations of individual techniques, yielding a more holistic and reliable interpretation of complex biological samples. As demonstrated in food chemistry research, combining LC-HRMS and 1H NMR through multi-omics data integration successfully classified Amarone wines based on processing parameters, achieving a lower classification error rate and revealing complementary metabolic profiles that single-platform analysis could not capture [19].
Hierarchical Framework of Data Fusion
Data fusion strategies can be systematically categorized into a three-level hierarchy based on the stage at which data from different platforms are combined. This progression mirrors a conceptual shift from parallel reporting to unified computational modeling.
Low-Level (Data-Level) Integration: This strategy involves the earliest possible combination of raw or pre-processed data from multiple analytical platforms. For HR-MS and NMR, this could mean aligning and concatenating binned NMR spectra with MS peak intensity vectors into a single, composite data matrix. The primary advantage is the preservation of all original information, allowing for the discovery of complex, cross-platform interactions. However, it is highly sensitive to technical noise, requires sophisticated normalization to handle the different scales and variances of MS and NMR data, and is computationally intensive.
Mid-Level (Feature-Level) Integration: In this approach, data from each platform are processed and distilled independently to extract significant features (e.g., statistically relevant peaks, identified compounds, or spectral bins). These selected feature sets are then fused into a combined matrix for subsequent multivariate analysis. This strategy offers a practical balance, reducing data dimensionality and noise while retaining the most biologically relevant information from each technique. It is the most commonly applied method in metabolomics, as it mitigates scaling issues and allows for platform-specific pre-processing.
High-Level (Decision-Level) Integration: This represents the most abstract level of fusion. Here, separate models or conclusions are generated independently from each data platform (e.g., a list of putative identifications from HR-MS and a validated list of compounds from NMR). These independent results are then merged or compared at the decision stage using consensus rules, voting systems, or meta-analysis. The strength of this strategy lies in its flexibility and robustness, as errors or uncertainties from one platform do not directly propagate into the analysis of the other. However, it can fail to capture lower-level correlations between the datasets.
Table 1: Comparison of Data Fusion Strategy Levels
| Integration Level | Stage of Fusion | Key Advantages | Primary Challenges |
|---|---|---|---|
| Low-Level (Data-Level) | Raw or pre-processed data matrices | Maximizes information retention; enables discovery of subtle, cross-platform patterns | Sensitive to noise and measurement artifacts; requires complex scaling/normalization. |
| Mid-Level (Feature-Level) | Selected, extracted features from each platform | Reduces dimensionality; balances information content with analytical tractability | Risk of losing informative features during selection; requires careful feature alignment. |
| High-Level (Decision-Level) | Results or decisions from independent models | Robust to platform-specific noise; flexible and modular | May overlook correlated patterns across platforms; dependent on quality of individual models. |
Application Notes: HR-MS and NMR in Metabolite Identification
The synergistic integration of HR-MS and NMR is a cornerstone of authoritative metabolite identification, aligning with the metabolomics standards initiative tiers of identification. The following application notes contextualize the fusion strategies within a practical research workflow.
Application Note 1: Expanding Metabolite Coverage and Validation HR-MS, particularly in liquid chromatography (LC) mode, is unparalleled for detecting thousands of metabolite features in a single run, providing accurate mass for formula prediction and fragmentation patterns for structural elucidation. However, co-elution, isobaric interferences, and the inability to distinguish isomers often lead to ambiguous identifications. 1H NMR directly addresses these limitations by providing unambiguous evidence for specific functional groups and stereochemistry. A mid-level fusion strategy is highly effective here: HR-MS spectral features are first matched against chemical databases to generate a list of candidate identifications. This list is subsequently validated against the NMR chemical shift and coupling constant data, which act as a orthogonal filter to confirm or reject proposed structures, dramatically increasing confidence in the final annotation.
Application Note 2: Quantitative Multi-Omics Profiling for Biomarker Discovery In differential analysis (e.g., case vs. control), the goal is to discover metabolites whose levels correlate with a phenotype. HR-MS provides sensitive detection of low-abundance potential biomarkers, while NMR offers absolute quantification and high technical reproducibility for a subset of central metabolites. A high-level fusion strategy is advantageous. Separate statistical models (e.g., t-tests, PLS-DA) are applied to the significant features from each platform. The final list of candidate biomarkers is a consensus from both models, weighted by factors such as statistical significance, fold-change, and platform-derived identification confidence. This approach was exemplified in a study classifying Amarone wines, where multi-omics integration using MCIA and sPLS-DA provided a broader characterization of the wine metabolome related to withering time and yeast strain than either technique alone [19].
Application Note 3: Dynamic Metabolic Flux Analysis Tracking the fate of isotopically labeled nutrients through metabolic pathways requires both broad detection (HR-MS) and specific positional enrichment information (2H or 13C NMR). A low-level or mid-level fusion strategy is necessary. Time-series data from HR-MS (showing total label incorporation per metabolite) and NMR (showing label position) can be integrated into a unified model to constrain and refine metabolic flux maps. The complementary data provides stronger constraints on network topology and reaction rates than either dataset could alone, enabling more accurate systems-level modeling.
Diagram 1: HR-MS/NMR Fusion Framework for Metabolomics
Detailed Experimental Protocols
The following protocols outline a standardized workflow for mid-level data fusion of LC-HRMS and 1H NMR data, as applied in recent metabolomics research [19].
Protocol 4.1: Sample Preparation and Multi-Platform Acquisition
Aim: To generate complementary HR-MS and NMR data from the same biological sample set.
Materials:
- Biological samples (e.g., plasma, urine, tissue extract, wine [19]).
- Appropriate internal standards for each platform (see The Scientist's Toolkit).
- NMR solvent (e.g., D₂O with 0.1-1 mM TSP or DSS for chemical shift referencing).
- LC-MS grade solvents (water, acetonitrile, methanol).
Procedure:
- Sample Processing: Prepare a single, homogeneous extract per sample using a suitable method (e.g., methanol/water precipitation for polar metabolites). Split the extract into two aliquots (e.g., 80% for HR-MS, 20% for NMR).
- LC-HRMS Analysis:
- LC Method: Use a reversed-phase (C18) or hydrophilic interaction liquid chromatography (HILIC) column. Employ a gradient elution (e.g., water/acetonitrile with 0.1% formic acid) optimized for metabolite separation.
- MS Method: Acquire data in data-dependent acquisition (DDA) mode on a high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap). Settings: positive/negative ionization switching, mass range m/z 50-1500, resolution > 30,000. Use lock mass calibration for high mass accuracy.
- 1H NMR Analysis:
- Sample Preparation: Combine the NMR aliquot with NMR buffer (e.g., phosphate buffer in D₂O, pH 7.4) and internal standard (TSP). Transfer to a standard 3 mm or 5 mm NMR tube.
- NMR Acquisition: Acquire 1D 1H NMR spectra on a spectrometer (≥ 600 MHz recommended). Use a standard NOESY-presat pulse sequence (noesygppr1d) for water suppression. Key parameters: spectral width 20 ppm, relaxation delay 4-5 s, number of scans 64-128, temperature 298 K.
Protocol 4.2: Data Pre-processing and Feature Extraction (Platform-Specific)
Aim: To independently convert raw instrument data into cleaned, aligned data matrices for each platform.
HR-MS Data Processing:
- Use specialized software (e.g., XCMS, MS-DIAL, Compound Discoverer) for peak picking, alignment, and gap filling.
- Perform retention time correction and group features across samples.
- Annotate features using accurate mass (± 5 ppm) and MS/MS libraries (e.g., GNPS, MassBank). Output a matrix where rows are samples, columns are m/z-RT features, and values are peak intensities. Apply probabilistic quotient normalization to correct for dilution effects.
1H NMR Data Processing:
- Process raw FIDs: Apply exponential line broadening (0.3-1.0 Hz), Fourier transformation, phase and baseline correction manually or via algorithms like NMRProcFlow.
- Reference the spectrum to the internal standard (TSP at δ 0.0 ppm).
- Perform spectral binning (bucket integration). Common approach: Divide the region δ 0.5-10.0 ppm into fixed-width bins (e.g., 0.04 ppm = 0.0025 ppm). Exclude the water region (δ 4.7-5.0 ppm).
- Normalize the binned data to the total spectral area or to an internal standard. Output a matrix where rows are samples and columns are spectral bins.
Protocol 4.3: Mid-Level Data Fusion and Multivariate Analysis
Aim: To statistically integrate the processed HR-MS and NMR feature matrices.
Procedure:
- Feature Selection & Scaling: For each platform's data matrix, apply univariate (e.g., ANOVA p-value < 0.05) or multivariate (e.g., VIP scores from a preliminary PLS-DA) feature selection to retain the most biologically relevant variables. Scale the selected data using unit variance scaling (UV) or Pareto scaling.
- Data Concatenation: Horizontally concatenate the scaled and selected HR-MS and NMR matrices to create a fused data matrix (X_fused). Ensure sample order is identical.
- Multivariate Modeling:
- Unsupervised Exploration: Apply Multi-Block Principal Component Analysis (MB-PCA) or Multiple Co-Inertia Analysis (MCIA) to explore the combined dataset and assess the correlation between blocks (HR-MS and NMR). An RV-coefficient (like the reported 16.4% [19]) can quantify this block consistency.
- Supervised Classification: Use sparse Partial Least Squares-Discriminant Analysis (sPLS-DA) to build a classification model (e.g., for disease states or treatment groups). The sparsity parameter forces the model to select the most discriminative variables from both platforms simultaneously, yielding a single, integrated biomarker panel.
- Model Validation: Validate the sPLS-DA model using permutation testing (e.g., 1000 permutations) and calculate the classification error rate (e.g., Balanced Error Rate). A lower error rate from the fused model compared to single-platform models demonstrates the added value of integration [19].
Diagram 2: Mid-Level Data Fusion Workflow for HR-MS/NMR
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagents and Materials for HR-MS/NMR Metabolomics Integration
| Item | Function/Description | Critical Application Notes |
|---|---|---|
| Deuterated Solvent (D₂O) with Buffer | Provides the NMR lock signal and constant pH environment. Commonly used with 0.1 M phosphate buffer, pH 7.4. | Ensures stable, reproducible NMR chemical shifts. The pH must be tightly controlled as it significantly affects the chemical shift of many metabolites (e.g., amino acids). |
| Internal Standard for NMR (TSP or DSS) | Provides a chemical shift reference (set to δ 0.0 ppm) and can be used for quantitative concentration determination. | Trimethylsilylpropanoic acid (TSP) is water-soluble and inert. It should be used at low concentration (0.1-1 mM). Note: It can bind to proteins, so alternatives like DSS (2,2-dimethyl-2-silapentane-5-sulfonate) may be preferred for complex samples. |
| Internal Standards for HR-MS | A cocktail of stable isotope-labeled compounds (e.g., 13C, 15N, 2H) covering various chemical classes. | Used for mass accuracy calibration, retention time alignment, and semi-quantitative normalization. They correct for instrument drift and matrix effects during LC-MS analysis. |
| LC-MS Grade Solvents | Ultra-pure water, acetonitrile, methanol, and additives (e.g., formic acid, ammonium acetate). | Essential for minimizing chemical noise and ion suppression in HR-MS. Contaminants can generate artefactual peaks and reduce sensitivity. |
| Solid Phase Extraction (SPE) Cartridges | For sample clean-up and metabolite fractionation prior to analysis (e.g., C18 for lipids, HILIC for polar metabolites). | Reduces sample complexity and matrix effects, improving detection of low-abundance metabolites in both HR-MS and NMR. |
| Standard 5 mm or 3 mm NMR Tubes | High-precision glassware for holding the sample within the NMR magnet. | Quality directly affects spectral resolution and lineshape. Use tubes matched to the spectrometer's probehead. 3 mm tubes allow for analysis with smaller sample volumes. |
Key Applications in Biomedical Research, Drug Discovery, and Clinical Metabolomics
1. Introduction: The Integrative Power of HR-MS and NMR in Metabolite Research
The comprehensive characterization of the metabolome is fundamental to advancing biomedical research, streamlining drug discovery, and realizing personalized clinical interventions. Within this context, the integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy has emerged as a transformative strategy, moving beyond the limitations of single-platform analyses [1]. HR-MS offers exceptional sensitivity and broad metabolite coverage, capable of detecting thousands of features in complex biological matrices [4]. NMR provides robust, quantitative, and reproducible structural elucidation with minimal sample preparation [20] [21]. While often viewed independently, these techniques are fundamentally complementary; their synergy enables more accurate metabolite identification, expands metabolome coverage, and increases confidence in biomarker discovery [2].
This article details specific application notes and experimental protocols for leveraging HR-MS/NMR integration. Framed within a broader thesis on analytical synergy, the content provides a roadmap for researchers and drug development professionals to implement these powerful, combined approaches across key domains. The subsequent sections outline practical workflows, from foundational data fusion strategies to domain-specific applications in disease research, drug development pipelines, and clinical diagnostics.
2. Foundational Data Integration Framework
The combined analysis of HR-MS and NMR data requires systematic integration strategies, commonly categorized by the level of data abstraction used [1].
Table 1: Data Fusion Strategies for HR-MS/NMR Integration
| Fusion Level | Description | Process | Advantages | Limitations |
|---|---|---|---|---|
| Low-Level | Concatenation of raw or pre-processed data matrices [1]. | NMR spectral bins and MS peak lists are scaled and merged into a single matrix for multivariate analysis. | Maximizes use of raw data information; simple conceptual framework. | High dimensionality; requires careful scaling to balance techniques; prone to technical noise [1]. |
| Mid-Level | Integration of features extracted from each dataset [1]. | Separate dimensionality reduction (e.g., PCA) on NMR and MS data, followed by concatenation of significant scores or loadings. | Reduces data dimensionality; focuses on most biologically relevant features from each platform. | Dependent on initial feature selection; may lose subtle interactions present in raw data. |
| High-Level | Fusion of model predictions or decisions [1]. | Independent classification or regression models are built for NMR and MS data; their outputs are combined via voting or meta-learners. | Flexible; allows use of optimal models for each data type; robust to platform-specific noise. | Complex to implement; requires separate model validation; final model can be less interpretable. |
An applied example of mid-level fusion is demonstrated in a study classifying Amarone wines, where NMR and LC-HRMS data were integrated using unsupervised and supervised methods. The multi-omics approach achieved a lower classification error rate (7.52%) compared to single-platform models, highlighting the improved predictive accuracy gained from complementarity [22].
Figure 1: Logical workflow for selecting and implementing data fusion strategies to integrate complementary HR-MS and NMR datasets [22] [1].
3. Application Note 1: Biomedical Research & Disease Mechanism Elucidation
3.1. Objective: To identify perturbed metabolic pathways and potential biomarkers in disease states by achieving comprehensive metabolome coverage through HR-MS/NMR integration.
3.2. Protocol: SYNHMET for Personalized Metabolic Profiling in Biofluids
This protocol is adapted from the SYnergic use of NMR and HRMS for METabolomics (SYNHMET) strategy for quantifying metabolites in human urine [4].
Sample Preparation:
- Collect urine samples following standardized clinical procedures. Centrifuge at 4°C to remove particulate matter.
- For NMR: Mix 540 µL of urine supernatant with 60 µL of phosphate buffer (1.5 M K₂HPO₄/NaH₂PO₄, pH 7.4, in D₂O containing 0.1% w/w sodium 3-(trimethylsilyl)propionate-2,2,3,3-d₄ (TSP) as chemical shift reference). Transfer to a 5 mm NMR tube [4].
- For HR-MS: Dilute urine supernatant 1:10 with a water/acetonitrile/isopropanol (1:1:1) solution containing a cocktail of stable isotope-labeled internal standards for quality control.
Data Acquisition:
- NMR: Acquire 1D ¹H NMR spectra at 600 MHz (or higher) using a standard NOESY-presaturation pulse sequence to suppress the water signal. Use 128 scans at 298K [4].
- HR-MS: Perform UHPLC-HRMS analysis in both positive and negative ionization modes. Use two chromatographic methods: Reversed-Phase (RP) for hydrophobic compounds and Hydrophilic Interaction Liquid Chromatography (HILIC) for polar compounds. Employ an Orbitrap or Q-TOF mass analyzer for high-resolution data [4].
Data Processing & Synergistic Quantification:
- Process NMR spectra (phase, baseline correction, reference to TSP). Deconvolute complex spectral regions using a database of reference chemical shifts (e.g., Chenomx) [4].
- Process MS data (peak picking, alignment, annotation). Generate a list of potential metabolites for each accurate mass (within 5 ppm tolerance).
- Integration Core: Use initial, approximate concentrations from NMR deconvolution as a seed. Correlate these values with intensities of candidate MS peaks across all samples. The MS feature showing the highest linear correlation is assigned to the metabolite.
- Convert the correlated MS intensity into an absolute concentration using the slope from the linear fit, thereby refining and validating the NMR-derived quantification.
3.3. Key Research Outputs: Application of this protocol enabled the absolute quantification of 165 metabolites in urine from healthy subjects, patients with chronic cystitis, and bladder cancer patients, establishing personalized metabolic profiles for disease monitoring [4].
Table 2: Key Metabolites Identified via Integrated HR-MS/NMR in Model Systems
| Disease/Model | Key Perturbed Pathways | Metabolites Uniquely Identified by NMR | Metabolites Uniquely Identified by HR-MS | Reference |
|---|---|---|---|---|
| Bladder Cancer (Human Urine) | TCA Cycle, Amino Acid Metabolism | 2-oxoglutarate, Glutamine, Succinate | Various low-abundance lipids, conjugates | [4] |
| Chlamydomonas reinhardtii (Treated) | Glycolysis, TCA Cycle, Amino Acid Biosynthesis | Glycine, Lysine, Valine, Acetate | Fructose-6-phosphate, Fumarate, Asparagine | [2] |
4. Application Note 2: Drug Discovery and Development
4.1. Objective: To employ metabolomics for target identification, mechanism of action (MoA) elucidation, and early toxicity screening of drug candidates.
4.2. Protocol: Cell-Based Metabolomics for MoA Deconvolution
This protocol outlines a cell-based assay to differentiate on-target from off-target metabolic effects of lead compounds [20].
Cell Treatment and Metabolite Extraction:
- Seed cancer cell lines (e.g., HeLa, primary AML cells) in 96-well plates. Treat with lead compounds, controls, and a reference inhibitor for 24-48 hours.
- Quench Metabolism & Extract: Rapidly remove media, wash with ice-cold saline, and quench metabolism by adding chilled methanol (-40°C). Add a mixture of methanol/chloroform (for polar and non-polar metabolites) containing internal standards (e.g., deuterated amino acids) [20] [23].
- Scrape cells, vortex, and centrifuge. Collect the supernatant. For biphasic separation, add water and chloroform, vortex, centrifuge, and collect both polar (upper) and non-polar (lower) layers.
Dual-Platform Analysis:
- NMR Analysis: Dry the polar extract and reconstitute in D₂O phosphate buffer. Acquire 1D ¹H CPMG spectra to suppress macromolecule signals and observe small molecules [20].
- HR-MS Analysis: Analyze both polar and non-polar extracts via UHPLC-HRMS in positive/negative modes. Use HILIC and RP columns for comprehensive coverage.
Data Integration and Interpretation:
- Process and normalize data from both platforms separately. Conduct multivariate statistical analysis (e.g., PCA, PLS-DA) on each dataset to identify treatment-induced metabolic perturbations.
- Use a mid-level fusion approach: Identify significant metabolic features (VIP > 1.0) from both NMR and MS models. Map these discriminating metabolites onto biochemical pathways (e.g., KEGG).
- MoA Assignment: A metabolic signature that matches the known effects of a selective reference inhibitor suggests on-target activity. Unique or additional perturbations indicate potential off-target effects or novel mechanisms [20] [24].
4.3. Key Research Outputs: This integrated approach can validate drug efficacy, predict toxicity via metabolic dysregulation (e.g., disrupted TCA cycle, redox imbalance), and help prioritize lead compounds with a cleaner on-target profile [20] [24].
Figure 2: The role of integrated HR-MS/NMR metabolomics at key stages of the modern drug discovery and development pipeline [20] [24].
5. Application Note 3: Clinical Metabolomics and Personalized Medicine
5.1. Objective: To discover and validate robust biomarkers for patient stratification, diagnosis, and treatment monitoring in clinical settings.
5.2. Protocol: Serum/Plasma Profiling for Biomarker Discovery
This protocol describes a standardized workflow for translational biomarker studies using blood-derived samples [21] [23].
Standardized Sample Collection & Biobanking:
- Draw blood into appropriate tubes (e.g., EDTA for plasma, serum tubes). Process samples (centrifuge, aliquot) within 30-60 minutes of collection to minimize ex vivo metabolic changes.
- Flash-freeze aliquots in liquid nitrogen and store at -80°C. Maintain consistent pre-analytical conditions across all patients and controls.
High-Throughput Metabolite Profiling:
- NMR for High-Level Profiling: Thaw plasma/serum on ice. Mix with phosphate buffer/D₂O. Use automated flow-injection NMR systems to acquire 1D ¹H spectra. NMR provides absolute quantification of ~40-60 major metabolites (lipoproteins, glucose, amino acids) with high reproducibility, ideal for large cohorts [21].
- HR-MS for Deep-Dive Profiling: Perform a targeted UHPLC-HRMS analysis focusing on predefined panels of metabolites (e.g., amines, organic acids, lipids) implicated in the disease of interest. Use isotopically labeled internal standards for precise quantification.
Biomarker Panel Validation:
- Integrate quantitative data from NMR and targeted MS. Use multivariate statistics to identify a multi-analyte biomarker panel that best discriminates patient groups.
- Validate the panel in an independent, blinded cohort. The combined use of NMR (for robust, absolute quantitation of core metabolites) and MS (for sensitive, specific measurement of lower-abundance markers) increases the panel's clinical validity and reliability [21].
5.3. Key Research Outputs: Integrated profiling moves beyond single biomarkers to define metabolic "phenotypes." For example, a combined signature of NMR-quantified branched-chain amino acids and MS-quantified specific acylcarnitines may provide a superior predictor of metabolic disease risk or treatment response than any single analyte [21].
6. The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for HR-MS/NMR Metabolomics
| Item | Function | Key Considerations | Primary Domain |
|---|---|---|---|
| Deuterated Solvents & NMR Standards (e.g., D₂O, TSP, DSS) | Provides lock signal, chemical shift reference, and quantitative standard for NMR spectroscopy. | High isotopic purity (>99.9% D) is essential for sensitive experiments. | All (NMR-based) |
| Stable Isotope-Labeled Internal Standards (e.g., ¹³C, ¹⁵N, ²H-labeled metabolites) | Enables precise quantification and corrects for matrix effects and instrument variability in HR-MS. | Should be added at the earliest possible stage of sample preparation (e.g., during quenching) [23]. | All (MS-based) |
| Dual Solvent Extraction Systems (e.g., Methanol/Chloroform/Water) | Simultaneously quenches metabolism and extracts a broad range of polar and non-polar metabolites. | The Bligh & Dyer (2:1:0.8) and Folch (2:1:0.75) methods are classic biphasic systems [23]. | Biomedical Research, Drug Discovery |
| Quality Control (QC) Pool Samples | A homogenized pool of all study samples run intermittently to monitor instrumental drift and reproducibility. | Essential for identifying and correcting systematic technical variation in large untargeted studies [23]. | All (Large Cohorts) |
| Chemical Shift Reference Buffer (e.g., Phosphate Buffer in D₂O, pH 7.4) | Standardizes pH, which critically affects NMR chemical shifts, especially for pH-sensitive metabolites like amines and acids. | Buffering capacity must be appropriate for the biofluid (e.g., urine) [4]. | Clinical Metabolomics, Biomarker Studies |
Methodological Workflows for Integrated HR-MS and NMR Metabolomics
Sample Preparation Protocols for Sequential NMR and Multi-Platform LC-MS Analysis
Within the framework of a thesis investigating High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) integration for comprehensive metabolite identification, the development of unified sample preparation protocols emerges as a foundational and non-trivial challenge. NMR spectroscopy and mass spectrometry (MS) are pivotal, complementary tools in metabolomics, yet they are traditionally employed in parallel with separate, platform-specific sample preparations [25] [26]. This divide necessitates larger sample volumes, increases analytical variability, and complicates the direct correlation of data. The integration of these techniques is driven by their complementary strengths: MS offers high sensitivity and broad metabolite coverage, while NMR provides non-destructive, reproducible analysis with unparalleled capabilities for structural elucidation and absolute quantification [26] [27]. Recent advancements underscore a paradigm shift towards sequential analysis from a single aliquot, a strategy that conserves precious biological material, enhances data alignment, and expands metabolome coverage [25] [28]. This application note details validated, robust protocols for preparing serum and tissue samples, enabling their sequential profiling by NMR and multiple LC-MS platforms, thereby facilitating a more holistic and efficient approach to metabolic phenotyping in research and drug development.
Pre-analytical Considerations and Sample Integrity
The reliability of any integrated metabolomics study is contingent upon rigorous control of pre-analytical variables. Biological confounders such as diet, circadian rhythm, age, and stress significantly influence the blood metabolome and must be documented and standardized where possible [27]. For biofluids like blood serum and plasma, collection protocols are critical. The use of standardized collection tubes, strict adherence to clotting times (for serum), rapid centrifugation, and immediate flash-freezing in liquid nitrogen are essential steps to halt enzymatic activity and preserve metabolic profiles [27]. Tissue samples require careful dissection, rinsing with saline to remove blood, snap-freezing, and storage at or below -80°C. Adherence to emerging standards, such as ISO 23118:2021 for pre-examination processes in metabolomics, is highly recommended to ensure inter-laboratory reproducibility and data comparability [27] [5].
Detailed Sequential Preparation Protocols
Protocol for Blood Serum/Plasma Samples
This protocol is designed to enable 1H-NMR spectroscopy followed by untargeted multi-platform LC-MS analysis (e.g., RPLC-HILIC, positive/negative ionization) from a single serum aliquot [25].
- Step 1: Initial Processing. Thaw frozen serum samples on ice. Gently vortex to ensure homogeneity.
- Step 2: Protein Removal for LC-MS Compatibility. Transfer a measured volume of serum (e.g., 200 µL) to a molecular weight cut-off (MWCO) filtration device (e.g., 3 kDa or 10 kDa). Centrifuge according to the manufacturer's specifications (typically 10-15 minutes at 4°C, 14,000 x g). Retain the filtrate (protein-free metabolite fraction). Note: Protein removal is a primary factor influencing downstream metabolite abundances and is essential for robust LC-MS analysis [25].
- Step 3: Preparation for NMR Analysis. Combine the protein-free filtrate with a deuterated phosphate buffer (e.g., 100 mM, pD 7.4, containing 0.5 mM TSP-d4 as a chemical shift reference) in a standard 1:1 ratio (e.g., 200 µL sample + 200 µL buffer). Transfer the mixture to a clean 3 mm or 5 mm NMR tube for data acquisition. Crucially, studies confirm that metabolites do not undergo deuterium incorporation when prepared in this manner, preserving their native mass for subsequent MS analysis [25].
- Step 4: Post-NMR Sample Recovery for LC-MS. Following NMR acquisition, the non-destructive nature of NMR allows for the complete recovery of the sample from the NMR tube. This sample can be directly injected for LC-MS analysis. Alternatively, for compatibility with specific LC-MS platforms or to concentrate metabolites, the sample can be lyophilized and reconstituted in an appropriate MS-compatible solvent (e.g., water/acetonitrile, 95:5 v/v). The NMR buffers are well-tolerated in LC-MS systems without suppressing ionization or causing interferences [25].
- Step 5: Quality Control (QC). Prepare a pooled QC sample from an aliquot of all study samples. Inject the QC repeatedly at the beginning of the analytical run and at regular intervals throughout to monitor instrument stability.
Protocol for Tissue Samples (e.g., Liver)
This biphasic/two-step extraction protocol maximizes the recovery of both polar metabolites and lipids from a single tissue specimen for sequential NMR and LC-MS lipidomics/untargeted profiling [28].
- Step 1: Tissue Homogenization. Weigh approximately 50 mg of frozen tissue. Add pre-chilled (-20°C) extraction solvent, typically methanol, at a ratio of 20 µL/mg tissue. Homogenize immediately using a bead mill or a mechanical homogenizer while keeping the sample on ice or in a cold room.
- Step 2: Sequential Metabolite Extraction.
- Polar and Lipid Metabolite Co-extraction: Add chilled chloroform and water to the homogenate to achieve a final solvent ratio of CHCl₃/MeOH/H₂O of 1:2:0.8 (v/v/v). Vortex vigorously.
- Phase Separation: Centrifuge (e.g., 10,000 x g, 10 min, 4°C) to achieve biphasic separation. The upper aqueous phase contains polar metabolites, while the lower organic phase contains lipids.
- Phase Transfer: Carefully collect and transfer the upper aqueous phase to a new tube. Transfer the lower organic phase to a separate tube.
- Step 3: Sample Processing for Sequential Analysis.
- Aqueous Phase (Polar Metabolites): Dry under a gentle stream of nitrogen or by vacuum centrifugation. Reconstitute a portion of the dried extract in deuterated phosphate buffer for NMR analysis. Following NMR, the sample can be recovered, further diluted if necessary, and analyzed by UHPLC-Q-Orbitrap MS for untargeted polar metabolomics [28].
- Organic Phase (Lipids): Dry the organic phase completely under nitrogen. Reconstitute the lipid extract in a suitable solvent (e.g., isopropanol/acetonitrile) for direct analysis by UHPLC-MS-based lipidomics [28].
- Step 4: Alternative Two-Step Extraction. For some tissues, a two-step sequential extraction may yield better results: first, extract with CHCl₃/MeOH for lipids, then re-extract the pellet with MeOH/H₂O for polar metabolites [28].
Key Quantitative Performance Metrics
The following table summarizes the performance outcomes of the described integrated protocols based on validation studies.
Table 1: Performance Metrics of Integrated NMR/LC-MS Sample Preparation Protocols
| Metric | Serum Protocol (MWCO Filtration) | Tissue Protocol (Biphasic Extraction) | Significance |
|---|---|---|---|
| Metabolite Recovery | High recovery of low-MW metabolites; proteins >10 kDa removed [25]. | Comprehensive coverage of polar metabolites (aqueous) and lipids (organic) [28]. | Enables detection of a broad chemical space from a single sample. |
| Reproducibility (CV) | LC-MS feature abundances show minimal variation post-NMR buffer preparation [25]. | High reproducibility for annotated metabolites in both phases [28]. | Essential for generating reliable, statistically powerful data. |
| Deuterium Artifact | No evidence of deuterium incorporation into metabolites [25]. | Not applicable (aqueous phase reconstituted in D₂O buffer for NMR). | Preserves native mass for accurate MS analysis. |
| Number of Annotated Metabolites | Enables detection of hundreds of compound-features across multiple LC-MS platforms [25]. | Allows generation of a comprehensive metabolic map for tissue [28]. | Maximizes biological information extracted from limited sample. |
| Primary Advantage | Single aliquot for sequential, complementary analysis; efficient sample use [25]. | Simultaneous profiling of polar metabolome and lipidome from one tissue piece [28]. | Overcomes the traditional sample volume limitation for multi-omics. |
Data Integration and Correlation Strategies
Acquiring data from multiple platforms creates the challenge and opportunity for integrated analysis. Data fusion strategies are classified by the level of abstraction at which integration occurs [26].
- Low-Level Data Fusion: Raw or pre-processed data matrices from NMR and LC-MS are concatenated into a single large matrix for multivariate analysis (e.g., PCA, PLS-DA). This approach retains all information but requires careful intra- and inter-block scaling (e.g., Pareto scaling) to balance the influence of each platform due to differing variances and numbers of variables [26].
- Mid-Level Data Fusion: Features (e.g., significant metabolites, spectral bins) are first selected or extracted from each platform's dataset independently. These selected features are then fused into a consolidated dataset for modeling. This reduces dimensionality and can focus analysis on the most biologically relevant signals from each technique [26].
- High-Level Data Fusion: Independent models (e.g., classifiers, regressions) are built from each analytical platform's data. The final prediction or decision is made by combining the outputs of these individual models, such as through majority voting or meta-classification [26].
- Direct Mathematical Correlation: For structural identification, approaches like the SCORE-metabolite-ID method can be employed. This involves fractionating a sample and acquiring NMR and DI-MS spectra for each fraction. The correlation of signal intensity profiles across fractions links specific NMR chemical shifts to associated MS m/z values, enabling confident metabolite identification directly from complex mixtures without isolation [29].
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Integrated Sample Preparation
| Item | Function / Purpose | Key Considerations & Recommendations |
|---|---|---|
| Deuterated Phosphate Buffer (e.g., 100-150 mM in D₂O, pD 7.4) | Provides a stable, isotopic lock signal for NMR; minimizes pH-induced chemical shift variation. Contains internal reference (TSP-d4). | Essential for NMR. Proven compatible with downstream LC-MS; does not cause deuterium exchange [25]. |
| Molecular Weight Cut-Off (MWCO) Filters (3 kDa or 10 kDa) | Removes high-molecular-weight proteins that interfere with LC-MS analysis and can foul chromatography columns. | A critical step influencing metabolite recovery. Filter material should be checked for non-specific binding of metabolites of interest. |
| Deuterium Oxide (D₂O), 99.9% | Solvent for NMR spectroscopy; provides the deuterium lock signal. | Standard reagent. Used in preparation of NMR buffers. |
| Methanol (MeOH), LC-MS Grade | Primary extraction solvent for tissue; protein precipitant for biofluids. | Use high-purity, LC-MS grade to avoid background ions and contaminants. |
| Chloroform (CHCl₃), HPLC Grade | Organic solvent for lipid extraction in biphasic separations. | Handle in a fume hood. Essential for comprehensive lipidomics from tissue [28]. |
| Internal Standards | For quantification and quality control. | NMR: TSP-d4 (chemical shift reference). MS: Stable isotope-labeled standards (e.g., for amino acids, lipids) added prior to extraction for absolute quantification. |
| Cryogenic Vials & Pre-chilled Storage Boxes | For snap-freezing and long-term storage of biofluids and tissue at -80°C. | Vital for preserving labile metabolites and ensuring sample integrity [27]. |
The integration of NMR and multi-platform LC-MS through sequential analysis of a single prepared sample represents a significant methodological advancement in metabolomics. The protocols detailed herein, validated for serum and tissue matrices, directly address the core challenge of sample volume limitation while harnessing the complementary analytical strengths of both platforms. This synergistic approach—facilitated by compatible preparation methods that avoid artifacts like deuterium exchange—enables broader metabolome coverage, more confident metabolite identification, and a stronger foundation for data fusion. For research in drug development and systems biology, where comprehensive metabolic phenotyping is paramount, adopting these integrated workflows enhances efficiency, conserves valuable specimens, and provides a more robust, multi-dimensional view of the biochemical state, ultimately driving more informed scientific conclusions.
The definitive identification of metabolites in complex biological matrices is a cornerstone of modern research in drug development, toxicology, and systems biology. This task presents a significant analytical challenge due to the vast chemical diversity, wide concentration ranges, and dynamic nature of the metabolome. No single analytical technique can provide a complete picture. Instead, the integration of two powerful platforms—Nuclear Magnetic Resonance (NMR) spectroscopy and High-Resolution Mass Spectrometry (HR-MS)—has emerged as the gold standard for confident metabolite structural elucidation and quantification [26].
The synergy between these techniques is rooted in their complementary strengths and weaknesses. HR-MS, particularly when coupled with liquid chromatography (LC), excels in sensitivity, capable of detecting metabolites at trace (attomolar) levels, and provides exact molecular mass and elemental composition data [26] [3]. However, it is a destructive technique and often cannot distinguish between structural isomers without extensive additional experimentation. Conversely, NMR spectroscopy is inherently non-destructive, offers exceptional reproducibility, and provides definitive information on molecular structure, functional groups, and atomic connectivity through parameters like chemical shift, coupling constants, and integration [26] [30]. Its principal limitation is lower sensitivity compared to MS.
The integration of these orthogonal data streams through Data Fusion (DF) strategies moves beyond simple parallel analysis. DF creates a unified, more informative dataset that offers a holistic view of the biochemical profile, enhancing the accuracy of biomarker discovery, pathway analysis, and the identification of unknown metabolites [26] [22]. This application note details standardized protocols for data acquisition from both platforms and frameworks for their integration, designed to support robust metabolite identification within a broader research thesis.
Experimental Protocols for Core Techniques
Protocol: HR-MS-Based Untargeted Metabolomics (LC-HRMS)
This protocol is optimized for profiling polar metabolites in biofluids (e.g., plasma, urine) using Hydrophilic Interaction Liquid Chromatography (HILIC) coupled to a high-resolution accurate mass spectrometer (e.g., Orbitrap or Q-ToF) [31].
Sample Preparation & Extraction:
- Thawing: Thaw biofluid samples slowly on ice.
- Aliquoting: Aliquot a precise volume (e.g., 50 µL of plasma) into a precooled microcentrifuge tube.
- Protein Precipitation/Extraction: Add 150 µL of ice-cold extraction solvent (Acetonitrile:Methanol:Formic Acid, 74.9:24.9:0.2, v/v/v) containing stable isotope-labeled internal standards (e.g., L-Phenylalanine-d8 at 0.1 µg/mL and L-Valine-d8 at 0.2 µg/mL) [31].
- Mixing & Incubation: Vortex vigorously for 30 seconds and incubate at -20°C for 30 minutes to ensure complete protein precipitation.
- Centrifugation: Centrifuge at >14,000 x g for 15 minutes at 4°C.
- Collection: Transfer the clear supernatant to a fresh LC-MS vial with insert. Keep samples at 4°C in the autosampler until analysis.
LC-HRMS Data Acquisition:
- Chromatography:
- Column: Atlantis HILIC Silica (2.1 x 150 mm, 3 µm) [31].
- Mobile Phase: A) 10 mM Ammonium Formate, 0.1% Formic Acid in Water; B) 0.1% Formic Acid in Acetonitrile [31].
- Gradient: Start at 85% B, ramp to 30% B over 15 min, hold for 3 min, re-equilibrate at 85% B for 7 min.
- Flow Rate: 0.3 mL/min. Column Temperature: 40°C.
- Mass Spectrometry (Orbitrap Example):
- Ionization: Heated Electrospray Ionization (HESI) in both positive and negative polarity modes.
- Resolution: ≥ 70,000 Full Width at Half Maximum (FWHM) at m/z 200.
- Scan Range: m/z 70-1050.
- Source Parameters: Sheath Gas: 40; Aux Gas: 10; Spray Voltage: ±3.5 kV; Capillary Temp: 320°C.
- Quality Control (QC): Inject a pooled QC sample (a mixture of all study samples) at the beginning of the run and after every 4-6 experimental samples to monitor instrument stability [31].
- Chromatography:
Data Processing & Feature Extraction:
- Convert raw files to an open format (e.g., .mzML).
- Use specialized software (e.g., Compound Discoverer, XCMS, MZmine) for peak picking, alignment, and deconvolution.
- For complex datasets, consider advanced feature extraction like the Region of Interest-Multivariate Curve Resolution (ROI-MCR) protocol, which effectively compresses data and resolves co-eluting signals [32].
- Annotate features using accurate mass (±5 ppm), isotopic patterns, and MS/MS spectral libraries (e.g., HMDB, METLIN, MassBank).
Protocol: NMR-Based Metabolomic Profiling
This protocol covers both standard solution-state NMR for extracts and High-Resolution Magic Angle Spinning (HR-MAS) NMR for intact tissues [33] [3].
A. Sample Preparation for Solution-State NMR (Biofluid/Extract):
- Buffer/D2O Addition: Mix 300 µL of sample (e.g., urine or reconstituted extract) with 300 µL of phosphate buffer (0.2 M, pH 7.4) containing 1 mM TSP (3-(trimethylsilyl)-2,2',3,3'-tetradeuteropropionic acid) as a chemical shift reference (δ 0.00 ppm) and 3 mM sodium azide [34].
- Centrifugation: Centrifuge at high speed to remove any particulate matter.
- Loading: Transfer 550 µL of the supernatant into a standard 5 mm NMR tube.
B. Sample Preparation for HR-MAS NMR (Intact Tissue):
- Tissue Handling: Rapidly harvest tissue (e.g., liver biopsy, plant leaf) and immediately freeze in liquid nitrogen. Store at -80°C [3].
- Loading: Place a small, weighed piece of tissue (5-15 mg) into a disposable MAS rotor insert.
- Reference Addition: Add 10 µL of D2O containing a known reference compound (e.g., TSP or DSS) for locking and referencing [3].
- Sealing: Cap the rotor tightly to ensure stable spinning.
NMR Data Acquisition:
- Standard 1D ¹H NMR:
- Experiment: 1D Nuclear Overhauser Effect Spectroscopy (NOESY) with pre-saturation for water suppression (for biofluids) or Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence to suppress broad macromolecular signals [3] [35].
- Parameters: Spectral Width: 12-14 ppm; Relaxation Delay (D1): 4 sec; Number of Scans: 64-128; Temperature: 298 K.
- HR-MAS ¹H NMR: Use similar pulse sequences with the magic angle spinning set to 4-5 kHz to average anisotropic interactions and achieve high-resolution spectra from semi-solid tissues [33] [3].
- 2D NMR for Structural Elucidation:
- Perform on samples of interest (e.g., purified metabolite, key sample fraction).
- ¹H-¹H COSY (Correlation Spectroscopy): Identifies scalar-coupled protons.
- ¹H-¹³C HSQC (Heteronuclear Single Quantum Coherence): Identifies direct proton-carbon bonds. Essential for structural assignment [30].
- Standard 1D ¹H NMR:
NMR Data Pre-processing:
- Fourier Transform raw FID data with exponential line broadening (0.3-1 Hz).
- Phase and baseline correct the spectrum manually or using automated algorithms [34] [35].
- Reference the spectrum to the internal standard (TSP/DSS at 0.0 ppm).
- Remove regions containing residual water (δ 4.7-4.9 ppm) and urea (if present).
- Perform spectral alignment (e.g., using the icoshift algorithm) to correct for small shifts.
- Integrate/Bin: Segment the spectrum into fixed (e.g., 0.01 ppm) or variable ("intelligent") bins. Alternatively, perform targeted integration of known metabolite peaks using tools like rDolphin or BATMAN [35].
- Normalize binned data using total sum normalization, probabilistic quotient normalization, or reference to an internal standard [35].
Data Integration: From Multiple Streams to Unified Insight
The true power of a multi-platform strategy lies in the formal integration of NMR and HR-MS datasets via Data Fusion (DF). DF strategies are classified by the level at which data are combined [26].
Table 1: Levels of Data Fusion for NMR and HR-MS Integration
| Fusion Level | Description | Process | Advantages | Challenges |
|---|---|---|---|---|
| Low-Level | Concatenation of raw or pre-processed data matrices. | NMR spectral buckets and MS peak intensities are scaled and merged into a single matrix [26]. | Maximizes information retention; uses all collected data. | High dimensionality; requires careful scaling to balance techniques; prone to technical noise. |
| Mid-Level | Fusion of extracted features from each dataset. | Features (e.g., identified metabolites from NMR, annotated compounds from MS) are combined into a new matrix for analysis [26]. | Reduces dimensionality; focuses on biologically relevant information. | Depends on accuracy of prior feature identification/annotation. |
| High-Level | Fusion of model predictions or decisions. | Separate statistical models (e.g., classification) are built for each platform, and their outputs are combined (e.g., by voting) [26]. | Robust to platform-specific noise; flexible. | Loses detail on variable-level interactions; interpretive complexity. |
A practical workflow often involves mid-level fusion. For example, a study classifying Amarone wines used unsupervised multi-platform integration (Multiple Co-inertia Analysis) to find complementary information between LC-HRMS and ¹H NMR datasets (RV-coefficient = 16.4%), followed by supervised modeling (sPLS-DA) on the fused dataset, achieving a low classification error rate (7.52%) [22].
Integrated Workflow from Sample to Biological Insight
Conceptual Framework for Multi-Level Data Fusion
The Scientist's Toolkit: Essential Reagents & Materials
Table 2: Key Research Reagent Solutions for Integrated Metabolomics
| Item | Function & Specification | Application Notes |
|---|---|---|
| Deuterated Solvent & NMR Reference | D2O (99.9% D) with TSP (trimethylsilylpropionic acid) or DSS (sodium trimethylsilylpropanesulfonate). Provides field-frequency lock and chemical shift reference (δ 0.00 ppm) [34] [3]. | Essential for all NMR experiments. TSP/DSS concentration should be precisely known if used for quantification. |
| Deuterated Chloroform (CDCl₃) | Organic solvent for NMR analysis of lipophilic extracts. Often includes TMS (tetramethylsilane) as an internal reference. | Used for reversed-phase LC-MS fractions or lipidomics samples. |
| LC-MS Grade Solvents | Water, Acetonitrile, Methanol, Formic Acid, Ammonium Formate/Acetate. Ultra-pure, low UV absorbance, minimal ion suppression [31]. | Critical for reproducible chromatography and MS sensitivity. Prepare mobile phases fresh weekly. |
| Stable Isotope-Labeled Internal Standards (IS) | e.g., L-Phenylalanine-d8, L-Valine-d8, or a broader mix. Used for quality control, monitoring extraction efficiency, and potential quantification in MS [31]. | Spiked into the extraction solvent at the start of sample prep to account for process variability. |
| Phosphate Buffer (for NMR) | 0.2 M potassium phosphate buffer in D2O, pH 7.4. Minimizes chemical shift variation due to pH differences between biofluid samples [35]. | Always include in biofluid NMR preparation for consistent metabolite chemical shifts. |
| MAS Rotor & Inserts | Zirconia rotor (e.g., 4 mm outer diameter) with disposable Kel-F or PCi inserts. Enables high-speed spinning of tissue samples for HR-MAS NMR [3]. | Disposable inserts prevent cross-contamination. Proper sealing is crucial for stable spinning. |
| Solid-Phase Extraction (SPE) Cartridges | Various chemistries (C18, HLB, Ion Exchange). For sample cleanup, fractionation, or metabolite concentration prior to analysis. | Useful for removing salts/interfering compounds or for isolating specific metabolite classes for deeper NMR analysis. |
Concluding Remarks
The systematic integration of HR-MS and NMR data acquisition protocols provides a powerful, orthogonal framework for comprehensive metabolite identification. While HR-MS offers the sensitivity to detect a wide breadth of metabolites, NMR delivers the definitive structural context required for unambiguous identification and absolute quantification. Implementing the standardized protocols for sample preparation, data acquisition, and processing outlined here ensures the generation of high-quality, interoperable datasets. Subsequent integration through appropriate data fusion strategies, particularly mid-level fusion of identified features, maximizes the analytical value of both platforms. This integrated approach is indispensable for advancing research in drug metabolism and safety (addressing MIST guidelines) [36], discovering robust biomarkers, and elucidating metabolic pathways in complex biological systems.
The comprehensive analysis of the metabolome, the complete set of low-molecular-weight metabolites in a biological system, is fundamental to advancing research in drug development, biomarker discovery, and systems biology [26]. Two analytical pillars support this field: High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy. Each technique offers distinct and complementary insights. HR-MS provides exceptional sensitivity, capable of detecting trace metabolites, and is ideal for complex sample profiling when coupled with chromatography. However, it is a destructive technique and can struggle with definitive structural elucidation and precise, absolute quantification. Conversely, NMR spectroscopy is non-destructive, highly reproducible, and excels at providing detailed structural information and unambiguous quantification, albeit with lower inherent sensitivity [26].
This dichotomy creates a significant analytical challenge. Relying on a single platform yields an incomplete picture of the metabolome, potentially missing critical biomarkers or misidentifying compounds. Consequently, data fusion (DF) strategies have emerged as a powerful paradigm to synergistically combine the multidimensional data from HR-MS and NMR. By integrating these datasets, researchers can construct more robust, accurate, and comprehensive models, leading to enhanced metabolite identification, superior sample classification (e.g., disease vs. healthy), and a more holistic understanding of biological pathways [26] [37]. This guide provides detailed application notes and protocols for implementing data fusion, framed within a broader thesis on HR-MS/NMR integration for metabolite identification.
Foundational Concepts: The Three Levels of Data Fusion
Data fusion methodologies are categorized by the level of abstraction at which data from different sources (e.g., HR-MS and NMR) are integrated. The choice of level balances analytical depth, interpretability, and computational complexity [26].
Low-Level Data Fusion (LLDF): Also known as early data fusion or block concatenation, this is the most straightforward approach. Raw or pre-processed data matrices from each platform are simply joined column-wise (variable-wise) to create a single, combined data matrix (X_fused = [X_NMR | X_MS]). While simple, it requires careful intra- and inter-block scaling to prevent the technique with higher variance (often MS) from dominating the model [26].
Mid-Level Data Fusion (MLDF): This two-step strategy first reduces the dimensionality of each dataset independently using feature extraction techniques like Principal Component Analysis (PCA). The extracted features (e.g., PCA scores) are then concatenated to form a fused matrix for final modeling. This approach effectively handles the "curse of dimensionality," especially when the number of variables (chemical shifts m/z peaks) far exceeds the number of samples [26].
High-Level Data Fusion (HLDF): Also called decision-level or late fusion, this is the most abstract approach. Separate models (e.g., classifiers or regressors) are built independently for each data block. Their outputs (predictions, probabilities, distances) are then combined using rules such as majority voting, weighted averaging, or a meta-classifier. This method is highly flexible and robust to heterogeneous data structures [26].
Table 1: Comparison of Data Fusion Levels for HR-MS/NMR Integration
| Fusion Level | Description | Key Advantages | Key Limitations | Best Use Case |
|---|---|---|---|---|
| Low-Level | Concatenation of raw/pre-processed data matrices. | Maximizes use of raw information; simple to implement. | Susceptible to scaling issues; high dimensionality; poor interpretability. | Preliminary exploration with well-scaled, similar-dimension datasets. |
| Mid-Level | Concatenation of features extracted from each block (e.g., PCA scores). | Reduces dimensionality; mitigates noise; good interpretability of block contributions. | Risk of losing informative variance during feature extraction. | Standard approach for combining high-dimensional MS with lower-dimensional NMR data. |
| High-Level | Combination of predictions from models built on each block. | Robust to heterogeneous data; allows different models per block; easy to update. | Loses granular variable-level information; complex to implement and interpret. | Integrating finalized, well-validated individual models for a consensus prediction. |
Detailed Experimental Protocols
Protocol: Sample Preparation for Coordinated HR-MS and NMR Analysis
Objective: To generate biologically paired and analytically compatible extracts for subsequent HR-MS and NMR profiling.
- Extraction: Perform a single-phase metabolite extraction from the biological sample (e.g., plasma, urine, tissue homogenate) using a solvent system compatible with both techniques (e.g., methanol:water 4:1 v/v with 0.1% formic acid for MS and deuterated buffer for NMR). Internal standards (e.g., DSS-d6 for NMR, isotope-labeled compounds for MS) must be added at this stage.
- Splitting: Immediately after extraction and thorough vortexing, split the homogeneous extract into two aliquots.
- MS Aliquot Preparation: Dry one aliquot under a gentle stream of nitrogen or in a vacuum concentrator. Reconstitute the dried extract in a solvent suitable for your chromatographic method (e.g., water:acetonitrile 95:5). Centrifuge and transfer the supernatant to an MS vial.
- NMR Aliquot Preparation: Take the second aliquot and dry it completely. Reconstitute the dried extract in a deuterated phosphate buffer (e.g., 100 mM potassium phosphate, pH 7.4, in D2O) containing 0.5 mM DSS-d6 as a chemical shift reference and quantification standard. Centrifuge and transfer 550-600 µL to a standard 5 mm NMR tube.
- Storage: Store all prepared samples at -80°C until analysis. Analyze MS and NMR samples in randomized order as closely in time as possible to minimize degradation.
Protocol: Low-Level Data Fusion Workflow
Objective: To create and analyze a fused dataset from pre-processed HR-MS and NMR data [26].
- Individual Data Pre-processing:
- NMR: Process FIDs (Fourier transformation, phasing, baseline correction). Align spectra to the DSS reference peak. Perform binning (e.g., 0.04 ppm buckets) or targeted integration of known metabolites. Normalize to the DSS signal or total spectral area.
- HR-MS: Process raw chromatograms (peak picking, alignment, gap filling). Annotate peaks with m/z and retention time. Normalize using internal standards or total ion count.
- Data Export & Formatting: Export both datasets as a sample-by-variable matrix (e.g., CSV files). Ensure sample order is identical.
- Intra-block Scaling: Scale variables within each technique to equalize their influence. Common methods include Pareto scaling (divide by the square root of the standard deviation) or Unit Variance (autoscaling) [26].
- Inter-block Scaling: Equalize the overall weight or contribution of the entire NMR block and the entire MS block to the fused model to prevent dominance by the noisier or higher-variance block. A common method is to scale each block so that the sum of the variances of its variables is equal [26].
- Concatenation: Join the scaled NMR and MS matrices horizontally to form the fused matrix
X_fused. - Modeling: Apply multivariate analysis (e.g., PCA for exploration, PLS-DA for classification, OPLS for regression) on
X_fused.
Diagram 1: Low-level data fusion workflow for NMR and MS data.
Protocol: Mid-Level Data Fusion with PCA Feature Extraction
Objective: To fuse HR-MS and NMR data at the feature level to manage high dimensionality [26] [37].
- Individual Pre-processing & Scaling: Complete steps 1-3 of the Low-Level Fusion Protocol for each dataset independently.
- Feature Extraction: Perform PCA separately on the scaled NMR matrix and the scaled MS matrix. Retain a sufficient number of principal components (PCs) to capture the majority of systematic variance (e.g., >70-80% cumulative R2X).
- Feature Matrix Creation: Extract the score matrices (
T_NMRandT_MS) for the selected PCs from each PCA model. - Feature Concatenation: Horizontally concatenate the score matrices to create the mid-level fused matrix:
T_fused = [T_NMR | T_MS]. - Supervised Modeling: Use the fused score matrix
T_fusedas the input for a supervised model like PLS-DA or classification SVM. The model will find the optimal combination of NMR- and MS-derived latent variables to predict the sample class.
Model Integration and Validation Strategies
Successful data fusion culminates in the creation and rigorous validation of integrated models. For High-Level Fusion, this involves training separate, optimized classifiers (e.g., Random Forest, SVM) on the NMR and MS datasets. Their prediction outputs (class labels or probabilities) are then integrated using a meta-strategy. A simple but effective method is weighted majority voting, where the final class is determined by the weighted sum of probabilities from each model, with weights proportional to the individual model's cross-validated accuracy [26].
A critical tool for validating any fused model is double-layer cross-validation. An outer loop handles data splitting into training and test sets. An inner loop, performed on the training set only, optimizes model parameters (e.g., number of latent variables in PLS-DA, number of PCs in mid-level fusion). This strict protocol prevents overfitting and provides a realistic estimate of the model's performance on new, unseen data. The performance of the fused model (e.g., accuracy, R2, Q2) must always be benchmarked against models built on the individual NMR and MS datasets to demonstrate the tangible added value of the fusion approach [37].
Diagram 2: Double-layer cross-validation for robust model validation.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for HR-MS/NMR Metabolomics
| Item | Function | Technical Notes |
|---|---|---|
| Deuterated NMR Solvent (e.g., D2O) | Provides a field-frequency lock for the NMR spectrometer; dissolves the analyte. | Must be ≥99.9% D. Buffered variants (e.g., in 100 mM phosphate, pD 7.4) are common for biological samples. |
| Chemical Shift Reference (e.g., DSS-d6) | Provides a known, internal reference signal (0.00 ppm) for spectral alignment and enables quantitative concentration determination. | Added in micromolar concentration. Should be chemically inert and resonate in a clear region of the spectrum. |
| MS-Grade Solvents & Additives | Used for chromatography (mobile phases) and sample reconstitution. Ultra-pure to minimize background noise and ion suppression. | Includes water, acetonitrile, methanol, and additives like formic acid or ammonium acetate. |
| Isotope-Labeled Internal Standards | Added pre-extraction to correct for variability in sample preparation, ionization efficiency, and instrument response in MS. | A mixture of standards covering different chemical classes improves quantification accuracy. |
| Quality Control (QC) Pool Sample | A pooled aliquot of all experimental samples run repeatedly throughout the analytical sequence. | Monitors instrument stability (retention time, signal intensity) and is used for data correction (e.g., batch effect removal). |
Diagram 3: Integrated decision pathway for metabolomics data fusion.
Implementation Considerations and Best Practices
- Data Quality is Paramount: The success of any fusion strategy depends entirely on the quality of the individual datasets. Rigorous quality control (QC) using the pooled sample is non-negotiable. Metrics like relative standard deviation (RSD%) of features in QC samples should be monitored, and signals with poor reproducibility (>20-30% RSD) should be filtered out.
- Metadata and Documentation: Maintain meticulous metadata, including sample information, extraction protocols, instrument parameters, and data processing steps. This is critical for reproducibility and for informing scaling/normalization choices.
- Start Simple, Then Iterate: Begin with a mid-level fusion approach (PCA + PLS-DA), as it is often the most robust starting point. Compare its performance directly against individual block models and a low-level fusion model to empirically determine the best strategy for your specific data.
- Interpretability vs. Performance: Low-level fusion may yield slightly better performance in some cases but at the cost of interpretability. Mid- and high-level fusion better preserve the identity of the source data block, allowing the researcher to understand which technique (NMR or MS) is driving specific classifications or correlations.
- Public Data Repositories: Upon publication, share raw and processed data in public repositories like MetaboLights, adhering to FAIR (Findable, Accessible, Interoperable, Reusable) principles to enhance the impact and utility of your research.
1. Introduction: The Integrated HR-MS/NMR Framework for Metabolite Identification
The comprehensive identification of metabolites in complex biological matrices is a cornerstone of modern biomedical research, driving advances in biomarker discovery, mechanistic toxicology, and precision medicine. Achieving this goal is challenged by the vast chemical diversity, wide concentration ranges, and matrix-specific interferences inherent to samples like urine, blood serum, and feces. No single analytical technique can overcome all these hurdles [38]. This article posits that the synergistic integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy is not merely beneficial but essential for robust, comprehensive metabolite identification research [2].
The inherent complementarity of the two platforms is clear: HR-MS offers superior sensitivity (detecting nanomolar to picomolar concentrations), high mass accuracy, and the ability to identify thousands of features in an untargeted manner. However, it can be hampered by ion suppression, requires extensive sample preparation, and often yields ambiguous identifications based on mass alone. Conversely, NMR provides an inherently quantitative, non-destructive, and reproducible profile of the most abundant metabolites (typically in the micromolar range) with minimal sample workup. Its principal strength lies in its unparalleled power for de novo structural elucidation and its robustness against matrix effects [38]. When combined, the platforms validate and contextualize each other's findings. NMR can provide definitive structural confirmation for putative identities from MS, while MS can delve into the low-abundance metabolome invisible to NMR, thereby creating a more complete metabolic snapshot [2]. This integrated approach forms the methodological thesis for the detailed application notes and protocols presented herein, focusing on three critical biological matrices.
2. Case Study I: Urine – Monitoring Drug-Induced Metabolic Perturbations
2.1 Experimental Context & Protocol This application note details a study designed to delineate the systemic metabolic effects of anti-tuberculosis drug treatment (Rifafour) in a murine model, using urine as a primary non-invasive diagnostic biofluid [39]. The goal was to identify metabolic signatures of drug exposure distinct from disease pathology.
Detailed Protocol:
- Animal Dosing & Sample Collection: C3HeB/FeJ mice received an oral gavage of Rifafour suspension (30 mg/mL in water) at 10 µL per gram body weight daily for 14 days. Control groups received water [39]. Urine was collected non-invasively over a defined period.
- Sample Preparation for NMR: Urine samples were centrifuged (10,000 x g, 10 min, 4°C) to remove particulates. A buffer solution was prepared with 1.5 M potassium phosphate (KH₂PO₄) in deuterium oxide (D₂O), adjusted to pH 7.4 using potassium deuteroxide (KOD). 540 µL of centrifuged urine was mixed with 60 µL of this buffer. The mixture was then centrifuged again. Finally, 550 µL of the supernatant was transferred to a 5 mm NMR tube for analysis [39].
- ¹H-NMR Spectroscopy: Spectra were acquired on a NMR spectrometer (e.g., 600 MHz) using a standard 1D pulse sequence with water suppression. Chemical shift was referenced internally to trimethylsilyl-2,2,3,3-tetradeuteropropionic acid (TSP-d₄) at 0.0 ppm [39]. Typical parameters included: 128 transients, spectral width of 20 ppm, acquisition time of 4 seconds, and a relaxation delay of 1 second.
- Data Processing & Analysis: Free Induction Decays (FIDs) were processed with exponential line broadening (0.3 Hz), Fourier transformation, and phase/baseline correction. Spectra were segmented into bins (e.g., 0.04 ppm width) and normalized (e.g., to total spectral area or creatinine signal). Multivariate statistical analysis (Principal Component Analysis - PCA, Orthogonal Partial Least Squares-Discriminant Analysis - OPLS-DA) was performed to differentiate treatment groups and identify significant spectral regions [39].
2.2 Key Findings & Metabolite Identification The NMR-based metabolomic analysis revealed a consistent and significant decrease in the urinary levels of several host-microbiota co-metabolites throughout the 14-day treatment period. Key identified metabolites included hippuric acid, indoxyl sulfate, phenylacetylglycine, and tryptophan (all with p < 0.05) [39]. The identification process for a metabolite like hippurate exemplifies the HR-MS/NMR integration:
- HR-MS Detection: In a parallel or subsequent LC-HRMS run, a chromatographic peak with an accurate mass matching [M+H]+ = 180.0655 (C9H9NO3) would be detected, suggesting hippurate or an isomer.
- NMR Verification: The ¹H-NMR spectrum shows a characteristic pattern: a singlet at ~3.97 ppm (2H, -CH2-), a complex multiplet between ~7.50-7.65 ppm (5H, aromatic), and a broad exchangeable proton from the carboxylic acid. This signature uniquely confirms the benzoyl-glycine structure of hippurate, eliminating isomeric possibilities suggested by MS alone [39].
- Biological Interpretation: The decline in these metabolites points to a Rifafour-induced alteration in gut microbiota composition or function, specifically affecting the metabolism of aromatic amino acids. This finding highlights a potential off-target effect of the drug regimen and underscores the utility of urine metabolomics in monitoring therapeutic interventions [39].
3. Case Study II: Blood Serum – Biomarker Discovery for Disease Diagnosis
3.1 Experimental Context & Protocol This protocol focuses on discovering diagnostic serum biomarkers for Generalized Ligamentous Laxity (GLL) using an untargeted UPLC-HRMS platform [40]. Serum provides a snapshot of systemic metabolism and is a standard matrix in clinical diagnostics.
Detailed Protocol:
- Sample Collection & Pre-processing: Venous blood was collected from GLL patients and healthy controls, allowed to clot, and centrifuged (e.g., 3000 x g, 10 min, 4°C) to obtain clear serum. Aliquots were immediately stored at -80°C [40].
- Metabolite Extraction: Serum samples were thawed on ice. A common protocol involves protein precipitation: a volume of serum (e.g., 100 µL) is mixed with a cold organic solvent like methanol or acetonitrile (e.g., 300-400 µL) in a 4:1 (v/v) ratio. The mixture is vortexed vigorously, incubated at -20°C for 1 hour, and then centrifuged (e.g., 15,000 x g, 15 min, 4°C). The supernatant containing the metabolome is carefully transferred and dried under a gentle stream of nitrogen or in a vacuum concentrator [40] [41].
- UPLC-HRMS Analysis: The dried extract is reconstituted in a mobile phase-compatible solvent (e.g., water/acetonitrile). Analysis is performed on a system like an ExionLC UPLC coupled to a TripleTOF 5600+ mass spectrometer.
- Chromatography: For positive ion mode, a reversed-phase column (e.g., Waters HSS T3) is used with a gradient of water (0.1% formic acid) and acetonitrile. For negative ion mode, a HILIC column (e.g., BEH Amide) may be used with a gradient of water (5 mM ammonium acetate) and acetonitrile [40].
- Mass Spectrometry: Data are acquired in information-dependent acquisition (IDA) mode. A full scan (e.g., m/z 50-1200) is performed at high resolution (>30,000 FWHM), followed by MS/MS scans on the most intense ions. Electrospray ionization (ESI) is used in both positive and negative modes [40].
- Data Processing: Raw data files are converted (e.g., using MSConvert) and processed with software like XCMS for peak picking, alignment, and integration. Metabolites are putatively annotated by matching accurate mass (often within 5 ppm) and MS/MS fragmentation patterns against public (e.g., HMDB, METLIN) or commercial libraries [40].
3.2 Key Findings & Integrated Identification Strategy The study identified 24 differentially expressed metabolites in GLL serum. A primary biomarker candidate was hexadecanamide (palmitamide), which showed high diagnostic power (AUC = 0.907) [40]. The integrated identification strategy for such a lipid mediator is critical:
- HR-MS Annotation: A prominent feature is detected with [M+H]+ = 256.2636 (C16H33NO). MS/MS fragments indicate an amide linkage (e.g., loss of NH3). This provides a confident putative annotation but cannot distinguish it from other fatty acid amides with similar fragmentation.
- NMR Structural Confirmation: ¹H-NMR analysis of the purified compound or a fraction enriched for this feature would reveal the specific structure: a long aliphatic chain (broad multiplet centered at ~1.25 ppm), methylene groups adjacent to the carbonyl (triplet at ~2.15 ppm), and the terminal methyl group (triplet at ~0.88 ppm). ¹³C-NMR would confirm the amide carbonyl carbon at ~175 ppm. This step is crucial to rule out isomers like oleamide or stearamide, which have identical molecular formulas but different double-bond positions or chain lengths [2].
- Pathway Analysis: The disturbance in lipid amide metabolism, alongside alterations in α-linolenic and linoleic acid pathways, points to systemic inflammatory and connective tissue remodeling processes underlying GLL pathology [40].
Table 1: Summary of Key Metabolites Identified in Case Studies
| Matrix | Study Context | Key Metabolites Identified | Associated Pathway/Biological Meaning | Primary Platform for Discovery |
|---|---|---|---|---|
| Urine | Anti-TB Drug Toxicity [39] | Hippurate, Indoxyl Sulfate, Phenylacetylglycine, Tryptophan | Gut Microbiota Metabolism, Aromatic Amino Acid Metabolism | NMR |
| Blood Serum | Glioblastoma [42] | Lactate, Choline, 2-Hydroxyglutarate, Pyruvate | Aerobic Glycolysis (Warburg Effect), Membrane Turnover | MS / NMR |
| Blood Serum | Generalized Ligamentous Laxity [40] | Hexadecanamide, α-Linolenic Acid, Linoleic Acid | Fatty Acid Amide Signaling, Inflammatory Lipid Metabolism | HR-MS |
| Feces | Anti-TB Drug Toxicity [39] | Choline, Succinate | Microbial Fermentation, Energy Metabolism | NMR |
4. Case Study III: Feces – Decoding the Host-Microbiome Interface
4.1 Experimental Context & Protocol Feces represent an exceptionally complex matrix, rich in microbial biomass, undigested food, host cells, and secreted compounds. Analyzing the fecal metabolome requires specialized protocols to manage this complexity and extract meaningful biochemical information [41].
Detailed Protocol for Multi-Platform Fecal Metabolomics:
- Sample Homogenization & Extraction: Fresh or frozen fecal samples are weighed. A standardized mass (e.g., 50 mg) is homogenized in an aqueous buffer (e.g., phosphate-buffered saline) or extraction solvent using bead-beating for mechanical lysis of microbial cells. For comprehensive coverage, a dual extraction is optimal:
- Aqueous Extract: Homogenize in water/methanol, centrifuge. The supernatant contains polar metabolites (sugars, amino acids, organic acids) for NMR and HILIC-MS.
- Organic Extract: The pellet or a separate aliquot is homogenized in a chloroform/methanol/water mixture (e.g., 2:2:1.8 ratio). After phase separation, the organic (lower) phase contains lipids and non-polar metabolites for reversed-phase (RP) LC-MS [41].
- Clean-up: Aqueous extracts may require filtration (e.g., 3 kDa molecular weight cut-off filters) to remove large proteins and polysaccharides before NMR analysis.
- Multi-Platform Analysis:
- NMR: The filtered aqueous extract is buffered in D₂O with phosphate buffer and TSP, as described for urine. ¹H-NMR provides a quantitative profile of major fermentation products (e.g., short-chain fatty acids like acetate, propionate, butyrate), amino acids, and bile acids [38] [41].
- HR-MS: Both aqueous (after dilution) and organic extracts are analyzed by UHPLC-HRMS. HILIC-MS is ideal for the polar aqueous metabolome, while RP-MS is used for the lipidome. This dual chromatography approach maximizes metabolite coverage [41].
4.2 Key Findings & Data Integration In the Rifafour study, fecal NMR analysis identified significant perturbations in choline and succinate levels [39]. These changes reflect alterations in microbial community structure and metabolic activity. Integrated identification is paramount in feces:
- Succinate Identification: An NMR signal at ~2.40 ppm (singlet, 4H) is a hallmark of succinate. In HR-MS, a feature with [M-H]- = 117.0193 (C4H5O4) would be detected. The correlation of the quantitative NMR data with the MS feature intensity across samples strengthens the validity of the finding.
- Biological Insight: Elevated succinate can indicate a shift in microbial fermentative pathways or a reduction in bacteria that consume it. Changes in choline, a substrate for microbial production of the pro-atherogenic metabolite trimethylamine-N-oxide (TMAO), link gut microbiome function directly to host systemic health [38] [39]. This demonstrates how fecal metabolomics, powered by integrated platforms, can provide direct functional readouts of microbiome activity.
5. Data Integration & Computational Tools for HR-MS/NMR Synergy
The true power of a multi-platform approach is realized through integrated data analysis. Statistical multiblock methods (e.g., Multi-Block PCA, OPLS) can combine datasets from NMR and MS to find latent variables that explain variance across both platforms [2]. Computational tools are essential for metabolite identification.
A prime example is the ROIAL-NMR program [43]. This Python-based tool automates the identification of metabolites from regions of interest (ROIs) in complex ¹H-NMR spectra (e.g., from serum, urine, feces) by querying the Human Metabolome Database (HMDB). It accounts for chemical shift variations and peak multiplicities, generating a list of candidate metabolites. This NMR-derived candidate list can then be used to guide the targeted interrogation of HR-MS data, searching for the exact masses and expected retention times of these candidates, thereby dramatically improving the efficiency and confidence of annotation across platforms [43].
Table 2: Complementary Strengths of HR-MS and NMR for Metabolite ID
| Feature | High-Resolution Mass Spectrometry (HR-MS) | Nuclear Magnetic Resonance (NMR) Spectroscopy | Synergistic Benefit of Integration |
|---|---|---|---|
| Sensitivity | Excellent (nM-pM) | Moderate (μM) | MS detects low-abundance signals; NMR quantifies core metabolites. |
| Throughput | High | Moderate | MS for rapid screening; NMR for definitive sub-set analysis. |
| Quantitation | Relative (requires standards) | Absolute (inherent) | NMR provides internal quantitative calibration for MS data. |
| Structural Insight | Molecular formula, fragments | Definitive bond connectivity, stereochemistry | MS suggests identity; NMR confirms it and resolves isomers. |
| Sample Prep | Complex (extraction, derivatization) | Minimal (often just buffer) | Single extract can be split for both analyses. |
| Matrix Effects | Susceptible (ion suppression) | Robust | NMR validates MS findings in complex matrices like feces [41]. |
| Metabolite ID | Putative annotation (mass, RT, MS/MS) | Definitive identification | Combined confidence level is vastly superior [2]. |
6. The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for Integrated Metabolomics
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Deuterium Oxide (D₂O) with TSP-d₄ | NMR solvent and internal chemical shift (δ=0 ppm) & quantification standard. | Used for preparing urine, serum, and fecal aqueous extracts for NMR [39]. |
| Potassium Phosphate Buffer (in D₂O) | Maintains constant pH (e.g., 7.4) in NMR samples to ensure reproducible chemical shifts. | Critical for biological sample analysis; prepared with KOD for pD adjustment [39]. |
| Methanol, Acetonitrile (LC-MS Grade) | Protein precipitation solvents for serum/plasma; mobile phase components for LC-MS. | Used cold for metabolite extraction to quench enzymatic activity [40] [41]. |
| Chloroform (HPLC Grade) | Organic solvent for lipid extraction from serum, tissues, or feces (e.g., Folch method). | Used in biphasic extraction to separate non-polar metabolites for lipidomics [41]. |
| Formic Acid / Ammonium Acetate (MS Grade) | Mobile phase additives for LC-MS to promote ionization in positive (FA) and negative (AmAc) modes. | Typically used at 0.1% in water and/or organic solvent [40]. |
| Molecular Weight Cut-Off (MWCO) Filters | Remove proteins and other large molecules from biofluid samples prior to analysis. | Essential for cleaning up serum or fecal extracts for NMR, preventing broad protein signals [41]. |
| Reference Metabolite Standards | Authentic chemical standards for validating metabolite identity and creating calibration curves. | Used to confirm retention time (LC-MS) and chemical shift (NMR) of identified biomarkers [40]. |
| Bead Beating Matrix & Homogenizer | Mechanically disrupts tough matrices (e.g., fecal matter, microbial pellets) for efficient metabolite extraction. | Critical for reproducible and comprehensive extraction from solid or semi-solid samples [41]. |
7. Visualizing Workflows, Pathways, and Methods
Integrated HR-MS/NMR Workflow for Metabolomics
Key Metabolic Pathways Altered in Glioblastoma
Matrix-Specific Sample Preparation Protocols
Troubleshooting and Optimizing Integrated HR-MS and NMR Workflows
The integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy represents a powerful synergistic approach for comprehensive metabolite identification and quantification in complex biological matrices [44] [45]. This integrated strategy leverages the high sensitivity and broad metabolome coverage of HR-MS with the unparalleled structural elucidation capabilities, quantitative accuracy, and reproducibility of NMR [44] [46]. However, the efficacy of this multi-platform analysis is fundamentally contingent upon rigorous and optimized sample preparation. Inconsistent or suboptimal preparation creates a critical bottleneck, leading to data that are not directly comparable between platforms and hindering the confident identification of metabolites [44] [47].
Two of the most pervasive challenges in this preparatory phase are solvent compatibility and efficient protein removal. The choice of solvent system must satisfy the distinct requirements of both analytical techniques—ensuring optimal solubility for NMR, compatibility with ionization for MS, and minimal spectral interference [48]. Concurrently, the presence of proteins in biofluids like plasma or serum can severely compromise analysis by attenuating metabolite signals in NMR, fouling LC columns and ion sources in MS, and introducing artefacts from metabolite-protein binding [44]. Addressing these challenges is not merely a procedural step but a foundational requirement for generating high-fidelity, multi-platform metabolomic data that can drive biomarker discovery and mechanistic biological insight [49] [45].
Core Challenge I: Solvent Compatibility for Multi-Platform Analysis
Selecting an appropriate solvent is a critical first decision that impacts every subsequent stage of analysis. The ideal solvent must serve the often-divergent needs of NMR and HR-MS while maintaining the integrity of the labile metabolome.
2.1 Key Considerations and Requirements For NMR spectroscopy, the primary requirements are the use of deuterated solvents to provide a lock signal and minimize the overwhelming solvent proton resonance [48]. The solvent must also provide excellent solubility for a broad range of metabolites and have a chemical shift that does not obscure regions of interest in the spectrum [48]. For HR-MS, particularly when coupled with liquid chromatography (LC-MS), the solvent must be compatible with the chromatographic separation (e.g., reversed-phase or HILIC) and facilitate efficient ionization in the source (electrospray ionization, ESI). Factors such as volatility, pH, and the presence of non-volatile salts or buffers are crucial, as they can cause ion suppression, source contamination, and adduct formation [46].
2.2 Protocol: Preparation of a Compatible Solvent System for Sequential HR-MS/NMR Analysis
A practical protocol for preparing a solvent system suitable for sequential analysis, beginning with LC-MS and followed by NMR, is outlined below.
- Objective: To extract metabolites from a cell pellet using a solvent system that is compatible with both reversed-phase LC-HR-MS analysis and subsequent 1H-NMR spectroscopy.
- Materials: Cell pellet (e.g., from E. coli culture), pre-chilled methanol (LC-MS grade), chloroform (LC-MS grade), deuterium oxide (D₂O, 99.9% D), phosphate buffer (50 mM, pH 7.0 in D₂O for NMR), liquid nitrogen, vortex mixer, centrifuge, speed-vac concentrator, lyophilizer.
- Procedure:
- Metabolite Extraction: Resuspend the cell pellet in ice-cold water. Subject the suspension to three cycles of rapid freezing in liquid nitrogen and thawing on ice to lyse cells.
- Protein Precipitation: Add pre-chilled methanol and chloroform sequentially to the lysate under vigorous vortexing to achieve a final H₂O:methanol:chloroform ratio of 1:1:1 (v/v/v) [15]. Incubate the mixture at -20°C for a minimum of 1 hour (or overnight) to complete protein precipitation and phase separation.
- Phase Separation: Centrifuge the mixture at 4,000 x g for 20 minutes at 4°C. Two distinct phases will form: a lower organic (chloroform) phase containing lipids and an upper aqueous phase containing polar metabolites.
- Aqueous Phase Recovery for Multi-Platform Analysis:
- For LC-HR-MS: Carefully collect the upper aqueous phase. Reduce the methanol content using a gentle stream of nitrogen or a speed-vac concentrator (avoid complete dryness for labile compounds). Reconstitute the residue in a solvent compatible with your LC-MS method (e.g., 98% H₂O / 2% acetonitrile with 0.1% formic acid). Centrifuge at 13,000 rpm for 5 minutes and transfer the supernatant to an LC vial [15].
- For NMR: Take an aliquot of the aqueous phase or the reconstituted LC-MS sample. Completely dry using a lyophilizer. Reconstitute the dried metabolite extract in 600 µL of a suitable NMR buffer (e.g., 50 mM phosphate buffer in D₂O, pH 7.0). Transfer to a 5 mm NMR tube, ensuring the sample height is between 40-50 mm for optimal shimming [48].
Table 1: Solvent Compatibility Considerations for HR-MS and NMR
| Solvent Property | HR-MS (ESI) Consideration | NMR Consideration | Recommended Compromise/Note |
|---|---|---|---|
| Deuteration | Not required; H₂O is acceptable. | Essential for frequency lock; D₂O required. | Use D₂O-based buffers for NMR prep; MS can tolerate low % D₂O. |
| Volatile Additives | Formic/acetic acid (0.1%) promote [M+H]+; ammonium salts promote [M+NH4]+. | Can cause pH-dependent shift changes; non-volatile buffers (phosphate) preferred. | Use volatile additives for MS; avoid in final NMR sample or use at minimal concentration. |
| Non-Volatile Salts | Cause severe ion suppression and source contamination. Must be avoided. | Phosphate buffer is standard for reproducibility and pH control in NMR. | Remove salts via SPE or dialysis prior to MS; use in NMR buffer after MS analysis is complete. |
| Organic Modifier | Acetonitrile/methanol essential for RPLC separation and ionization efficiency. | High concentrations can denature proteins or precipitate metabolites; can obscure NMR regions. | Use standard LC gradients for MS; ensure organic solvent is removed before NMR analysis. |
Core Challenge II: Protein Removal for Unbiased Metabolite Profiling
The analysis of protein-rich biofluids like blood serum or plasma is particularly challenging. Proteins can bind metabolites, rendering them "NMR-invisible," broaden NMR signals via slow tumbling, and interfere with chromatographic separation and ionization in MS [44].
3.1 Quantitative Comparison of Protein Removal Methods A critical evaluation of protein removal methods is essential. While ultrafiltration is gentle, it can be inefficient due to metabolite binding to the filter membrane or trapped proteins. Protein precipitation with organic solvents is generally more effective at recovering a broader range of metabolites [44].
Table 2: Efficacy of Protein Precipitation Methods for Blood Serum/Plasma Analysis [44]
| Precipitation Agent | Typical Protocol (Serum:Solvent) | Key Advantages | Key Disadvantages | Metabolite Recovery & NMR Suitability |
|---|---|---|---|---|
| Acetonitrile | 1:2 (v/v) | Excellent protein removal; clear supernatants; minimal residue. | Can precipitate some hydrophilic metabolites. | Superior. High recovery for many acids, alcohols. Excellent NMR spectral quality. |
| Methanol | 1:2 or 1:3 (v/v) | Good precipitation efficiency; common in multi-omics. | Can be less efficient than ACN; may leave more soluble proteins. | High. Good recovery for broad range. Good NMR spectra. |
| Perchloric Acid | ~6% (v/v) final conc. | Effective for acid-stable metabolites; common for phosphorylated compounds. | Extremely low pH can degrade labile metabolites; requires neutralization. | Moderate. Selective recovery. Salt from neutralization can interfere with MS/NMR. |
| Ultrafiltration | N/A (Molecular weight cut-off filter) | Gentle; no solvent addition. | Low throughput; metabolite loss via binding; incomplete removal of small proteins. | Variable/Lower. Often lower recovery compared to solvent precipitation [44]. |
3.2 Protocol: Optimized Protein Precipitation for Serum/Plasma Prior to HR-MS/NMR
This protocol is designed for maximal protein removal and metabolite recovery from blood-derived samples.
- Objective: To deproteinize human serum or plasma for subsequent untargeted metabolomic profiling by LC-HR-MS and quantitative NMR.
- Materials: Human serum or plasma, pre-chilled acetonitrile (LC-MS grade), internal standard mixture (e.g., DSS-d6 for NMR, isotope-labeled compounds for MS), vortex mixer, centrifuge, vacuum concentrator.
- Procedure:
- Sample Preparation: Thaw serum/plasma samples on ice. Centrifuge briefly to remove any particulates.
- Precipitation: Aliquot 100 µL of serum into a pre-chilled microcentrifuge tube. Add a suitable internal standard mix. Rapidly add 300 µL of ice-cold acetonitrile (1:3 ratio) [44].
- Vortexing and Incubation: Vortex vigorously for 60 seconds. Incubate on ice for 10 minutes to ensure complete protein precipitation.
- Pellet Removal: Centrifuge at 14,000 x g for 15 minutes at 4°C. A compact protein pellet should form.
- Supernatant Recovery and Division: Carefully transfer the clear supernatant to two fresh, labeled tubes:
- Tube A (For HR-MS): Transfer ~150 µL. Dry completely in a vacuum concentrator without heat. Reconstitute in 50 µL of starting LC-MS mobile phase (e.g., 98% H₂O, 2% ACN, 0.1% FA). Centrifuge and analyze.
- Tube B (For NMR): Transfer the remaining supernatant (~250 µL). Dry completely. Reconstitute in 600 µL of NMR buffer (e.g., 75 mM phosphate in D₂O, pH 7.4, containing 0.5 mM DSS-d6). Transfer to an NMR tube.
Advanced Integrated Workflows for Metabolite Identification
Overcoming the "dark matter" of metabolomics—the vast number of unidentifiable spectral features—requires moving beyond simple parallel analysis to true integration [47] [45]. Advanced workflows systematically combine the strengths of HR-MS and NMR.
4.1 The SYNHMET Approach: MS-Assisted NMR Deconvolution The SYnergic use of NMR and HRMS for METabolomics (SYNHMET) strategy uses HR-MS data to guide the deconvolution of complex, overlapping NMR signals [46]. The process begins with an initial NMR profile and a list of candidate metabolites from databases. HR-MS data (exact mass, retention time) are then used to correlate specific MS features with the NMR-derived concentration estimates of these candidates. This correlation helps to unambiguously assign MS peaks to specific metabolites, which in turn provides more accurate concentration constraints to refine the NMR spectral deconvolution iteratively. This loop significantly increases the number of metabolites that can be accurately quantified in absolute terms from complex samples like urine [46].
4.2 Protocol: The SUMMIT MS/NMR Strategy for De Novo Identification The SUMMIT MS/NMR (Structure of Unknown Metabolomic Mixture components by MS/NMR) approach is designed for identifying completely unknown metabolites without pure standards [15].
- HR-MS Analysis: A high-resolution mass spectrum of the mixture is acquired to determine the exact mass(es) of unknown component(s).
- Formula Generation: All chemically plausible molecular formulas are generated for the exact mass.
- Structure Manifold Creation: For each candidate formula, all possible constitutional isomers (structural manifold) are retrieved from chemical structure databases (e.g., PubChem, ChemSpider).
- NMR Prediction and Matching: Theoretical 1H and/or 13C NMR spectra are predicted for every structure in the manifold.
- Structure Selection: The predicted NMR spectra are systematically compared to the experimental NMR spectrum of the mixture. The structure(s) whose predicted spectra best match the experimental data are assigned as the correct identification [15].
4.3 The UHPLC-MS/SPE-NMR Integrated Platform For higher-throughput confident identification, physical integration of platforms is the gold standard. In a UHPLC-MS/SPE-NMR system, the chromatographic eluent is split post-column [45].
- A small fraction (~5%) is directed to the MS for real-time detection and triggering.
- The majority (~95%) is directed to a solid-phase extraction (SPE) cartridge station. When the MS detects a peak of interest (e.g., an unknown), the corresponding HPLC fraction is automatically captured on a dedicated SPE cartridge.
- After repeated injections to accumulate sufficient material (typically nanomole amounts), the trapped metabolite is automatically eluted from the SPE cartridge with a minimal volume of deuterated solvent directly into an NMR tube or a flow NMR probe for structure elucidation [45]. This platform automates the purification and concentration of metabolites of interest for definitive NMR characterization, bridging the gap between screening (MS) and confirmation (NMR).
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Sample Preparation
| Item | Function & Purpose | Key Considerations |
|---|---|---|
| Deuterated Solvents (D₂O, CD₃OD) | Provides the lock signal for stable NMR acquisition; minimizes large solvent proton peak [48]. | Purity level (99.9% D); store with molecular sieves to prevent H₂O absorption; choose grade (e.g., CD₃OD vs. CD₃OH) to control exchangeable protons [48]. |
| Internal Standards for NMR (e.g., DSS-d6, TSP-d4) | Provides a chemical shift reference (0 ppm) and enables absolute quantitation via a single known concentration [44]. | Must be chemically inert and produce a singlet resonance in an uncrowded spectral region. DSS is preferred over TSP for biological samples as it does not bind proteins. |
| Isotope-Labeled Internal Standards for MS | Enables accurate quantitation in MS by correcting for matrix-induced ion suppression/enhancement. | Should be chemically identical to the target analyte (e.g., 13C, 15N-labeled); essential for targeted quantification assays. |
| Protein Precipitation Solvents (ACN, MeOH) | Denatures and precipitates proteins from biofluids, freeing metabolites and preventing analytical interference [44]. | Pre-chill for efficiency; LC-MS grade purity minimizes background ions; acetonitrile often provides superior pellet formation [44]. |
| SPE Cartridges (for cleanup or trapping) | Remove salts, contaminants, or concentrate specific metabolite classes. In integrated systems, trap HPLC fractions for NMR [45]. | Select phase (C18, HILIC, Ion Exchange) based on analyte chemistry; condition carefully for reproducible recovery. |
| High-Quality NMR Tubes | Holds sample within the magnetic field; quality directly impacts spectral resolution [48]. | Use tubes with good concentricity and camber (e.g., not "economy" grade) [48]; match tube diameter (5mm, 3mm) to available probe. |
| Cryogenically Cooled NMR Probes | Increase sensitivity by cooling the detector coil and preamplifiers, reducing thermal noise. | Essential for analyzing mass-limited samples or low-concentration metabolites; can reduce experiment time by >10-fold [45]. |
| UHPLC Columns (RP & HILIC) | Provide high-resolution chromatographic separation of metabolites prior to MS detection, reducing ion suppression. | Column chemistry (C18, phenyl, amide) defines metabolome coverage; use dedicated columns for biological samples to prevent contamination. |
Abstract
Within the broader thesis on integrating High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) for definitive metabolite identification, optimizing core NMR parameters is a critical prerequisite. This application note details two fundamental pillars for maximizing data quality: advanced phase correction algorithms for spectral fidelity and modern pulse sequence engineering for sensitivity enhancement. We provide validated experimental protocols and quantitative comparisons of methodologies, framing these technical optimizations as essential steps that ensure NMR-derived structural information is robust and synergistic with HR-MS data in multi-omics workflows [50] [22] [51].
1. Phase Correction Algorithms: Ensuring Spectral Fidelity for Accurate Integration
Accurate phasing of NMR spectra is non-negotiable for correct peak integration, which directly impacts metabolite quantification and subsequent statistical analysis. Automated, optimized algorithms are superior to manual phasing, eliminating subjective bias and enabling high-throughput reproducibility [50] [52].
1.1 Core Algorithm Comparison and Performance Data Five principal linear-model-based algorithms were evaluated for their optimization potential. The following table summarizes their core principles and quantified performance gains after implementation of the proposed tuning strategies [52].
Table 1: Comparison of Optimized Phase Correction Algorithms for ¹H NMR Spectra
| Algorithm | Core Principle | Optimization Strategy | Performance Gain (Relative Phase Error Reduction) |
|---|---|---|---|
| Automics | Estimates φ₀ and φ₁ from mean phase at spectrum tails [52]. | Adaptive interval length determination using statistical trend testing [52]. | From 10.25% to 2.40% (low noise); from 12.45% to 2.66% (high noise) [52]. |
| Shannon's Entropy Minimization | Minimizes entropy of absorption spectrum, assuming ideal spectrum is positive-definite [52]. | Application of Nelder-Mead solver with intelligent initial conditions (e.g., from water peak phase) [52]. | Significant increase in correction accuracy and computational speed [52]. |
| Ernst's Method | Minimizes the integral of the dispersion spectrum [50] [52]. | Use of integral global optimization algorithms [52]. | Improved accuracy in parameter estimation [52]. |
| Dispa | Maximizes a symmetry-based functional (Q) derived from the absorption spectrum [52]. | Not specified in detail [52]. | Tuning possible via parameter adjustment [52]. |
| eDispa | Maximizes a normalized, enhanced version (η) of the Dispa functional [52]. | Application of Nelder-Mead solver with intelligent initial conditions [52]. | Significant increase in correction accuracy and computational speed [52]. |
1.2 Protocol: Optimized Automated Phase Correction for ¹H NMR Metabolomics Data
- Sample Preparation: Prepare biofluid (e.g., urine, plasma) or tissue extract sample using standard metabolomics protocols with a deuterated buffer containing a reference compound (e.g., TSP or DSS for chemical shift referencing at 0 ppm) [50] [51].
- Data Acquisition: Acquire a 1D ¹H NMR spectrum using a standard presaturation pulse sequence (e.g., NOESYPR1D) on a spectrometer (500 MHz or higher). Key parameters: spectral width ~20 ppm, acquisition time ≥2 s, relaxation delay 4-5 s, 64-128 transients [50] [51].
- Pre-processing (Prior to Phasing): Apply exponential line broadening (0.3-1.0 Hz), zero-filling to at least 64k points, and Fourier Transform [50].
- Optimized Automatic Phasing Protocol:
- Load Spectrum: Import the real and imaginary components of the FID after FT.
- Select Algorithm: Implement the tuned version of the chosen algorithm (e.g., adaptive-interval Automics).
- Set Parameters: For tuned Automics, set statistical significance level α=0.05 for interval growth. For tuned Shannon's or eDispa, set initial φ₀ based on the phase at the maximum of the dominant water peak [52].
- Execute & Validate: Run the optimization. Visually validate the baseline flatness in the real part (absorption mode) and check that the imaginary part (dispersion mode) is centered around zero.
- Reference: Reference the spectrum to the known internal standard (TSP at 0.0 ppm or DSS at 0.0 ppm) [50].
- Finalize: Proceed with baseline correction and peak picking/integration.
2. Sensitivity Enhancement via Advanced Pulse Sequences
Sensitivity, a key limitation in NMR metabolomics, can be significantly improved by replacing error-prone hard π pulses with compensated pulses, directly increasing the signal-to-noise ratio (SNR) for low-abundance metabolites [53].
2.1 Triply-Compensated G5 Pulse: Principles and Efficacy The G5 pulse is a π pulse optimized via a genetic algorithm to compensate simultaneously for (i) RF inhomogeneity/miscalibration, (ii) off-resonance effects, and (iii) J-coupling evolution during the pulse. Its universal design makes it effective for inversion, refocusing, and decoupling on ¹H, ¹³C, and ¹⁵N channels [53].
Table 2: Performance of the G5 Triply-Compensated π Pulse in Biomolecular NMR
| Parameter | Hard π Pulse | G5 Compensated Pulse | Experimental Outcome |
|---|---|---|---|
| Compensation | Sensitive to RF errors, offset, J-evolution. | Triply-compensated: RF (≥±10%), offset, J-coupling [53]. | Robust performance across sample conditions. |
| Operational Bandwidth | Limited, signal loss at edges. | Wide: ~23.5 ppm (¹H), ~105 ppm (¹⁵N), ~116 ppm (¹³C) at 900 MHz [53]. | Uniform sensitivity across full spectral width. |
| Duration | 1 x τₚ (π). | 5 x τₚ (π) [53]. | Marginally longer but offset by large SNR gain. |
| Sensitivity Gain | Baseline (often with significant losses). | 80% to 240% signal enhancement in 2D/3D experiments (e.g., HSQC, TROSY-HNCA) [53]. | Enables detection of weaker signals or reduced acquisition time. |
2.2 Protocol: Implementing G5 Pulses in a 2D ¹H-¹³C HSQC Experiment
- Prerequisite - G5 Pulse Calibration: On the target sample (or a standard), calibrate the π/2 pulse width for the nucleus of interest (e.g., ¹³C). The G5 pulse amplitude is set to the same RF field strength (ν₁), with its duration set to 5 times the calibrated hard π pulse duration [53].
- Sample: 0.3-1.0 mM ¹³C-labeled protein or a complex metabolite mixture in appropriate buffer [53].
- Pulse Sequence Modification: Modify a standard
hsqcetgpsisp2.2or equivalent pulse sequence [53].- Identify Target Pulses: Locate all hard π pulses on the ¹H and ¹³C channels used for refocusing and decoupling within the INEPT and reverse-INEPT transfer periods.
- Substitution: Replace these hard π pulses with the shaped G5 pulse element. Ensure the pulse power and phase cycling are correctly configured.
- Adjust Delays: Account for the increased duration of the G5 pulse (5τₚ vs. 1τₚ) by slightly shortening the adjacent delays to maintain total transfer periods for optimal J-coupling evolution.
- Data Acquisition: Acquire the modified 2D ¹H-¹³C HSQC. Keep all acquisition parameters (spectral widths, points, number of scans) identical to a standard experiment for direct comparison.
- Processing and Analysis: Process both standard and G5-enhanced datasets identically. Quantify the sensitivity enhancement by comparing the SNR or peak volume of well-resolved signals, particularly those in spectral regions prone to offset effects.
3. Integrated HR-MS/NMR Workflow for Metabolite Identification
The optimization of NMR parameters directly enhances its contribution to a complementary multi-omics workflow. HR-MS provides high sensitivity and putative identification, while optimized NMR delivers definitive structural validation and quantification in complex mixtures [50] [22] [51].
Diagram 1: Integrated HR-MS/NMR Metabolite Identification Workflow.
4. The Scientist's Toolkit: Key Reagents & Materials
Table 3: Essential Research Reagent Solutions for NMR-based Metabolomics
| Item | Function in Protocol | Key Consideration |
|---|---|---|
| Deuterated Solvent (D₂O) | Provides field-frequency lock for the NMR spectrometer; dissolves polar metabolites [50] [51]. | Use 99.9% atom D or higher. May include buffers like phosphate. |
| Internal Chemical Shift Reference | Provides a known signal (0 ppm) for accurate chemical shift referencing essential for database matching [50]. | TSP (for protein-free biofluids) or DSS (better for samples with proteins) [50]. |
| Deuterated Buffer Salts | Maintains constant pH in the NMR sample, stabilizing chemical shifts of pH-sensitive metabolites (e.g., citrate) [50]. | Use sodium/potassium phosphate, TRIS, etc., prepared in D₂O. |
| Cryogenically Cooled Probe (CryoProbe) | Increases sensitivity by cooling the receiver coils, reducing thermal noise. Critical for detecting low-concentration metabolites [53]. | Standard equipment on modern metabolomics spectrometers. |
| Pulse Sequence Library | Contains standard and optimized pulse programs (e.g., 1D NOESY, 2D HSQC with G5 pulses) for data acquisition [53]. | Vendor-provided (Bruker, Varian) or open-source (NMRPipe). |
| Spectral Databases | Reference repositories of known metabolite NMR chemical shifts for identification [50]. | HMDB (Human Metabolome Database), BMRB (Biological Magnetic Resonance Bank) [50] [54]. |
Abstract The integration of High-Resolution Mass Spectrometry (HRMS) with Nuclear Magnetic Resonance (NMR) spectroscopy represents a transformative strategy in metabolomics, synergistically enhancing metabolite identification, quantification, and coverage [55] [4]. This article details application notes and protocols focused on optimizing three pillars of MS performance—ionization efficiency, chromatographic separation, and metabolite coverage—within the framework of HR-MS/NMR integration. We present quantitative comparisons of ionization sources, evaluate high-throughput chromatographic methods, and demonstrate how combined NMR/MS workflows like the NMR/MS Translator and SYNHMET protocols can expand the measurable metabolome and yield highly confident, quantitative profiles for biomedical and pharmaceutical research [56] [57] [4].
Ionization Efficiency and Source Evaluation
Electrospray ionization (ESI) performance is critical for metabolite coverage and signal robustness in untargeted metabolomics. Systematic evaluation of ion source configurations ensures optimal sensitivity and reproducibility for large-scale studies.
- Comparative Performance of ESI Setups: A study comparing a standard ESI source (REF) against a high-temperature "IonBooster" interface (ALT) using a biosample dilution series demonstrated significant performance differences. Feature intensity-based evaluation showed the ALT setup provided an average 4.3-fold higher signal in HILIC mode and a 2.3-fold increase in RPC mode [56].
- Selectivity and In-Source Fragmentation: While the ALT setup showed higher overall sensitivity, 17–24% of features were more intense in the REF setup, indicating selectivity differences [56]. Furthermore, evaluating in-source fragmentation through reconstructed MS1 spectra is vital, as excessive fragmentation can distort feature counts and complicate annotation [56].
Table 1: Performance Evaluation of Alternative vs. Reference ESI Ion Source Setups [56]
| Evaluation Metric | HILIC Mode | Reversed-Phase (RPC) Mode | Key Implication |
|---|---|---|---|
| Avg. Fold-Change (ALT/REF) | 4.3-fold (log₂FC=2.11) | 2.3-fold | ALT offers significant sensitivity gain. |
| Features with Higher Response in ALT | 83% of features | 76% of features | Majority of metabolome benefits from ALT. |
| Features with Higher Response in REF | 17% of features | 24% of features | Source selectivity affects a compound subset. |
| In-Source Fragmentation Analysis | Required via compound spectra | Required via compound spectra | Prevents overestimation of analyte numbers. |
Chromatographic Separation and High-Throughput Methods
Chromatographic strategy directly impacts metabolome coverage, analysis time, and suitability for epidemiological-scale studies.
- UPLC-HRMS vs. Direct Infusion-nESI-HRMS: A comparative study of urine metabolomics evaluated ultra-performance liquid chromatography (UPLC)-HRMS against direct infusion-nanoelectrospray (DI-nESI)-HRMS [57]. UPLC-HRMS provides superior chromatographic separation, reducing ion suppression and enabling isomer distinction. In contrast, DI-nESI-HRMS offers extreme throughput, analyzing a sample set in 9 hours versus 5 days for UPLC-HRMS [57].
- Quantitative Correlation and Applications: Despite lacking separation, DI-nESI showed strong quantitative correlation (Pearson’s r > 0.9) with UPLC for 10 metabolites, making it suitable for high-throughput quantification of predefined analytes [57]. For complex separations, such as resolving hemoglobin variants with mass differences <1 Da, specialized LC methods using C4 reversed-phase columns are essential [58].
Table 2: Comparison of UPLC-HRMS and DI-nESI-HRMS for Metabolic Profiling [57]
| Parameter | UPLC-HRMS | DI-nESI-HRMS | Contextual Recommendation |
|---|---|---|---|
| Total Run Time (132 samples) | ~5 days | ~9 hours | DI-nESI is superior for rapid, high-throughput screening. |
| Metabolite Identification Specificity | High (orthogonal RT + m/z) | Low (m/z only) | UPLC is essential for novel compound ID or complex matrices. |
| Quantitative Correlation (Strong) | Reference Method | 10 metabolites (r > 0.9) | DI-nESI valid for targeted quant. of stable, known analytes. |
| Quantitative Correlation (Weak) | Reference Method | 5 metabolites (r < 0.4) | UPLC required for analytes prone to ion suppression. |
| Primary Advantage | Separation, specificity, coverage | Speed, simplicity, throughput | Choice depends on study goals: discovery vs. screening. |
Expanding Metabolite Coverage via HR-MS and NMR Integration
The synergistic integration of NMR and HRMS overcomes the inherent limitations of each standalone technique, significantly expanding reliable metabolite identification and quantification.
- The NMR/MS Translator Protocol: This automated strategy uses identified metabolites from 1D/2D NMR spectra as input. It calculates the m/z of their possible ions, adducts, and fragments, then matches these against the experimental MS1 spectrum. This directly assigns MS signals to NMR-identified metabolites with high confidence and validates the NMR-derived identifications [55].
- The SYNHMET Protocol for Personalized Metabolic Profiles: This synergic approach uses statistical correlation between preliminary NMR quantifications and MS feature intensities to guide accurate MS peak assignment. These assigned MS features then refine and improve the quantitative deconvolution of the NMR spectrum, yielding a final, expanded set of absolute concentrations [4]. Applied to urine, SYNHMET quantified 165 metabolites with minimal missing values across healthy and disease cohorts [4].
- Coverage in Untargeted Diagnostic Screening: In clinical contexts, untargeted LC-HRMS can profile thousands of features. A feasibility study for untargeted diagnostic screening (UDS) successfully revealed spiked xenobiotics and endogenous compounds in human serum by comparing a single patient's profile against controls, demonstrating the potential to uncover novel diagnostic hypotheses [59].
Detailed Experimental Protocols
Objective: To unambiguously assign MS1 signals by leveraging prior metabolite identification from NMR. Workflow Summary:
- Sample Preparation: Prepare sample in appropriate buffer (e.g., 20 mM phosphate in D2O for NMR). For MS, dilute sample in 50/50 ACN/H2O with 0.1% formic acid.
- NMR Data Acquisition & Query: Acquire 2D 13C-1H HSQC spectrum. Query spectrum against a database (e.g., COLMAR) using standard chemical shift tolerances (0.03 ppm for 1H, 0.3 ppm for 13C).
- Theoretical m/z Calculation: For each database-identified metabolite, calculate the monoisotopic m/z for common ions: [M+H]+, [M+Na]+, [M-H]-, [M+FA-H]-, [M+NH4]+, and common fragments.
- MS1 Data Acquisition: Acquire high-resolution MS1 spectrum via direct infusion or LC-MS, ensuring mass accuracy < 5 ppm.
- Translator Matching: Match theoretical m/z list to experimental MS1 spectrum within a defined error window (e.g., < 30 ppm). Assign matched MS peaks to the corresponding metabolite.
- Validation: Use the matched MS signals to cross-validate the NMR-based identifications.
Objective: To obtain an expanded set of accurate absolute metabolite concentrations by iteratively refining NMR and MS data. Workflow Summary:
- Multi-platform Data Acquisition: For the same sample, acquire a quantitative 1H-NMR spectrum. Separately, perform comprehensive UHPLC-HRMS analysis in both positive and negative ionization modes, using two chromatographic methods (e.g., RPC and HILIC).
- Initial NMR Deconvolution: Deconvolve the NMR spectrum using a database of chemical shifts, obtaining a first-pass list of metabolite concentrations.
- Correlation-based MS Feature Assignment: For each NMR-quantified metabolite, search HRMS data for all features with accurate mass matching its theoretical mass (e.g., < 5 ppm). Correlate the intensities of these candidate MS features across samples with the preliminary NMR concentrations. Select the MS feature with the highest correlation coefficient for the metabolite.
- MS-assisted NMR Refinement: Use the intensity of the confidently assigned MS feature (converted to concentration via the correlation slope) to constrain and improve the quantitative deconvolution of the overlapping NMR signals.
- Final Integrated Dataset: Output a final concentration table containing all metabolites quantified by the refined NMR deconvolution, enhanced by the selectivity of MS.
Objective: To compare two ESI ion source setups in an unbiased manner using a non-targeted dilution series experiment. Workflow Summary:
- Dilution Series Preparation: Prepare a series of 8 sequential one-in-four dilutions of a representative pooled biosample (e.g., from 1:1 to 1:16,384).
- Data Acquisition: Analyze the complete dilution series on the LC-HRMS systems equipped with the two ion sources (REF and ALT) to be compared, using identical chromatographic methods.
- Feature Detection & Alignment: Process raw data to extract features (unique m/z-RT pairs). Align features across all dilution levels and instrumental setups.
- Robust Fold-Change Calculation: For each feature, model the intensity response across the dilution series for both setups. Calculate a robust fold-change (ALT/REF) from the linear or log-linear ranges of the dilution curves, avoiding saturation and low-signal regions.
- Statistical & Chemical Evaluation: Analyze the distribution of fold-changes. Perform chemical interpretation by classifying features or conducting targeted validation with standards to confirm source-dependent selectivity differences.
Diagram Title: NMR/MS Translator Integrated Identification Workflow [55]
Diagram Title: Non-Targeted Ion Source Performance Evaluation [56]
Diagram Title: Decision Workflow for Chromatographic Method Selection [57]
The Scientist's Toolkit: Key Reagent Solutions
Table 3: Essential Research Reagents and Materials for HR-MS/NMR Metabolomics
| Reagent/Material | Typical Specification/Example | Function in Workflow |
|---|---|---|
| Deuterated NMR Solvent with Buffer | D2O with 20-50 mM phosphate buffer (pH 7.4), 0.1-1 mM DSS [55] [4] | Provides a stable, locked chemical shift reference for NMR spectroscopy and controls pH. |
| MS Ionization Additive | Formic Acid (0.1%) or Ammonium Acetate/Formate (5-10 mM) [55] [57] | Enhances protonation/deprotonation in ESI source, improving ionization efficiency and stability. |
| Chromatography Column (Reversed-Phase) | C18 or C8 column (e.g., 2.1 x 100 mm, 1.7-1.8 μm) [57] | Standard workhorse for separating medium-to-nonpolar metabolites in UPLC-HRMS. |
| Chromatography Column (HILIC) | Silica or Amide column (e.g., 2.1 x 150 mm, 1.7 μm) [56] [4] | Separates polar and hydrophilic metabolites that are poorly retained on reversed-phase columns. |
| Chromatography Column (Specialized RP) | C4 Reversed-Phase Column [58] | Used for separating intact proteins and large peptides (e.g., hemoglobin subunits). |
| Internal Standard Mix (Isotope-Labeled) | 13C- or 2H-labeled metabolites spanning chemical classes [57] | Corrects for variability in sample preparation, ionization, and instrument response. |
| Mass Calibrant | Low-concentration tuning mix (e.g., Agilent G1969-85000) [55] | Ensures high mass accuracy (< 5 ppm) is maintained on the HRMS instrument. |
| Sample Diluent (MS) | 50/50 Acetonitrile/Water with 0.1% Formic Acid [55] | Standard solvent for reconstituting or diluting samples prior to LC-MS or direct infusion MS. |
Strategies for Handling Data Integration Issues and Complex Biological Matrices
The comprehensive identification and quantification of metabolites in biological systems present a dual challenge: the intrinsic complexity of the sample matrices and the multidimensional nature of the data generated by complementary analytical platforms. Biological matrices such as urine, blood plasma, and tissues contain thousands of metabolites across a wide concentration range within a dense, interfering background [4]. No single analytical technique can capture this full complexity. While High-Resolution Mass Spectrometry (HR-MS) offers exceptional sensitivity and the ability to detect thousands of features, it can be destructive, suffer from variable ionization efficiencies, and provide limited structural information [1]. Nuclear Magnetic Resonance (NMR) spectroscopy, in contrast, is non-destructive, highly reproducible, and provides definitive structural elucidation and absolute quantification, but has lower sensitivity [4] [60].
This complementary relationship makes the integration of HR-MS and NMR data a powerful strategy for advanced metabolite identification research [1]. However, combining these disparate datasets introduces significant data integration issues, including differences in data structure, scale, noise characteristics, and biological coverage. Successfully navigating these issues is essential to move from simply acquiring data from two sources to generating a coherent, more complete biochemical profile than either method could provide alone [4] [61].
Integrated Analytical Approaches for Complex Matrices
Addressing matrix complexity often begins at the sample preparation and analysis stage. The choice of technique can minimize preparatory steps and maximize the information retrieved from challenging samples.
High-Resolution Magic Angle Spinning (HR-MAS) NMR is a critical tool for analyzing semi-solid and intact tissue samples without the need for metabolite extraction. By spinning the sample at the "magic angle" (54.7°), it averages out anisotropic interactions (like dipole-dipole couplings), resulting in high-resolution, liquid-like NMR spectra from complex matrices like plant leaves, biopsies, or cell clusters [3]. This allows for in-situ metabolic profiling, preserving the native state of the sample and eliminating artifacts introduced by extraction procedures.
For broader metabolite coverage, synergistic HR-MS and NMR protocols are employed. A leading strategy is the SYnergic use of NMR and HRMS for METabolomics (SYNHMET) [4]. This approach does not merely run the techniques in parallel but uses data from one to inform and refine the analysis of the other. In SYNHMET, an initial NMR spectral deconvolution provides approximate concentrations for a set of metabolites. HR-MS data, with its high sensitivity, is then used to resolve ambiguities in this initial NMR model by identifying specific MS chromatographic peaks whose intensities correlate with the NMR-derived concentrations. Finally, the accurately quantified MS peaks are fed back to refine and correct the NMR deconvolution, yielding a final set of absolute concentrations for a large number of metabolites without the need for individual analytical standards [4].
The complementary strengths and limitations of the core analytical platforms are summarized in the table below.
Table 1: Comparative Analysis of NMR and MS Platforms for Metabolomics [4] [3] [1]
| Feature | NMR Spectroscopy | Mass Spectrometry (HR-MS) |
|---|---|---|
| Sensitivity | Lower (μM-mM range). Improves with higher field strength. | Very high (pM-nM range). |
| Sample Preparation | Minimal for liquids; none for HR-MAS on tissues. Often non-destructive. | Extensive; typically requires extraction, chromatography. Destructive. |
| Reproducibility | Excellent; highly quantitative over a wide dynamic range. | Moderate; affected by ionization suppression, matrix effects, instrument tuning. |
| Structural Elucidation | Excellent for novel compound identification and stereochemistry. | Limited; requires MS/MS fragmentation and comparison to libraries. |
| Quantification | Absolute, using a single internal standard. | Relative or semi-quantitative; requires isotope-labeled standards for absolute quantitation. |
| Throughput | Moderate to high (minutes per sample). | Variable, often lower due to chromatographic separation. |
| Key Strength | Quantitative, reproducible, structural, non-destructive. | Sensitive, broad metabolite coverage. |
Detailed Experimental Protocols
This protocol enables metabolic profiling of intact plant or animal tissue, preserving in-vivo metabolic states.
1. Sample Harvesting and Preparation:
- Harvest material under controlled, consistent conditions (time of day, light exposure) to minimize biological variation.
- Immediately flash-freeze tissue in liquid nitrogen.
- For analysis, place ~10-20 mg of frozen tissue into a 4 mm zirconium oxide HR-MAS rotor.
- Add 20 μL of a deuterated buffer (e.g., D₂O phosphate buffer, pH 7.0) containing 0.5 mM 3-(trimethylsilyl)-2,2',3,3'-tetradeuteropropionic acid (TSP-d₄) as a chemical shift reference (δ 0.0 ppm) and quantification standard.
- Securely seal the rotor with a Kel-F cap to prevent leakage during spinning.
2. Data Acquisition:
- Insert the rotor into a dedicated HR-MAS probe on a spectrometer (preferably ≥ 500 MHz).
- Set the sample temperature to 4°C to slow enzymatic activity.
- Set the magic angle spinning speed to 4-6 kHz.
- Acquire a 1D ¹H-NMR spectrum using a pulse sequence with water suppression, such as the 1D Nuclear Overhauser Effect Spectroscopy (NOESY) preset sequence.
- Typical parameters: Spectral width: 12-14 ppm; Number of scans: 128-256; Relaxation delay: 4-5 seconds; Acquisition time: ~3 seconds.
3. Data Pre-processing:
- Process free induction decays (FIDs): Apply an exponential line-broadening function of 0.3-1.0 Hz, followed by Fourier transformation.
- Manually phase and baseline correct the spectrum.
- Reference the spectrum to the TSP-d₄ signal (0.0 ppm).
- For multivariate analysis, segment the spectrum into small regions ("buckets" or "bins") of equal width (e.g., 0.04 ppm) and integrate the signal within each bucket. Exclude regions containing the water and buffer residue signals.
This protocol details a sequential integration workflow to obtain absolute concentrations from complex biofluids like urine.
1. Parallel Sample Analysis:
- NMR Sample Prep: Mix 400 μL of biofluid (e.g., urine) with 200 μL of a phosphate buffer (pH 7.4) in D₂O containing 1 mM TSP. Transfer to a 5 mm NMR tube.
- NMR Data Acquisition: Acquire 1D ¹H-NMR spectra at 600 MHz or higher. Use a water-presaturation pulse sequence (e.g., NOESY-presat) for biofluids.
- HR-MS Sample Prep: Dilute the same biofluid sample appropriately with a solvent compatible with the chromatography mode (e.g., water for HILIC, organic solvent for RP).
- HR-MS Data Acquisition: Analyze using a UHPLC system coupled to a high-resolution mass spectrometer (e.g., Orbitrap, Q-TOF). Employ both Reverse-Phase (RP) and Hydrophilic Interaction Liquid Chromatography (HILIC) in positive and negative ionization modes to maximize metabolite coverage.
2. Initial NMR Deconvolution:
- Using commercial (e.g., Chenomx) or in-house software, perform a lineshape fitting (deconvolution) of the 1D NMR spectrum.
- Start with a library of 150-200 metabolites known to be present in the matrix. The software iteratively adjusts the concentration and chemical shift of each compound to minimize the difference between the calculated sum of all signals and the experimental spectrum. This yields a first-estimate concentration list (
C_NMR_est).
3. MS-Assisted Peak Assignment and Refinement:
- For each metabolite in
C_NMR_est, search the HR-MS dataset (all four modes: RP+/-, HILIC+/-) for chromatographic peaks with an accurate mass within 5 ppm of the metabolite's theoretical monoisotopic mass. - Correlate the intensity of each candidate MS peak across all samples with the
C_NMR_estfor that metabolite. The correct MS feature will show a high linear correlation. - Use the slope of this correlation to convert the MS feature intensity into an MS-derived concentration (
C_MS).
4. Final NMR Deconvolution with MS Constraints:
- Re-run the NMR spectral deconvolution, but this time fix the concentrations of the metabolites with high-confidence
C_MSvalues. - Allow the algorithm to adjust only the remaining, uncertain metabolites. This "MS-assisted" deconvolution dramatically improves the accuracy and number of quantified metabolites in the final output.
Diagram 1: The SYNHMET synergistic workflow.
Data Fusion and Integration Strategies
The fusion of NMR and MS data can be implemented at different levels of abstraction, each with specific advantages and computational requirements [1] [61].
Low-Level Data Fusion (LLDF): Also called early fusion or concatenation. Raw or pre-processed data matrices from NMR spectra and MS chromatograms are simply joined together (concatenated) side-by-side to create a single, large data matrix for statistical analysis. This retains all original information but is vulnerable to dominance by the technique with more variables (typically MS) and requires careful scaling to equalize their contributions [1].
Mid-Level Data Fusion (MLDF): Also called feature-level fusion. Features are first extracted separately from each dataset using dimensionality reduction techniques like Principal Component Analysis (PCA) or by selecting identified metabolites and their concentrations. The extracted features (e.g., PCA scores, concentration lists) are then fused into a joint matrix for analysis. This reduces noise and data size while preserving the most relevant information from each platform [1].
High-Level Data Fusion (HLDF): Also called decision-level or late fusion. Separate models (e.g., classification or regression) are built independently for the NMR and MS datasets. Their predictions or decisions are then combined using rules (e.g., voting, averaging) or meta-learners. This is flexible and allows for technique-specific modeling but may fail to capture subtle inter-platform interactions [1].
These strategies can be mapped to broader multi-omics integration frameworks [62] [61]:
- Vertical Integration: Applied to matched samples analyzed by both NMR and MS (the same sample analyzed by both techniques). The sample is the "anchor."
- Diagonal/Mosaic Integration: Applied when integrating data from different studies or cohorts where not all samples have been analyzed by both platforms. Requires advanced computational methods to align the datasets in a shared latent space.
Table 2: Data Fusion Strategies for NMR and MS Integration [1] [61]
| Strategy | Description | Advantages | Disadvantages & Challenges |
|---|---|---|---|
| Low-Level (LLDF) | Concatenation of raw/pre-processed data. | Maximum information retention; conceptually simple. | High dimensionality; prone to technical noise dominance; requires careful scaling. |
| Mid-Level (MLDF) | Fusion of extracted features (e.g., PCA scores, metabolite concentrations). | Reduces noise and dimensionality; focuses on biologically relevant features. | Risk of losing important information during feature extraction; dependent on extraction method. |
| High-Level (HLDF) | Combination of independent model predictions. | Allows for platform-specific modeling; modular and flexible. | Does not model inter-platform correlations; depends heavily on performance of individual models. |
Diagram 2: Conceptual framework for multi-level data fusion.
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Research Reagent Solutions for Integrated NMR/MS Metabolomics
| Item | Function/Application | Key Characteristics |
|---|---|---|
| Deuterated Solvents & Buffers (D₂O, CD₃OD, buffer salts) | Provides a lock signal for NMR spectrometers; minimizes interfering proton signals in ¹H-NMR. | High isotopic purity (99.9% D or higher); pH-adjusted for biological relevance. |
| Internal NMR Standards (TSP-d₄, DSS-d₆) | Chemical shift reference (0.0 ppm) and quantitative concentration standard for NMR. | Chemically inert, soluble, gives a single, sharp resonance in a clear spectral region. |
| Zirconium Oxide HR-MAS Rotors | Holds intact tissue samples for magic angle spinning NMR. | Biologically inert, withstands high spinning speeds (kHz), compatible with Kel-F caps. |
| UHPLC Columns (C18 RP, HILIC) | Separates metabolites by polarity prior to MS detection to reduce ion suppression and complexity. | High reproducibility, sub-2μm particle size for high resolution, stable under wide pH ranges. |
| Mass Calibration Solutions | Calibrates the mass accuracy of the HR-MS instrument before and during analysis. | Contains known ions across a broad m/z range (e.g., sodium formate clusters). |
| Stable Isotope-Labeled Internal Standards (¹³C, ¹⁵N) | Enables precise absolute quantification in MS, corrects for matrix effects and recovery losses. | Should be chemically identical to the target analyte except for isotopic composition. |
| Solid Phase Extraction (SPE) Cartridges | Pre-fractionates complex samples to reduce matrix interference and concentrate metabolites of interest. | Various chemistries (C18, ion exchange, mixed-mode) for selective metabolite capture. |
Application in Disease Research: A Case Study
The power of integrated strategies is illustrated in clinical metabolomics. In a study profiling urine from healthy controls, chronic cystitis patients, and bladder cancer patients, the SYNHMET protocol quantified 165 metabolites with minimal missing values [4].
Workflow & Integration: Initial NMR deconvolution provided a baseline. HR-MS data (from RP and HILIC in both ionization modes) was used to resolve ambiguities for overlapping NMR signals, such as in the crowded 2.37-2.47 ppm spectral region containing 11 metabolites. The intensity of specific MS features was linearly correlated with the initial NMR estimates to assign the correct MS peak to 2-oxoglutarate and other metabolites. These MS-derived concentrations were then used to constrain the final NMR deconvolution.
Outcome: This synergistic, mid-level fusion approach generated a comprehensive, quantitative personalized metabolic profile for each subject. It allowed for precise comparison against known normal ranges and identified distinct metabolic signatures between disease groups, demonstrating the strategy's potential for biomarker discovery and understanding pathophysiology with a level of accuracy and coverage unattainable by either technique alone [4].
Validation and Comparative Analysis of Integrated Metabolite Identification
Validation Frameworks for Metabolite Identification and Absolute Quantification
In the context of a broader thesis on High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) integration, establishing robust validation frameworks is paramount for advancing metabolite identification and absolute quantification research. These frameworks are critical for transforming raw analytical data into reliable biological insights, particularly in drug development where decisions hinge on precise metabolic phenotyping [20] [24]. The inherent complementarity of HR-MS and NMR spectroscopy provides a powerful foundation for such validation; HR-MS delivers high sensitivity and broad metabolite coverage, while NMR offers unmatched structural elucidation, reproducibility, and non-destructive analysis [4] [63]. However, the complexity of biological matrices, instrument variability, and data integration challenges necessitate systematic validation protocols. This document outlines detailed application notes and experimental protocols for validation frameworks, leveraging integrated HR-MS/NMR approaches to enhance accuracy, ensure data quality, and support the translation of metabolomic findings into actionable research and clinical outcomes.
Core Principles and Workflows for Integrated Validation
The validation of metabolomic data relies on a synergistic framework that leverages the complementary strengths of NMR and MS. NMR spectroscopy provides highly reproducible, quantitative data with definitive structural information but has lower sensitivity [63]. In contrast, MS, particularly HR-MS, offers high sensitivity and the ability to detect thousands of features but can struggle with definitive identification and accurate quantification without authentic standards [4] [1]. A core principle of integrated validation is the use of data from one platform to guide and verify results from the other.
The SYNHMET (SYnergic use of NMR and HRMS for METabolomics) strategy exemplifies this principle [4]. Its workflow begins with the deconvolution of complex ¹H-NMR spectra to obtain an initial, approximate concentration profile for a set of metabolites. Concurrently, untargeted HR-MS analysis generates a list of chromatographic peaks with accurate masses. Crucially, the tentative concentration data from NMR are correlated with the intensity data of MS features sharing a plausible mass match. Strong linear correlations help unambiguously assign MS peaks to specific metabolites. Subsequently, the accurately quantified intensities from the validated MS features are used to refine and correct the initial NMR-derived concentrations, leading to a final dataset of absolute concentrations achieved without exhaustive use of analytical standards [4]. This iterative, cross-validating workflow significantly increases both the number of accurately quantified metabolites and confidence in their identities.
For subcellular metabolomics, such as profiling the mitochondrial matrix, validation requires additional layers of methodological rigor due to the need for rapid, specific organelle isolation. A developed protocol uses immunocapture of epitope-tagged mitochondria followed by LC-MS analysis, enabling absolute quantification of over 100 matrix metabolites [64] [65]. Key to validation here is assessing isolation specificity and metabolite stability. This is achieved by using congruent yield calculations from both a protein marker (citrate synthase) and a metabolite marker (coenzyme A), and by confirming the absence of markers from contaminating compartments like lysosomes [64]. The integration of a predicted mitochondrial metabolite database ("MITObolome") further validates findings by focusing the analysis on a physiologically relevant chemical space [65].
Validation Framework: SYNHMET & Subcellular Analysis
Beyond wet-lab protocols, computational data fusion strategies form a critical pillar of the modern validation framework. These strategies integrate datasets from NMR and MS at different levels of abstraction to improve model performance and biological interpretation [22] [1]. Low-level fusion concatenates raw or pre-processed data matrices, requiring careful scaling to equalize the contribution of each platform. Mid-level fusion first reduces the dimensionality of each dataset separately (e.g., via PCA), then concatenates the extracted features. High-level (decision-level) fusion combines the final predictions or classifications from models built on each dataset independently [1]. The choice of strategy involves a trade-off between data detail, model complexity, and interpretability, and must be validated for each specific application.
Experimental Protocols for Key Validation Experiments
Protocol: The SYNHMET Workflow for Cross-Platform Quantification
This protocol details the synergistic use of ¹H-NMR and UHPLC-HRMS to obtain absolute concentrations of metabolites in complex biofluids like urine, without requiring a calibration curve for every compound [4].
Materials & Sample Preparation:
- Biofluid Sample: e.g., Human urine. Centrifuge at 4°C to remove particulate matter.
- NMR Buffer: Phosphate buffer (e.g., 1.5 M K₂HPO₄/NaH₂PO₄, pH 7.4) in D₂O containing 0.1% w/w sodium trimethylsilylpropanesulfonate (DSS-d6) as a chemical shift and quantitative reference. Note: TSP is not recommended for samples with proteins as it can bind and broaden [63].
- NMR Sample: Mix 350 μL of clarified urine with 250 μL of NMR buffer. Transfer to a 5 mm NMR tube.
- MS Solvents: LC-MS grade water, methanol, and acetonitrile. Formic acid or ammonium acetate for mobile phase modifiers.
Instrumental Analysis:
- ¹H-NMR Spectroscopy:
- Acquire spectra on a high-field spectrometer (e.g., 600 MHz) using a standard 1D NOESY-presaturation pulse sequence to suppress the water signal.
- Parameters: Spectral width 20 ppm, acquisition time ~4 seconds, relaxation delay 5-6 seconds, 128-256 transients, temperature 298 K.
- Process spectra: Apply exponential apodization (0.3 Hz line broadening), zero-filling, Fourier transformation, phase and baseline correction. Calibrate spectra to the DSS methyl peak at 0.0 ppm.
- UHPLC-HRMS Analysis:
- Perform in both positive and negative ionization modes to maximize coverage.
- Use complementary chromatographic separations: Reverse-Phase (RP) for hydrophobic compounds and Hydrophilic Interaction Liquid Chromatography (HILIC) for polar compounds [4].
- Column: e.g., C18 for RP; amide or silica for HILIC.
- Mass Spectrometer: High-resolution instrument (e.g., Q-Exactive Orbitrap). Set resolution > 70,000 (at m/z 200), scan range m/z 70-1050.
- Data-Dependent Acquisition (DDA): Use full MS scan followed by MS/MS scans of top N ions for annotation support.
Data Processing & Validation Workflow:
- Initial NMR Deconvolution:
- Using commercial (e.g., Chenomx) or custom software, fit the NMR spectrum with a library of reference metabolite signatures.
- This generates List A: Metabolite identities and approximate concentrations (CNMRapprox).
- HRMS Feature Extraction:
- Process raw files with software (e.g., XCMS, MS-DIAL) for peak picking, alignment, and annotation.
- Generate List B: Features with m/z, retention time (RT), and intensity (I_MS).
- Cross-Platform Correlation & Assignment:
- For each metabolite in List A, search List B for all features with a measured m/z within 5 ppm of its theoretical monoisotopic mass.
- For each candidate MS feature, plot its intensity (IMS) across all samples against CNMR_approx for the metabolite.
- Identify the MS feature showing the highest linear correlation coefficient (R²). This strong correlation validates the assignment of that MS peak to the metabolite.
- Concentration Refinement:
- For the validated metabolite-MS feature pair, use the slope of the correlation plot (IMS vs. CNMRapprox) as a response factor.
- Convert all MS intensities for that feature into refined, absolute concentrations (Cfinal).
- These Cfinal values, traceable to the quantitative NMR reference, replace the initial CNMR_approx estimates.
Protocol: Absolute Quantification of Mitochondrial Matrix Metabolites
This protocol enables the specific, rapid isolation of mitochondria and absolute quantification of their matrix metabolite concentrations, critical for understanding compartmentalized metabolism [64] [65].
Materials & Cell Culture:
- Cell Line: HeLa or other mammalian cells stably expressing an outer mitochondrial membrane (OMM) tagged protein (e.g., 3xHA-EGFP-OMP25).
- Isolation Buffer (LC-MS Compatible): 150 mM KCl, 25 mM KH₂PO₄, pH 7.2 (KPBS). Avoid sucrose/HEPES which interfere with LC-MS [64].
- Antibody & Beads: Anti-HA (or appropriate epitope) antibody conjugated to magnetic beads (~1 μm diameter, non-porous).
- Quenching & Extraction Solvent: Cold (-20°C or -80°C) 80% methanol/20% water or acetonitrile/methanol/water mixtures. Include internal standards for quantification.
Mitochondrial Immunoisolation:
- Harvest & Homogenize: Wash cells (2-4 x 10⁷) in ice-cold KPBS. Resuspend in KPBS with protease inhibitors and homogenize with a ball-bearing homogenizer (clearance ~10-12 μm) until >90% cell lysis is achieved (~10 passes). Keep samples at 0-4°C.
- Clear Lysate: Centrifuge homogenate at 1,000 x g for 5 min at 4°C to remove nuclei and unbroken cells. Transfer post-nuclear supernatant (PNS) to a fresh tube.
- Immunocapture: Incubate the PNS with anti-HA magnetic beads for 3-5 minutes with gentle rotation.
- Wash: Place tube on a magnetic stand. Discard supernatant. Rapidly wash beads 2x with 1 mL of ice-cold KPBS (complete within 60 seconds total).
Metabolite Extraction & Analysis:
- Extract: To the bead-bound mitochondria, immediately add 500 μL of cold quenching/extraction solvent. Vortex vigorously and incubate at -20°C for 20 min.
- Pellet Debris: Centrifuge at 16,000 x g for 10 min at 4°C. Transfer supernatant (metabolite extract) to a new tube. Dry under vacuum or nitrogen stream.
- LC-MS Analysis: Reconstitute in LC-MS compatible solvent. Analyze using a targeted or semi-targeted HILIC-LC-MS method optimized for polar metabolites.
- Absolute Quantification: Use calibration curves from authentic standards spiked into a mitochondrial extract matrix. Normalize peak areas to internal standards.
Validation of Isolation Fidelity:
- Yield & Purity: Perform western blot analysis on PNS, bead flow-through, and bead eluate for markers of mitochondria (e.g., ATP5A, COX IV), cytosol (e.g., LDH), lysosomes (e.g., LAMP2, Cathepsin C), and other organelles.
- Metabolite Integrity Check: Quantify a metabolite known to be highly enriched in the matrix (e.g., Coenzyme A) and compare its yield to that of a mitochondrial protein (Citrate Synthase). Similar yields indicate intact organelles without metabolite leakage [64].
- Matrix Volume Determination: Use confocal microscopy to measure the average volume of mitochondria in the cell line. Combine this with the yield (from step 1) to estimate the total matrix volume in the isolate, enabling the conversion of moles to molar concentration [64].
Protocol: Data Fusion for Classification Validation
This protocol describes a mid-level data fusion approach to integrate ¹H-NMR and LC-HRMS datasets for improved sample classification, as applied in foodomics and clinical phenotyping [22] [1].
Materials & Data:
- Pre-processed Datasets: ¹H-NMR data matrix (bucketed or peak-integrated) and LC-HRMS feature intensity matrix. Both matrices must be aligned to the same set of samples.
Data Pre-processing:
- Intra-Block Scaling: Scale each dataset (block) separately to account for different variances. Common methods include Pareto scaling (
1/√σ) or unit variance scaling (autoscaling) [1]. - Dimensionality Reduction (Per Block): Apply Principal Component Analysis (PCA) to each scaled data block. Retain a defined number of principal components (PCs) that explain most of the variance (e.g., >70-80%). This step extracts the most informative latent variables from each platform.
- Inter-Block Concatenation: Create a new fused data matrix by concatenating the scores from the selected PCs of the NMR model with the scores from the selected PCs of the MS model.
Model Building & Validation:
- Supervised Classification: Apply a supervised method like Partial Least Squares-Discriminant Analysis (PLS-DA) or sparse PLS-DA (sPLS-DA) to the fused scores matrix, using the sample classes (e.g., disease/control, wine type) as the Y-variable [22].
- Cross-Validation: Validate the model using rigorous cross-validation (e.g., k-fold, leave-one-out) to avoid overfitting. Calculate performance metrics (accuracy, R², Q²).
- Comparison: Build and validate separate PLS-DA models on the NMR-only and MS-only datasets. Compare their classification error rates to the fused model. A superior performance (lower error rate) of the fused model demonstrates the validated added value of data integration [22].
Table 1: Key Research Reagent Solutions for Integrated Metabolomics Validation
| Item | Function & Role in Validation | Key Considerations |
|---|---|---|
| Quantitative NMR Reference (e.g., DSS-d6) | Provides chemical shift reference (0 ppm) and serves as an internal standard for absolute quantification in NMR. Integral of its singlet is used as calibrant [63]. | Use DSS over TSP for biofluids; it interacts less with proteins. Must be stable and non-volatile. |
| Stable Isotope-Labeled Internal Standards (for MS) | Enable absolute quantification by standard addition, correct for ion suppression/enhancement, and monitor extraction efficiency. | Ideal is ¹³C or ¹⁵N labeled analog of the target analyte. Used in targeted or semi-targeted MS assays. |
| LC-MS Compatible Isolation Buffers (e.g., KCl/KH₂PO₄) | Enable metabolite profiling of isolated organelles without introducing polymeric contaminants (e.g., sucrose) that suppress ionization or co-elute in LC-MS [64]. | Must maintain organelle integrity while being compatible with MS detection. |
| Epitope Tagging System (e.g., 3xHA-OMP25 construct) | Enables rapid, specific immunocapture of organelles (e.g., mitochondria) for compartment-specific metabolomics, validating spatial metabolic regulation [64] [65]. | Requires generation of stable cell line. Choice of tag (HA, FLAG) and bead size (~1 μm) is critical for yield. |
| Authenticated Chemical Standards | Essential for validating metabolite identities (by matching RT and MS/MS or NMR spectrum) and for constructing calibration curves for absolute quantification. | Purity must be certified. Stored appropriately to prevent degradation. |
| Quality Control (QC) Pool Sample | A pooled mixture of all study samples, run repeatedly throughout the analytical sequence. Monitors instrument stability, validates data quality, and is used for signal correction in MS [66]. | Should be representative of the entire sample set. |
Data Management, Statistical Validation, and Reporting Standards
Machine Learning for Enhanced Validation: Machine learning (ML) algorithms have become integral to validating and interpreting complex, integrated metabolomics data. They assist in key validation steps: processing raw spectra (peak picking, alignment), imputing missing values, and, most importantly, performing robust classification and biomarker selection [67] [66]. For instance, Random Forest (RF) provides high interpretability for identifying critical metabolites that discriminate between sample classes, while Support Vector Machines (SVM) can handle high-dimensional data well. Artificial Neural Networks (ANNs) and deep learning models excel at finding complex, non-linear patterns in fused datasets [66]. Validating the output of these models requires strict protocols: splitting data into independent training, validation, and test sets; using nested cross-validation to tune hyperparameters; and reporting performance metrics on the held-out test set to prevent overfitting and ensure generalizability.
Standardization and Reporting: For a validation framework to be credible and reproducible, adherence to community reporting standards is non-negotiable. The Metabolomics Standards Initiative (MSI) outlines minimum reporting requirements for chemical analysis, data processing, and context [24]. When reporting integrated HR-MS/NMR studies, the following must be explicitly documented:
- Sample Preparation: Detailed protocols for extraction, derivatization, and internal standard addition.
- Instrumental Parameters: Complete NMR pulse sequences and parameters; LC column, gradient, and MS ionization settings.
- Data Processing: Software and algorithms used for NMR phasing, baseline correction, bucketing; and for MS peak picking, alignment, and annotation (with tolerance windows, e.g., 5 ppm for mass accuracy).
- Identification Levels: Metabolites must be reported with a clear level of confidence (e.g., Level 1: confirmed by authentic standard; Level 2: putative annotation based on spectral library; Level 3: tentative class) [66].
- Quantification Method: Stating whether quantification is absolute or relative, and the basis for it (e.g., internal standard, calibration curve, cross-platform normalization as in SYNHMET).
- Statistical & Model Validation: Detailed description of data fusion level and method, ML model parameters, cross-validation strategy, and full performance results.
Table 2: Summary of Quantitative Performance from Integrated Methodologies
| Study & Technique | Quantification Output | Key Performance Metrics & Validation Outcome | Reference |
|---|---|---|---|
| SYNHMET (NMR + UHPLC-HRMS) on Human Urine | Absolute concentrations for 165 metabolites. | Quantification achieved without individual calibration curves. Validation via cross-correlation of platforms increased accuracy and reduced missing values. Applied to discriminate bladder cancer patients. | [4] |
| Immunocapture + LC-MS for Mitochondrial Matrix | Absolute concentrations for >100 metabolites in HeLa cell mitochondria. | Validation of isolation specificity via immunoblotting (low cytosolic contamination). Metabolite yield matched enzyme yield, confirming integrity. Revealed compartmentalized metabolic dynamics. | [64] [65] |
| Data Fusion (NMR + LC-HRMS) for Wine Classification | Improved classification model of wines by withering time and yeast strain. | Mid-level data fusion (PCA-scores concatenation) followed by sPLS-DA achieved a lower classification error rate (7.52%) compared to single-platform models. Validated complementarity of platforms. | [22] |
| Machine Learning (RF, SVM) for Biomarker Discovery | Identification of predictive metabolite panels for disease states (e.g., cardiometabolic risk). | ML models validated on independent test sets provide quantitative performance metrics (AUC, accuracy). Highlight the need for rigorous validation to ensure clinical translatability. | [67] [66] |
Conclusion: Towards a Unified Framework The integration of HR-MS and NMR, supported by robust experimental protocols, computational data fusion, and stringent reporting standards, constitutes a powerful validation framework for modern metabolomics. This framework moves beyond simple instrument calibration to a systems-level validation of biological findings. By cross-verifying identities and quantities across platforms, ensuring spatial specificity in subcellular studies, and applying robust statistical and machine learning models to fused data, researchers can generate metabolite data of the highest confidence. This rigorous approach is essential for advancing the role of metabolomics in fundamental biochemical research, reliable biomarker discovery, and informed drug development, ensuring that conclusions are not artifacts of the method but true reflections of biological state.
This work is framed within a broader thesis on the strategic integration of High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy for advanced metabolite identification research. While MS-based platforms dominate the field in terms of publication volume and sensitivity, NMR spectroscopy remains the undisputed gold standard for quantitative accuracy, structural elucidation, and methodological reproducibility [68] [69]. The erroneous perspective that metabolomics is better served by MS alone limits metabolome coverage and can diminish research quality [2]. This article argues that the future of robust, actionable metabolomics, particularly in drug discovery, lies not in choosing one platform over the other but in implementing integrated workflows that leverage their complementary strengths. We present application notes and protocols to guide researchers in harnessing the combined power of NMR's gold-standard validation and MS's expansive detection capabilities.
Performance Comparison: Core Capabilities and Metrics
The selection between NMR and MS is guided by their fundamental, complementary technical profiles. The table below summarizes their core characteristics.
Table 1: Core Technical Comparison of NMR and MS Platforms
| Characteristic | Nuclear Magnetic Resonance (NMR) | Mass Spectrometry (MS), primarily LC-MS/GC-MS |
|---|---|---|
| Key Principle | Detection of nuclear spin transitions in a magnetic field [68]. | Measurement of mass-to-charge ratio (m/z) of ionized molecules [26]. |
| Sensitivity | Lower (typically ≥1 μM) [2] [69]. | High (pM to nM range) [26] [69]. |
| Quantitation | Inherently and highly quantitative; excellent linearity without need for compound-specific standards [5] [69]. | Semi-quantitative; requires isotope-labeled internal standards for precise quantitation [70] [71]. |
| Structural Insight | High. Provides direct information on atomic connectivity, functional groups, and stereochemistry [68]. | Limited. Infers structure from fragmentation patterns and accurate mass [26]. |
| Reproducibility | Exceptionally high (CV ≤ 5%); ideal for longitudinal studies [5] [69]. | Moderate; susceptible to ion suppression and matrix effects [26]. |
| Sample Throughput | Moderate. | High. |
| Sample Destructiveness | Non-destructive; sample can be recovered for further analysis [68] [25]. | Destructive. |
| Metabolite Identification Confidence | Gold Standard. Direct structural confirmation [68]. | Requires orthogonal validation (e.g., NMR or standards) for highest confidence [71]. |
The practical outcome of these technical differences is evidenced in metabolomics studies. A comparative study on Chlamydomonas reinhardtii treated with lipid modulators provides clear quantitative data on detection overlap and uniqueness [2].
Table 2: Metabolite Detection in a Comparative Study (C. reinhardtii) [2]
| Detection Category | Number of Metabolites | Key Implications |
|---|---|---|
| Total Detected | 102 | Combined approach maximizes coverage. |
| Detected by GC-MS only | 82 | MS excels at detecting low-abundance, ionizable metabolites. |
| Detected by NMR only | 20 | NMR captures metabolites missed by MS (e.g., non-ionizable, under-derivatized). |
| Detected by both techniques | 22 | Provides high-confidence identifications with complementary data. |
| Of interest (perturbed by treatment) | 47 | Integrated analysis informs more complete biological interpretation. |
| Uniquely identified by NMR | 14 | e.g., Glycine, valine, acetate, malate. |
| Uniquely identified by GC-MS | 16 | e.g., Fructose-6-phosphate, fumarate, asparagine. |
| Identified by both | 17 | High-confidence core set (e.g., adenosine, succinate). |
Application Notes and Detailed Protocols
Protocol A: Sequential Untargeted Analysis of Blood Serum via NMR and Multi-LC-MS
This protocol enables comprehensive profiling from a single serum aliquot, addressing compatibility challenges [25].
1. Sample Preparation:
- Starting Material: 100-150 μL of human blood serum.
- Protein Removal & NMR Buffer Addition: Add 300-450 μL of ice-cold methanol:acetonitrile (1:1, v/v) to the serum. Vortex vigorously and incubate at -20°C for 1 hour. Centrifuge at 14,000 g for 15 minutes at 4°C. Split the supernatant (~400 μL) into two equal parts.
- NMR Sample: Transfer one part to a 5 mm NMR tube. Add 200 μL of deuterated phosphate buffer (100 mM, pD 7.4) containing 0.5 mM TSP-d4 as a chemical shift and quantitation reference [25].
- LC-MS Sample: Dry the second part under a gentle stream of nitrogen or vacuum. Reconstitute the dried extract in 100 μL of water:acetonitrile (95:5, v/v) with 0.1% formic acid for positive ionization mode, or in water:methanol for negative mode. Centrifuge prior to injection [25].
2. Data Acquisition:
- NMR: Acquire 1D 1H NMR spectra at 25°C using a NOESYGPPR1D presaturation pulse sequence to suppress the water signal. Use a sufficiently long relaxation delay (e.g., 4 s) to ensure quantitative accuracy [5] [25].
- LC-MS: Perform analysis using reversed-phase (C18) chromatography coupled to a high-resolution accurate mass (HRAM) spectrometer. Use both positive and negative electrospray ionization (ESI) modes. A typical gradient for a C18 column involves water and acetonitrile, both with 0.1% formic acid [70].
3. Data Processing & Analysis:
- NMR: Process spectra (Fourier transformation, phase, baseline correction). Reference the TSP methyl signal to 0.0 ppm. Integrate regions or perform spectral deconvolution for quantification [2].
- LC-MS: Process raw files using software (e.g., CompoundDiscoverer, XCMS, MS-DIAL) for feature detection, alignment, and annotation against databases (HMDB, METLIN) [70] [71]. Use Level 1 identification (matching m/z, RT, and MS/MS to an authentic standard) where possible for highest confidence [71].
Protocol B: Metabolite Identification (MetID) in Drug Discovery using HR-MS
This protocol outlines a standard in vitro MetID workflow to identify metabolic soft spots of drug candidates [70].
1. Hepatocyte Incubation:
- Preparation: Thaw cryopreserved pooled human hepatocytes (BioIVT) and suspend in Leibovitz L-15 buffer at 1.0 million viable cells/mL [70].
- Incubation: Pre-incubate 245 μL of cell suspension in a deep-well plate at 37°C for 15 min. Initiate the reaction by adding 5 μL of a 200 μM substrate solution (in DMSO/ACN/water) to achieve a final test compound concentration of 4 μM. Incubate at 37°C with shaking [70].
- Quenching: At designated time points (e.g., 0, 40, 120 min), remove a 50 μL aliquot and quench with 200 μL of ice-cold acetonitrile:methanol (1:1, v/v). Centrifuge to pellet proteins [70].
2. LC-HRMS Analysis:
- Chromatography: Use UHPLC with a C18 column. Employ a water/acetonitrile gradient, both phases containing 0.1% formic acid [70].
- Mass Spectrometry: Acquire data in data-dependent acquisition (DDA) mode on a high-resolution mass spectrometer. First, collect a full MS scan. Then, automatically select the most intense ions (including those from a predicted metabolite list) for MS/MS fragmentation [70].
3. Data Interpretation:
- Use software (e.g., MetabolitePilot, MassMetaSite, CompoundDiscoverer) to process data. The software compares the chromatographic and spectral data of metabolites to the parent drug, identifying potential biotransformations (e.g., +16 Da for oxidation, -14 Da for demethylation) [70].
- Structural Proposal: Review extracted ion chromatograms (XICs) and MS/MS spectra to propose metabolite structures. Critical Note: MS peak areas are only semi-quantitative due to differential ionization efficiencies; they indicate relative abundance but require NMR or synthetic standards for definitive confirmation and absolute quantification [70] [71].
Integrated Data Analysis Strategies
The synergy of NMR and MS is realized through formal Data Fusion (DF) strategies, which are classified by the level of data integration [26] [1].
1. Low-Level Data Fusion (LLDF):
- Process: Raw or pre-processed data matrices from NMR and MS are concatenated column-wise into a single "super-matrix" [26] [1].
- Pre-processing Requirement: Critical. Intra-block scaling (e.g., Pareto scaling per platform) and inter-block weighting (e.g., scaling to equal sum of standard deviations) must be applied to prevent one platform's variance from dominating the model [26] [1].
- Analysis: Multiblock PCA or PLS models are applied to the fused matrix to find correlations across the entire dataset [2] [26].
2. Mid-Level Data Fusion (MLDF):
- Process: Features are first extracted and dimensionality is reduced separately for each platform (e.g., via PCA). The resulting scores or selected features are then fused for final modeling [26] [1].
- Advantage: Reduces the high dimensionality and noise of raw MS data before fusion, improving model robustness.
3. High-Level Data Fusion (HLDF):
- Process: Separate statistical models (e.g., classifiers) are built for each analytical platform. The final predictions or decisions from these independent models are then combined using rules (e.g., voting, Bayesian consensus) [26] [1].
- Application: Useful for classification tasks (e.g., disease diagnosis) where platform-specific models contribute to a final, consensus outcome.
The Scientist's Toolkit: Essential Reagents & Materials
Table 3: Key Research Reagent Solutions for Integrated Metabolomics
| Item | Function & Specification | Key Application/Note |
|---|---|---|
| Deuterated Solvents & Buffers (D₂O, Phosphate buffer in D₂O) | Provides field-frequency lock for NMR without significant ¹H background signal. Contains a chemical shift reference (e.g., TSP-d4) [5] [25]. | NMR sample preparation. Compatibility studies show they do not interfere with subsequent LC-MS analysis [25]. |
| Deuterated Internal Standard (TSP-d4, DSS-d6) | Serves as primary quantitative and chemical shift (0.0 ppm) reference in ¹H NMR due to its inert and sharp singlet signal [5]. | Quantitative NMR metabolomics. Added at known concentration to enable absolute quantification of all detected metabolites [69]. |
| Protein Precipitation Solvents (Methanol, Acetonitrile) | Efficiently denatures and precipitates proteins from biofluids (serum, plasma) to protect analytical instrumentation and release metabolites [70] [25]. | Sample preparation for MS and integrated workflows. Methanol:acetonitrile (1:1) is common. The supernatant is used for analysis [25]. |
| LC-MS Grade Solvents & Additives (Water, ACN, MeOH, Formic Acid) | High-purity solvents minimize background ions and noise. Acidifiers (e.g., 0.1% formic acid) promote protonation in positive ESI mode [70]. | Mobile phase for LC-HRMS. Critical for reproducible chromatography and stable ionization. |
| Pooled Cryopreserved Hepatocytes | Biologically relevant in vitro system containing the full complement of drug-metabolizing enzymes (CYPs, UGTs, etc.) [70]. | Drug metabolism (MetID) studies. Used to generate phase I and II metabolites for identification [70] [72]. |
| Authenticated Chemical Standards | Pure compounds with confirmed structure. Used to build in-house spectral libraries [71]. | Gold-standard metabolite identification (Level 1). Matching of m/z, RT, and MS/MS fragmentation pattern to a standard run on the same platform provides highest confidence ID [71]. |
NMR spectroscopy maintains its role as the gold standard in metabolomics for definitive structural identification and absolute quantification, providing the validation backbone for the field [68] [69]. Mass spectrometry, particularly HR-MS, is the engine for broad, sensitive discovery. The future of metabolite identification research, as evidenced by growing trends in literature and data fusion methodologies, is inherently integrated [26] [1] [69].
Successful integration requires careful experimental design, such as sequential analysis from a single sample aliquot [25], and the application of robust chemometric data fusion strategies to extract coherent biological insight from multimodal data [2] [26]. For drug development professionals, this integrated approach translates to higher-confidence biomarker discovery, more reliable assessment of metabolic soft spots and toxic metabolites, and ultimately, a stronger data package for regulatory submission and clinical decision-making [70] [71].
Abstract Integrating data from High-Resolution Mass Spectrometry (HR-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy is a cornerstone of comprehensive metabolomic analysis within a broader thesis on metabolite identification. This integration is critical due to the complementary analytical strengths of each platform: HR-MS offers high sensitivity for detecting trace metabolites, while NMR provides robust, quantitative data and detailed structural elucidation [26]. The efficacy of this integration hinges on the chosen data fusion strategy, which directly impacts the accuracy, biological interpretability, and reproducibility of research outcomes. This article presents detailed application notes and protocols for benchmarking three primary data fusion levels—low-, mid-, and high-level—framed within the context of rigorous experimental design and standardized reporting [5]. We provide a comparative analysis of the quantitative performance of these strategies, including benchmarked metrics from next-generation software tools like MassCube [73], and detail step-by-step experimental methodologies. Furthermore, we introduce a validated, integrated HR-MS/NMR workflow and a curated "Scientist's Toolkit" of essential reagents and resources, offering researchers and drug development professionals a practical framework to implement and evaluate fusion strategies for enhanced metabolomic discovery.
Metabolomics, the comprehensive study of low-molecular-weight metabolites, is indispensable for understanding biochemical mechanisms in health, disease, and drug response. No single analytical technique can fully capture the complexity of the metabolome. High-Resolution Mass Spectrometry (HR-MS), particularly when coupled with liquid chromatography (LC), excels in sensitivity, enabling the detection and relative quantification of thousands of metabolites in complex biological matrices [26]. Conversely, Nuclear Magnetic Resonance (NMR) spectroscopy, while less sensitive, is inherently quantitative, non-destructive, and provides unparalleled structural information and high analytical reproducibility [26] [74].
The integration of HR-MS and NMR data is therefore not merely additive but synergistic, aiming to construct a more complete and reliable biochemical profile. However, combining these heterogeneous data streams—differing in scale, dimensionality, and noise structure—presents significant computational and statistical challenges. The choice of data fusion strategy is a critical methodological decision that influences downstream analysis, from feature selection to biological inference. This article establishes a framework for benchmarking these strategies, with a dedicated focus on their impact on accuracy (the correctness of metabolite identification and quantification) and reproducibility (the consistency of results across experiments and laboratories)—two pillars of credible metabolomic science [5].
CORE DATA FUSION STRATEGIES: A THREE-LEVEL ARCHITECTURE
Data fusion strategies are systematically categorized into three levels based on the stage at which data from different platforms are integrated. The choice of level involves a fundamental trade-off between leveraging raw information content and managing computational complexity and model interpretability [26].
2.1 Low-Level (Early) Data Fusion Low-level data fusion (LLDF) involves the direct concatenation of pre-processed raw data matrices from HR-MS and NMR before any feature reduction or modeling. This approach retains the maximum amount of original information.
- Process: NMR spectral bins (e.g., δ 0.05 ppm intervals) and MS feature tables (m/z and retention time pairs with intensities) are normalized, scaled, and combined into a single composite matrix [26].
- Advantages: Maximizes theoretical information usage; allows the model to discover interactions between variables from different platforms.
- Challenges: Creates very high-dimensional data with a high risk of overfitting; requires sophisticated scaling to balance the inherent variance differences between MS and NMR data; model interpretation can be difficult [26] [75].
- Typical Use Case: Exploratory studies where all potential interactions are of interest, supported by robust cross-validation.
2.2 Mid-Level (Intermediate) Data Fusion Mid-level data fusion (MLDF) is a two-step strategy that reduces data dimensionality within each platform first, then fuses the selected features.
- Process: Discriminative features are selected independently from each dataset using methods like Principal Component Analysis (PCA), Partial Least Squares (PLS), or machine learning-based selection (e.g., VIP scores). The selected features (e.g., PCA scores or key metabolites) are then concatenated for final modeling [26] [75].
- Advantages: Reduces data noise and dimensionality, mitigating overfitting; improves model performance and interpretability by focusing on the most relevant variables from each platform; highly effective for sample classification [75].
- Challenges: Risk of losing informative but weak signals during the initial feature selection; fusion is performed after feature extraction, potentially missing inter-platform correlations present in raw data.
- Typical Use Case: Targeted hypothesis testing or sample classification (e.g., disease vs. control, authenticity verification), as demonstrated in distinguishing Green and Ripe Forsythiae Fructus [75].
2.3 High-Level (Late) Data Fusion High-level data fusion (HLDF) operates at the decision or prediction level, where separate models are built for each analytical platform and their outputs are combined.
- Process: Independent classification or regression models (e.g., PLS-DA, Random Forest) are developed for HR-MS and NMR datasets. The final prediction is made by aggregating the results from these models, often using methods like majority voting, weighted averaging, or meta-classifiers [26].
- Advantages: Allows for the use of optimal, platform-specific preprocessing and modeling techniques; highly modular and flexible; can improve robustness by leveraging consensus.
- Challenges: Does not model interactions between MS and NMR variables directly; performance depends heavily on the strength of the individual models.
- Typical Use Case: Integrating results from highly standardized, platform-specific pipelines or when models must be developed independently.
Table 1: Comparative Characteristics of Data Fusion Levels for HR-MS/NMR Integration
| Fusion Level | Stage of Integration | Key Advantages | Primary Challenges | Best-Suited Application |
|---|---|---|---|---|
| Low-Level (Early) | Concatenation of pre-processed raw data [26] | Maximizes use of raw information; can capture inter-platform interactions | High dimensionality; overfitting risk; complex scaling needed [75] | Exploratory, hypothesis-generating studies |
| Mid-Level (Intermediate) | Concatenation of selected features from each platform [26] [75] | Reduces noise & overfitting; improves model performance & interpretability | May discard informative weak signals; post-feature extraction | Sample classification, targeted hypothesis testing [75] |
| High-Level (Late) | Combination of predictions from platform-specific models [26] | Modular; allows platform-optimal modeling; can enhance robustness | No modeling of inter-platform variable interactions | Integrating standardized, independent analyses |
EXPERIMENTAL PROTOCOLS FOR BENCHMARKING FUSION STRATEGIES
To objectively benchmark fusion strategies, a rigorous experimental protocol encompassing sample preparation, data acquisition, processing, and analysis is required.
3.1 Protocol: Designing a Benchmarking Study with a Standard Reference Material
- Objective: To evaluate the accuracy and reproducibility of low-, mid-, and high-level fusion strategies in identifying and quantifying metabolites in a complex biological matrix.
- Sample Preparation:
- Standard Reference Material: Use a commercially available, well-characterized sample like NIST SRM 1950 (Metabolites in Human Plasma) or a pooled sample from your biological model system. This provides a ground truth for benchmarking.
- Experimental Design: Prepare a minimum of 30 aliquots. Include at least 6 replicate preparations from the same stock to assess technical variability. For a more robust benchmark, introduce controlled, biologically relevant variance (e.g., spike-in experiments with known concentrations of specific metabolites) [5].
- Sample Prep for HR-MS: Follow standard protocols for protein precipitation (e.g., using cold methanol/acetonitrile). Use a pooled Quality Control (QC) sample, created from an aliquot of all samples, which is injected repeatedly throughout the LC-MS sequence to monitor instrument stability [73].
- Sample Prep for NMR: For biofluids, mix an aliquot (e.g., 300 µL) with a standardized buffer (e.g., phosphate buffer in D₂O, pH 7.4) containing a chemical shift reference (e.g., 0.5 mM DSS or TSP). For tissues, use a standardized solvent extraction method [5].
- Data Acquisition:
- HR-MS Parameters: Acquire data in both positive and negative ionization modes using a high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap). Use data-dependent acquisition (DDA) or data-independent acquisition (DIA) to collect MS/MS spectra for annotation. The QC sample should be analyzed every 5-10 injections [73].
- NMR Parameters: Acquire 1D ¹H NMR spectra (e.g., NOESY-presat or CPMG pulse sequences for biofluids) at a consistent temperature (e.g., 298 K) on a spectrometer with a field strength of ≥500 MHz. Standardize the number of scans, acquisition time, and relaxation delay across all samples [5] [74].
- Data Processing & Fusion Analysis:
- HR-MS Processing: Process raw files using benchmarked software (e.g., MassCube [73], MS-DIAL, XCMS) for peak picking, alignment, and annotation. Export a feature intensity table.
- NMR Processing: Process FIDs (Fourier transform, phase correction, baseline correction) using standardized parameters. Reference spectra (e.g., to DSS at 0.0 ppm). Perform spectral binning (e.g., 0.04 ppm buckets) and integrate regions for quantified metabolites. Export a bucket table or a quantified metabolite concentration table [5].
- Fusion Implementation:
- Low-Level: Normalize and scale (e.g., Pareto scaling) MS and NMR data matrices separately. Concatenate them column-wise. Analyze with multivariate tools (e.g., PCA, PLS-DA).
- Mid-Level: Perform feature selection on each dataset independently (e.g., using PLS-DA VIP scores > 1.5). Concatenate the selected features. Build a final classification/regression model.
- High-Level: Build a separate classifier (e.g., PLS-DA) for the MS data and the NMR data. Use a simple majority vote or a supervised meta-model (e.g., logistic regression) to combine the class predictions.
- Benchmarking Metrics:
- Accuracy: For spike-in compounds, calculate the recovery rate and correlation between measured and expected concentrations. Assess correct classification rates for known sample groups.
- Reproducibility: Calculate the coefficient of variation (CV%) for technical replicates across the entire workflow for key metabolites. Monitor QC sample variation in both MS and NMR data [5] [73].
- Model Performance: Use repeated cross-validation to report robust metrics: R²X/R²Y (explained variance), Q² (predictive ability), and AUC (Area Under the ROC Curve) for classification models [75].
QUANTITATIVE PERFORMANCE ANALYSIS
Benchmarking studies reveal clear performance trade-offs between fusion strategies. Software tool performance is also a critical variable.
4.1 Performance of Fusion Strategies A study on Forsythiae Fructus demonstrated that a mid-level fusion model (OPLS-DA) integrating UPLC-MS and HS-GC-MS data achieved superior predictive metrics (R²Y = 0.986, Q² = 0.974) compared to single-platform models [75]. It also streamlined the identification of differential metabolites from 61 to 30, reducing noise. This highlights mid-level fusion's strength in enhancing classification and focus. However, low-level fusion might be more appropriate for discovery-based studies where unknown platform interactions are sought.
4.2 Benchmarking Software Tools: The MassCube Example The performance of the data processing software itself is a major factor in accuracy. A 2025 benchmark of the open-source tool MassCube demonstrated significant advantages [73]:
- Peak Detection Accuracy: Achieved 96.4% accuracy on a synthetic dataset designed to test challenging peak shapes and noise, outperforming other common tools.
- Processing Speed: Processed 105 GB of high-resolution MS data in 64 minutes on a laptop, which was 8-24 times faster than other software [73].
- Comprehensive Coverage: Achieved 100% signal coverage by clustering all MS1 signals, minimizing information loss during peak picking [73].
Table 2: Benchmarking Key Metrics for Data Fusion Strategies and Tools
| Benchmark Dimension | Metric | Typical Outcome/Performance | Implication for Accuracy/Reproducibility |
|---|---|---|---|
| Mid-Level Fusion Model [75] | Predictive ability (Q²) | Q² = 0.974 (fusion) vs. ≤0.930 (single-platform) | Higher predictive robustness and model reliability. |
| Mid-Level Fusion Model [75] | Number of Differential Metabolites | 30 (fusion) vs. 61 (single-platform) | Reduces false positives and focuses on high-confidence biomarkers. |
| MS Processing Tool (MassCube) [73] | Peak Detection Accuracy | 96.4% on synthetic benchmark data | Minimizes mis-identification and missing peaks, enhancing data quality. |
| MS Processing Tool (MassCube) [73] | Data Processing Speed | 8-24x faster than other tools | Enables rapid re-analysis and validation, improving workflow reproducibility. |
| QC-Based Reproducibility [5] | Coefficient of Variation (CV%) | NMR CV% ≤ 5% for technical replicates | Ensures quantitative consistency across sample preparation and analysis. |
VISUALIZATION OF WORKFLOWS AND LOGICAL RELATIONSHIPS
Three-Level Data Fusion Framework for HR-MS/NMR
Integrated HR-MS/NMR Metabolomics Benchmarking Workflow
A successful, reproducible fusion study requires standardized materials and tools. This toolkit lists key items for implementing the protocols described.
Table 3: Essential Research Reagent Solutions and Resources
| Category | Item/Resource | Function & Purpose | Example/Specification |
|---|---|---|---|
| Reference Standards | Certified Reference Material (CRM) | Provides a ground truth for benchmarking method accuracy and recovery rates. | NIST SRM 1950 (Metabolites in Human Plasma). |
| Internal Standards | Isotope-Labeled Internal Standards (for MS) | Corrects for matrix effects and ionization efficiency variance during MS sample prep. | ¹³C, ¹⁵N-labeled amino acid mix for cell culture; deuterated standards for biofluids. |
| Internal Standards | Quantitative NMR (qNMR) Standard | Provides chemical shift reference and enables absolute quantification in NMR. | DSS-d₆ (4,4-dimethyl-4-silapentane-1-sulfonic acid) or TSP-d₄ (sodium trimethylsilylpropanesulfonate). |
| QC Materials | Pooled Quality Control (QC) Sample | Monitors instrument stability and data reproducibility throughout the analytical sequence. | A homogeneous pool comprising an aliquot of every experimental sample [73]. |
| Software Tools | MS Data Processing Software | Converts raw instrument data into aligned, annotated feature tables. Critical for accuracy. | MassCube [73] (open-source), MS-DIAL, or vendor-specific software. |
| Software Tools | NMR Processing Software | Processes FID data, performs phasing, binning, and quantification. | MestReNova, TopSpin, Chenomx NMR Suite. |
| Software Tools | Statistical & Fusion Analysis Platform | Performs data normalization, scaling, fusion, and multivariate statistical modeling. | R/Python (with mixOmics, ropls packages), SIMCA, Matlab. |
| Databases | Metabolite Databases | Essential for annotating MS/MS spectra and assigning NMR signals. | HMDB, MassBank, GNPS for MS; BMRB, HMDB for NMR. |
Impact on Biomarker Discovery, Clinical Translation, and Personalized Metabolic Profiling
The fields of biomarker discovery and personalized medicine are being fundamentally reshaped by advances in metabolomics, which provides a dynamic, functional readout of an organism's physiological state. The metabolome, comprising all small-molecule metabolites, sits downstream of genomic, transcriptomic, and proteomic variations, making it exquisitely sensitive to disease processes, drug interventions, and environmental influences [76]. However, the immense chemical diversity and wide concentration range of metabolites in biological samples pose a significant analytical challenge. No single technology can comprehensively capture the entire metabolome with high confidence.
This limitation has driven the strategic integration of complementary analytical platforms. Nuclear Magnetic Resonance (NMR) spectroscopy and High-Resolution Mass Spectrometry (HR-MS) have emerged as the cornerstones of modern metabolomics [69]. NMR offers exceptional quantitative accuracy, high reproducibility, and non-destructive analysis, providing robust structural information with minimal sample preparation [21]. Conversely, HR-MS delivers superior sensitivity, enabling the detection of thousands of metabolite features at very low concentrations [46]. The synergistic combination of these techniques leverages the quantitative rigor and structural elucidation power of NMR with the expansive metabolite coverage of HR-MS. This integrated approach is transforming research paradigms, enhancing the accuracy of metabolite identification and quantification, and accelerating the translation of metabolic biomarkers from discovery to clinical application and personalized health profiling [77] [69].
Advancing Biomarker Discovery Through Enhanced Metabolite Identification and Quantification
The integrated use of HR-MS and NMR significantly augments the biomarker discovery pipeline by improving both the coverage and confidence of metabolite annotation. This synergy addresses critical weaknesses inherent to each technique when used in isolation.
Complementary Strengths and a Synergistic Workflow
NMR spectroscopy excels at providing unambiguous structural identification for medium-to-high abundance metabolites in complex mixtures. It is inherently quantitative, as the signal intensity is directly proportional to the number of resonant nuclei, allowing for concentration determination using a single internal or external standard [69]. However, its relatively low sensitivity means many clinically relevant, low-abundance metabolites remain undetected. HR-MS, particularly when coupled with liquid chromatography (LC), offers parts-per-billion sensitivity and can detect a vastly larger number of metabolic features. Yet, metabolite identification by MS alone can be ambiguous, relying on matching measured mass-to-charge ratios and fragmentation patterns to databases, which can lead to false positives [46]. Quantification by LC-MS can also be less reproducible due to ion suppression effects and requires compound-specific calibration curves.
The synergistic workflow, exemplified by the SYNHMET (SYnergic use of NMR and HRMS for METabolomics) strategy, merges these datasets to overcome individual limitations [46]. The process begins with an initial quantitative profile from NMR. Signals from co-eluting or low-abundance metabolites are often poorly defined. HR-MS data is then used to guide the deconvolution of the NMR spectrum: MS peaks correlating with the tentative NMR concentrations help pinpoint the correct chromatographic feature for a given metabolite. This MS-assisted refinement leads to a more accurate NMR-based concentration, which in turn validates the MS identification. This iterative cross-validation results in a final dataset where metabolites are identified with high confidence and quantified accurately, without the need for synthetic standards for every compound [46] [77].
Quantitative Outcomes from Integrated Studies
Integrated studies consistently yield more comprehensive and reliable quantitative datasets. The SYNHMET approach applied to human urine, a notoriously complex and variable biofluid, quantified 165 metabolites across healthy controls, chronic cystitis patients, and bladder cancer patients with minimal missing values [46]. This number surpasses what is typically achievable by NMR alone for urine. The study demonstrated that cross-validation between techniques is particularly crucial for metabolites with overlapping NMR signals or low concentrations, dramatically improving quantification accuracy [77].
Automated tools are further streamlining discovery. The ROIAL-NMR (Region Of Interest Assessment of Liquids by NMR) Python program, for instance, automates the identification of potential metabolites from pre-defined regions of interest in NMR spectra by querying databases like the Human Metabolome Database (HMDB) [43]. In a study of serum from lung cancer patients, ROIAL-NMR rapidly identified 88 potential metabolites, with 66 differentiating cancer from controls and 80 distinguishing cancer patients with and without Alzheimer's disease-related dementia [43]. This demonstrates how computational tools, when fed high-quality data from integrated platforms, can rapidly generate robust candidate biomarker lists.
Table 1: Key Quantitative Outcomes from Integrated HR-MS/NMR Metabolomics Studies
| Study Focus | Technique Used | Key Quantitative Outcome | Impact on Biomarker Discovery |
|---|---|---|---|
| Bladder Cancer & Cystitis [46] | SYNHMET (NMR + UHPLC-HRMS) | Quantified 165 metabolites in urine across 46 subjects. | Generated a comprehensive, quantitative profile for a difficult biofluid, identifying disease-specific metabolic disturbances. |
| Lung Cancer & ADRD [43] | NMR + ROIAL-NMR Algorithm | Identified 88 metabolites; 66 differentiated LC from control. | Enabled rapid, automated screening of NMR data against HMDB to prioritize biomarker candidates for complex comorbidities. |
| Multiple Sclerosis Subtyping [78] | Targeted ¹H-NMR | Identified Leucine as a significant differentiator (AUC=0.74, p=0.025 FDR-adjusted). | Provided a proof-of-concept for a serum-based, non-invasive biomarker to stratify early-stage MS patients. |
Diagram 1: Integrated HR-MS/NMR Workflow for Biomarker Discovery - This diagram shows the parallel analysis of a sample by NMR and HR-MS, followed by data integration to produce a refined, high-confidence quantitative profile for biomarker prioritization.
Detailed Experimental Protocols
Protocol: The SYNHMET Workflow for Quantitative Urinary Metabolite Profiling
This protocol details the steps for synergistic NMR and HR-MS analysis to achieve absolute quantification of metabolites in human urine [46].
I. Sample Preparation
- Collection & Storage: Collect mid-stream urine samples. Immediately aliquot and freeze at -80°C.
- NMR Sample Prep: Thaw an aliquot on ice. Centrifuge at 14,000 x g for 10 min at 4°C to remove debris. Mix 540 µL of supernatant with 60 µL of NMR buffer (1.5 M potassium phosphate, pH 7.4, in D₂O containing 0.1% w/w sodium trimethylsilylpropanesulfonate [DSS] as a chemical shift and quantitation reference). Transfer to a 5 mm NMR tube.
- HR-MS Sample Prep: Thaw a separate aliquot. Centrifuge as above. Dilute supernatant 1:10 with LC-MS grade water. Filter through a 0.2 µm centrifugal filter. Transfer to an LC vial.
II. Instrumental Analysis
- ¹H-NMR Spectroscopy:
- Instrument: 600 MHz spectrometer equipped with a cryoprobe.
- Experiment: 1D NOESY-presat pulse sequence for water suppression.
- Parameters: Spectral width 20 ppm, acquisition time 4 s, relaxation delay 4 s, 128 transients, temperature 298 K.
- Processing: Fourier transformation with 0.3 Hz line broadening. Reference the DSS methyl signal to 0 ppm.
- UHPLC-HRMS Analysis:
- Chromatography: Utilize two complementary methods: Reverse-Phase (RP) and Hydrophilic Interaction Liquid Chromatography (HILIC). Use a Vanquish Neo UHPLC system or equivalent [79].
- MS Detection: Orbitrap Q-Exactive or Orbitrap Astral mass spectrometer with a HESI ion source [46] [79].
- Acquisition: Run in both positive and negative ionization modes. Use a full scan range of m/z 70-1050 at a resolution of 70,000. Include data-dependent MS/MS scans for top ions.
III. Data Processing & SYNHMET Integration
- Initial NMR Quantification: Deconvolute the 1D NMR spectrum using commercial (e.g., Chenomx) or custom software. Use a starting library of ~180 known urinary metabolites. Obtain a first-approximation concentration list.
- HR-MS Feature Alignment: Process raw MS files using software like MZmine or XCMS. Align peaks across samples, annotate with putative formulae using exact mass (<5 ppm error).
- Correlative Integration:
- For each metabolite from the NMR list, extract all MS chromatographic peaks whose accurate mass matches its monoisotopic mass (within 5 ppm).
- Plot the first-approximation NMR concentration against the intensity of each candidate MS peak across all samples.
- The MS peak showing the highest correlation coefficient is assigned to that metabolite, resolving MS identification ambiguity.
- Use this confident MS assignment to refine the NMR spectral deconvolution, particularly for regions of signal overlap.
- Final Quantification: The refined NMR deconvolution yields the final absolute concentration (in µM or mM) for each confirmed metabolite, traceable to the DSS reference standard.
Protocol: Automated Metabolite Identification with ROIAL-NMR
This protocol uses the ROIAL-NMR Python program to identify metabolites from regions of interest in 1H-NMR spectra of serum [43].
I. Prerequisite: NMR Data Acquisition and ROI Definition
- Acquire 1H-NMR spectra of serum samples (e.g., from healthy and disease cohorts) under standardized conditions (pH, temperature, field strength).
- Process spectra (Fourier transform, phasing, baseline correction, referencing).
- Perform statistical analysis (univariate or multivariate) to identify spectral regions (ROIs) where signal intensities differ significantly between cohort groups (e.g., p < 0.05 after FDR correction). Define each ROI by its chemical shift range (e.g., 1.31-1.34 ppm).
II. ROIAL-NMR Execution
- Environment Setup: Ensure Python 3 is installed. Clone the ROIAL-NMR repository from GitHub (
https://github.com/Leo-Cheng-Lab/ROIAL-NMR.git). - Prepare Input File: Create a CSV file listing all ROIs. Required columns: ROIID, ChemicalShiftMin (ppm), ChemicalShift_Max (ppm), and optionally, statistical trend (Increased/Decreased in disease) and p-value.
- Run Identification:
- Interpret Output: The program outputs a table listing all metabolites with assigned peaks falling within the input ROIs. It calculates a "match ratio" (number of a metabolite's spectral regions found / total number of its regions). High-match-ratio metabolites are high-confidence identifications linking spectral changes to biology.
Translating Discoveries to Clinical Applications
The transition from biomarker discovery to clinical utility requires robust validation and demonstration of clear clinical value. The quantitative accuracy and high confidence in metabolite identity afforded by integrated HR-MS/NMR approaches provide a stronger foundation for this translation.
From Discovery to Validation: A Structured Pipeline
The path to clinical adoption involves several stages beyond initial discovery. The integrated profile serves as the discovery engine, identifying a panel of candidate biomarkers. These must then be validated in larger, independent cohorts. Targeted assays, often using more rapid and cost-effective techniques like tandem mass spectrometry (MS/MS) or benchtop NMR, are developed for these specific metabolites [69]. The ultimate goal is to deploy these assays in clinical settings to aid in diagnosis, prognosis, or treatment selection.
A study on multiple sclerosis (MS) exemplifies this principle. While cerebrospinal fluid analysis is diagnostic, it is invasive. A targeted ¹H-NMR serum metabolomics study sought a non-invasive alternative [78]. By comparing patients with different oligoclonal band patterns, researchers identified leucine as a serum metabolite significantly elevated in patients with definite intrathecal antibody synthesis (OCB Type 2). After age-adjustment and multiple testing correction, leucine showed an AUC of 0.74, indicating good diagnostic potential, and correlated with the established IgG index [78]. This study demonstrates how a targeted, quantitative NMR assay can identify a single, clinically actionable biomarker derived from broader discovery work.
Case Study: Stratification in Bladder Cancer
The SYNHMET study on bladder cancer (BC), chronic cystitis (CC), and healthy controls (CTRL) provides a template for clinical stratification [46]. By quantifying 165 urinary metabolites, the method created detailed, individualized metabolic profiles. Comparing a patient's profile to established reference ranges for healthy and disease states allows for precise monitoring. This approach can potentially distinguish between inflammatory (CC) and malignant (BC) conditions based on metabolic signatures, which is a common clinical challenge. The accurate, standard-free quantification inherent to the SYNHMET method makes such profiles reliable and transferable, a key requirement for clinical adoption.
Table 2: Promising Biomarkers from Integrated & Targeted Metabolomics Studies
| Disease Area | Key Metabolite Biomarker(s) | Biological/Clinical Implication | Stage of Development |
|---|---|---|---|
| Multiple Sclerosis [78] | Leucine (elevated in OCB Type 2) | Marker of intrathecal immune activity; correlates with IgG index. Non-invasive serum alternative. | Pilot Validation (AUC=0.74, requires larger cohort validation). |
| Bladder Cancer [46] | Panel of 165 quantified metabolites | Enables creation of personalized metabolic profiles for distinguishing cancer from inflammation and monitoring progression. | Discovery & Proof-of-Concept for stratification. |
| Lung Cancer & ADRD [43] | Panels of 66 and 80 metabolites | Differentiates lung cancer from controls and lung cancer with/without dementia, suggesting metabolic links between comorbidities. | Discovery (algorithm-driven candidate identification). |
Diagram 2: Biomarker Translation Pipeline from Discovery to Clinic - This diagram outlines the critical pathway from initial biomarker discovery using integrated omics to the development and deployment of a clinical-grade diagnostic or monitoring assay.
Enabling Personalized Metabolic Profiling and Pharmacometabolomics
The ultimate promise of integrated metabolomics is the move from population-level biomarkers to individualized metabolic phenotypes—or "metabotypes"—that guide personalized healthcare.
Constructing the Personalized Metabolic Profile
A personalized metabolic profile is a quantitative snapshot of an individual's metabolite concentrations at a given time. As shown in the SYNHMET approach, integrating NMR and HR-MS builds a highly accurate and comprehensive profile [77]. This profile can be compared to dynamic reference intervals (healthy ranges, disease-state ranges) to identify specific metabolic deviations. Over time, longitudinal profiling of an individual can track the progression of a chronic disease, the resolution of an acute condition, or the metabolic response to a therapeutic intervention, offering a powerful tool for personalized monitoring.
Pharmacometabolomics: Informing Drug Development and Therapy
Pharmacometabolomics applies metabolomics to predict or assess an individual's response to a drug [76]. By analyzing the pre-treatment metabotype, researchers can stratify patients into likely responders and non-responders. Monitoring post-treatment metabolic changes reveals drug mechanism of action, efficacy, and off-target effects. This is crucial in drug development, where over 60% of Phase III failures are due to lack of efficacy, and adverse drug reactions (ADRs) are a major concern [76]. Integrating metabolomics with other omics data, as seen in large-scale proteomics studies with GLP-1 agonists, provides a systems-level view of drug effects and helps establish causality [80]. The quantitative robustness of NMR-integrated data is particularly valuable for the longitudinal studies required in clinical trials, ensuring that observed metabolic changes are reliable and actionable.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Integrated HR-MS/NMR Metabolomics
| Category | Item / Reagent | Function & Specification | Example / Note |
|---|---|---|---|
| NMR Analysis | Deuterated Buffer & Reference | Provides a field-frequency lock for NMR and a chemical shift/quantitation reference. Contains a known concentration of a reference compound like DSS or TSP. | 1.5 M Potassium Phosphate buffer in D₂O, pD 7.4, 0.1% DSS [46]. |
| NMR Analysis | Standardized NMR Tubes | High-quality, matched tubes ensure consistent spectral line shape and quality, critical for quantification. | 5 mm outer diameter, 7-inch length, matched for high-resolution studies. |
| HR-MS Analysis | Chromatography Columns (Dual-Mode) | Provides orthogonal separation to maximize metabolite coverage. RP for hydrophobic metabolites; HILIC for polar metabolites. | e.g., C18 RP column and Amide HILIC column [46]. |
| HR-MS Analysis | Mobile Phase Additives | Enhance ionization and chromatographic separation. Must be MS-grade to avoid background interference. | e.g., Formic acid (positive mode), ammonium acetate or ammonium hydroxide (negative mode). |
| Sample Prep | Protein Precipitation Solvents | Remove proteins from biofluids like serum/plasma prior to analysis. | Cold methanol, acetonitrile, or combination solvents (e.g., 2:1:1 methanol:acetonitrile:water) [69]. |
| Sample Prep | Internal Standards (for MS) | Correct for variability in sample prep and ionization efficiency. Should cover different chemical classes. | Stable isotope-labeled metabolites (e.g., ¹³C, ¹⁵N-labeled amino acids, fatty acids). |
| Data Processing | Spectral Databases | Essential for metabolite identification by matching spectral patterns. | HMDB (NMR & MS) [43], Chenomx NMR Suite (NMR), GNPS (MS/MS) [81]. |
| Data Processing | Specialized Software | For spectral deconvolution, statistical correlation, and data integration. | ROIAL-NMR [43] (automated NMR ID), MzMine/XCMS (MS data processing), custom scripts for SYNHMET-type integration. |
The integration of HR-MS and NMR spectroscopy represents a paradigm shift in metabolomics, directly addressing the core challenges of biomarker discovery, validation, and translation. By marrying the quantitative rigor and structural insight of NMR with the sensitive, expansive coverage of HR-MS, this synergy generates data of unprecedented confidence and comprehensiveness. As demonstrated in studies from bladder cancer to multiple sclerosis, this approach is already yielding robust biomarker candidates and frameworks for personalized metabolic monitoring.
The future trajectory of the field points toward deeper integration and technological innovation. The rise of benchtop NMR spectrometers promises to make quantitative metabolic profiling more accessible for clinical validation and point-of-care testing [69]. Advances in hyperpolarization techniques like DNP could dramatically enhance NMR sensitivity, narrowing the gap with MS [69]. Furthermore, the integration of metabolomics with other omics layers—proteomics, genomics, and transcriptomics—within a systems biology framework will be essential to move from correlation to causation, unravel complex disease mechanisms, and fully realize the potential of personalized and precision medicine [76] [80]. The continued development of automated tools, standardized protocols, and shared databases will be critical in translating the powerful research presented here into routine clinical practice.
Conclusion
The integration of HR-MS and NMR spectroscopy represents a transformative paradigm in metabolomics, leveraging the complementary strengths of both techniques for superior metabolite identification and quantification. Key takeaways include the critical role of data fusion strategies, optimized sample preparation protocols, and robust validation frameworks in enhancing analytical coverage and reliability. Future directions should focus on developing standardized, automated workflows, advancing computational tools for multi-omics data integration, and expanding applications in precision medicine, therapeutic monitoring, and large-scale cohort studies to drive innovations in biomedical and clinical research.
References
- 1. Integrating NMR and MS for Improved Metabolomic Analysis [https://www.mdpi.com/1420-3049/30/12/2624]
- 2. Combining Mass Spectrometry and NMR Improves Metabolite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6668615/]
- 3. HR-MAS NMR Applications in Plant Metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7916392/]
- 4. Personalized Metabolic Profile by Synergic Use of NMR and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8304707/]
- 5. Securing the Future of NMR Metabolomics Reproducibility [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489887/]
- 6. High-resolution magic angle spinning NMR spectroscopy [https://www.sciencedirect.com/science/article/abs/pii/S0079656508000708]
- 7. Integration of LC-HRMS and 1H NMR metabolomics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11279979/]
- 8. A multilevel LC-HRMS and NMR correlation workflow ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914024010208]
- 9. 1H NMR-MS-based heterocovariance as a drug discovery ... [https://www.nature.com/articles/s41598-019-47434-8]
- 10. Combining mass spectrometry and nuclear magnetic ... [https://www.sciencedirect.com/science/article/abs/pii/S0079656516300656]
- 11. Recent Advances in Mass Spectrometry-Based Structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9572214/]
- 12. Quantitative NMR-Based Biomedical Metabolomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC7662776/]
- 13. High-field and benchtop NMR spectroscopy for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0379073821000384]
- 14. NMR and Metabolomics—A Roadmap for the Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9394421/]
- 15. Metabolomics Beyond Spectroscopic Databases: A Combined MS/NMR Strategy for the Rapid Identification of New Metabolites in Complex Mixtures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5035699/]
- 16. Advantages of HRMS [https://resolvemass.ca/advantages-of-hrms/]
- 17. NMR spectroscopy in drug discovery and development [https://pmc.ncbi.nlm.nih.gov/articles/PMC8963582/]
- 18. High Resolution Mass Spectrometry (HRMS) - an overview [https://www.sciencedirect.com/topics/chemistry/high-resolution-mass-spectrometry-hrms]
- 19. Integration of LC-HRMS and 1 H NMR metabolomics data ... [https://pubmed.ncbi.nlm.nih.gov/39071933/]
- 20. NMR Metabolomics Protocols for Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7025395/]
- 21. Nuclear Magnetic Resonance Spectroscopy in Clinical ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.698337/full]
- 22. Integration of LC-HRMS and 1H NMR metabolomics data ... [https://www.sciencedirect.com/science/article/pii/S2590157524004954]
- 23. Mass-spectrometry based metabolomics: an overview of ... [https://proteomesci.biomedcentral.com/articles/10.1186/s12953-025-00241-8]
- 24. Metabolomics in drug research and development [https://www.sciencedirect.com/science/article/pii/S2211383523001661]
- 25. Integrating NMR and multi-LC-MS-based untargeted ... [https://www.sciencedirect.com/science/article/pii/S0003267025003733]
- 26. Integrating NMR and MS for Improved Metabolomic Analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12196070/]
- 27. Recommendations for sample selection, collection and ... [https://link.springer.com/article/10.1007/s11306-025-02259-7]
- 28. Sample preparation optimization for metabolomics and ... [https://pubmed.ncbi.nlm.nih.gov/41075457/]
- 29. Identification of metabolites from complex mixtures by 3D ... [https://www.nature.com/articles/s41598-023-43056-3]
- 30. High-Resolution NMR for Complex Molecule Analysis [https://www.creative-biostructure.com/resource-high-resolution-nmr-complex-molecule-analysis.htm?srsltid=AfmBOoqi8nVcrUT2LzTmSt4tAU04GPjWURJ9sPBzjHy3lzaJbdG8N3-K]
- 31. A Protocol for Untargeted Metabolomic Analysis: From Sample ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9284939/]
- 32. Comparative analysis of features extraction protocols for ... [https://www.sciencedirect.com/science/article/pii/S0026265X24019751]
- 33. Metabolomics using high-resolution magic angle spinning ... [https://www.sciencedirect.com/science/article/pii/S1056871925008020]
- 34. Optimizing NMR Processing: Techniques and Best Practices [https://www.jeolusa.com/NEWS-EVENTS/Blog/optimizing-nmr-processing-techniques-and-best-practices]
- 35. 2.3 NMR data handling and (pre-)processing [https://rformassspectrometry.github.io/metaRbolomics-book/0203-R_packages_for_metabolomics-NMR_data_handling_and_pre_processing.html]
- 36. Metabolite Profiling and Identification (MetID) | Cyprotex [https://www.evotec.com/solutions/drug-discovery-preclinical-development/cyprotex-adme-tox-solutions/adme-pk/drug-metabolism/metabolite-profiling-and-identification]
- 37. between high resolution (1) H- Data and fusion ... NMR mass [https://pubmed.ncbi.nlm.nih.gov/27086012/]
- 38. The Application of NMR-Based Metabolomics in the Field ... [https://www.mdpi.com/2673-8392/5/4/174]
- 39. 1 H-NMR-based metabolomics study of Rifafour in ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1661938/full]
- 40. Clinical metabolomics reveals potential diagnostic ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1554936/full]
- 41. Optimization of Sample Preparation for Metabolomics ... [https://www.semanticscholar.org/paper/Optimization-of-Sample-Preparation-for-Metabolomics-Martias-Baroukh/6e9dc3979e7be1fef23bc698c77ddf6c9e9bdcac]
- 42. From Biomarker Discovery to Clinical Applications of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12112835/]
- 43. Automatic Identification of Potential Cellular Metabolites for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445015/]
- 44. Can NMR solve some significant challenges in metabolomics? [https://pmc.ncbi.nlm.nih.gov/articles/PMC4646661/]
- 45. Recent Developments Toward Integrated Metabolomics ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.720955/full]
- 46. Personalized Metabolic Profile by Synergic Use of NMR ... [https://www.mdpi.com/1420-3049/26/14/4167]
- 47. Challenges in Identifying the Dark Molecules of Life - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6716371/]
- 48. NMR Sample Preparation - NMR and Chemistry MS Facilities [https://nmr.chem.cornell.edu/new-users/training/nmr-sample-preparation/]
- 49. Can NMR solve some significant challenges in ... [https://www.sciencedirect.com/science/article/abs/pii/S1090780715001731]
- 50. A guide to the identification of metabolites in NMR-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4821453/]
- 51. NMR-based plant metabolomics protocols: a step-by- ... [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2024.1414506/full]
- 52. Strategies for optimizing the phase correction algorithms in ... [https://biomedical-engineering-online.biomedcentral.com/articles/10.1186/1475-925X-14-S2-S5]
- 53. Enhancing the Sensitivity of Multidimensional NMR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5738291/]
- 54. Rapid protein assignments and structures from raw NMR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9579175/]
- 55. NMR/MS Translator for the Enhanced Simultaneous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5035700/]
- 56. Towards Unbiased Evaluation of Ionization Performance in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9144264/]
- 57. Ultra-Performance Liquid Chromatography–High-Resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6184476/]
- 58. An LC–HRMS Method for Separation and Identification of ... [https://www.chromatographyonline.com/view/an-lc-hrms-method-for-separation-and-identification-of-hemoglobin-variant-subunits]
- 59. LC-HRMS Metabolomics for Untargeted Diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6027396/]
- 60. The future of NMR-based metabolomics [https://www.sciencedirect.com/science/article/pii/S0958166916301768]
- 61. Omics data integration in computational biology viewed ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2023.1191961/full]
- 62. A Guide to Multi-omics Integration Strategies [https://frontlinegenomics.com/a-guide-to-multi-omics-integration-strategies/]
- 63. Quantitative NMR Methods in Metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9992124/]
- 64. Absolute quantification of matrix metabolites reveals the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5030821/]
- 65. Absolute Quantification of Matrix Metabolites Reveals the ... [https://www.sciencedirect.com/science/article/pii/S0092867416309904]
- 66. Machine Learning Applications for Mass Spectrometry-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7345470/]
- 67. Integration of metabolomics and machine learning for ... [https://www.sciencedirect.com/science/article/pii/S0261561425001396]
- 68. NMR as a “Gold Standard” Method in Drug Design and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7594251/]
- 69. NMR-based metabolomics: Where are we now and where ... [https://www.sciencedirect.com/science/article/abs/pii/S0079656525000081]
- 70. Metabolite Identification Data in Drug Discovery, Part 1 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12587390/]
- 71. Metabolon: The Gold Standard for Actionable Metabolomics [https://www.metabolon.com/why-metabolon/identification/]
- 72. Metabolite Identification Data in Drug Discovery [https://chemrxiv.org/engage/chemrxiv/article-details/682204bfe561f77ed450ef24]
- 73. MassCube improves accuracy for metabolomics data ... [https://www.nature.com/articles/s41467-025-60640-5]
- 74. Validating NMR-based non-targeted methods and fostering ... [https://www.sciencedirect.com/science/article/abs/pii/S0079656525000068]
- 75. Mid-Level Data Fusion Techniques of LC-MS and HS-GC ... [https://www.mdpi.com/1420-3049/30/7/1404]
- 76. Metabolomics and Pharmacometabolomics - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12654014/]
- 77. Personalized Metabolic Profile by Synergic Use of NMR ... [https://pubmed.ncbi.nlm.nih.gov/34299442/]
- 78. A Targeted 1H-NMR Comparison of OCB Type 1 and ... [https://pubmed.ncbi.nlm.nih.gov/40954342/]
- 79. US HUPO 2025 [https://www.thermofisher.com/us/en/home/about-us/events/industrial/us-hupo.html]
- 80. 2025 Proteomics Breakthroughs [https://www.genengnews.com/topics/drug-discovery/2025-proteomics-breakthroughs/]
- 81. HPLC-ESI-HRMS/MS-Based Metabolite Profiling and ... [https://www.mdpi.com/2223-7747/14/15/2395]