Structure-Activity Relationship (SAR) Directed Optimization of Natural Leads: Strategies for Modern Drug Discovery
This article provides a comprehensive guide for researchers and drug development professionals on leveraging Structure-Activity Relationship (SAR) studies to optimize natural product leads into viable drug candidates.
Structure-Activity Relationship (SAR) Directed Optimization of Natural Leads: Strategies for Modern Drug Discovery
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on leveraging Structure-Activity Relationship (SAR) studies to optimize natural product leads into viable drug candidates. It covers the foundational principles of SAR in natural products chemistry, explores advanced methodological approaches including build-up libraries and in situ screening, and addresses key challenges in multi-parameter optimization. The content also examines validation strategies through case studies on successful anticancer and antibacterial drug development, highlighting the integration of computational tools, AI/ML, and contemporary data analysis platforms to streamline the optimization workflow and bridge the gap from natural leads to clinical candidates.
The Foundation of SAR: Unlocking Nature's Chemical Blueprint for Drug Discovery
Defining SAR in the Context of Natural Products Chemistry
The structure–activity relationship (SAR) is defined as the relationship between the chemical structure of a molecule and its biological activity [1]. This concept, first presented by Alexander Crum Brown and Thomas Richard Fraser as early as 1868, enables the determination of the chemical groups responsible for evoking a target biological effect in an organism [1]. In the context of natural products chemistry, SAR analysis provides a powerful framework for understanding how the complex chemical scaffolds found in nature interact with biological targets, thereby guiding the optimization of these compounds for therapeutic applications.
Natural products (NPs) play an indispensable role in modern drug discovery, accounting for a significant percentage of FDA-approved drugs. Between 1981 and 2019, natural products and botanical mixtures accounted for 4.6% of new drug approvals, while NP derivatives constituted an additional 18.9% [2]. This utility stems from the evolutionary refinement of NPs to target specific proteins, making them valuable as starting points for drug development against homologous targets in human diseases [2]. However, most natural products require optimization before clinical use due to insufficient activity, selectivity, or unfavorable pharmacokinetic properties, making SAR studies essential for rational design of improved analogs.
Conceptual Framework of SAR Analysis
Fundamental Principles
At its core, SAR analysis involves systematically modifying a compound's structure and measuring the resulting changes in biological activity. This enables medicinal chemists to identify which chemical features are essential for activity (pharmacophore elements), which modifications enhance or diminish potency, and which alterations improve drug-like properties. For natural products, this process is particularly challenging due to their complex molecular architectures, but modern approaches have developed specialized methodologies to address these challenges [2].
SAR is typically evaluated in table format (SAR tables), which organize compounds, their physical properties, and activities, allowing experts to identify patterns through sorting, graphing, and structural feature scanning [3]. This systematic approach facilitates the recognition of which structural characteristics correlate with chemical and biological reactivity, enabling predictions about uncharacterized compounds based on structural similarities to molecules with known activities [3].
From Qualitative SAR to Quantitative SAR (QSAR)
While SAR traditionally provides qualitative assessments of how structural changes affect bioactivity, the field has evolved to include quantitative structure-activity relationships (QSAR), which build mathematical models to correlate chemical structure with biological activity [1]. QSAR models use numerical descriptors of chemical structures to predict activities quantitatively, enabling more precise optimization campaigns [4]. A related term, structure affinity relationship (SAFIR), focuses specifically on binding affinity measurements [1].
Table 1: Comparison of SAR and QSAR Approaches
| Feature | Qualitative SAR | Quantitative SAR (QSAR) |
|---|---|---|
| Foundation | Chemical intuition and pattern recognition | Mathematical models and statistical analysis |
| Data Output | Relative rankings (e.g., high, medium, low activity) | Numerical predictions of activity (e.g., IC50, Ki values) |
| Application | Initial optimization direction | Precise potency optimization |
| Complexity | Accessible to medicinal chemists | Requires specialized computational expertise |
| Visualization | Structural alerts and pharmacophore maps | Coefficient plots and descriptor importance charts |
Experimental Protocols for SAR Elucidation of Natural Products
Diverted Total Synthesis for Analog Production
Purpose: To generate structural analogs of natural products for systematic SAR studies through chemical synthesis.
Background: Total synthesis of natural products represents a powerful approach for accessing complex scaffolds and their analogs. The diverted total synthesis strategy (also referred to as collective total synthesis) involves identifying points on the natural product structure suitable for diversification and designing synthetic routes that allow systematic variation at these positions from common intermediates [2].
Protocol Steps:
- Retrosynthetic Analysis: Deconstruct the target natural product to identify potential branch points for diversification. These should be positions hypothesized to influence biological activity based on preliminary data or structural analogy.
- Common Intermediate Identification: Design synthetic routes that converge on key intermediates capable of being transformed into multiple analogs.
- Library Synthesis: Employ parallel synthesis techniques to generate analog libraries with systematic variations at the targeted positions.
- Purification and Characterization: Purify all analogs to >95% purity and characterize structures using NMR, MS, and HPLC.
- Biological Evaluation: Test all analogs in relevant biological assays to determine potency, selectivity, and other pharmacological properties.
Application Example: The Danishefsky group applied diverted total synthesis to produce migrastatin analogs, resulting in compounds with improved antitumor activity and plasma stability compared to the natural product [2].
Late-Stage Derivatization of Natural Product Isolates
Purpose: To efficiently generate analogs through chemical modification of isolated natural products.
Background: For natural products that can be isolated in sufficient quantities from their natural sources, late-stage derivatization provides a more efficient route to analogs than total synthesis. This approach preserves the complex core structure while enabling modification at functional groups amenable to chemical transformation.
Protocol Steps:
- Natural Product Isolation: Purify the parent natural product to homogeneity from biological sources (microbial fermentation, plant material, or marine organisms).
- Functional Group Analysis: Identify chemically accessible functional groups (hydroxyl, amino, carboxyl, carbonyl groups) that can be selectively modified.
- Derivatization Reactions:
- Acylation: Treat with acid chlorides or anhydrides to create esters and amides
- Alkylation: Use alkyl halides to create ethers or alkylated amines
- Oxidation/Reduction: Selectively modify hydroxyl groups or carbonyls
- Cross-coupling: Employ transition metal-catalyzed reactions for C-C bond formation
- Purification and Characterization: Isulate and characterize all derivatives as described in protocol 3.1.
- SAR Analysis: Correlate structural modifications with changes in biological activity to identify key structural requirements.
Key Consideration: This approach is limited to naturally occurring functional groups and may not access all regions of the molecule, particularly the core scaffold.
Diagram 1: Late-stage derivatization workflow for SAR studies
Biochemical Assays for SAR Profiling
Purpose: To quantitatively measure the effects of structural modifications on biological activity.
Background: Comprehensive SAR studies require testing compounds in multiple assays to evaluate different aspects of biological activity, including potency, efficacy, selectivity, and mechanisms of action.
Protocol for Multi-Parameter SAR Profiling:
Primary Target Assay:
- Prepare dilution series of each analog (typically ½-log or log dilutions)
- Incubate with molecular target (enzyme, receptor) under optimized conditions
- Measure activity (inhibition, activation, binding)
- Calculate IC50, EC50, or Ki values from dose-response curves
Selectivity Profiling:
- Test compounds against related targets and common antitargets
- Include kinases, GPCRs, ion channels, or cytochrome P450s as relevant
- Calculate selectivity ratios (primary target IC50/off-target IC50)
Cellular Activity Assay:
- Evaluate activity in cell-based systems measuring functional responses
- Determine cellular potency (IC50, EC50) and efficacy (% of control)
Early ADMET Assessment:
- Measure metabolic stability in liver microsomes
- Assess membrane permeability (Caco-2, PAMPA)
- Evaluate cytotoxicity against relevant cell lines
Data Integration:
- Compile all data into SAR tables
- Identify structural features associated with optimal profiles
Application Note: Modern SAR analysis often uses automated platforms like the PULSAR application, which enables systematic, data-driven analysis that integrates multiple SAR parameters simultaneously, significantly reducing analysis time from days to hours [5].
Computational Approaches for SAR Analysis
Molecular Docking for Structure-Based SAR
Purpose: To predict binding modes of natural product analogs and rationalize observed SAR.
Background: Molecular docking simulations predict how small molecules interact with protein targets at the atomic level, providing structural insights to explain SAR observations.
Protocol Steps:
Protein Preparation:
- Obtain 3D structure from Protein Data Bank (resolution <2.5 Å recommended)
- Add hydrogen atoms, assign protonation states
- Remove water molecules except those involved in key interactions
- Define binding site around co-crystallized ligand or known active site
Ligand Preparation:
- Generate 3D structures of natural product analogs
- Assign proper bond orders, charges, and tautomeric states
- Perform conformational sampling for flexible molecules
Docking Simulation:
- Select appropriate docking algorithm (e.g., genetic algorithm, Monte Carlo)
- Define search parameters and scoring function
- Generate multiple binding poses for each compound
Pose Analysis and Validation:
- Cluster similar binding poses
- Analyze protein-ligand interactions (H-bonds, hydrophobic contacts, π-effects)
- Compare binding modes across analog series
- Validate method by re-docking known crystallographic ligands
SAR Interpretation:
- Correlate computed binding energies with experimental activities
- Identify key interactions responsible for potency differences
- Suggest favorable modifications to improve binding
Key Consideration: Docking results should be interpreted cautiously, as scoring functions have limitations in accurately predicting binding affinities, particularly for complex natural product scaffolds [6].
Pharmacophore Modeling for SAR Analysis
Purpose: To identify essential molecular features responsible for biological activity across natural product analogs.
Background: Pharmacophore models abstract ligands into essential functional features (hydrogen bond donors/acceptors, hydrophobic areas, charged groups) common to active compounds.
Protocol Steps:
Compound Selection:
- Collect structures with measured activities (include both active and inactive analogs)
- Ensure structural diversity while maintaining some common features
Conformational Sampling:
- Generate representative conformational ensembles for each compound
- Consider biologically relevant conformations
Model Generation:
- Identify common chemical features among active compounds
- Exclude features present in inactive analogs
- Define spatial relationships between features (distances, angles)
Model Validation:
- Test ability to discriminate known actives from inactives
- Assess predictive performance through cross-validation
- Refine model based on validation results
Virtual Screening:
- Use validated model to search compound databases
- Prioritize hits for experimental testing
Application: Pharmacophore models are particularly valuable for natural products with conformational flexibility, as they capture the essential 3D arrangement of features required for activity without strict structural constraints.
Diagram 2: Pharmacophore modeling workflow for SAR analysis
Machine Learning and Explainable AI for SAR
Purpose: To uncover complex, non-linear SAR patterns in natural product datasets using advanced computational approaches.
Background: Modern machine learning (ML) methods can identify complex structure-activity relationships that may not be apparent through traditional approaches. Explainable AI (XAI) techniques make these "black box" models interpretable to medicinal chemists.
Protocol Steps:
Data Curation:
- Collect comprehensive dataset of natural product analogs with associated activities
- Apply data cleaning (remove duplicates, correct errors, standardize representations)
- Divide data into training, validation, and test sets
Descriptor Calculation:
- Compute molecular descriptors capturing structural features
- Include constitutional, topological, electronic, and physicochemical descriptors
- Consider 3D descriptors for conformation-dependent activities
Model Training:
- Select appropriate ML algorithms (random forest, neural networks, support vector machines)
- Train models to predict activity from structural descriptors
- Optimize hyperparameters through cross-validation
Model Interpretation:
- Apply XAI techniques (SHAP, LIME) to identify important structural features
- Generate visualizations highlighting substructures influencing activity
- Extract actionable SAR insights from model interpretations
Prospective Prediction:
- Use trained models to predict activities of proposed analogs
- Prioritize synthetic targets based on predicted activities and confidence estimates
- Iteratively refine models as new data becomes available
Application Note: ML approaches are particularly valuable for natural products due to their ability to handle complex, multi-parameter optimization challenges and identify non-intuitive structure-activity relationships [2].
Research Reagent Solutions for SAR Studies
Table 2: Essential Research Reagents and Tools for Natural Product SAR Studies
| Reagent/Tool | Function in SAR Studies | Application Notes |
|---|---|---|
| ChEMBL Database | Public database of bioactive molecules with curated SAR data [7] | Source of reference activities and compound structures for comparative analysis |
| GUSAR Software | (Q)SAR modeling platform for antitarget prediction and activity forecasting [7] | Uses MNA and QNA descriptors; validated for prediction of drug-antitarget interactions |
| PULSAR Application | Integrated platform for multi-parameter SAR analysis and visualization [5] | Combines Matched Molecular Pairs and SAR Slides modules for comprehensive analysis |
| Matched Molecular Pairs (MMPs) | Algorithm to identify and analyze systematic structural changes [5] | Identifies conserved structural transformations and their effects on multiple properties |
| Protein Data Bank (PDB) | Repository of 3D protein structures for structure-based design [6] | Source of target structures for molecular docking and structure-based SAR analysis |
| VEGA Platform | (Q)SAR platform for environmental fate and toxicity prediction [8] | Useful for predicting biodegradability, bioaccumulation, and environmental persistence |
Data Presentation and Analysis in SAR Studies
SAR Table Construction and Interpretation
Purpose: To systematically organize and visualize SAR data for pattern recognition and hypothesis generation.
Protocol Steps:
Table Organization:
- List compounds with increasing structural complexity or systematic variations
- Include key structural representations (2D diagrams or highlighted modifications)
- Organize biological data in adjacent columns for easy comparison
Data Annotation:
- Highlight significant improvements in potency or properties
- Indicate structural features associated with favorable or unfavorable changes
- Include statistical measures of data quality where appropriate
Pattern Recognition:
- Identify consistent trends across multiple analogs
- Note abrupt changes in activity that may indicate key interactions
- Correlate multiple parameters (potency, selectivity, properties) to identify optimal balanced compounds
Application Note: Modern software platforms can automate SAR table generation and provide interactive visualization capabilities, significantly enhancing efficiency in large optimization campaigns [5].
SAR Landscape Visualization
Purpose: To visualize complex SAR data in an intuitive format that captures both structural and activity relationships.
Background: The SAR landscape paradigm views chemical structure and bioactivity simultaneously in a 3D representation, with structure represented in the X-Y plane and activity along the Z-axis [4]. This approach reveals the "topography" of SAR datasets, with smooth regions indicating gradual activity changes with structural modifications, and cliffs representing dramatic activity changes from small structural changes.
Protocol Steps:
Structural Similarity Calculation:
- Compute pairwise molecular similarities using appropriate metrics
- Select descriptors that capture relevant structural features
Dimensionality Reduction:
- Apply methods like multi-dimensional scaling (MDS) or t-SNE to project structures into 2D space
- Preserve similarity relationships as much as possible
Activity Mapping:
- Represent activity as color or elevation in the landscape visualization
- Use contour lines or color gradients to indicate activity levels
Landscape Analysis:
- Identify smooth regions suitable for gradual optimization
- Note SAR cliffs that may indicate key interactions or mechanism changes
- Highlight activity peaks representing local optima in chemical space
Application: SAR landscape visualization is particularly valuable for understanding the optimization potential of natural product series and planning efficient synthetic strategies.
SAR analysis provides an essential framework for optimizing natural products into viable therapeutic agents. By combining sophisticated experimental approaches for analog generation with advanced computational methods for data analysis, researchers can efficiently navigate the complex chemical space of natural product derivatives. The integration of diverted synthesis, late-stage functionalization, structural biology, and machine learning creates a powerful feedback loop for SAR elucidation. As these methodologies continue to evolve, they will undoubtedly accelerate the transformation of natural product leads into clinically valuable drugs, fully realizing the potential of nature's chemical diversity in addressing human disease.
Historical Evolution and Key Milestones in SAR Studies
The Structure-Activity Relationship (SAR) is a fundamental concept in medicinal chemistry that describes the relationship between a molecule's chemical structure and its biological activity. Within the context of natural product research, SAR-directed optimization is the systematic process of modifying a natural lead compound to improve its properties as a potential drug candidate [9]. Natural products have been a predominant source of anticancer drugs, with approximately 80% of anticancer drugs approved between 1981 and 2010 originating from natural products [9]. However, these natural molecules often require optimization to address limitations in drug efficacy, ADMET profiles (Absorption, Distribution, Metabolism, Excretion, and Toxicity), and chemical accessibility [9].
SAR analysis depends on recognizing which structural characteristics correlate with chemical and biological reactivity. This enables researchers to draw conclusions about uncharacterized compounds based on their structural features and comparisons against databases of known molecules [3]. When combined with professional judgment, SAR becomes a powerful method for understanding the functional implications of structural changes, particularly for sensitive toxicological endpoints like carcinogenicity or cardiotoxicity [3].
Historical Evolution of SAR Studies
The evolution of SAR methodologies parallels key developments in drug discovery paradigms, moving from empirical observation to increasingly rational and data-driven approaches.
Early Foundations and Classical Approaches
The roots of SAR can be traced back over a century to the pioneering work of Langmuir, who explored the effects of altering functional groups while maintaining essential physicochemical properties [10]. The formalization of rational drug design (RDD) in the 1950s enabled theoretical insights into drug-receptor interactions to reinforce practical drug testing [10]. This approach matured in the 1970s and 1980s with successful developments like lovastatin and captopril, which remain in clinical use today [10].
Early SAR studies on natural products primarily involved direct chemical manipulation of functional groups through derivation or substitution, alteration of ring systems, and isosteric replacement [9]. These efforts were largely empirical and intuition-guided, particularly in phenotypic approaches. The paclitaxel discovery exemplifies this era—its identification and the revelation of its novel mechanism of action (tubulin-assembly promotion) marked a milestone in anticancer drug discovery [9].
The Rise of Modern Screening and Analysis
The introduction of high-throughput screening (HTS) in the 1990s created increased demand for large, diverse compound libraries [11]. Early collections came from in-house archives or combinatorial chemistry, though purely combinatorial approaches often lacked the complexity and relevance needed for clinical success [11]. This period saw SAR methodology evolve from simple functional group analysis to SAR table evaluation, where experts review compounds, their physical properties, and activities by sorting, graphing, and scanning structural features to identify relationships [3].
Library design shifted from quantity-driven to quality-focused, incorporating guidelines like Lipinski's Rule of Five and additional filters for toxicity and assay interference to define 'drug-likeness' [11]. Screening collections became increasingly curated with attention to molecular properties, scaffold diversity, natural product-inspired motifs, and target-class relevance [11].
Contemporary Data-Driven Approaches
In recent years, artificial intelligence (AI) and machine learning (ML) have transformed SAR studies [11] [10]. Predictive models can now virtually screen massive chemical spaces and rank compounds by likelihood of activity, allowing researchers to focus physical screening on enriched, higher-probability subsets [11]. The concept of the "informacophore" has emerged, extending traditional pharmacophore models by incorporating data-driven insights from computed molecular descriptors, fingerprints, and machine-learned representations of chemical structure [10].
The development of ultra-large "make-on-demand" virtual libraries has significantly expanded accessible chemical space, with suppliers like Enamine and OTAVA offering 65 and 55 billion novel make-on-demand molecules respectively [10]. Screening such vast chemical spaces requires ultra-large-scale virtual screening, as direct empirical screening of billions of molecules remains infeasible [10].
Table: Historical Milestones in SAR Studies
| Time Period | Key Developments | Primary Approaches | Representative Technologies |
|---|---|---|---|
| Pre-1950s | Early structure-activity observations; Functional group manipulation | Empirical observation; Chemical intuition | Basic chemical synthesis; Physiological testing |
| 1950s-1980s | Formalization of rational drug design; Natural product drug discovery | Structure-activity relationship (SAR) establishment; Bioisosteric replacement | Molecular modeling; X-ray crystallography |
| 1990s-2000s | High-throughput screening; Computational chemistry | SAR table analysis; Library filtering; Target-focused design | HTS robotics; Combinatorial chemistry; Rule-of-5 |
| 2010s-Present | AI and machine learning; Ultra-large libraries | Informatics-driven optimization; Multi-parameter design | Machine learning; Cloud computing; Make-on-demand libraries |
Key Methodologies and Experimental Protocols
SAR Establishment Through Systematic Modification
The initial phase of SAR studies involves systematic modification of the natural lead compound to explore how structural changes affect biological activity. As illustrated in the optimization of natural leads to anticancer agents, this typically proceeds through three progressive levels [9]:
- Direct Chemical Manipulation: The most straightforward approach involves derivation or substitution of functional groups, alteration of ring systems, and isosteric replacement.
- SAR-Directed Optimization: With accumulation of chemical and biological data, meaningful SAR can be established to guide more rational optimization while generally maintaining the basic structural core.
- Pharmacophore-Oriented Design: The core structures may be significantly changed based on identified pharmacophores, often to solve chemical accessibility issues while maintaining key interaction elements.
Table: Common Structural Modifications in Natural Lead Optimization
| Modification Type | Objective | Typical Methods | Impact on Drug Properties |
|---|---|---|---|
| Functional Group Replacement | Enhance target binding; Improve solubility; Reduce toxicity | Bioisosteric replacement; Chemical derivation | Alters polarity, hydrogen bonding, molecular interactions |
| Scaffold Hopping | Maintain activity while improving synthetic accessibility or intellectual property position | Molecular modeling; Structure-based design | May significantly change physicochemical properties while maintaining key interactions |
| Ring System Alteration | Modulate conformational flexibility; Improve metabolic stability | Ring expansion/contraction; Heteroatom introduction | Affects molecular rigidity, spatial orientation, and metabolic sites |
| Side Chain Optimization | Fine-tune potency, selectivity, and pharmacokinetics | Alanine scanning; Functional group variation | Directly influences binding affinity and ADMET properties |
Computational SAR Evaluation Protocol
Recent advances in computational methods have created robust protocols for SAR evaluation, as demonstrated in studies of natural compound analogs targeting SARS-CoV-2 proteases [12]:
Step 1: Analog Identification and Library Creation
- Apply chemical similarity algorithms to natural product scaffolds using databases like ChEMBL to retrieve structurally related analogs.
- Curate a focused library (e.g., 600+ candidates) for virtual screening.
Step 2: Molecular Docking and Binding Assessment
- Perform automated docking against therapeutic targets (e.g., viral proteases).
- Evaluate binding scores and interaction patterns with key residues.
- Compare results to parent natural scaffolds and positive controls.
Step 3: Interaction Pattern Analysis
- Identify specific interactions: hydrogen bonds, hydrophobic contacts, ionic interactions.
- Map binding orientations and key residue contacts.
Step 4: ADMET Profiling
- Compute comprehensive absorption, distribution, metabolism, excretion, and toxicity profiles.
- Apply drug-likeness filters (e.g., Lipinski's Rule of Five).
- Predict potential toxicity endpoints.
Step 5: Gene Expression Analysis
- Utilize tools like DIGEP-Pred to predict pathway influences.
- Assess potential effects on disease-relevant biological processes.
Step 6: Multi-Criteria Optimization and Hit Prioritization
- Integrate binding affinity, interaction quality, ADMET properties, and pathway relevance.
- Select top candidates for experimental validation.
Computational SAR Evaluation Workflow
SAR Table Analysis Protocol
SAR is typically evaluated in table format, which forms the basis for rational decision-making in lead optimization [3]:
Experimental Protocol:
Data Compilation
- Create a comprehensive table containing compound structures, physical properties, and biological activities.
- Include both qualitative and quantitative data where available.
Structural Feature Identification
- Identify common structural features among active compounds.
- Note features associated with inactivity or toxicity.
Data Sorting and Trend Analysis
- Sort compounds by specific structural modifications.
- Graph activity versus property relationships.
- Identify optimal ranges for physicochemical properties.
Hypothesis Generation
- Formulate testable hypotheses about which structural elements drive activity.
- Design new analogs to probe specific SAR questions.
Iterative Optimization
- Synthesize proposed analogs.
- Test and incorporate results into expanded SAR table.
- Refine hypotheses and repeat cycle.
Modern SAR studies rely on a sophisticated infrastructure of chemical, computational, and biological resources.
Table: Essential Research Reagent Solutions for SAR Studies
| Resource Category | Specific Examples | Function in SAR Studies | Key Characteristics |
|---|---|---|---|
| Compound Libraries | Natural product collections; Fragment libraries; Targeted screening sets | Provide starting points and analogs for SAR exploration | Diversity; Drug-likeness; Structural novelty; Synthetic tractability |
| Chemical Suppliers | Enamine; OTAVA; Molport | Source for purchaseable compounds and make-on-demand libraries | Breadth of inventory; Quality control; Reliability |
| Computational Platforms | Molecular docking software; ADMET prediction tools; Machine learning frameworks | Enable virtual screening and property prediction | Accuracy; Speed; User-friendliness; Interpretability |
| Structural Biology Resources | Protein Data Bank (PDB); Crystallization kits; Cryo-EM facilities | Provide structural insights for structure-based design | Resolution; Relevance to human biology; Completeness |
| Biological Assays | High-throughput screening; Enzymatic assays; Cell-based phenotypic assays | Generate experimental data for SAR tables | Relevance; Reproducibility; Throughput; Cost-effectiveness |
| Chemical Synthesis Tools | Automated synthesizers; Flow chemistry systems; Purification equipment | Enable rapid analog synthesis and testing | Efficiency; Versatility; Scalability |
Case Study: SAR-Driven Optimization of Natural Anticancer Agents
The optimization of natural products into approved anticancer drugs provides compelling case studies of successful SAR application. As noted in natural product research, derivatives of natural products account for approximately one-third of small-molecule anticancer drugs [9]. These optimization efforts typically address three main purposes: enhancing drug efficacy, optimizing ADMET profiles, and improving chemical accessibility [9].
Recent research on ginger-derived compounds against SARS-CoV-2 proteases demonstrates modern SAR principles in action. Studies identified CHEMBL1720210 (a shogaol-derived analog) with strong interaction with PLpro (-9.34 kcal/mol), and CHEMBL1495225 (a 6-gingerol derivative) showing high affinity for 3CLpro (-8.04 kcal/mol) [12]. Molecular interaction analysis revealed specific residue interactions: CHEMBL1720210 forms hydrogen bonds with key PLpro residues including GLY163, LEU162, GLN269, TYR265, and TYR273, complemented by hydrophobic interactions with TYR268 and PRO248 [12]. This level of detailed structural insight enables rational optimization of natural leads.
Natural Lead Optimization Framework
Future Directions in SAR Studies
The field of SAR studies continues to evolve with emerging technologies and methodologies. Artificial intelligence and machine learning are playing increasingly transformative roles in how compound libraries are designed, prioritized, and exploited [11]. Predictive models can virtually screen massive chemical spaces and rank compounds by likelihood of activity, allowing researchers to focus physical screening on enriched, higher-probability subsets [11].
The concept of the informacophore represents a significant evolution from traditional pharmacophore approaches. By incorporating data-driven insights derived from computed molecular descriptors, fingerprints, and machine-learned representations of chemical structure, informacophores enable a more systematic and bias-resistant strategy for scaffold modification and optimization [10]. However, this approach also raises challenges of model interpretability, as machine-learned informacophores can be challenging to interpret directly compared to traditional pharmacophore models rooted in human expertise [10].
The development of ultra-large, "make-on-demand" virtual libraries has dramatically expanded the accessible chemical space for SAR exploration [10]. With suppliers offering tens of billions of novel make-on-demand molecules, researchers can explore SAR relationships across unprecedented chemical diversity. This expansion necessitates advanced computational approaches, as direct empirical screening of such vast libraries remains impractical [10].
As these technologies mature, the integration of AI-driven insights with medicinal chemistry expertise will likely define the next era of SAR studies. The role of experienced medicinal chemists remains essential to oversee the process, validate AI-generated suggestions, select appropriate building blocks, and critically review retrosynthetic approaches to ensure proposed molecules are both synthetically feasible and aligned with project goals [11]. This synergistic combination of human expertise and computational power holds significant promise for accelerating the optimization of natural leads into effective therapeutics.
Structure-Activity Relationship (SAR) analysis is a fundamental methodology in medicinal chemistry and drug discovery that investigates the relationship between a molecule's chemical structure and its biological activity [13]. The core principle is that the biological activity of a compound is a function of its molecular structure and physicochemical properties [14]. By systematically modifying a compound's structure and observing the resulting changes in biological activity, researchers can identify which molecular features are essential for its biological function [13].
SAR techniques are employed across various applications, including in-silico design of virtual chemical libraries, screening databases for lead discovery, and mining gene expression data for target identification [13]. The basic assumption underlying SAR analysis is that similar molecules have similar activities, though this comes with the challenge of defining meaningful molecular similarities that correlate with biological function—a concept known as the SAR paradox [15]. When these relationships are quantified mathematically, the approach is termed Quantitative Structure-Activity Relationship (QSAR) modeling [15] [14].
Natural products serve as particularly valuable starting points for SAR studies due to their inherent biological relevance, structural complexity, and diversity [9]. Historically, natural products have made significant contributions to drug discovery, especially in oncology, where approximately 79.8% of anticancer drugs approved from 1981 to 2010 were natural products or derivatives thereof [9]. However, these complex molecules often require optimization to improve their drug-like properties, efficacy, and synthetic accessibility [9].
Key Methodologies in SAR Analysis
SAR and QSAR Modeling Approaches
SAR analysis has evolved into several specialized methodologies, each with distinct advantages for different applications:
- Fragment-Based (GQSAR): This approach analyzes contributions of molecular fragments or substituents to biological activity. It allows researchers to study various molecular fragments of interest in relation to biological response variation, including cross-term fragment descriptors that identify key fragment interactions [15].
- 3D-QSAR: These methods utilize three-dimensional molecular structures and force field calculations. Comparative Molecular Field Analysis (CoMFA) examines steric and electrostatic fields around molecules, correlating them with biological activity through statistical methods like partial least squares regression [15].
- Chemical Descriptor-Based: This methodology computes descriptors quantifying electronic, geometric, or steric properties of entire molecules rather than individual fragments [15].
- q-RASAR: An emerging hybrid approach that merges QSAR with similarity-based read-across techniques, potentially offering improved predictive capability [15].
Statistical and Computational Tools
SAR modeling employs various statistical and machine learning methods to correlate structural features with biological activity:
- Multiple Linear Regression (MLR): Correlates molecular descriptors with biological activity through linear relationships [13] [14].
- Principal Component Analysis (PCA): Reduces dimensionality of complex datasets to identify underlying patterns in SAR data [13].
- Partial Least Squares (PLS): Addresses issues with MLR and PCA by abstracting both descriptors and biological activities into new variables to enhance correlations [14].
- Artificial Neural Networks (ANN): Models complex, non-linear relationships between molecular structure and biological activity [13] [14].
- Support Vector Machine (SVM): Used for classification and regression tasks within SAR analysis [13].
Specialized software including MATLAB, Python libraries, ChemDraw, and Molecular Operating Environment (MOE) are typically employed for implementing these statistical models and visualizing results [13].
SAR-Driven Optimization of Natural Products
Strategic Framework for Natural Lead Optimization
The optimization of natural products through SAR studies typically addresses three primary objectives: enhancing drug efficacy, optimizing ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiles, and improving chemical accessibility [9]. These efforts can be implemented at different levels of structural modification:
- Direct Chemical Manipulation: The most straightforward approach involving derivation or substitution of functional groups, alteration of ring systems, and isosteric replacement [9]. These efforts may be empirical or guided by structure-based design when target structures are available.
- SAR-Directed Optimization: This approach involves establishing comprehensive structure-activity relationships through systematic modification, followed by rational optimization based on these insights [9].
- Pharmacophore-Oriented Molecular Design: This strategy may significantly alter the core structures of natural products to solve chemical accessibility issues while maintaining key pharmacological features [9].
Table 1: Optimization Strategies for Natural Product-Based Drug Discovery
| Optimization Strategy | Key Features | Primary Applications | Representative Examples |
|---|---|---|---|
| Direct Chemical Manipulation [9] | Functional group derivation, ring system alteration, isosteric replacement | Improving potency, addressing reactive functional groups | Rh(II)-catalyzed C–H amination of eupalmerin acetate [16] |
| SAR-Directed Optimization [9] | Systematic modification, establishment of structure-activity relationships | Enhancing efficacy, optimizing ADMET profiles | Dibromoacetophenones as mIDH1 inhibitors [17] |
| Pharmacophore-Oriented Design [9] | Focus on essential features for activity, scaffold hopping | Improving synthetic accessibility, creating novel analogs | MraY inhibitor build-up library [18] |
| Build-Up Library Approach [18] | Fragment ligation, in situ screening, minimal purification | Rapid exploration of chemical space, natural product optimization | Hydrazone-based MraY inhibitors [18] |
Advanced Techniques: Build-Up Library Approach
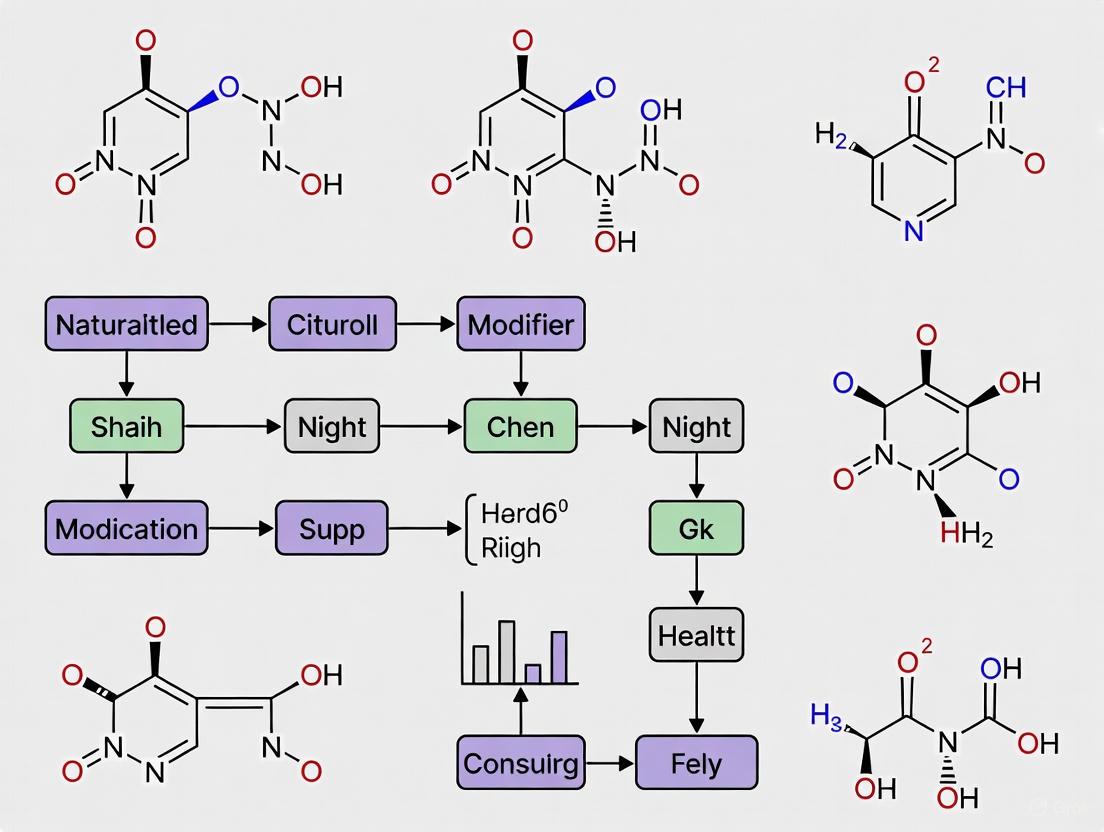
A recent innovative strategy for SAR studies of natural products involves the construction of "build-up libraries" through fragment ligation [18]. This approach divides natural products into core fragments (responsible for target binding) and accessory fragments (modulating binding affinity, selectivity, and disposition properties) [18]. These fragments are ligated using high-yielding, chemoselective reactions such as hydrazone formation, which produces only water as a by-product, enabling direct biological evaluation without purification [18].
This method was successfully applied to MraY inhibitory natural products, using 7 core structures and 98 accessory fragments to generate a 686-compound library [18]. The approach allowed simultaneous optimization of multiple natural product classes, leading to identification of promising analogues with potent and broad-spectrum antibacterial activity against drug-resistant strains, both in vitro and in vivo [18].
Diagram 1: Build-up library workflow for natural product optimization. This approach enables rapid generation and screening of analog libraries through fragment ligation and in situ evaluation [18].
Experimental Protocols for SAR Studies
Protocol 1: Rh(II)-Catalyzed C–H Amination for Natural Product Functionalization
Purpose: To functionalize natural products at 'unfunctionalized' positions via Rh(II)-catalyzed amination, enabling simultaneous SAR studies and arming (alkynylation) of natural products for subsequent conjugation to cellular probes [16].
Materials:
- Natural product substrate (≤ 1 mg)
- Trichloroethyl sulfamate nitrene precursor (1.0 equiv)
- Rh₂(esp)₂ or Rh₂(OAc)₄ catalyst (Du Bois catalyst system)
- PhI(O₂CtBu)₂ as oxidant
- Anhydrous benzene, dichloromethane, or α,α,α-trifluorotoluene as solvent
Procedure:
- Dissolve the natural product substrate (≤ 1 mg) and sulfamate nitrene precursor (1.0 equiv) in anhydrous benzene (∼0.13 M concentration) under inert atmosphere.
- Add Rh(II) catalyst (Du Bois catalyst system) to the reaction mixture.
- Add solid PhI(O₂CtBu)₂ oxidant in one portion to initiate the reaction.
- Monitor reaction progress by TLC or LC-MS. For carvone as a model substrate, typical conversions yield 23-26% with excellent mass recovery.
- To alter chemoselectivity:
- Add Brønsted acids (e.g., HOAc) or Lewis acids (e.g., In(OTf)₃) to favor C–H amination
- Add inorganic bases (e.g., K₂CO₃) to favor alkene aziridination
- Purify the product using standard chromatographic techniques.
- The alkynylated derivatives can be subsequently conjugated to reporter tags (e.g., biotin or fluorophores) for mechanism of action studies.
Applications: This protocol was successfully applied to the marine-derived anticancer diterpene eupalmerin acetate (EPA), enabling quantitative proteome profiling that identified several protein targets in HL-60 cells associated with cancer proliferation [16].
Protocol 2: In Situ Build-Up Library Synthesis and Screening
Purpose: To rapidly generate and screen natural product analogues via hydrazone formation between aldehyde cores and hydrazine accessory fragments, enabling direct biological evaluation without purification [18].
Materials:
- Aldehyde-functionalized natural product cores (e.g., MraY inhibitors)
- Library of hydrazine accessory fragments (98 diverse chemotypes)
- Anhydrous DMSO
- 96-well assay plates
- Centrifugal concentrator
Procedure:
- Prepare 10 mM DMSO solutions of aldehyde core and hydrazine fragments.
- Mix aldehyde core and hydrazine fragments in approximately 1:1 stoichiometry in 96-well plates (total volume 31 μL).
- Incubate at room temperature for 30 minutes without any additives.
- Remove DMSO using centrifugal concentration under vacuum at room temperature overnight.
- Dissolve the resulting residues in 30 µL of DMSO to prepare 5 mM library solutions.
- Confirm hydrazone formation by LC-MS analysis (typically ≥80% yield).
- Directly evaluate MraY inhibitory activity and antibacterial activity without purification, assuming 100% conversion for concentration calculations.
Applications: This protocol enabled identification of promising MraY inhibitor analogues with potent and broad-spectrum antibacterial activity against drug-resistant strains, validated in an acute thigh infection model [18].
Table 2: Key Research Reagent Solutions for SAR Studies
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Rh(II) Catalysts [16] | Catalyze C-H amination/aziridination | Rh₂(esp)₂, Rh₂(OAc)₄, Rh₂(OCOC8H15)₄, Rh₂(TPA)₄ |
| Sulfamate Nitrene Precursors [16] | Source of metal nitrenoid for C-H functionalization | Trichloroethyl sulfamate with terminal alkyne (e.g., compound 9) |
| Aldehyde Core Fragments [18] | Core structures of natural products for library synthesis | MraY inhibitory natural product cores with aldehyde handle |
| Hydrazine Accessory Fragments [18] | Variable fragments for diversity-oriented synthesis | Aromatic (BZ, PA), alkyl (AC), amino acid (AA, LA) hydrazides |
| Molecular Descriptors [15] [14] | Quantitative parameters for QSAR modeling | Hydrophobicity, electronic properties, steric effects, topological indices |
| Validation Software [13] | Statistical validation of SAR/QSAR models | Cross-validation, external validation, Y-scrambling techniques |
Case Study: SAR Optimization of mIDH1 Inhibitors
Lead Identification and Optimization
A recent case study demonstrates the power of systematic SAR optimization in developing inhibitors against mutant isocitrate dehydrogenase 1 (mIDH1), an important anticancer target [17]. Researchers screened an in-house library of 109 compounds and identified a dibromoacetophenone lead compound (1-1) that showed 73.6% inhibition of IDH1 R132H at 2 μM [17].
Through iterative structure-activity relationship optimization, the team developed a series of potent compounds inhibiting both IDH1 R132H and R132C mutants [17]. Key structural modifications included:
- Phenyl Ring Substitutions: Introducing electron-donating groups (e.g., -OH, -OCH₃) at specific positions significantly enhanced inhibitory activity.
- Acetophenone Core Modifications: Systematic variation of substituents on the acetophenone core to optimize binding interactions.
- Side Chain Optimization: Fine-tuning side chain properties to improve potency and selectivity.
Table 3: SAR Data for Selected mIDH1 Inhibitors [17]
| Compound | R¹ Substituent | R² Substituent | IDH1 R132H IC₅₀ (μM) | IDH1 R132C IC₅₀ (μM) | Key Structural Features |
|---|---|---|---|---|---|
| Lead 1-1 | 2,4-dibromo | -CH₃ | 0.92 | 1.35 | Initial lead from screening |
| Analog 2 | 2-bromo-4-hydroxy | -CH₂CH₃ | 0.15 | 0.21 | Electron-donating group enhances activity |
| Analog 5 | 2,4-dihydroxy | -C₃H₇ | 0.08 | 0.13 | Dual hydroxylation maximizes potency |
| Analog 8 | 2-methoxy-4-hydroxy | -C₂H₅ | 0.11 | 0.16 | Mixed ether/phenol optimal for selectivity |
The most promising compounds exhibited IC₅₀ values in the nanomolar range against both IDH1 R132H and R132C mutants, demonstrating the success of the SAR-guided optimization approach [17]. This case study illustrates how systematic structural modification based on biological evaluation data can significantly enhance compound potency and develop structure-activity relationship trends for further optimization.
Diagram 2: Iterative SAR optimization cycle for lead development. This feedback-driven process systematically improves compound properties through design, synthesis, and testing iterations [17] [10].
SAR analysis provides a powerful framework for linking molecular structure to biological activity, serving as an indispensable tool in modern drug discovery, particularly in the optimization of natural product leads. By applying systematic structural modifications and analyzing resulting changes in biological activity, researchers can identify key molecular features responsible for pharmacological effects. The integration of traditional SAR studies with advanced methodologies such as build-up library approaches, computational QSAR modeling, and innovative chemical biology techniques like Rh(II)-catalyzed C-H amination continues to advance our ability to rationally optimize natural products into therapeutic agents. As these methodologies evolve, they will undoubtedly continue to accelerate the discovery and development of novel drugs from natural product starting points.
Identifying Key Functional Groups and Their Pharmacological Roles
Within the context of structure-activity relationship (SAR) directed optimization of natural product leads, the strategic identification and manipulation of functional groups is a cornerstone of medicinal chemistry. Functional groups are specific arrangements of atoms or moieties that confer predictable chemical and physical properties to a molecule, thereby dictating its biological activity and pharmacological behavior [19] [20]. In the hit-to-lead optimization phase, understanding the role of these groups is paramount for improving the potency, selectivity, and drug-like properties of a compound while minimizing adverse effects [19] [10]. This application note provides a structured overview of key functional groups, their associated pharmacological roles, and practical protocols for their study in a natural product lead optimization program.
Key Functional Groups: Properties and Pharmacological Roles
The following tables summarize the core functional groups, their defining characteristics, and their strategic importance in drug discovery.
Table 1: Fundamental Hydrocarbon and Halogen Functional Groups
| Functional Group | Structural Formula | Key Properties | Pharmacological Role & Impact |
|---|---|---|---|
| Alcohol | R–OH | Polar; H-bond donor & acceptor; increases water solubility [20] [21] | Enhances target binding via H-bonds; improves solubility; metabolically vulnerable to oxidation [19] [21] |
| Aromatic Ring | C₆H₅–R | Planar; hydrophobic; electron-rich system [20] | Facilitates π-π stacking and cation-π interactions with protein targets; contributes to van der Waals interactions [21] [22] |
| Alkyl Halide | R–X (X = F, Cl, Br, I) | Polar C–X bond; serves as an electrophile [20] [21] | Chlorine/Bromine/Iodine can be metabolic liabilities; Fluorine is used as a bioisostere for hydrogen or to block metabolic sites [21] |
Table 2: Carbonyl-Derived and Nitrogen-Containing Functional Groups
| Functional Group | Structural Formula | Key Properties | Pharmacological Role & Impact |
|---|---|---|---|
| Carboxylic Acid | R–COOH | Acidic; ionizable at physiological pH; strong H-bond donor/acceptor [20] [21] | Can form strong ionic bonds with basic residues in targets; high prevalence in drugs [21] [23] |
| Ester | R–COOR' | Polar; H-bond acceptor only [20] | Used as a prodrug strategy to mask carboxylic acids or alcohols, improving absorption [21] |
| Amine | R–NH₂, R₂NH, R₃N | Basic; ionizable; H-bond donor & acceptor (if N-H present) [20] | Critical for forming ionic bonds with acidic residues; common in active transport; influences distribution [19] [20] |
| Amide | R–CONR₂ | Polar; planar conformation; excellent H-bond donor & acceptor [20] | Forms stable H-bonds with targets; cornerstone of peptide and protein structure; high metabolic stability [20] [22] |
Table 3: Frequency of Key Functional Groups and Ring Systems in Marketed Drugs [22]
| Structural Group | Example(s) | Approximate Frequency in Drugs | Common Therapeutic Associations |
|---|---|---|---|
| Benzene Ring | Benzene | Very High | Ubiquitous; present in a vast majority of drug categories [22] |
| Saturated Heterocycles | Piperidine, Piperazine, Azetidine | High | Common scaffolds providing three-dimensional structure and nitrogen for salt formation [22] |
| Unsaturated Heterocycles | Pyridine, Imidazole, Indole | High | Found in targets like kinases and GPCRs; can act as H-bond acceptors or donors [22] |
| Carboxylic Acid | Acetate | High | Prevalent in anti-inflammatory, cardiovascular, and antibiotic drugs [22] [24] |
Experimental Protocols for SAR Analysis
The following protocols outline a systematic approach for evaluating the role of functional groups in natural product analogs.
Protocol: Systematic Functional Group Interconversion for SAR
Objective: To establish the contribution of a specific functional group to biological activity and pharmacokinetic properties through targeted synthetic modification.
Materials:
- Research Reagent Solutions: See Table 4.
- Equipment: Standard synthetic chemistry laboratory equipment (reactors, purification systems), analytical HPLC, NMR spectrometer.
Table 4: Key Research Reagent Solutions for SAR Exploration
| Reagent / Material | Function in SAR Studies |
|---|---|
| Bioisosteric Replacement Libraries | Collections of reagents for replacing functional groups with moieties of similar physicochemical properties (e.g., carboxylic acid with tetrazole) to optimize ADMET [10]. |
| Click Chemistry Toolkits (e.g., CuAAC) | Enables rapid, modular assembly of diverse compound libraries for initial SAR screening, using reactions like copper-catalyzed azide-alkyne cycloaddition [25]. |
| Metabolic Enzyme Assay Kits (e.g., CYP450) | Used to assess the metabolic stability of lead compounds and identify vulnerable functional groups [19]. |
| Computational Chemistry Software | For molecular docking, QSAR analysis, and predicting the binding affinity of analogs before synthesis [10] [26]. |
Procedure:
- Lead Identification: Begin with a purified natural product lead compound with confirmed biological activity.
- Structural Analysis: Identify all potential functional groups and sites for chemical modification.
- Analog Design & Synthesis:
- Design a series of analogs where a single functional group is systematically modified.
- Common modifications include: a) Removal (e.g., -OH to -H). b) Conversion (e.g., -COOH to -CONH₂). c) Bioisosteric replacement (e.g., ester to amide). d) Addition (e.g., -H to -F) [19] [21].
- Synthesize the planned analogs using appropriate organic synthesis techniques.
- Biological Evaluation: Test all synthesized analogs in the relevant biological assay (e.g., enzyme inhibition, cell-based viability assay) to determine IC₅₀ or EC₅₀ values.
- Data Analysis: Correlate the structural changes with changes in biological potency to define the SAR.
Protocol: In Vitro ADMET Profiling of Functional Group Modifications
Objective: To determine the effect of functional group changes on the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profile of lead compounds.
Materials:
- Caco-2 cell lines
- Human liver microsomes (HLM) or hepatocytes
- CYP450 isoform inhibition assay kits
- Phosphate Buffered Saline (PBS), Hanks' Balanced Salt Solution (HBSS)
- LC-MS/MS system for analytical quantification
Procedure:
- Passive Permeability (Papp) Assay:
- Culture Caco-2 cells on semi-permeable membranes until they form a confluent monolayer.
- Add the test compound to the donor compartment (e.g., apical side) and monitor its appearance in the receiver compartment (basolateral side) over time using LC-MS/MS.
- Calculate the apparent permeability (Papp). Functional groups like esters (prodrugs) can significantly improve Papp for polar acids [21].
- Metabolic Stability in HLM:
- Incubate the test compound with HLM in the presence of NADPH cofactor.
- Take samples at time points (e.g., 0, 5, 15, 30, 60 min) and quench the reaction.
- Analyze the remaining parent compound to determine half-life (t₁/₂) and intrinsic clearance (CLint). Replacing a metabolically soft spot (e.g., hydrogen with fluorine) can improve t₁/₂ [21].
- CYP450 Inhibition Screening:
- Incubate CYP450 isoforms with specific probe substrates in the presence of the test compound.
- Measure the formation of the metabolite specific to each isoform.
- Calculate the % inhibition of enzyme activity. Certain functional groups (e.g., alkyl amines) can form reactive metabolites that lead to CYP inhibition [24].
Data Analysis and Visualization
The workflow for SAR-driven optimization of natural leads, from functional group analysis to candidate selection, is a cyclical process of design, testing, and learning. The following diagram illustrates this integrated workflow, highlighting how functional group manipulation is central to both activity and ADMET optimization.
Diagram 1: SAR-Driven Optimization Workflow. This diagram outlines the iterative cycle of functional group analysis, analog design, synthesis, and testing that defines SAR-driven optimization of natural product leads.
The interaction of functional groups with a biological target is a key determinant of efficacy. The following diagram maps the logical relationship between common functional groups present in a drug molecule and their corresponding interactions with amino acid residues in a protein binding pocket.
Diagram 2: Functional Group - Target Interaction Map. This diagram visualizes how specific functional groups on a drug molecule mediate binding to key residues in a protein target through defined chemical interactions.
The Critical Importance of SAR in Modern Drug Discovery Pipelines
Structure-Activity Relationship (SAR) analysis represents a fundamental pillar in modern drug discovery, providing the critical framework for understanding how chemical modifications influence biological activity. SAR depends on the systematic characterization of structural features and their correlation with biological reactivity, enabling researchers to draw meaningful conclusions about uncharacterized compounds by comparing them against established molecular databases [3]. In the specific context of natural product optimization, SAR-guided approaches transform complex natural scaffolds into refined therapeutic candidates with enhanced pharmacological profiles.
The evolution from traditional SAR to quantitative SAR (QSAR) and the integration of sophisticated artificial intelligence (AI) tools has dramatically accelerated the drug discovery process. These advancements allow research teams to navigate the vast chemical space more efficiently, identifying optimal structural modifications that enhance desired activities while minimizing undesirable properties [27]. For natural products, which often serve as excellent starting points but frequently require optimization for drug-like properties, SAR-driven optimization has become indispensable for successful clinical translation.
SAR Methodologies and Experimental Protocols
Integrated Computational-Experimental Workflow for Natural Product Optimization
The contemporary SAR analysis process follows a systematic workflow that integrates both computational predictions and experimental validation. Figure 1 illustrates this integrated approach, which has become standard practice in modern drug discovery pipelines for natural product optimization.
Figure 1. Integrated SAR Workflow for Natural Product Optimization - This diagram illustrates the systematic approach combining computational predictions and experimental validation in modern SAR-driven drug discovery.
Core Experimental Protocols in SAR Analysis
Molecular Descriptor Calculation and QSAR Modeling Protocol
Purpose: To quantify molecular properties and establish predictive models linking structure to activity.
- Procedure:
- Calculate comprehensive molecular descriptors (electronic, hydrophobic, steric, topological) using software such as alvaDesc or Dragon.
- Perform descriptor reduction via Principal Component Analysis (PCA) to eliminate redundancy and reduce dimensionality.
- Develop QSAR models using multiple linear regression (MLR), partial least squares (PLS), or principal component regression (PCR).
- Validate models using internal cross-validation and external test sets to ensure predictive capability.
- Key Parameters: Model performance metrics (R², RMSE, Q²), descriptor importance, applicability domain definition.
- Application Note: In shikonin derivative optimization, the PCR model demonstrated superior predictive performance (R² = 0.912, RMSE = 0.119), with electronic and hydrophobic descriptors identified as most significant for cytotoxic activity [28].
Build-Up Library Synthesis and In Situ Screening Protocol
Purpose: To rapidly generate and screen analogue libraries for natural product optimization.
- Procedure:
- Design core fragments (retaining key pharmacophores) and diverse accessory fragments.
- Employ high-yielding, chemoselective ligation reactions (e.g., hydrazone formation) in 96-well plate format.
- Concentrate reaction mixtures without purification using centrifugal evaporation.
- Directly evaluate biological activity in both enzymatic and cell-based assays without intermediate purification.
- Confirm hit structures and synthetic yields through LC-MS analysis of library samples.
- Key Parameters: Reaction conversion (>80%), concentration accuracy, assay reproducibility, minimal byproduct interference.
- Application Note: This approach enabled efficient evaluation of 686 MraY inhibitor analogues, identifying promising antibacterial candidates with potent activity against drug-resistant strains [18].
Comprehensive Biological Profiling Protocol
Purpose: To evaluate compound efficacy across multiple therapeutic endpoints and establish structure-activity correlations.
- Procedure:
- Conduct target-based assays (e.g., enzyme inhibition, receptor binding) to determine primary mechanism of action.
- Perform cell-based viability assays (e.g., MTT, ATP-lite) to establish cellular potency.
- Evaluate secondary pharmacological activities (e.g., anti-aggregation, mitochondrial effects) for polypharmacology assessment.
- Determine physicochemical properties (logD, solubility, stability) to correlate with biological activity.
- Assess in vivo efficacy in disease-relevant animal models for selected lead compounds.
- Key Parameters: IC50/EC50 values, selectivity indices, correlation coefficients between properties and activity.
- Application Note: Ferrocene-curcumin analogue analysis revealed distinct structure-dependent effects on both Aβ fibrillogenesis inhibition and glioblastoma cell cytotoxicity, enabling identification of optimal substituents for dual functionality [29].
SAR Applications in Natural Product Optimization
Case Studies in Diverse Therapeutic Areas
Table 1. SAR-Driven Optimization of Natural Product-Derived Therapeutics
| Natural Product Scaffold | Therapeutic Area | Key Structural Modifications | Optimized Activity Profile | Citation |
|---|---|---|---|---|
| Shikonin derivatives | Cancer | Acylation of hydroxynaphthoquinone core | Enhanced cytotoxic activity (PCR R² = 0.912); improved target binding to 4ZAU | [28] |
| SERCA2a activators | Cardiovascular | Indoline, benzofuran, benzodioxole analogs | 57% increase in ATPase activity (EC50 = 0.7-9 μM); reduced Ca²⁺ affinity | [30] |
| Ferrocene-curcumin hybrids | Neurodegenerative/Oncology | Pyrazole vs pyrimidine ring variations; substituent optimization | Dual Aβ aggregation inhibition & glioblastoma cytotoxicity; structure-dependent effects | [29] |
| MraY inhibitors | Anti-bacterial | Hydrazone accessory fragment diversification | Potent broad-spectrum activity against drug-resistant strains; improved in vivo efficacy | [18] |
| Fenarimol analogs | Anti-fungal | Core ring substituent optimization targeting logD < 2.5 | Enhanced in vivo activity in larval survival assay; improved therapeutic window | [31] |
Advanced SAR Methodologies
Cross-Structure-Activity Relationship (C-SAR) Approach
The C-SAR methodology represents a significant advancement beyond traditional SAR analysis by extracting pharmacophoric substitution patterns from diverse chemotypes targeting the same biological entity. This approach utilizes matched molecular pair (MMP) analysis to identify critical structural modifications that enhance or diminish activity across multiple chemical series [32]. For natural product optimization, C-SAR enables knowledge transfer between structurally distinct scaffolds, accelerating the identification of optimal substituents without being constrained to a single parent structure.
Protocol: C-SAR Analysis Implementation
- Data Curation: Compile diverse compound set with consistent biological activity data from public databases (ChEMBL) or proprietary sources.
- MMP Identification: Identify matched molecular pairs differing only at specific substitution sites using tools like DataWarrior.
- Pattern Extraction: Analyze activity cliffs (significant potency changes from minor structural modifications) across different chemotypes.
- Knowledge Application: Apply identified favorable substitutions to natural product scaffolds during optimization cycles.
Covalent Inhibitor SAR Analysis
For targeted covalent inhibitors (TCIs), SAR analysis requires special consideration of both noncovalent interactions and covalent bonding potential. The SCARdock protocol integrates quantum chemistry-based warhead reactivity calculations with traditional docking scores to predict TCI efficacy [33].
Application Note: In developing nonsubstrate-based covalent inhibitors of S-adenosylmethionine decarboxylase, this approach achieved a 70% hit rate, successfully identifying 12 new inhibitors through careful analysis of both noncovalent interactions and covalent bonding contributions [33].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2. Key Research Reagent Solutions for SAR Studies
| Reagent/Solution | Function in SAR Analysis | Application Context | |
|---|---|---|---|
| alvaDesc Molecular Descriptors | Comprehensive molecular property calculation for QSAR modeling | Shikonin derivative optimization; physicochemical property correlation | [28] |
| CDD Vault SAR Table | Visualization of structural features versus biological activity | Collaborative SAR data management and trend analysis across compound series | [3] |
| Hydrazone Building Blocks | Diverse accessory fragments for build-up library synthesis | MraY inhibitor optimization; rapid analogue generation | [18] |
| Matched Molecular Pairs (MMPs) | Identification of activity cliffs across diverse chemotypes | C-SAR analysis of HDAC6 inhibitors; knowledge transfer between series | [32] |
| SCARdock Protocol | Integrated covalent/noncovalent docking and reactivity assessment | Targeted covalent inhibitor discovery for AdoMetDC | [33] |
AI-Enhanced SAR Implementation
Modern Molecular Representation Methods
Traditional molecular representations like Simplified Molecular-Input Line-Entry System (SMILES) and molecular fingerprints are increasingly being supplemented by AI-driven approaches that learn continuous feature embeddings directly from molecular data [27]. These advanced representations include:
- Graph Neural Networks (GNNs): Capture both local and global molecular features through graph-based learning.
- Transformer Models: Treat molecular structures as chemical language, enabling sophisticated pattern recognition.
- Multimodal Learning: Integrate multiple representation types for enhanced predictive capability.
These AI-driven representations have proven particularly valuable for scaffold hopping - identifying structurally distinct cores that maintain similar biological activity - which is essential for natural product optimization to overcome limitations of original scaffolds [27].
Automated SAR Workflows
The implementation of Scientific Data Management Platforms (SDMPs) such as CDD Vault has become critical for supporting AI-ready SAR workflows. These platforms provide:
- Structured Data Capture: Ensures consistent formatting of chemical structures and associated biological data.
- Advanced Search Capabilities: Enables substructure, similarity, and pattern recognition across compound series.
- Bioisosteric Suggestion Tools: Facilitates strategic molecular modifications during optimization cycles.
- Integration with ML Models: Allows direct application of predictive algorithms to structured SAR data [34].
Figure 2 illustrates the build-up library synthesis approach, which exemplifies modern high-efficiency SAR exploration for natural products.
Figure 2. Build-Up Library Synthesis for SAR Exploration - This workflow demonstrates the efficient generation and screening of natural product analogues using fragment ligation approaches.
SAR analysis continues to evolve as an indispensable component of modern drug discovery, particularly in the optimization of natural product leads. The integration of computational predictions, efficient library synthesis approaches, and AI-enhanced molecular representations has created a powerful paradigm for accelerating therapeutic development. The critical importance of SAR is reflected in its successful application across diverse therapeutic areas, from oncology and infectious diseases to neurodegenerative disorders and cardiovascular conditions.
Future advancements in SAR methodologies will likely include increased incorporation of multimodal AI approaches that integrate structural, biochemical, and cellular data; enhanced predictive ADMET modeling early in the optimization process; and more sophisticated scaffold hopping algorithms that leverage large-scale chemical and biological data. For research teams working with natural products, establishing robust SAR workflows supported by AI-ready data management platforms will be essential for translating complex natural scaffolds into clinically viable therapeutics.
The continued refinement of SAR-guided optimization strategies ensures that natural products will remain a vital source of inspiration and starting points for drug discovery, with modern approaches overcoming traditional limitations through systematic, data-driven structural elaboration.
Strategic Methodologies: From Library Synthesis to Lead Optimization
Fragment-Based Drug Design (FBDD) has established itself as a premier strategy for discovering small molecule therapeutics, particularly for challenging targets such as protein-protein interactions [35] [36]. This approach utilizes low molecular weight compounds (typically ≤300 Da) as starting points, which despite their weak initial binding affinity, efficiently sample chemical space and can be optimized into high-quality drug leads [37]. When integrated with natural product research, FBDD offers powerful strategies to navigate the complex chemical space of natural extracts and address the inherent challenges of structural redundancy and bioactive rediscovery [38] [39].
This application note details practical build-up library strategies that combine fragment-based design with in-situ screening techniques, framed within a structure-activity relationship (SAR) directed optimization workflow for natural product research. We provide validated protocols, quantitative performance data, and essential toolkits to enable researchers to implement these approaches effectively.
Strategic Framework and Rationale
The Synergy Between Natural Products and FBDD
Natural products provide privileged scaffolds with evolved biological relevance and high structural diversity [39]. However, their structural complexity often hampers rapid SAR studies. Fragment-based approaches address this by deconstructing complex natural products into simpler structural units or using natural product-derived fragments as starting points for de novo design [39]. This strategy combines the bioactive relevance of natural architectures with the systematic optimizability of fragment libraries.
Rational library design significantly enhances screening efficiency. Recent studies demonstrate that leveraging liquid chromatography-tandem mass spectrometry (LC-MS/MS) spectral similarity to reduce library size can achieve an 84.9% reduction in resources needed while increasing bioassay hit rates against microbial targets [38]. For instance, in a library of 1,439 fungal extracts, a rationally designed subset of only 50 extracts captured 80% of the scaffold diversity present in the full library, a 28.8-fold size reduction [38].
Key Methodological Advantages
- Efficient Chemical Space Sampling: Fragment libraries cover broader chemical space with fewer compounds due to their small size and structural simplicity [37].
- Enhanced Hit Rate: Reducing chemical redundancy through rational library design increases the probability of identifying unique bioactive scaffolds [38].
- SAR-Driven Optimization: Fragment hits with weak but efficient binding provide ideal starting points for systematic SAR exploration through iterative structural elaboration [36] [37].
Table 1: Comparative Performance of Rational vs. Random Library Design
| Screening Parameter | Full Library (1,439 extracts) | 80% Diversity Rational Library (50 extracts) | Random Selection of 50 Extracts |
|---|---|---|---|
| Anti-P. falciparum Hit Rate | 11.26% | 22.00% | 8.00-14.00% (quartile range) |
| Anti-T. vaginalis Hit Rate | 7.64% | 18.00% | 4.00-10.00% (quartile range) |
| Neuraminidase Inhibition Hit Rate | 2.57% | 8.00% | 0.00-2.00% (quartile range) |
| Scaffold Diversity Level | 100% | 80% | 40-60% (estimated) |
| Features Correlated with Anti-P. falciparum Activity Retained | 10 | 8 | Variable |
Experimental Protocols
Protocol 1: LC-MS/MS-Based Library Rationalization
Purpose: To dramatically reduce natural product library size while minimizing bioactive loss and increasing screening hit rates.
Materials:
- Natural product extract library
- Liquid chromatography-tandem mass spectrometry system
- GNPS classical molecular networking software
- Custom R code for diversity-based selection
Procedure:
- LC-MS/MS Data Acquisition: Perform untargeted LC-MS/MS analysis on all library extracts using standardized conditions.
- Molecular Networking: Process MS/MS fragmentation patterns through GNPS to group spectra into structural scaffolds based on fragmentation similarity [38].
- Scaffold Diversity Assessment: Calculate scaffold diversity across the library using custom algorithms.
- Iterative Library Design:
- Select the extract with the greatest scaffold diversity.
- Iteratively add extracts containing the most scaffolds not yet represented.
- Continue until desired diversity threshold is reached (typically 80-100% of maximal diversity) [38].
- Bioactivity Validation: Screen rationalized library against relevant targets; compare hit rates with full library and randomly selected subsets.
Typical Results: Implementation of this protocol on a fungal extract library achieved 80% scaffold diversity with only 50 extracts (versus 109 needed with random selection) and increased hit rates from 11.26% to 22.00% for anti-Plasmodium activity [38].
Protocol 2: Affinity Selection Mass Spectrometry (AS-MS) Screening
Purpose: To identify ligand-target interactions directly from complex natural product mixtures without labeling.
Materials:
- Biological target (soluble protein, membrane receptor, nucleic acid)
- Natural product library or extracts
- Ultrafiltration devices (molecular weight cut-off appropriate for target)
- LC-MS system with electrospray ionization
- Appropriate buffers and denaturing solvents
Procedure:
- Equilibrium Establishment: Incubate target protein with natural product library at micromolar concentrations optimal for detecting high-affinity ligands [40].
- Complex Separation: Apply ultrafiltration to separate ligand-protein complexes from unbound molecules. Centrifugal force, vacuum, or pressure can drive separation.
- Ligand Dissociation: Disrupt non-covalent bonds using denaturing conditions (e.g., methanol or acetonitrile with volatile organic acid like formic acid) [40].
- Ligand Identification: Analyze dissociated ligands via LC-MS. For natural product libraries, perform fragmentation experiments and use spectral libraries with molecular networking for structural annotation [40].
- Hit Validation: Confirm binding through dose-response experiments and functional assays.
Applications: This protocol has been successfully applied to identify 5-lipoxygenase ligands from Inonotus obliquus, leading to the discovery of botulin, lanosterol, and quercetin as potential inhibitors [40].
Protocol 3:In-SituClick Chemistry Screening
Purpose: To generate potent inhibitors directly within the target's binding pocket through in-situ click chemistry.
Materials:
- Target protein (enzyme or receptor)
- Azide and alkyne-functionalized fragment libraries
- Copper catalyst (for CuAAC) or appropriate strain-promoted reagents
- LC-MS or HPLC systems for reaction monitoring
- Biophysical validation tools (SPR, ITC, X-ray crystallography)
Procedure:
- Fragment Library Design: Create fragment libraries containing azide and alkyne functional groups for copper-catalyzed azide-alkyne cycloaddition (CuAAC) or strain-promoted variants [41] [25].
- In-Situ Reaction: Incubate target protein with complementary fragment libraries under physiological conditions to allow templated synthesis of triazole products within the binding pocket [25].
- Product Detection: Monitor reaction progress via LC-MS. Identify triazole products formed through specific target-templated reactions.
- Hit Characterization: Isplicate and characterize products for binding affinity and functional activity.
- SAR Exploration: Use the triazole core as a scaffold for further optimization through synthetic elaboration.
Advantages: This protocol leverages the target protein itself to select and synthesize its own inhibitors from complementary fragment pairs, often resulting in high-affinity ligands with optimized binding characteristics [25].
Research Reagent Solutions
Table 2: Essential Research Reagents for Fragment-Based Natural Product Screening
| Reagent/Technology | Function | Application Notes |
|---|---|---|
| GNPS Classical Molecular Networking | Groups MS/MS spectra into structural scaffolds based on fragmentation similarity | Enables scaffold-based diversity assessment; critical for rational library design [38] |
| Microporous Fixed-Target Sample Holders | High-throughput serial crystallography for fragment screening | Enables room-temperature data collection capturing physiologically relevant conformations [42] |
| Ultrafiltration Devices | Separation of protein-ligand complexes from unbound molecules | MWCO should be appropriate for target size; enables AS-MS screening [40] |
| Cu(I) Catalysts | Catalyzes azide-alkyne cycloaddition for in-situ click chemistry | Essential for CuAAC reactions in in-situ screening protocols [41] [25] |
| Fragment Libraries with Poised Functionality | Provides starting points with defined derivatization sites | Contains analogues around each core for rapid SAR assessment [37] |
| RECAP Algorithm | Retrosynthetic fragmentation of natural products into building blocks | Generates fragment libraries from natural product databases [39] |
| Biacore Systems with SPR | Label-free detection of fragment binding | Enables high-throughput screening on target arrays; reveals selectivity patterns [36] |
Workflow Visualization
Diagram 1: Integrated workflow for fragment-based design from natural product libraries.
Diagram 2: AS-MS workflow for ligand discovery from complex mixtures.
Performance Metrics and Data Analysis
Quantitative Assessment of Protocol Efficiency
Table 3: Retention of Bioactive Compounds in Rationally Designed Libraries
| Bioactivity Assay | Significant Features in Full Library | Retained in 80% Diversity Library | Retained in 100% Diversity Library |
|---|---|---|---|
| Anti-P. falciparum Activity | 10 | 8 (80%) | 10 (100%) |
| Anti-T. vaginalis Activity | 5 | 5 (100%) | 5 (100%) |
| Neuraminidase Inhibition | 17 | 16 (94%) | 17 (100%) |
The data demonstrate that rational library design preserves most bioactive components while dramatically reducing library size. Even at 80% diversity coverage, the protocol retains 80-100% of bioactive features correlated with target activity [38]. This conservation of bioactivity combined with reduced screening burden underscores the efficiency of the approach.
Temperature conditions during screening significantly impact results. Recent systematic comparisons of fragment screening at room temperature versus cryogenic conditions reveal that room-temperature serial crystallography captures previously unobserved conformational states of active sites, offering additional starting points for drug design [42]. However, cryogenic screening typically identifies more binders overall, though some may represent non-physiological interactions [42].
Concluding Remarks
The integration of fragment-based design with in-situ screening strategies provides a powerful framework for natural product-based drug discovery. The protocols outlined herein enable researchers to efficiently navigate the chemical complexity of natural extracts while maintaining focus on structurally diverse, biologically relevant scaffolds. By applying these build-up library strategies within an SAR-driven optimization paradigm, research teams can accelerate the transformation of natural product leads into developable therapeutic candidates.
The continued advancement of these approaches, particularly through the incorporation of cutting-edge computational methods, structural biology, and AI-assisted design, promises to further enhance their impact on natural product research and drug development pipelines [35] [41] [36].
The optimization of natural products through Structure-Activity Relationship (SAR) studies represents a cornerstone of modern drug discovery. Natural products provide privileged scaffolds with evolved biological activity, yet they often require optimization to enhance potency, selectivity, and pharmacokinetic properties for therapeutic application. This application note details practical methodologies for two fundamental optimization strategies: functional group manipulation and bioisosteric replacement, framed within the context of SAR-directed natural product research.
Functional group manipulation through systematic SAR studies enables researchers to determine the importance of specific structural elements for biological activity [43]. When a new active compound is discovered, chemists create analogs through alterations in its molecular structure to produce new compounds with similar or improved activity [43]. The approach proceeds with successive iterations, starting with the analysis of the structure of the initial lead molecule [43].
Bioisosterism serves as a qualitative technique for the rational modification and optimization of bioactive compounds, providing several beneficial effects including increased potency, enhanced selectivity, improvements in pharmacokinetic properties, and elimination of toxicity [44]. The installation of a bioisostere leads to structural changes in molecular size, shape, electronic distribution, polarity, pKa, dipole, or polarizability, which can be beneficial or detrimental to biological activity [44].
Fundamental Principles and Definitions
Structure-Activity Relationships (SAR) in Natural Product Optimization
SAR analysis involves relating biological activities to molecular structures to maximize the knowledge that can be extracted from raw data in molecular terms [43]. This knowledge is exploited to identify which molecules should be synthesized and to select lead compounds for additional modifications or further pre-clinical studies [43]. For natural products, this is particularly valuable as it helps identify key structural features contributing to their biological activity [45].
Data of high informational content is obtained when derived from single structural modifications of an initial lead structure [43]. The introduction of multiple changes should be avoided because of the difficulties created for the correct interpretation of the biological results [43]. Each novel molecule synthesized is expected to yield useful knowledge about the structural requirements for activity [43].
Bioisosteres: Classical and Nonclassical
Bioisosteres are classified into two main categories: classical and nonclassical [46] [44].
Classical bioisosteres satisfy Grimm's hydride displacement law and Erlenmeyer's concepts of isosteres and can be subdivided into five categories [44]:
- Monovalent atoms or groups (e.g., F, H, NH₂, OH, CH₃, CF₃)
- Divalent atoms or groups (e.g., -O-, -S-, -CH₂-)
- Trivalent atoms or groups (e.g., -N=, -P=, -CH=)
- Tetravalent atoms or groups (e.g., =C=, =Si=)
- Ring equivalents (e.g., benzene, thiophene, pyridine)
Nonclassical bioisosteres possess more advanced mimicry of their emulated counterparts and do not fulfill the criteria of steric and electronic factors required for classical isosteres [44]. These include exchangeable groups, carbonyl group bioisosteres, amide group bioisosteres, and ring versus acyclic structures [44].
Table 1: Classical Bioisosteres Categories and Examples
| Category | Description | Examples |
|---|---|---|
| Monovalent | Atoms or groups with similar binding properties | F and H; NH and OH; CH₃ and CF₃ |
| Divalent | Atoms or groups with two binding sites | -O-; -S-; -CH₂-; -C=O |
| Trivalent | Atoms or groups with three binding sites | -N=; -P=; -CH= |
| Tetravalent | Atoms or groups with four binding sites | =C=; =Si=; =N+= |
| Ring Equivalents | Aromatic or aliphatic ring replacements | Benzene vs. pyridine vs. thiophene |
Experimental Protocols for SAR Determination
Protocol 1: Probing Hydrogen Bond Interactions
Principle: This method determines the existence and role of hydrogen bond interactions with specific functional groups by preparing analogs where the functional group is unable to form hydrogen bonds [43].
Procedure:
- Identify potential H-bonding groups: Screen the natural product structure for hydroxyl (-OH), carbonyl (C=O), and amine (-NH-) groups.
- Design synthetic analogs:
- For hydroxyl groups: Prepare O-alkylated (ether) derivatives or replace with hydrogen or methyl groups.
- For carbonyl groups: Replace C=O with C=CH₂, CH₂, or reduce to alcohol.
- For amine groups: Alkylate to form tertiary amines or replace with non-hydrogen bonding groups.
- Evaluate biological activity of analogs compared to parent compound.
- Interpret results:
- If activity drops significantly, the original group likely participates in critical H-bonding.
- If activity remains, the group is not essential for binding.
Case Example: Pyrazolopyrimidines Replacing a phenolic OH with a methoxy group led to complete loss of biological activity, indicating the hydroxyl forms a crucial hydrogen bond with the receptor as a hydrogen bond donor [43]. Similarly, in benzimidazoles, replacement of a phenolic OH with both methoxy and hydrogen atoms confirmed the importance of this group for activity [43].
Protocol 2: Amide Bond Bioisosteric Replacement
Principle: Amide bonds are enzymatically labile in vivo; replacement with bioisosteres can improve metabolic stability while maintaining biological activity [44].
Procedure:
- Identify amide bonds in the natural product structure.
- Select appropriate bioisosteres based on:
- Similar geometry and electronics
- Synthetic accessibility
- Potential to maintain key interactions
- Common amide bioisosteres include:
- 1,2,3-triazoles
- Oxadiazoles
- Imidazoles
- Tetrazoles
- Reverse amides
- Sulfonamides
- Thioamides
- Carbamates
- Synthesize analogs incorporating selected bioisosteres.
- Evaluate:
- Biological potency
- Metabolic stability in liver microsomes
- Selectivity profile
- Physicochemical properties
Case Example: Peptidomimetics The U.S. FDA has approved over 60 peptide drugs, but therapeutic peptides often suffer from poor metabolic stability due to rapid degradation of amide bonds by proteases [44]. Replacement of amide bonds with bioisosteres such as 1,2,3-triazoles, oxadiazoles, or sulfonamides has yielded new peptidomimetics with improved biological properties and retention of therapeutic effect [44].
Protocol 3: Metabolic Soft Spot Identification and Stabilization
Principle: Identify metabolically vulnerable sites in natural products and stabilize them through strategic modification using bioisosteres [46].
Procedure:
- Conduct metabolic stability assays:
- Incubate compound with liver microsomes or hepatocytes
- Identify metabolites using LC-MS/MS
- Locate metabolic soft spots
- Implement stabilization strategies:
- For vulnerable C-H bonds: Incorporate deuterium (deuterium kinetic isotope effect) or fluorine
- For susceptible functional groups: Employ bioisosteric replacement
- Evaluate stabilized analogs:
- Metabolic stability in liver microsomes
- CYP enzyme inhibition potential
- In vivo pharmacokinetics
Case Example: Deuterium Replacement Deucravacitinib, a selective Janus kinase (JAK) inhibitor of TYK2, contains a deuterated "magic-methyl" moiety critical for activity [46]. Deuterium blocks metabolism at the sp³ site since the C-D bond is ~5-10-fold stronger than the C-H bond due to lower vibration frequencies [46].
Table 2: Common Metabolic Soft Spots and Bioisosteric Solutions
| Metabolic Soft Spot | Bioisostere Solution | Effect |
|---|---|---|
| Aromatic C-H | Fluorine substitution | Blocks oxidation; similar bond length |
| Aliphatic C-H | Deuterium substitution | Slows metabolism via kinetic isotope effect |
| Ester group | Amide, reverse ester, heterocycle | Improved enzymatic stability |
| Phenolic OH | Fluorine, methoxy, bioisosteric rings | Blocks glucuronidation and sulfation |
| N-Dealkylation | Cyclization, conformational constraint | Prefers oxidative metabolism |
Computational and AI-Enhanced Approaches
Modern SAR analysis has been revolutionized by computational methods and artificial intelligence. The new computational Cross-Structure-Activity Relationship (C-SAR) approach accelerates structure development by examining a library of molecules with diverse chemotypes for pharmacophoric substituents exhibiting distinct substitution patterns and their associated biological activities [32].
C-SAR facilitates the generation of novel compounds by eliminating the necessity for SAR studies to be conducted solely on a single parent chemical [32]. This strategy uses matched molecular pairs (MMP) analysis – molecules with the same parent structure – to extract SAR from the examined series [32].
AI-powered approaches are revolutionizing pharmacology by enhancing compound optimization, predictive analytics, and molecular modeling [47]. Machine learning (ML), deep learning (DL), and generative models integrate with traditional computational methods such as molecular docking, quantum mechanics, and molecular dynamics simulations [47]. Core AI algorithms including support vector machines, random forests, graph neural networks, and transformers have applications in molecular representation, virtual screening, and ADMET property prediction [47].
Figure 1: Integrated Workflow for Natural Product Optimization
Case Studies in Natural Product Optimization
Anti-Tuberculosis Natural Products
Natural products have played a crucial role in the discovery of anti-TB drugs, with four of the first-line anti-TB drugs (ethambutol, isoniazid, pyrazinamide, and rifampicin) developed from natural sources [45]. SAR studies have been essential for identifying novel structural features that enhance drug efficacy against Mycobacterium tuberculosis [45].
Tryptanthrin Optimization: Tryptanthrin is an indoloquinazoline alkaloid with antimycobacterial activity against M. tuberculosis H37Rv [45]. Optimization through functional group manipulation demonstrated:
- Fluorination in the benzene ring increased MIC value from 1.00 to 0.06 mg/L
- Chlorination also improved activity but slightly less than fluorination
- The most optimal structure was achieved with OCF₃ substitution at R8 with MIC value of 0.03 mg/L [45]
This case demonstrates how systematic SAR through halogen substitution significantly enhanced natural product potency.
OTB-658 Development Through SAR
The development of OTB-658 represents a successful case of comprehensive SAR study leading to a preclinical candidate [45]:
- The journey began with fluoro-benzoxazinyl-oxazolidinone (compound A) incorporating an acetylaminomethyl unit from linezolid
- Compound A demonstrated good anti-TB activity (MIC90 of 0.48 µg/mL) with low cytotoxicity
- Optimization led to compound B with a hydroxyacetyl moiety replacing the dihydroxypropanoyl group
- Compound B showed enhanced activity (MIC90 of 0.39 µg/mL) and improved pharmacokinetic profile
- Further optimization yielded OTB-658 with MIC90 of 0.03 µg/mL against both drug-susceptible and drug-resistant strains [45]
This case highlights the iterative nature of SAR-driven optimization in natural product-based drug discovery.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for SAR Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Liver Microsomes | Metabolic stability assessment | Identifying metabolic soft spots |
| Coupling Reagents (e.g., carbodiimides) | Amide bond formation | Synthesis of analogs |
| Deuterated Solvents (e.g., DMSO-d6) | NMR spectroscopy | Structural characterization |
| HPLC-MS System | Compound purity and identity | Analytical characterization |
- Molecular Docking Software (e.g., Molecular Operating Environment): Predicts binding modes and interactions between compounds and target proteins [32].
- Data Analysis Software (e.g., DataWarrior): Cheminformatics data configuration, visualization, and graphical presentations of datasets [32].
- AI/ML Platforms (e.g., Random Forest, XGBoost): Predictive modeling of compound activity and properties [45] [47].
- Pharmacophore Modeling Software (e.g., BIOVIA Discovery Studio): Construction of pharmacophore models for ligand-based drug design [48].
Figure 2: Essential Research Tools for SAR Studies
Functional group manipulation and bioisosteric replacement remain fundamental strategies in the SAR-directed optimization of natural products. The iterative process of synthesizing analogs, testing biological activity, and interpreting results continues to drive the development of improved therapeutic agents from natural leads.
Future directions in the field include:
- AI-enhanced SAR analysis: The integration of deep learning and knowledge graphs significantly improves the accuracy of target prediction and builds interdisciplinary collaboration networks across chemical informatics, systems biology, and clinical medicine [48].
- C-SAR methodologies: Cross-structure-activity relationship approaches enable SAR expansion beyond single chemical series, applying existing knowledge of various compounds targeting the same biological entity to new chemotypes [32].
- Advanced bioisostere libraries: Expanding collections of characterized bioisosteres with known ADMET properties and synthetic accessibility.
These advances, combined with the fundamental protocols outlined in this application note, provide researchers with a comprehensive toolkit for optimizing natural product leads into viable therapeutic candidates through rational SAR-driven design.
Application Notes
The integration of Quantitative Structure-Activity Relationship (QSAR) modeling, molecular docking, and pharmacophore modeling has become a cornerstone in modern drug discovery, providing a powerful computational framework for the structure-activity relationship (SAR)-directed optimization of natural product leads. These methods are particularly valuable for optimizing the complex chemical scaffolds of natural products, which often exhibit high structural diversity but are hampered by limited bioavailability or insufficient potency [49]. The following application notes detail the specific roles and recent advancements of these computational strategies.
QSAR for Predictive Bioactivity Modeling: QSAR models are employed to predict the biological activity of natural product analogs based on their chemical descriptors, thereby guiding the selection of promising candidates for synthesis. A significant recent advancement is the use of Multi-Task Learning (MTL), which leverages bioactivity data across related biological targets to improve prediction accuracy, especially when experimental data for a specific target is scarce. For natural product activity prediction, the integration of evolutionary relatedness metrics of protein targets (Instance-Based MTL) has been shown to outperform traditional single-task learning, with notable success in kinase and cytochrome P450 protein families [50]. Furthermore, the challenge of predicting Activity Cliffs (ACs)—pairs of structurally similar compounds with large potency differences—is a key focus. Studies confirm that while ACs present a major source of prediction error for standard QSAR models, the use of graph isomorphism networks (GINs) shows promise in improving AC classification and, consequently, lead optimization [51].
Molecular Docking for Binding Mode Analysis: Molecular docking is a well-established method for predicting the preferred orientation (pose) of a small molecule within a target protein's binding site. This provides atomic-level insights into key intermolecular interactions, such as hydrogen bonding and hydrophobic contacts, which are critical for understanding SAR [52]. Docking is extensively used in lead optimization to rationalize the potency of analogs and suggest structural modifications that could enhance binding affinity or selectivity. For example, the SCARdock protocol, which integrates quantum chemistry-based warhead reactivity calculations with non-covalent docking scores, has proven highly effective in discovering targeted covalent inhibitors, achieving a 70% hit ratio for S-adenosylmethionine decarboxylase inhibitors [33].
Pharmacophore Modeling for Feature-Based Screening: A pharmacophore model abstractly represents the spatial arrangement of essential chemical features (e.g., hydrogen bond donors/acceptors, hydrophobic regions, aromatic rings) necessary for a molecule's biological activity. These models can be derived from the alignment of active compounds (ligand-based) or from the 3D structure of a protein-ligand complex (structure-based). They are particularly useful for the virtual screening of compound libraries to identify novel chemotypes that fulfill the same pharmacophoric requirements, a process known as scaffold hopping [52] [32]. This approach directly supports SAR expansion by identifying diverse structural motifs that can modulate activity.
Integrated and Emerging Workflows: The synergy of these methods creates robust workflows for natural product optimization. For instance, high-throughput crystallographic screening of crude reaction mixtures can now directly yield crystallographic SAR (xSAR) data. This purification-agnostic approach uses a rule-based scoring scheme to identify conserved chemical features linked to binding, enabling rapid SAR model building and hit optimization without the need for initial compound purification [53]. Another emerging methodology is the Cross-Structure-Activity Relationship (C-SAR) approach. C-SAR analyzes Matched Molecular Pairs (MMPs) across diverse chemotypes to extract general rules for converting inactive compounds into active ones, thereby accelerating SAR development beyond a single parent structure [32].
Table 1: Summary of Key Computational Approaches for SAR-Driven Optimization
| Computational Method | Primary Role in SAR Optimization | Key Recent Advancements |
|---|---|---|
| QSAR Modeling | Predicts bioactivity from chemical structure to prioritize analogs. | Multi-Task Learning (MTL) with evolutionary metrics [50]; Graph Isomorphism Networks (GINs) for Activity Cliff prediction [51]. |
| Molecular Docking | Visualizes binding modes and identifies key protein-ligand interactions. | Integrated protocols like SCARdock for covalent inhibitor discovery [33]; High-throughput docking for fragment-based optimization. |
| Pharmacophore Modeling | Identifies essential chemical features for activity to guide scaffold design. | Use in scaffold hopping and virtual screening to explore diverse chemical space [32]. |
| Integrated / Emerging Methods | Combines multiple data sources for accelerated SAR analysis. | Crystallographic SAR (xSAR) from crude reaction mixtures [53]; Cross-SAR (C-SAR) analysis [32]. |
Experimental Protocols
Protocol: Multi-Task Learning QSAR for Natural Product Activity Prediction
This protocol describes the development of a QSAR model enhanced with Multi-Task Learning to predict the bioactivity of natural product derivatives, leveraging data from evolutionarily related protein targets to improve performance in data-scarce scenarios [50].
Step 1: Data Curation and Protein Classification
- Curate a dataset of natural products and their bioactivity data (e.g., IC₅₀, Ki) from public databases like ChEMBL.
- Classify the target proteins using a hierarchical system (e.g., ChEMBL's 6-level classification).
- Group proteins based on evolutionary relatedness, for instance, at the "target parent" level.
Step 2: Molecular Featurization
Step 3: Model Training with Instance-Based MTL
- Baseline: Train a Single-Task Learning (STL) model for each target protein independently.
- Feature-Based MTL (FBMTL): Train a model that shares knowledge across all proteins within a pre-defined group.
- Instance-Based MTL (IBMTL): Implement a model that weights the contribution of data from other proteins based on their evolutionary relatedness to the primary target. This optimizes the trade-off between relatedness and dataset size.
Step 4: Model Validation and Activity Prediction
- Validate model performance using cross-validation and an external test set. Key metrics include AUC-ROC, precision, and recall.
- Use the validated IBMTL model to predict the bioactivity of novel natural product analogs, prioritizing those with predicted high potency for synthesis and testing.
Protocol: Integrated Docking and Pharmacophore Modeling for Lead Optimization
This protocol outlines an integrated approach using molecular docking and pharmacophore modeling to guide the optimization of a natural product lead compound [52] [32] [55].
Step 1: Protein Preparation
- Obtain the 3D structure of the target protein (e.g., from the Protein Data Bank, PDB).
- Perform essential pre-processing steps: add hydrogen atoms, assign partial charges, and remove crystallographic water molecules unless they are part of a key binding interaction.
Step 2: Ligand Preparation and Docking
- Prepare the natural product lead and its analogs: generate 3D conformations, minimize energy, and assign correct protonation states at biological pH.
- Define the docking search space (grid) around the known binding site or via blind docking.
- Execute molecular docking simulations using software such as Molecular Operating Environment (MOE) or AutoDock. Analyze the top-ranked poses to identify critical binding interactions (hydrogen bonds, pi-pi stacking, hydrophobic contacts).
Step 3: Pharmacophore Model Generation
- Structure-based: Based on the docking pose of the most active lead, define the pharmacophore features that interact with the protein.
- Ligand-based: Align multiple active analogs and extract common chemical features to create a ligand-based pharmacophore model.
Step 4: Virtual Screening and Analog Design
- Use the validated pharmacophore model as a 3D query to screen in-house compound libraries or virtual databases for novel hits.
- Analyze the results to propose new analogs of the natural product. Focus on modifications that reinforce key pharmacophore features or improve physicochemical properties, guided by the docking-derived interaction map.
Protocol: Cross-SAR (C-SAR) Analysis for SAR Expansion
This protocol utilizes the C-SAR methodology to extract generalizable SAR rules from a diverse set of active compounds, enabling the transformation of inactive chemotypes into active ones [32].
Step 1: Construction of a Matched Molecular Pairs (MMPs) Dataset
- From a chemical database (e.g., ChEMBL), curate a set of compounds with confirmed activity and known chemotypes for a specific target (e.g., HDAC6 inhibitors).
- Fragment the compounds to generate Matched Molecular Pairs (MMPs)—pairs of compounds that differ only at a single site (a specific R-group or substituent).
Step 2: C-SAR Analysis and Rule Extraction
- For each MMP, calculate the activity difference (ΔActivity). Identify "activity cliffs" where a small structural change causes a large activity shift.
- Analyze the dataset to find recurring substitution patterns that consistently lead to increased or decreased activity across different parent scaffolds. These patterns form the basis of C-SAR rules (e.g., "replacing a methyl group with a methoxy group at position R₁ consistently boosts potency").
Step 3: Application of C-SAR Rules for Molecular Transformation
- To optimize a new chemotype, apply the extracted C-SAR rules to suggest specific chemical modifications.
- The rules provide a direct strategy for converting an inactive compound into an active one by incorporating a substituent pattern that has proven beneficial across multiple scaffolds.
Table 2: Key Research Reagent Solutions
| Research Reagent / Software | Function in Protocol | Application Context |
|---|---|---|
| ChEMBL Database | Public repository of bioactive molecules with drug-like properties. | Primary source for curating bioactivity data and building MMPs for QSAR and C-SAR analysis [32] [51]. |
| OECD QSAR Toolbox | Software for grouping chemicals, filling data gaps, and (Q)SAR model development. | Used for chemical category formation, profiling, and mechanism-based read-across in regulatory contexts [54]. |
| Molecular Operating Environment (MOE) | Integrated software for molecular modeling, simulation, and drug design. | Used for molecular docking, pharmacophore modeling, and MMP analysis in lead optimization workflows [32] [55]. |
| DataWarrior | Open-source program for data visualization and analysis. | Used for cheminformatics data configuration, visualization, and graphical presentation of chemical datasets and MMPs [32]. |
| SCARdock Protocol | Computational method combining quantum chemistry and docking for covalent inhibitors. | Specifically used for the high-efficiency discovery of targeted covalent inhibitors [33]. |
Integrating AI and Machine Learning for Predictive SAR Modeling
The integration of artificial intelligence (AI) and machine learning (ML) into Structure-Activity Relationship (SAR) modeling is revolutionizing the optimization of natural product leads in modern drug discovery. SAR, which establishes the critical relationship between a compound's chemical structure and its biological activity, has evolved from traditional qualitative analysis to sophisticated quantitative and predictive computational models [3]. This transformation is particularly valuable for natural products research, where complex chemical scaffolds present both unique opportunities and challenges for optimization. AI and ML algorithms can analyze vast chemical datasets to identify subtle patterns and predict how structural modifications will affect potency, selectivity, and other pharmacological properties, thereby accelerating the journey from natural product hits to viable drug candidates [56].
The application of AI-driven SAR modeling addresses several inherent challenges in natural product optimization, including chemical complexity, limited synthetic accessibility, and the need to maintain favorable pharmacokinetic profiles. By leveraging techniques from supervised learning to deep neural networks, researchers can now navigate the complex chemical space of natural product derivatives more efficiently, reducing the traditionally time-consuming and costly trial-and-error approach [57] [58]. This document provides detailed application notes and protocols for implementing these cutting-edge computational methodologies within the context of natural product lead optimization.
Computational Foundations for Predictive SAR
AI and ML Algorithms in SAR Modeling
The successful application of AI and ML to SAR modeling relies on selecting appropriate algorithms based on dataset characteristics and research objectives. The following table summarizes the key algorithms and their specific applications in natural product optimization:
Table 1: AI/ML Algorithms for SAR Modeling in Natural Product Research
| Algorithm Category | Specific Methods | Natural Product Applications | Key Advantages |
|---|---|---|---|
| Classical ML | Random Forests, SVM, k-NN | Preliminary screening, toxicity prediction | Handles noisy data; built-in feature selection [56] |
| Deep Learning | Graph Neural Networks, Transformers | De novo design of natural product analogs | Captures complex nonlinear structure-activity patterns [34] [56] |
| Generative Models | VAEs, GANs, Diffusion Models | Scaffold hopping and bioisostere replacement [32] | Creates novel structures with desired properties |
| Interpretability Methods | SHAP, LIME | Rationalizing natural product SAR | Explains model predictions; identifies key structural features [56] |
The transition from classical to contemporary ML approaches has significantly enhanced SAR predictive capabilities. While classical methods like Multiple Linear Regression and Partial Least Squares remain valuable for interpretable, linear relationships, they often falter with complex natural product datasets exhibiting nonlinear patterns [56]. Modern deep learning architectures, particularly graph neural networks that operate directly on molecular graph structures, can automatically learn relevant features from natural product structures without manual descriptor engineering, capturing intricate structure-activity relationships that would be difficult to discern through traditional methods [56].
Molecular Descriptors and Representations
The predictive power of AI-driven SAR models fundamentally depends on how molecular structures are numerically represented. For natural products, which often contain complex ring systems, stereochemistry, and diverse functional groups, selecting appropriate molecular descriptors is particularly important:
- 1D Descriptors: Simple molecular properties (molecular weight, logP, rotatable bonds) useful for preliminary filtering and QSAR modeling [56]
- 2D Descriptors: Topological indices, fingerprint-based representations (ECFP, MACCS) that encode molecular connectivity and substructures [56]
- 3D Descriptors: Geometric and shape-based descriptors derived from molecular conformations, crucial for natural products with specific spatial arrangements [56]
- Quantum Chemical Descriptors: HOMO-LUMO gaps, electrostatic potential surfaces, and other electronic properties that influence binding interactions [56]
- Learned Representations: Deep learning-derived embeddings that automatically capture relevant features from molecular graphs or SMILES strings [56]
For natural product optimization, combining multiple descriptor types often yields the most robust models, as this approach captures both structural and electronic properties relevant to biological activity.
Integrated Protocol for AI-Driven SAR Optimization
Protocol: AI-Enhanced SAR Expansion for Natural Product Leads
Objective: Systematically optimize natural product leads using AI-driven SAR analysis to improve potency, selectivity, and drug-like properties.
Workflow Overview:
AI-Driven SAR Optimization Workflow
Step 1: Data Curation and Preparation
- Collect all available structural and bioactivity data for the natural product and known analogs
- Apply rigorous data quality control: remove duplicates, standardize activity measurements (convert to uniform units, e.g., IC50, Ki), and verify structural integrity [56]
- For natural products with limited analog data, employ data augmentation strategies using matched molecular pairs (MMP) analysis from public databases like ChEMBL to identify relevant structural transformations [32]
- Divide data into training (80%), validation (10%), and test sets (10%) using time-split or structural clustering to ensure representative chemical space coverage
Step 2: Molecular Featurization
- Compute comprehensive molecular descriptors using tools like RDKit, DRAGON, or PaDEL [56]
- Generate 2D molecular fingerprints (ECFP6, MACCS) for similarity-based methods
- Calculate 3D descriptors for conformation-dependent properties using molecular mechanics or quantum chemical methods
- For deep learning approaches, use graph-based representations that preserve atom and bond information
Step 3: Model Training and Validation
- Train multiple AI/ML models using different algorithms (Random Forest, XGBoost, Graph Neural Networks)
- Implement rigorous cross-validation with chemical domain applicability [56]
- Apply model interpretation techniques (SHAP, LIME) to identify structural features driving activity [56]
- Validate models against external test sets and decoy compounds to assess predictive power
Step 4: Virtual Compound Generation and Prioritization
- Generate analog structures using bioisosteric replacement, scaffold hopping, and generative AI methods [34] [32]
- Predict activity and ADMET properties for all generated analogs using trained models
- Apply multi-parameter optimization to balance potency, selectivity, and drug-like properties
- Select top candidates for synthesis based on predicted activity, synthetic accessibility, and structural novelty
Step 5: Experimental Validation and Model Refinement
- Synthesize and test prioritized analogs using appropriate biological assays
- Incorporate new experimental data into the training set
- Retrain models with expanded data to improve predictive accuracy
- Iterate the process until lead optimization goals are achieved
Protocol: Cross-Structure-Activity Relationship (C-SAR) Analysis
Objective: Leverage structural and activity data across multiple chemotypes to guide natural product optimization, particularly useful when working with structurally unique natural products with limited analog data.
Methodology:
C-SAR Analysis Methodology
- Compound Library Assembly: Curate a diverse collection of compounds with demonstrated activity against the target, including both natural products and synthetic compounds from databases like ChEMBL [32]
- Matched Molecular Pair (MMP) Analysis: Identify pairs of compounds that differ only by a single structural transformation at a specific site, along with their corresponding activity changes [32]
- C-SAR Pattern Extraction: Analyze MMPs to identify structural transformations that consistently increase or decrease activity, regardless of the core scaffold [32]
- Transformational Rule Application: Apply the most promising C-SAR-derived transformations to the natural product scaffold, prioritizing modifications with demonstrated positive effects across multiple chemotypes
- Synthesis and Validation: Prepare selected analogs and validate predictions experimentally, focusing on transformations with the highest confidence scores from C-SAR analysis
The C-SAR approach is particularly valuable for natural product optimization as it leverages existing structure-activity information across multiple chemical classes, effectively expanding the available SAR knowledge beyond what would be possible studying the natural product scaffold in isolation [32].
Advanced Applications and Integration with Structural Biology
AI-Powered Structure-Based SAR
The integration of AI-predicted protein structures with SAR analysis has created new opportunities for natural product optimization, particularly for targets with limited structural information:
Table 2: Structure-Based AI Approaches for SAR Modeling
| Method | Application in Natural Product SAR | Protocol Requirements | Key Outputs |
|---|---|---|---|
| AlphaFold2 Prediction | Generate 3D target structures for natural product targets [59] | Target sequence, multiple sequence alignment | Predicted structure with confidence metrics (pLDDT) [59] |
| Ensemble Docking | Account for target flexibility in natural product binding [60] | Multiple receptor conformations, compound library | Binding poses and scores across conformations |
| Free Energy Perturbation | Precise prediction of activity changes for natural product analogs [61] | High-quality protein-ligand structures, MD setup | Relative binding free energies for designed analogs |
| AI-Augmented Molecular Dynamics | Understand binding kinetics and mechanism of action [61] | Specialized hardware (GPUs), extended simulation time | Binding/unbinding rates, conformational changes |
Advanced structural AI approaches are particularly valuable for natural product optimization against challenging targets like GPCRs, where traditional structure-based drug design has been difficult to apply. For these targets, AI-generated structural models (e.g., from AlphaFold2) provide reliable starting points for understanding natural product binding modes and rationalizing SAR observations [59]. When combined with molecular dynamics simulations and free energy calculations, researchers can create accurate models that predict how structural modifications to natural product scaffolds will affect target binding and selectivity [61].
Protocol: Structure-Based AI Workflow for Natural Product SAR
Objective: Integrate AI-predicted protein structures and computational docking to guide natural product optimization.
Step 1: Target Structure Preparation
- Obtain target protein structure from PDB or generate using AlphaFold2 for targets without experimental structures [59]
- For AlphaFold2 models, assess quality using pLDDT scores, with particular attention to binding site residues (pLDDT >80 recommended) [59]
- Generate conformational ensembles using molecular dynamics or specialized sampling to account for binding site flexibility
Step 2: Binding Mode Analysis
- Dock natural product and known analogs to identify consensus binding modes
- Perform binding site analysis to identify key interactions and potential modification sites
- Use interaction fingerprints to quantify and compare binding modes across analogs
Step 3: Structure-Based Analog Design
- Design analogs that optimize interactions with key binding site residues
- Use bioisostere replacement to maintain critical interactions while improving properties
- Apply generative AI methods constrained by binding site geometry to propose novel analogs
Step 4: Binding Affinity Prediction
- Use free energy perturbation or ML-scoring functions to predict binding affinity changes for designed analogs [61]
- Prioritize analogs with improved predicted affinity and maintained favorable interactions
- Validate top candidates through synthesis and experimental testing
Essential Research Tools and Platforms
The Scientist's Toolkit: AI-SAR Research Reagent Solutions
Table 3: Essential Resources for AI-Driven SAR Research
| Resource Category | Specific Tools/Platforms | Key Functionality | Application in Natural Product SAR |
|---|---|---|---|
| Chemical Databases | ChEMBL [32], ZINC, NPASS | Source of structural and activity data for model training | Provides analog structures and activity data for C-SAR analysis [32] |
| Cheminformatics | RDKit [56], OpenBabel, PaDEL [56] | Molecular descriptor calculation, fingerprint generation | Featurization of natural product structures for ML models |
| AI/ML Platforms | scikit-learn [56], DeepChem, PyTorch Geometric | Model development and training | Implementation of custom SAR models for natural product optimization |
| Commercial SDMPs | CDD Vault [34], Dotmatics, Benchling | Structured data management for AI readiness [34] | Centralized storage of natural product structures and assay data |
| Structure Prediction | AlphaFold2 [59], RoseTTAFold | Protein structure prediction | Generate target structures for natural product docking [59] |
| Molecular Modeling | Schrödinger, MOE [32], AutoDock | Docking, free energy calculations | Structure-based design of natural product analogs |
| Cloud Platforms | Google Cloud AI, AWS SageMaker | Scalable computing for training large models | Handling computational demands of deep learning on natural product libraries |
When selecting platforms for AI-driven natural product SAR, key considerations include support for both small molecules and biologics, role-based accessibility, bioisosteric suggestion tools, and advanced search capabilities [34]. Cloud-based platforms currently dominate due to their ability to handle large datasets and facilitate collaboration, though hybrid deployment options are emerging to balance computational power with data security requirements [57].
Validation and Best Practices
Model Validation Framework
Rigorous validation is essential for AI-driven SAR models to ensure reliable predictions in natural product optimization:
- Internal Validation: Apply k-fold cross-validation with stratified splits to maintain activity distribution, monitoring both R² (coefficient of determination) and Q² (cross-validated R²) metrics [56]
- External Validation: Reserve temporally or structurally distinct test sets, particularly including natural product analogs not represented in training data
- Domain of Applicability: Define the chemical space where models make reliable predictions using similarity-based metrics or descriptor range analysis
- Prospective Validation: Synthesize and test compounds selected based on model predictions to assess real-world performance
Addressing Common Challenges in Natural Product SAR
Natural products present specific challenges for AI-driven SAR modeling that require specialized approaches:
- Data Scarcity: Employ transfer learning from larger synthetic compound datasets, using fine-tuning techniques to adapt models to natural product chemical space
- Structural Complexity: Use graph-based representations that capture stereochemistry and complex ring systems more effectively than traditional fingerprints
- Synthetic Accessibility: Integrate synthetic complexity scores into the candidate prioritization process to ensure designed analogs are practically feasible
- Conformational Flexibility: Employ multi-conformer representations and ensemble docking to account for the structural flexibility common in natural products
The integration of AI and ML into SAR modeling represents a paradigm shift in natural product optimization, enabling more efficient navigation of complex chemical space and data-driven decision making. By implementing the protocols and best practices outlined in this document, researchers can accelerate the transformation of promising natural product hits into optimized lead candidates with improved potency, selectivity, and drug-like properties.
The escalating global health crisis of antimicrobial resistance (AMR) necessitates the development of antibiotics with novel mechanisms of action. MraY (phospho-N-acetylmuramoyl-pentapeptide-transferase), a bacterial membrane enzyme essential for peptidoglycan biosynthesis, represents a highly promising yet underexplored antibacterial target [18] [62]. It catalyzes the first membrane step of peptidoglycan synthesis, transferring the phospho-MurNAc-pentapeptide from UDP-MurNAc-pentapeptide to the lipid carrier undecaprenyl phosphate, yielding Lipid I [62]. As MraY is universally present in bacteria and has no mammalian homolog, its inhibition offers a broad-spectrum therapeutic strategy against drug-resistant pathogens such as MRSA and VRE [18] [62].
Natural products have historically been a rich source of MraY inhibitors, yielding five primary classes: muraymycins, tunicamycins, capuramycins, mureidomycins, and caprazamycins [62]. These nucleoside antibiotics share a common uridine moiety critical for binding the enzyme's active site but diverge in their accessory structures, which govern their antibacterial spectrum and potency [18]. However, the clinical translation of these natural leads has been hampered by their intrinsic structural complexity, poor pharmaceutical properties, and limited in vivo efficacy [62] [63].
This Application Note details a Structure-Activity Relationship (SAR)-driven strategy for optimizing MraY inhibitory natural products. We present a novel "build-up library" approach that streamlines analogue synthesis and evaluation, alongside key protocols for assessing compound efficacy. The methodologies described herein are designed to accelerate the transformation of complex natural product scaffolds into viable antibacterial drug candidates with enhanced potency and drug-like properties.
MraY as a Therapeutic Target and Optimization Challenge
Biological Rationale and Significance
MraY functions at a critical juncture in the cytoplasmic membrane, linking the soluble, cytoplasmic phase of peptidoglycan synthesis with the membrane-associated lipid-linked steps [62]. Inhibiting MraY disrupts the production of Lipid I, an indispensable precursor for cell wall biosynthesis, leading to bacterial cell lysis and death. Its position as a gatekeeper enzyme and high conservation across bacterial species make it an attractive target for countering multidrug-resistant infections [62].
Recent structural biology breakthroughs have illuminated the challenges and opportunities in MraY inhibitor design. The catalytic site of MraY resides on the cytoplasmic side of the membrane, requiring inhibitors to possess adequate membrane permeability to reach their target [18]. Co-crystal structures reveal that MraY is a conformationally dynamic enzyme that undergoes significant induced fit upon inhibitor binding [18]. This flexibility complicates rational design, as clear interaction pockets are often absent in the apo enzyme structure. Furthermore, MraY belongs to the polyprenyl-phosphate N-acetylhexosamine-1-phosphate transferase (PNPT) superfamily, which includes the human enzyme GPT. Achieving selectivity over GPT is crucial to avoid off-target cytotoxicity, as demonstrated by the toxicity profile of tunicamycins [62].
Key Challenges in Natural Product Optimization
Optimizing natural MraY inhibitors presents a multi-faceted challenge that integrates medicinal chemistry, structural biology, and microbiology.
- Structural Complexity: Natural MraY inhibitors often feature intricate architectures, such as nucleoside-peptide hybrids, requiring multi-step, low-yielding synthetic routes that hinder large-scale analogue production and comprehensive SAR exploration [18] [63].
- Balancing Potency and Permeability: The common uridine moiety is essential for high-affinity MraY binding but contributes to high polarity, which can limit bacterial cell penetration and overall whole-cell activity [18] [62]. Optimizing accessory fragments to improve bacterial accumulation without compromising target binding is a primary SAR objective.
- Selectivity and Cytotoxicity: Designing inhibitors that selectively target bacterial MraY over human GPT is paramount. SAR studies must carefully monitor for off-target effects, as the structural similarity between these enzymes can lead to cytotoxicity [62].
The following diagram illustrates the core challenges and optimization cycle in developing MraY inhibitors.
SAR-Driven Optimization Strategy: The Build-up Library Approach
Core Concept and Workflow
A transformative strategy for the rapid optimization of MraY inhibitors involves the construction and in situ evaluation of a "build-up library" [18]. This approach circumvents the bottleneck of traditional multi-step synthesis by dividing the natural product structure into two key fragments:
- Core Fragment: A synthetically accessible moiety containing the essential uridine pharmacophore required for binding to MraY's conserved uridine pocket.
- Accessory Fragment: A diverse collection of chemical groups designed to modulate binding affinity, bacterial accumulation, and selectivity.
These fragments are ligated using a high-yielding, chemoselective hydrazone formation reaction between an aldehyde-functionalized core and hydrazine-containing accessory fragments [18]. This reaction is ideal for in situ screening as it proceeds without reagents or by-products that could interfere with biological assays. The entire library is synthesized in a microplate format, concentrated, and directly subjected to biological evaluation without purification, dramatically accelerating the SAR cycle.
The workflow for this build-up library approach is detailed below.
Library Design and Synthesis Protocol
Protocol: Build-up Library Synthesis via Hydrazone Formation
Principle: This protocol describes the construction of a 686-member hydrazone library from 7 core aldehydes (derived from MraY inhibitory natural products) and 98 accessory hydrazines for the rapid identification of potent antibacterial agents [18].
Materials:
- Aldehyde Core Fragments (10 mM in DMSO): Derived from capuramycin, muraymycin, or other MraY inhibitors [18].
- Accessory Hydrazine Fragments (10 mM in DMSO): Include aromatic (benzoyl-type, phenyl acetyl-type), alkyl (acyl-type), and N-acyl aminoacyl hydrazides [18].
- Equipment: 96-well plates, centrifugal concentrator, LC-MS system.
Procedure:
- Fragment Preparation: Prepare stock solutions (10 mM in DMSO) of all aldehyde cores and hydrazine accessory fragments.
- Library Assembly: In a 96-well plate, combine each aldehyde core with each hydrazine fragment in a 1:1 stoichiometry. The total reaction volume per well is 31 µL [18].
- Ligation Reaction: Allow the plate to incubate at room temperature for 30 minutes to facilitate hydrazone formation.
- Solvent Removal: Place the plate in a centrifugal concentrator and remove DMSO under vacuum at room temperature overnight.
- Library Reconstitution: Add 30 µL of DMSO to each well to dissolve the reaction residues, resulting in a nominal 5 mM library solution for screening [18].
- Reaction Validation: Analyze a representative subset of reactions by LC-MS to confirm hydrazone formation. Yields typically exceed 80% [18].
Critical Notes:
- The hydrazone formation reaction is selected for its clean conversion and absence of cytotoxic by-products, making it suitable for direct cell-based assays [18].
- Biological activity is evaluated assuming 100% conversion for initial screening. Confirm potency of active hits with purified compounds.
Experimental Protocols for Biological Evaluation
MraY Enzymatic Inhibition Assay
Protocol: Radioactive MraY Inhibition Assay
Principle: This assay measures the ability of test compounds to inhibit the conversion of the UDP-MurNAc-pentapeptide substrate to Lipid I, using a radioactive tracer and scintillation proximity technology [18].
Materials:
- Particulate MraY: Membrane preparations from Staphylococcus aureus or Escherichia coli overexpressing MraY [18].
- Substrate: UDP-N-acetyl[³H]muramyl-pentapeptide (UDP-[³H]MurNAc-pp).
- Cofactor: Undecaprenyl phosphate (C55-P).
- Reaction Buffer: 50 mM HEPES (pH 7.5), 10 mM MgCl₂, 0.1% (w/v) Brij-58 [18].
- Equipment: Scintillation counter, 96-well filter plates.
Procedure:
- Reaction Setup: In a 96-well plate, add 10 µL of test compound (serially diluted in assay buffer) and 10 µL of MraY membrane preparation.
- Initiate Reaction: Add 30 µL of a substrate/cofactor mixture containing UDP-[³H]MurNAc-pp and undecaprenyl phosphate in reaction buffer.
- Incubation: Incubate the reaction mixture at 30°C for 30 minutes.
- Reaction Termination: Stop the reaction by adding 50 µL of a 5% (v/v) phosphoric acid solution.
- Product Capture and Detection:
- Transfer the entire reaction mixture to a 96-well filter plate.
- Wash the filter extensively with water to remove unincorporated radioactive substrate.
- Quantify the bound radioactive Lipid I product by scintillation counting [18].
- Data Analysis: Calculate percentage inhibition relative to a DMSO control (0% inhibition) and a no-enzyme background (100% inhibition). Determine IC₅₀ values using nonlinear regression analysis.
Whole-Cell Antibacterial Susceptibility Testing
Protocol: Determination of Minimum Inhibitory Concentration (MIC)
Principle: The MIC is the lowest concentration of an antimicrobial agent that prevents visible growth of a microorganism. This protocol standardizes the assessment of whole-cell antibacterial activity for MraY inhibitors [64] [45].
Materials:
- Bacterial Strains: Include reference strains (e.g., S. aureus ATCC 29213) and drug-resistant clinical isolates (e.g., MRSA, VRE) [18] [64].
- Culture Media: Cation-adjusted Mueller-Hinton Broth (CAMHB).
- Equipment: 96-well round-bottom microtiter plates, plate reader.
Procedure:
- Inoculum Preparation: Adjust the turbidity of logarithmically-phase bacterial cultures to a 0.5 McFarland standard (~1-2 x 10⁸ CFU/mL). Further dilute the suspension in CAMHB to achieve a final inoculum of ~5 x 10⁵ CFU/mL in the assay well.
- Compound Dilution: Serially dilute the test compound (e.g., two-fold dilutions) in CAMHB directly in the microtiter plate.
- Inoculation: Add the prepared bacterial inoculum to each well containing the diluted compound.
- Incubation: Incubate the plate at 37°C for 16-20 hours under static conditions.
- Endpoint Determination: The MIC is the lowest compound concentration that completely inhibits visible bacterial growth, as observed visually or by measuring optical density at 600 nm [45].
Critical Notes:
- Include quality control strains and reference antibiotics in each assay run.
- For in situ build-up library screening, the assay is performed directly from the DMSO library solutions [18].
Key Research Reagent Solutions
The following table catalogs essential reagents and materials required for the synthesis and evaluation of MraY inhibitors as described in the featured protocols.
Table 1: Essential Research Reagents for MraY Inhibitor Development
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Aldehyde Core Fragments [18] | Core building block for build-up library; provides essential uridine pharmacophore for MraY binding. | Synthetically derived from natural product scaffolds (e.g., capuramycin). Conjugated aldehydes enhance hydrazone stability [18]. |
| Accessory Hydrazine Fragments (BZ, PA, AC, AA, LA types) [18] | Modulates biological activity, permeability, and selectivity in build-up libraries. | Diverse chemotypes (aromatic, alkyl, amino acid-based) are crucial for probing SAR and improving bacterial accumulation [18]. |
| Particulate MraY Enzyme | Biological target for in vitro enzymatic inhibition assays. | Sourced from membrane preparations of S. aureus or E. coli. Maintain activity by avoiding repeated freeze-thaw cycles [18]. |
| UDP-N-acetyl[³H]MurNAc-pentapeptide | Radiolabeled substrate for the MraY enzymatic assay. | Enables sensitive quantification of Lipid I production. Requires specific handling and disposal procedures for radioactive materials [18]. |
| Undecaprenyl Phosphate (C55-P) [62] | Native lipid carrier substrate for MraY in the enzymatic assay. | Critical for replicating the physiological reaction condition. Solubilize in appropriate detergents (e.g., Brij-58) [18]. |
Data Analysis and SAR Interpretation
Quantitative Analysis of Inhibitor Potency
Data from biological evaluations must be systematically analyzed to establish meaningful SAR. The table below summarizes representative quantitative data for different classes of MraY inhibitors, illustrating the spectrum of achievable potency.
Table 2: Potency Profile of MraY Inhibitors from Natural and Synthetic Origins
| Compound / Class | MraY IC₅₀ | Antibacterial MIC (S. aureus MRSA) | Key Structural Feature | Reference |
|---|---|---|---|---|
| Muraymycin D2 (Natural Product) | Low nM range | ~0.5 - 1 µg/mL | Lipophilic sidechain | [62] |
| Capuramycin Analog (SQ641) | Not Reported | Sub-µg/mL (M. tuberculosis) | Aromatic substituent | [62] |
| Triazinedione 7j (Synthetic) | N/A | 2 - 4 µg/mL | 4-CF₃ aryl, guanidine, n-octyl chain [64] | [64] |
| 1,2,4-Triazole 12a (Synthetic) | 25 µM (MraY⁽ˢᵃ⁾) | Active vs. MRSA & VRE | Non-nucleoside scaffold [63] | [63] |
| Build-up Library Hit 2 | Potent Inhibition | Potent and broad-spectrum in vitro & in vivo | Optimized hydrazone [18] | [18] |
SAR Insights and Design Principles
Analysis of the biological data from build-up libraries and other synthetic campaigns reveals critical SAR trends for MraY inhibitor optimization:
- Uridine Core is Essential but Insufficient: The core uridine fragment is necessary for binding but exhibits MraY inhibitory activity 100 to 1000-fold lower than the full natural product structure. The accessory fragment is critical for high potency [18].
- Accessory Fragment Dictates Antibacterial Spectrum: The accessory fragment's structure profoundly influences the antibacterial spectrum and potency. For instance, muraymycins often require long, lipophilic substituents for effective bacterial accumulation, whereas capuramycins can utilize aromatic groups for the same purpose [18].
- Balancing Lipophilicity for Permeability and Solubility: Introducing heteroatoms (N, O, S) into accessory fragments can ameliorate the poor aqueous solubility often associated with nucleoside inhibitors, improving their drug-like properties [62].
- Achieving Selectivity over Human GPT: Structural insights into the differences between bacterial MraY and human GPT are guiding the design of inhibitors that minimize off-target cytotoxicity [62].
The SAR-driven "build-up library" strategy represents a powerful and efficient framework for optimizing complex natural product-based MraY inhibitors. By integrating streamlined chemical synthesis with direct biological evaluation, this approach rapidly generates critical SAR data, accelerating the identification of lead compounds with potent enzymatic inhibition and whole-cell antibacterial activity. The experimental protocols outlined provide a standardized workflow for synthesizing analogue libraries and rigorously assessing their biological activity. As structural insights into MraY-inhibitor complexes continue to grow, these methodologies will be instrumental in designing novel, potent, and selective antibacterial agents to combat the escalating threat of antimicrobial resistance.
Navigating Complex Challenges in Multi-Parameter Optimization
Overcoming Synthetic Bottlenecks and Library Production Challenges
The journey from a natural product lead to a pre-clinical drug candidate is paved with the systematic exploration of structure-activity relationships (SAR). However, this critical optimization phase is often hampered by significant synthetic bottlenecks, particularly when creating the diverse compound libraries required to establish a robust SAR. These challenges are exacerbated by the increasing molecular complexity of modern therapeutic modalities, including longer oligonucleotides, complex peptides, and chemically modified scaffolds derived from natural products [65] [66]. This Application Note provides detailed protocols and data-driven strategies to overcome these hurdles, enabling the accelerated SAR-directed optimization of natural leads.
Key Bottlenecks and Strategic Solutions
The production of high-quality libraries for SAR is frequently impeded by several interrelated challenges. The table below summarizes the primary bottlenecks and the corresponding strategic solutions adopted from industry and academic research.
Table 1: Common Synthetic Bottlenecks and Strategic Solutions in Library Production
| Bottleneck Category | Specific Challenge | Proposed Solution | Key Benefit |
|---|---|---|---|
| Synthesis & Purification | Low yields & aggregation in long peptide sequences [66] | Use of pseudoproline building blocks, high-swelling resins, and fragment condensation [66] | Mitigates aggregation, improves yield and purity |
| Synthesis & Purification | Time-consuming impurity profiling in oligonucleotides [65] | Automated LC-UV-MS data processing workflows [65] | Reduces analysis time from ~6 hours to 30 minutes |
| Synthesis & Purification | Challenging purification of structurally similar impurities [66] | Advanced techniques like Supercritical Fluid Chromatography (SFC) & Centrifugal Partition Chromatography (CPC) [66] | Offers scalable, high-resolution alternatives to RP-HPLC |
| Analysis & Data Management | High volume of complex, siloed data [65] | Implementation of unified software platforms for data analysis [65] | Standardizes workflows, enables high-throughput decision-making |
| Analysis & Data Management | Reliance on slow, manual data processing [65] | Automation of routine data analysis and characterization tasks [65] | Frees expert resources for higher-value strategic work |
| Library Screening | Physical binding data lacking functional context [67] | Application of DNA-Encoded Libraries (DELs) in functional or phenotypic assays [67] | Provides richer biological insight during hit identification |
Case Study: SAR-Driven Optimization of a Novel mIDH1 Inhibitor
The following section details a practical example of overcoming synthetic challenges to perform a successful SAR study, leading to the identification of a potent dual inhibitor.
Lead Identification and Library Synthesis
An in-house library of 109 compounds, sourced from ongoing projects in antitumor drug discovery and natural product synthesis, was screened at a single 2 µM concentration for inhibitory activity against the IDH1 R132H mutant [17] [68]. This led to the identification of a dibromoacetophenone derivative, compound 1-1, which showed 73.6% inhibition and served as the lead for SAR exploration [68].
Systematic structural modifications were undertaken to establish the SAR. The synthesis involved acylation of appropriate amine precursors with bromoacetyl bromide, followed by further functionalization to produce a focused library of analogs [17]. This iterative "synthesize-and-test" cycle required a reliable and reproducible synthesis protocol to rapidly generate the necessary compounds.
Key SAR Findings and Potency Optimization
The SAR investigation focused on several regions of the lead compound 1-1, with key findings summarized in the table below.
Table 2: Summary of SAR and In Vitro Potency for Optimized mIDH1 Inhibitors
| Compound | R Group | IC50 vs IDH1 R132H (nM) | IC50 vs IDH1 R132C (nM) | EC50 (2-HG in cells) | Key Finding |
|---|---|---|---|---|---|
| Lead 1-1 | (Initial lead) | Not reported (73.6% inhib. at 2 µM) | Not reported | Not reported | Starting point, also a PDK1 inhibitor [68] |
| Compound 7a | Phenethyl | 115.0 | 102.0 | Not reported | Demonstrated importance of lipophilic group [17] |
| Compound 15j | 4-Fluorophenethyl | 93.0 | 87.0 | Not reported | Fluorine substitution slightly improves potency [17] |
| Compound 27j | 4-(Methylsulfonyl)phenethyl | 80.0 | 58.0 | 69.0 nM (U87-MG cells) | Optimal compound; subnanomolar potency, minimal wt-IDH1/2 activity, dual mIDH1/PDK1 inhibition (PDK1 IC50 = 0.61 µM) [17] [68] |
Through this SAR campaign, the 4-(methylsulfonyl)phenethyl analog (27j) was identified as a highly potent and selective dual inhibitor of mIDH1 and PDK1, demonstrating significant cellular activity by reducing the oncometabolite 2-HG and inhibiting the proliferation of mIDH1-driven cancer cells [17] [68].
SAR Workflow Diagram
Detailed Experimental Protocols
Protocol: Enzymatic Inhibition Assay for mIDH1 R132H
This protocol is adapted from the methodology used to identify and optimize the dibromoacetophenone inhibitors [17] [68].
4.1.1 Principle The assay measures the NADPH-dependent production of the oncometabolite 2-hydroxyglutarate (2-HG) by the mutant IDH1 R132H enzyme. Test compounds are evaluated for their ability to inhibit this reaction.
4.1.2 Reagents and Materials
- Recombinant mutant IDH1 R132H protein
- Substrate solution: α-Ketoglutarate (α-KG) and NADPH in assay buffer
- Assay Buffer: Tris-HCl (pH 7.5), MgCl₂, NaCl
- Test compounds (e.g., 10 mM stock in DMSO)
- Detection reagent for 2-HG (e.g., fluorescent or luminescent)
4.1.3 Procedure
- Reaction Setup: In a 96-well plate, prepare a 50 µL reaction mixture containing:
- Assay Buffer
- 10 µM α-KG
- 100 µM NADPH
- 50 nM IDH1 R132H enzyme
- Test compound at desired concentration (e.g., 2 µM for single-point screening; a serial dilution for IC50 determination).
- Incubation: Incubate the reaction plate at 37°C for 1 hour.
- Reaction Termination: Stop the reaction by adding 50 µL of the detection reagent.
- Detection: Incubate for 10 minutes at room temperature and measure the signal (e.g., fluorescence/ luminescence) using a plate reader.
- Data Analysis: Calculate percent inhibition relative to a DMSO control (0% inhibition) and a no-enzyme control (100% inhibition). Fit dose-response data to determine IC50 values.
Protocol: Automated LC-MS Data Processing for Oligonucleotide Impurity Profiling
This protocol outlines the automated workflow implemented to overcome analytical bottlenecks in oligonucleotide analysis, as demonstrated by Roche and Genedata [65].
4.2.1 Principle Liquid Chromatography with Ultraviolet and Mass Spectrometry detection (LC-UV-MS) coupled with automated data processing software is used to rapidly identify and quantify oligonucleotide impurities and truncated sequences.
4.2.2 Equipment and Software
- HPLC system coupled with UV detector and mass spectrometer
- Automated data processing platform (e.g., Genedata Expressionist)
- Computer workstation
4.2.3 Procedure
- Sample Preparation: Reconstitute crude oligonucleotide samples in appropriate solvent (e.g., water).
- LC-UV-MS Analysis:
- Column: C18 or ion-exchange column suitable for oligonucleotides.
- Gradient: Optimized water/acetonitrile gradient with ion-pairing reagents.
- MS Detection: Electrospray Ionization (ESI) in negative mode.
- Data Automation Workflow:
- Data Ingestion: Raw LC-UV-MS data files are automatically imported into the processing platform.
- Peak Picking & Deconvolution: The software automatically identifies UV peaks and deconvolutes MS spectra to determine the molecular weight of each component.
- Impurity Identification: Truncated (n-1, n-2) and other impurity sequences are automatically flagged by comparing observed masses to theoretical values.
- Quantification: The relative abundance of each impurity is calculated based on UV peak area.
- Report Generation: A comprehensive report detailing sample purity and impurity profile is automatically generated.
4.2.4 Notes
- Implementation of this automated solution has been shown to reduce analysis time from 5-6 hours to approximately 30 minutes per sample [65].
- This allows for high-throughput characterization of synthesis outcomes, directly feeding back into the SAR-driven optimization cycle by quickly informing chemists on reaction purity and success.
mIDH1 Inhibition Assay
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and materials critical for executing the protocols and overcoming the synthetic bottlenecks described in this note.
Table 3: Essential Research Reagents and Their Functions in SAR-Driven Optimization
| Reagent / Material | Function / Application | Key Consideration |
|---|---|---|
| Pseudoproline Dipeptides | Prevents aggregation during solid-phase peptide synthesis (SPPS) of difficult sequences, enabling longer, more complex peptides [66]. | Critical for improving crude yield and purity, directly impacting library quality. |
| High-Swelling Low-Loading Resins | Provides a better reaction environment for SPPS, minimizing intermolecular interactions and deletion sequences [66]. | Essential for synthesizing hydrophobic or structured peptides. |
| Fragment Condensation Building Blocks | Enables chemical synthesis of long peptides (50-150 AA) by coupling pre-purified, shorter fragments [66]. | Overcomes length limitations of linear SPPS. |
| DNA-Encoded Library (DEL) Kits | Facilitates the creation of ultra-large combinatorial libraries (billions of members) for high-throughput screening against purified targets [67] [69]. | Provides a bridge between synthetic chemistry and biological screening. |
| CleanCap Capping Reagents | Co-transcriptional capping for in vitro transcribed (IVT) RNA, enhancing stability and translational efficiency [70]. | Key for producing high-quality mRNA for therapeutics and vaccines. |
| Lipid Nanoparticle (LNP) Formulation Kits | Encapsulates and protects nucleic acid payloads (RNA, oligonucleotides) for efficient cellular delivery [70]. | Enables functional cellular and in vivo testing of nucleic acid-based leads. |
| Stable Cell Lines Overexpressing Target Protein | Provides a consistent and reproducible source of protein for enzymatic assays and compound screening [17]. | Crucial for reliable and scalable bioactivity assessment. |
Within the context of structure-activity relationship (SAR) directed optimization of natural leads, achieving a balance between high biological potency and desirable absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties is a critical challenge. Natural products often serve as excellent starting points for drug discovery due to their structural complexity and bioactivity, but their optimization requires a meticulous approach to maintain efficacy while ensuring viable pharmacokinetics and safety [71] [72]. Undesirable ADMET profiles remain a principal cause of failure during drug development; it has been estimated that such deficiencies contribute to up to 50% of attrition [71]. This Application Note provides a practical framework and detailed protocols for the integrated evaluation and optimization of ADMET properties alongside potency, leveraging modern in silico tools and experimental assays.
Core ADMET Properties and Their Impact on Drug Viability
A successful drug must achieve a finely tuned combination of biochemical activity, pharmacokinetics, and safety. An ideal candidate is appropriately absorbed, distributed to target tissues, metabolized in a manner that does not immediately negate its activity, suitably eliminated, and confirmed to be non-toxic [71]. The following properties are particularly crucial during the optimization of natural product leads.
Key ADMET Properties for Natural Lead Optimization:
- Absorption: Governed by properties such as human intestinal absorption (HIA), Caco-2 permeability, and solubility. A compound must be sufficiently lipophilic to cross cell membranes but not so lipophilic that it becomes insoluble or stuck in membranes [72].
- Distribution: Key parameters include plasma protein binding (PPB) and blood-brain barrier (BBB) penetration. Only the unbound drug fraction is pharmacologically active, and BBB penetration must be carefully controlled—desired for CNS targets but avoided for others to prevent side effects [72].
- Metabolism: Stability against cytochrome P450 (CYP450) enzymes is critical, as this superfamily metabolizes 75-90% of hepatically cleared drugs. Understanding metabolic soft spots helps avoid rapid inactivation and potential drug-drug interactions [72].
- Toxicity: Screening for hERG channel inhibition (a biomarker for cardiotoxicity), mutagenicity, and other toxic endpoints is essential early in development to mitigate safety risks [72].
A Practical Framework for Integrated SAR and ADMET Optimization
The following workflow outlines a systematic, iterative process for balancing potency and ADMET properties. It begins with the initial natural product lead and cycles through prediction, synthesis, and testing until a balanced candidate is identified.
Phase 1: In Silico Prediction and Profiling
Computational tools provide a cost-effective and rapid means for early ADMET assessment, allowing researchers to prioritize which analogs to synthesize [73].
Protocol 1.1: Utilizing Web-Based ADMET Prediction Platforms
- Objective: To obtain a computational ADMET profile for a natural lead compound and its proposed analogs.
- Materials:
- Procedure:
- Input Preparation: Generate the canonical SMILES string for your compound. Alternatively, use the integrated molecular editor to draw the 2D structure.
- Platform Access: Navigate to the web server (e.g., https://admetmesh.scbdd.com/ for ADMETlab 2.0 or http://lmmd.ecust.edu.cn/admetsar3/ for admetSAR3.0).
- Job Submission: Submit the single molecule or a batch file (e.g., a .txt or .sdf file containing multiple SMILES) for evaluation.
- Result Interpretation: Review the output, which is typically displayed in a tabular format. For classification models, predictions are often given as probability ranges (e.g., 0.9-1.0(+++) for high likelihood). Pay close attention to endpoints critical for your project, such as solubility, hERG inhibition, and CYP450 interactions [71] [74].
- Notes: ADMETlab 2.0 computes approximately 88 ADMET-related endpoints and provides results for batch screening of up to 1000 compounds [71]. admetSAR3.0 covers an even broader range of 119 endpoints, including environmental and cosmetic risk assessments [74].
Protocol 1.2: Applying QSAR and Multi-Task Graph Learning
- Objective: To build or use predictive Quantitative Structure-Activity Relationship (QSAR) models for specific ADMET endpoints.
- Materials:
- Dataset of compounds with known ADMET properties for training.
- Software for molecular descriptor calculation (e.g., RDKit) and machine learning (e.g., Python with PyTorch/DGL libraries) [71].
- Procedure:
- Data Curation: Collect a high-quality, non-redundant dataset. Filter out organometallics, mixtures, and neutralize salts. Standardize SMILES notations [71].
- Descriptor Calculation & Feature Selection: Compute molecular descriptors or use graph-based representations where atoms are nodes and bonds are edges. Employ feature selection methods (filter, wrapper, or embedded) to identify the most relevant descriptors and avoid overfitting [73].
- Model Training: Implement a multi-task graph neural network framework. This approach allows for shared learning across related ADMET tasks, improving model robustness and accuracy, especially with limited data [71] [75].
- Validation: Validate models using rigorous methods such as k-fold cross-validation and an external test set. Always define the model's applicability domain [15] [73].
Table 1: Essential In Silico Tools for ADMET Prediction
| Tool Name | Key Features | Number of Endpoints | Best Used For |
|---|---|---|---|
| ADMETlab 2.0 [71] | Multi-task graph attention framework; batch screening; free, no login. | ~88 endpoints (17 physicochemical, 13 medicinal chemistry, 23 ADME, 27 toxicity) | High-throughput screening of compound libraries during early lead optimization. |
| admetSAR3.0 [74] | Comprehensive platform with search, prediction, and optimization modules; includes environmental and cosmetic risk. | 119 endpoints | A one-stop shop for extensive ADMET profiling, including niche endpoints. |
| StarDrop [72] | Integrated with MPO and "Glowing Molecule" visualization for SAR; proprietary data usage. | Select key ADMET and toxicity endpoints | Guiding SAR by highlighting structural features influencing properties. |
Phase 2: SAR-Guided Design and Structural Optimization
This phase uses insights from in silico predictions and biological assays to design new analogs with improved ADMET properties without sacrificing potency.
Protocol 2.1: Matched Molecular Pair Analysis (MMPA) for Property Optimization
- Objective: To identify specific structural transformations that reliably improve a given ADMET property.
- Materials: A database of chemical transformations and their associated property changes (e.g., as implemented in admetSAR3.0's ADMETopt2 module) [74].
- Procedure:
- Identify Desired Change: Pinpoint the ADMET property that needs improvement (e.g., low solubility).
- Query Transformation Rules: Use the MMPA library to find molecular pairs where a small structural change led to the desired improvement in the target property.
- Apply Rule: Propose synthesizing an analog of your lead compound that incorporates this beneficial transformation.
- Validate: Re-run in silico predictions on the proposed analog to confirm the expected improvement.
Protocol 2.2: Scaffold Hopping to Mitigate Toxicity
- Objective: To replace a problematic substructure (toxicophore) in the natural lead with a bioisostere that retains potency but eliminates the toxicity.
- Materials: Toxicophore rule libraries (e.g., the 8 toxicophore rules with 751 substructures in ADMETlab 2.0 [71]); scaffold databases (e.g., ChEMBL, Enamine [74]).
- Procedure:
- Toxicophore Identification: Use the in silico tools to identify undesirable substructures in your lead compound (e.g., PAINS, SureChEMBL rules).
- Scaffold Search: Search scaffold databases for structurally similar but distinct scaffolds that do not contain the toxicophore.
- Design & Synthesize: Design a new analog based on the identified scaffold, incorporating key functional groups necessary for potency.
- Test: Evaluate the new analog for both potency and the previously flagged toxicity.
Phase 3: Experimental Validation and Multi-Parameter Optimization
Predictions must be confirmed experimentally. This phase details key assays for validating critical ADMET properties.
Protocol 3.1: Key In Vitro ADMET Assays
- Objective: To experimentally determine critical ADMET parameters for prioritized compounds.
- Materials: See Table 2 for essential research reagents and assay systems.
Table 2: Key Research Reagent Solutions for In Vitro ADMET Profiling
| Reagent/Assay System | Function | Application in ADMET |
|---|---|---|
| Caco-2 Cell Line | Model of human intestinal epithelium. | Prediction of oral absorption and permeability [72]. |
| P-glycoprotein (P-gp) Assay | Determines interaction with the efflux transporter. | Assesses potential for transporter-mediated drug resistance and altered absorption [72]. |
| Human Liver Microsomes (HLM) | Contains major CYP450 and other metabolizing enzymes. | Evaluation of metabolic stability and metabolite identification [72]. |
| hERG Potassium Channel Assay (e.g., patch-clamp, binding) | Measures inhibition of the hERG channel. | Critical screening for cardiotoxicity risk (Torsade de Pointes) [72]. |
| Plasma Protein Binding (PPB) Assay (e.g., equilibrium dialysis) | Measures the fraction of drug bound to plasma proteins. | Determines the pharmacologically active, unbound drug concentration [72]. |
Protocol 3.2: Data Integration and Multi-Parameter Optimization (MPO)
- Objective: To integrate all potency and ADMET data to select the most promising overall candidate.
- Materials: Experimental and predicted data for all key properties; MPO software (e.g., StarDrop's Probabilistic Scoring).
- Procedure:
- Define Ideal Profile: For each property (e.g., IC50, solubility, hERG IC50), define the desired target value and an acceptable range.
- Weight Properties: Assign a relative importance weight to each property based on project goals.
- Calculate MPO Score: Use a scoring function that combines all properties according to their targets and weights, generating a single composite score (e.g., from 0 to 1) for each compound.
- Select Leads: Prioritize compounds with the highest MPO scores, indicating the best balance of all attributes [72].
The following diagram summarizes the core computational workflow, from data input to optimized design.
Balancing potency with ADMET properties is not a sequential process but an integrated, iterative cycle. By employing the practical framework outlined here—leveraging powerful in silico predictions for early triaging, applying SAR-guided design strategies like MMPA and scaffold hopping, and validating findings with targeted experimental assays—researchers can significantly de-risk the development of natural product-derived leads. This structured approach increases the likelihood of identifying candidates with a optimal balance of efficacy and safety, thereby improving the efficiency of the entire drug discovery pipeline.
Advanced Tools for Multi-Parameter SAR Analysis and Data Visualization
Structure-Activity Relationship (SAR) analysis serves as a fundamental pillar in modern drug discovery, particularly in the optimization of bioactive natural products. The systematic exploration of how modifications to a molecule's structure affect its biological activity allows researchers to navigate the vast chemical space from initial natural product "hits" to well-optimized drug candidates [76]. In the context of natural products, which have contributed to approximately 79.8% of anticancer drugs approved between 1981 and 2010, SAR studies address critical optimization challenges including enhancing drug efficacy, optimizing ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles, and improving chemical accessibility [9]. The core premise of SAR is that specific arrangements of atoms and functional groups within a molecule dictate its properties and interactions with biological systems, meaning even small structural changes can significantly alter biological activity, potency, selectivity, metabolic stability, and toxicity [76].
Multi-parameter SAR analysis has evolved beyond simple potency optimization to encompass sophisticated computational and experimental methodologies that simultaneously evaluate multiple compound properties. This holistic approach is essential for natural product optimization, where the structural complexity of native compounds often requires strategic modification to overcome pharmacological limitations while maintaining desirable bioactivity [9]. The integration of state-of-the-art computational tools with high-throughput experimental validation now enables researchers to deconvolute intricate structure-activity relationships and accelerate the development of viable drug candidates from natural product leads.
Computational Framework for Multi-Parameter SAR Analysis
Core Computational Platforms and Capabilities
Modern SAR analysis leverages advanced computational chemistry platforms that integrate multiple methodologies to accelerate and de-risk drug discovery projects. These platforms enable researchers to systematically explore the structure-activity relationships of natural product derivatives and build predictive models that guide optimization efforts [76].
Table 1: Key Computational Platforms for Multi-Parameter SAR Analysis
| Platform/Software | Primary Function | Key Features | Application in Natural Product SAR |
|---|---|---|---|
| Molecular Operating Environment (MOE) | Integrated drug design | Combines Structure-Based (SBDD) and Ligand-Based Drug Design (LBDD) | SAR/QSAR analysis, molecular modeling, and scaffold hopping for natural product analogs |
| KNIME (Konstanz Information Miner) | Workflow automation | Automates computational LBDD and SBDD workflows | Enables high-throughput, reproducible in silico screening of natural product libraries |
| NAMD | Molecular dynamics simulations | Explores dynamic behavior and stability of ligand-protein complexes | Investigates natural product binding mechanisms and complex stability at atomic resolution |
| QSAR Modeling | Predictive activity modeling | Uses mathematical models to relate structural properties to biological activity | Predicts activity of novel natural product analogs based on physicochemical descriptors |
These computational tools facilitate both fundamental medicinal chemistry principles (e.g., bio-isosterism) and state-of-the-art rational drug design techniques (e.g., structure-based design) to optimize natural leads [9]. The Molecular Operating Environment (MOE) serves as a central platform that seamlessly integrates Structure-Based Drug Design (SBDD) and Ligand-Based Drug Design (LBDD) approaches, providing comprehensive solutions for SAR exploration [76]. Workflow automation through KNIME further enhances efficiency, enabling researchers to manage the complex multi-parameter optimization challenges presented by natural product scaffolds.
Quantitative Structure-Activity Relationship (QSAR) Methodologies
Quantitative Structure-Activity Relationship (QSAR) modeling represents a more advanced approach that uses mathematical models to quantitatively relate specific physicochemical properties of compounds to their biological activity [76]. This methodology involves two critical steps: descriptor calculation and statistical modeling. Descriptor calculation quantifies various structural and physicochemical properties of molecules (e.g., hydrophobicity, electronic properties, steric bulk), while statistical modeling employs regression analysis and machine learning methods to build predictive equations based on these descriptors [76].
The application of QSAR in natural product optimization allows for more precise predictions and a deeper understanding of the factors governing activity, enabling researchers to prioritize synthetic targets before engaging in resource-intensive laboratory work. Recent advances have incorporated machine learning models to predict the biological activity of new compounds based on their chemical structure, with these models being developed using data from experimental SAR studies [76]. The integration of artificial intelligence models in QSAR has significantly enhanced the predictive power and applicability of these approaches for complex natural product scaffolds.
Experimental Protocols for SAR Determination
Comprehensive SAR Workflow for Natural Product Optimization
The SAR study workflow for natural product optimization follows an iterative "Design-Make-Test-Analyze" cycle that systematically progresses from initial natural product identification to optimized lead candidates [76]. This structured approach enables researchers to efficiently explore the chemical space around promising natural product scaffolds.
Diagram 1: SAR Optimization Workflow for Natural Products
Protocol 1: SAR-Based Analog Design and Synthesis
Purpose: To systematically design and synthesize natural product analogs for establishing comprehensive structure-activity relationships.
Materials and Reagents:
- Parent natural product compound
- Chemical reagents for derivatization (e.g., acylating agents, alkylating agents, protecting groups)
- Solvents for chemical synthesis (anhydrous where required)
- Catalysts for specific chemical transformations
- Chromatography materials for purification (silica gel, HPLC columns)
- Analytical standards for quality control
Procedure:
- Structural Analysis: Conduct comprehensive computational analysis of the natural product scaffold to identify sites amenable to chemical modification, including functional groups, stereocenters, and ring systems.
- Analog Design Strategy: Design analogs based on one or more of the following approaches:
- Functional group manipulation through derivation or substitution
- Alteration of ring systems (expansion, contraction, or replacement)
- Bio-isosteric replacement of specific molecular fragments
- Stereochemical modifications where chiral centers are present
- Synthetic Planning: Develop synthetic routes that allow for efficient generation of analog libraries, prioritizing convergent syntheses and protecting group strategies that maximize diversity.
- Compound Synthesis: Execute synthetic protocols with careful attention to reaction conditions, purification methods, and characterization techniques.
- Quality Control: Verify purity and structural identity of all synthesized analogs using appropriate analytical methods (NMR, LC-MS, HPLC).
Key Considerations: The design strategy should balance structural diversity with synthetic feasibility, focusing on modifications that probe specific hypotheses about structure-activity relationships. Special attention should be given to maintaining or improving drug-like properties during analog design [9].
Protocol 2: Multi-Parameter Biological Profiling
Purpose: To comprehensively evaluate the biological activity and ADMET properties of natural product analogs to establish correlations between structural features and pharmacological profiles.
Materials and Reagents:
- Target-specific assay reagents (enzymes, substrates, buffers)
- Cell-based assay systems relevant to therapeutic area
- ADMET screening platforms (e.g., metabolic stability assays, permeability models)
- Selectivity profiling panels (kinase panels, receptor panels)
- High-throughput screening instrumentation
- Data analysis software for dose-response calculations
Procedure:
- Primary Activity Screening:
- Test all analogs in target-specific assays at multiple concentrations (typically 10-point dose response)
- Calculate IC50/EC50 values and efficacy parameters relative to reference compounds
- Include parent natural product as internal control in all assays
Selectivity Profiling:
- Screen promising analogs (potency <1 μM) against related off-targets and anti-targets
- Utilize panel-based screening platforms for efficiency
- Calculate selectivity indices for key targets
ADMET Property Assessment:
- Evaluate metabolic stability in liver microsomes or hepatocytes
- Determine membrane permeability using Caco-2 or PAMPA assays
- Assess solubility in physiologically relevant media
- Screen for cytochrome P450 inhibition potential
- Evaluate plasma protein binding where appropriate
Cellular Phenotypic Assessment:
- Test selected analogs in disease-relevant cellular models
- Evaluate functional responses beyond primary target engagement
- Assess cytotoxicity in relevant cell lines
Key Considerations: Implement standardized assay protocols across all compounds to ensure data comparability. Include appropriate reference compounds in each experiment and maintain consistent data quality thresholds throughout the profiling cascade [76].
Protocol 3: SAR Data Integration and Model Building
Purpose: To integrate experimental data from biological profiling and build predictive SAR models that guide subsequent optimization cycles.
Materials and Reagents:
- Computational chemistry software (MOE, Schrodinger Suite, etc.)
- Chemical structure databases with associated biological data
- Statistical analysis software (R, Python with relevant packages)
- Molecular descriptor calculation tools
- Machine learning platforms for QSAR modeling
Procedure:
- Data Curation: Compile all chemical structures and associated experimental data into a searchable database, applying appropriate data normalization and quality checks.
Molecular Descriptor Calculation:
- Compute comprehensive sets of molecular descriptors (topological, electronic, steric, thermodynamic)
- Generate 3D molecular fields and pharmacophore features
- Calculate physicochemical properties (logP, PSA, HBD/HBA counts)
SAR Pattern Recognition:
- Identify activity cliffs (small structural changes that cause large activity differences)
- Map key structural features to specific biological outcomes
- Establish correlations between substituent properties and activity
Predictive Model Development:
- Apply machine learning algorithms (random forest, support vector machines, neural networks) to build QSAR models
- Validate models using appropriate cross-validation techniques
- Apply domain of applicability analysis to define model boundaries
Visualization and Hypothesis Generation:
- Create chemical space maps using dimensionality reduction techniques
- Develop structure-activity landscape models to guide analog design
- Generate testable hypotheses for next design cycle
Key Considerations: Focus on building interpretable models that provide actionable insights for medicinal chemistry optimization. Balance model complexity with predictive power and ensure rigorous validation to avoid overfitting [76].
Advanced Data Visualization in SAR Studies
Structure-Activity Landscape Visualization
Effective visualization of SAR data enables researchers to identify critical structure-activity trends and prioritize optimization strategies. Advanced visualization techniques transform complex multi-parameter data into actionable insights for natural product optimization.
Diagram 2: SAR Data Visualization Workflow
Multi-Parameter Optimization Visualization
The complexity of natural product optimization requires simultaneous consideration of multiple parameters, including potency, selectivity, and ADMET properties. Radar plots and parallel coordinate plots effectively communicate these complex relationships and enable identification of compounds with balanced profiles.
Table 2: Key Parameters for Multi-Parameter SAR Visualization
| Parameter Category | Specific Metrics | Visualization Methods | Optimization Goal |
|---|---|---|---|
| Potency | IC50, EC50, Ki | Dose-response curves, heat maps | Lower nM range for primary target |
| Selectivity | Selectivity indices, panel screening results | Radar plots, dendrograms | >30-fold vs. key anti-targets |
| ADMET Properties | Metabolic stability, permeability, solubility | Parallel coordinates, property landscapes | Balanced profile meeting criteria |
| Physicochemical Properties | logP, PSA, HBD/HBA | 2D property space maps | Drug-like space adherence |
| Structural Features | Substituent characteristics, scaffold topology | Structure-activity landscapes | Identify optimal chemical space |
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of multi-parameter SAR studies requires access to specialized research reagents and platforms that enable comprehensive compound profiling. The following toolkit outlines essential materials for establishing robust SAR capabilities in natural product research.
Table 3: Essential Research Reagents for SAR Studies
| Category | Specific Reagents/Platforms | Function in SAR Studies | Key Suppliers |
|---|---|---|---|
| Chemical Biology Tools | Functional group modification kits, bioisostere libraries | Enable systematic structural modification of natural product scaffolds | Sigma-Aldrich, ComGenex, Enamine |
| Target Assay Systems | Recombinant enzymes, cell lines with target expression, receptor binding assay kits | Quantify compound potency and selectivity against primary targets and anti-targets | Reaction Biology, Eurofins, BPS Bioscience |
| ADMET Screening Platforms | Caco-2 cells, liver microsomes, plasma protein binding assays | Evaluate pharmacokinetic properties and identify potential toxicity issues | BD Biosciences, Thermo Fisher, Corning |
| Computational Software | MOE, Schrodinger Suite, KNIME, R/Python with chemoinformatics packages | Facilitate molecular modeling, QSAR analysis, and data visualization | Chemical Computing Group, Schrodinger, KNIME |
| Data Integration Platforms | CDD Vault, Dotmatics, Benchling | Manage chemical and biological data, enable collaborative SAR analysis | Collaborative Drug Discovery, Dotmatics, Benchling |
Implementation Case Study: Natural Product Lead Optimization
The practical application of these advanced tools and protocols can be illustrated through a case study of optimizing a hypothetical natural product lead with initial activity but suboptimal drug properties. This integrated approach demonstrates how multi-parameter SAR analysis accelerates the development of viable drug candidates from natural product starting points.
Initial Natural Product Profile:
- Moderate target potency (IC50 = 1.2 μM)
- Poor metabolic stability (t1/2 < 10 min in liver microsomes)
- Low solubility (<5 μg/mL in aqueous buffer)
- Complex structure with limited synthetic accessibility
SAR-Driven Optimization Strategy:
- Initial SAR Exploration: Synthesize focused library of 30 analogs exploring modifications at three strategic positions on the natural product scaffold.
- Multi-Parameter Profiling: Test all analogs in primary potency assay, metabolic stability assay, and solubility measurement.
- QSAR Model Development: Build predictive models correlating structural features with key properties using random forest algorithm.
- Design Cycle 2: Apply QSAR predictions to design 15 analogs with improved predicted properties.
- Iterative Optimization: Repeat testing and model refinement through two additional design cycles.
Optimization Outcomes:
- 45-fold improvement in target potency (IC50 = 27 nM)
- 8-fold improvement in metabolic stability (t1/2 = 78 min)
- 12-fold improvement in aqueous solubility (62 μg/mL)
- Simplified chemical structure with improved synthetic accessibility
This case study demonstrates the power of integrated multi-parameter SAR analysis in addressing the complex optimization challenges typically presented by natural product leads. The systematic application of computational tools, experimental profiling, and data visualization enables efficient navigation of the chemical space to identify analogs with balanced drug-like properties.
Future Perspectives in SAR Analysis
The field of SAR analysis continues to evolve with emerging technologies that promise to further accelerate natural product optimization. Machine learning and artificial intelligence are increasingly being applied to predict the biological activity of new compounds based on their chemical structure [76]. These models, developed using data from experimental SAR studies, can identify promising lead compounds and optimize their structure for improved activity and selectivity [76].
Advanced methods such as molecular dynamics simulations using platforms like NAMD allow researchers to explore the dynamic behavior and stability of ligand-protein complexes, providing deeper insights into molecular mechanisms at atomic resolution [76]. The integration of these sophisticated computational approaches with high-throughput experimental data creates a powerful framework for addressing the unique challenges of natural product drug discovery.
As these technologies mature, the implementation of robust, multi-parameter SAR analysis will become increasingly essential for unlocking the therapeutic potential of natural products and addressing the ongoing challenges of drug discovery in areas of unmet medical need.
The structure-activity relationship (SAR) directed optimization of natural product leads is a cornerstone of modern drug discovery. This process, however, generates immense and complex datasets, creating a significant challenge of data overload for researchers. Modern High-Throughput Screening (HTS) campaigns, for instance, can test hundreds of thousands of compounds, yet often yield hit rates below 2%, generating a vast amount of data that is challenging to interpret efficiently [77]. The traditional solution—increasing manpower—is often impractical and inefficient. This Application Note details structured digital protocols and informatics tools designed to manage this data deluge, enabling researchers to accelerate the identification of promising drug candidates from natural sources.
The Computational Toolkit for SAR Analysis
A range of sophisticated software and algorithms is available to dissect complex SAR data. The selection of a specific tool depends on the research question, the nature of the data, and the desired output, ranging from simple data visualization to the generation of novel molecular structures.
Table 1: Digital Solutions for SAR Data Analysis
| Solution Category | Specific Tool/Technique | Key Function in SAR Analysis | Application Context |
|---|---|---|---|
| Cheminformatics Platforms | CDD Vault Visualization [78] | Enables in-depth SAR analysis with features like R-group pattern display, on-the-fly calculations, and color-coded data columns. | Analysis of HTS and medium-throughput screening (MTS) data. |
| Machine Learning (ML) Models | Random Forest [79] [45] | A robust ML algorithm used to build predictive models correlating molecular structures with biological activity (e.g., pChEMBL values, MIC values). | SAR exploration and prediction of compound activity for targets like adenosine receptors or anti-tuberculosis agents. |
| Deep Learning (DL) Models | Stacked LSTM [79] | A generative model that uses SMILES representations to create novel, drug-like molecular structures. | De novo molecular design for natural drug candidates. |
| Matched Molecular Pair (MMP) Analysis | DataWarrior [32] | Identifies activity cliffs by systematically comparing pairs of compounds that differ only by a single structural change. | Cross-SAR (C-SAR) analysis to extract transformative rules from diverse chemotypes. |
| Multi-objective Optimization | Pareto Optimization [79] | Balances multiple, often competing, objectives (e.g., potency, selectivity, synthetic accessibility) during molecular design. | Optimization of natural product leads against multiple parameters simultaneously. |
Application Note: A Multi-Protocol Workflow for Natural Lead Optimization
This section integrates various digital solutions into a cohesive, end-to-end workflow for the SAR-driven optimization of a natural product lead.
The following diagram illustrates the integrated digital workflow for efficient SAR trend analysis, from initial data management to final candidate selection.
Detailed Experimental Protocols
Protocol 1: Data Curation and Cross-SAR (C-SAR) Setup with MMP Analysis
Objective: To transform a raw HTS dataset into a structured format suitable for trend analysis and identify critical structural transformations using the C-SAR approach [32].
- Data Sourcing: Publicly available databases like ChEMBL are primary sources for building a library of bioactive compounds [32] [79]. For a study on selective HDAC6 inhibitors, over 23,000 ligands with associated bioactivity data (e.g., pChEMBL values) were extracted [32].
- Data Preprocessing: Using cheminformatics toolkits (e.g., RDKit), standardize molecular structures by removing charges, eliminating metals and salts, and deduplicating the dataset [79].
- MMP Analysis: Utilize software like DataWarrior to fragment molecules and generate Matched Molecular Pairs (MMPs)—pairs of compounds that differ only at a single site by a single substructure [32].
- C-SAR Data Extraction: Analyze the MMPs to identify "activity cliffs," where a small structural change results in a significant change in biological activity. These cliffs form the basis for rules on how to transform an inactive compound into an active one, even across different parent scaffolds [32].
Protocol 2: Building a Predictive Machine Learning Model for SAR
Objective: To train a machine learning model that can predict the biological activity of novel natural product analogs.
- Feature Representation: Encode the chemical structures of the curated dataset. Common methods include using SMILES (Simplified Molecular-Input Line-Entry System) strings or classical cheminformatics descriptors [79].
- Model Training: Employ a Random Forest algorithm to build a quantitative structure-activity relationship (QSAR) model. The model is trained to correlate the structural features (input) with the biological activity data (e.g., pChEMBL value, MIC value) from the dataset [79] [45].
- Model Validation: Validate the model's predictive accuracy using standard techniques such as k-fold cross-validation or a separate hold-out test set not used during training.
Protocol 3: Generative Design and Multi-Objective Optimization of Analogs
Objective: To create novel natural product-derived compounds with optimized properties.
- Molecular Generation: Implement a stacked Long Short-Term Memory (LSTM) neural network, a type of deep learning model. This model, pre-trained on large chemical databases (e.g., ChEMBL) and fine-tuned on the project-specific dataset, can generate new, valid molecular structures in SMILES format [79].
- Activity and Property Prediction: Screen the generated molecules using the trained Random Forest model from Protocol 2 to predict their biological activity.
- Multi-Objective Optimization: Apply Pareto optimization to balance multiple critical parameters. The goal is to find a set of candidate molecules that offer the best compromise between high predicted activity, low affinity for the hERG channel (a key toxicity risk), favorable drug-likeness, and good synthetic accessibility [79].
The Scientist's Toolkit: Essential Research Reagents & Materials
Successful implementation of the digital protocols above relies on a foundation of specific reagents, software, and data resources.
Table 2: Key Research Reagents and Solutions for Digital SAR Workflows
| Category | Item | Function in SAR Analysis |
|---|---|---|
| Software & Informatics | CDD Vault [78] | A centralized platform for managing biological and chemical data, featuring visualization tools for SAR trend analysis. |
| DataWarrior [32] | An open-source cheminformatics program used for data analysis, visualization, and MMP analysis. | |
| Genedata Screener [80] | A robust enterprise platform for processing and analyzing large-scale HTS data, ensuring data fidelity and efficiency. | |
| Computational Libraries | RDKit [79] | An open-source toolkit for cheminformatics and machine learning, used for data preprocessing, descriptor calculation, and more. |
| Data Resources | ChEMBL Database [32] [79] | A manually curated database of bioactive molecules with drug-like properties, providing essential data for model training. |
| LeadFinder Diversity Library [80] | A commercially available, expertly designed compound library (150,000 compounds) used for HTS and follow-up SAR studies. | |
| AI/ML Frameworks | Random Forest [45] | A versatile machine learning algorithm used for building predictive QSAR models and analyzing feature importance in SAR. |
| LSTM Neural Network [79] | A type of deep learning model adept at processing sequential data like SMILES strings for generative molecular design. |
The integration of advanced digital solutions—from C-SAR and MMP analysis to machine learning and multi-objective optimization—provides a powerful and necessary strategy for overcoming data overload in natural product drug discovery. The protocols outlined herein equip researchers with a structured framework to not only manage large datasets but to extract profound, actionable insights. By adopting these computational methodologies, research teams can significantly accelerate the SAR optimization cycle, increasing the efficiency and probability of success in delivering novel therapeutics derived from nature's chemical arsenal.
In the Structure-Activity Relationship (SAR) directed optimization of natural product leads, the Domain of Applicability (DSA) is a critical concept that defines the chemical space over which a predictive model is reliable. It establishes the boundaries within which the model's predictions for compound activity and properties can be trusted. For research teams, accurately defining the DSA prevents misdirection by flagging when novel compounds fall outside the model's trained experience, thereby reducing the risk of costly experimental follow-up on unreliable predictions. This protocol outlines the methodologies for DSA assessment and its application in natural product optimization projects.
Quantitative Assessment of the DSA
A model's DSA is not a binary state but a quantified measure of reliability. The following metrics, when calculated for a new compound, help determine its position relative to the model's established domain.
Table 1: Key Metrics for Quantifying the Domain of Applicability
| Metric | Description | Calculation Method | Interpretation Threshold |
|---|---|---|---|
| Leverage (hᵢ) | Measures a compound's distance to the centroid of the training set chemical space. [32] | Based on the hat matrix from PCA or PLS of the training set descriptors. | hᵢ > h* = 3p/n (where p=descriptors, n=compounds) indicates high leverage and potential unreliability. |
| Standardized Residual | Quantifies how well the model predicts the activity of a compound. | Difference between observed and predicted activity, divided by the standard error of the prediction. | Absolute value > 2-3 standard deviations suggests the model cannot accurately predict for this structure. |
| Similarity Distance | Assesses the nearest-neighbor similarity between a new compound and the training set. | Euclidean or Tanimoto distance to the k-nearest neighbors in the training set. | Distance > predefined percentile (e.g., 95th) of training set distances indicates an outlier. |
| DSA Consensus Score | A unified score combining multiple metrics for a holistic assessment. | Normalized and weighted sum of leverage, residual, and similarity scores. | Score > 0.7-0.8 typically flags a compound for careful manual inspection before trust is placed in its prediction. |
Protocol for DSA Calculation and Model Application
This protocol provides a step-by-step guide for integrating DSA assessment into a natural lead optimization workflow using a Matched Molecular Pairs (MMP) analysis approach. [32]
Experimental Setup and Reagent Solutions
Table 2: Essential Research Reagent Solutions for DSA Workflow
| Research Reagent / Tool | Function in DSA Workflow | Application Example |
|---|---|---|
| DataWarrior Software | Open-source tool for cheminformatics data configuration, visualization, and graphical presentation. [32] | Used to plot chemical structures against activity data to visualize dataset diversity and identify clusters and outliers. |
| Matched Molecular Pairs (MMP) Analysis | A method to extract SAR data from compound series by identifying pairs that differ only at a single site. [32] | Core to the Cross-SAR (C-SAR) strategy; enables extraction of pharmacophoric substitution patterns from diverse chemotypes. |
| Neural Network Potentials (NNPs) | Pre-trained models (e.g., eSEN, UMA) for fast, accurate computation of molecular potential energy surfaces. [81] | Provides high-accuracy molecular energy and property predictions on massive, diverse datasets like OMol25 to inform model reliability. |
| Molecular Operating Environment (MOE) | Software platform for molecular docking, simulations, and QSAR model development. [32] | Used to perform molecular docking studies and calculate molecular descriptors for training set characterization. |
| Random Forest / XGBoost Algorithms | Machine learning models for SAR exploration and enhancing predictive accuracy of docking scores. [45] | Applied to analyze structural characteristics and re-rank docking results to identify key features for anti-TB activity. |
Step-by-Step Procedure
Define the Chemical Space of the Training Set
- Input: A curated library of compounds with known biological activity, such as selective HDAC6 inhibitors from the ChEMBL database. [32]
- Calculation: Compute a set of molecular descriptors (e.g., topological, electronic, steric) for every compound in the training set.
- Dimensionality Reduction: Perform Principal Component Analysis (PCA) on the descriptor matrix. The resulting first two or three principal components define the major axes of the model's chemical space.
Calculate DSA Decision Boundaries
- Leverage Threshold (h*): Calculate the critical leverage,
h*, using the formulah* = 3p/n, wherepis the number of model descriptors andnis the number of training compounds. [32] - Similarity Threshold: Calculate the Euclidean distance between every possible pair of compounds in the training set. The 95th percentile of this distribution is defined as the similarity distance threshold.
- Record these threshold values for use in subsequent prediction steps.
- Leverage Threshold (h*): Calculate the critical leverage,
Profile a New Compound and Assess its DSA Fit
- Descriptor Calculation: Compute the same molecular descriptors for the new, uncharacterized natural product derivative.
- Projection: Project the new compound into the PCA space defined by the training set in Step 1.
- Metric Calculation:
- Calculate its Leverage (hᵢ) relative to the training set.
- Calculate its Similarity Distance to its k-nearest neighbors in the training set.
- DSA Classification: Compare the calculated metrics to the thresholds from Step 2. A compound is considered within the DSA only if
hᵢ < h*ANDSimilarity Distance < Threshold.
Interpret Prediction and Guide Optimization
- Within DSA: Proceed with high confidence in the model's prediction (e.g., predicted IC50, selectivity). The result can be used to prioritize compounds for synthesis.
- Outside DSA: Flag the prediction as unreliable. This compound represents an opportunity for SAR expansion. Consider synthesizing it as a probe to expand the chemical space of the model, then re-train the model with the new data.
Workflow Visualization
The following diagram illustrates the logical workflow for applying the Domain of Applicability in a natural product lead optimization project.
DSA Assessment Workflow
Case Study: Application in Anti-TB Natural Product Optimization
A 2025 review on anti-tuberculosis natural products exemplifies the DSA concept in practice. [45] Researchers built a machine learning model using a dataset of potent natural products (MIC < 5 µg mL–1). The model's DSA was defined by the structural features of these compounds, including marine organisms, terrestrial plants, and microorganisms.
When the model was applied to a newly isolated fluorinated derivative of tryptanthrin, the compound fell within the DSA because the core indoloquinazoline alkaloid scaffold and the type of fluorine substitution were well-represented in the training set. The model correctly predicted its enhanced potency (MIC of 0.06 mg L−1), a prediction later confirmed by experiment. [45] This successful application within the DSA gave researchers high confidence in the result.
In contrast, if a compound with a completely novel scaffold, unlike any in the training set, were to be screened, it would be flagged as outside the DSA. Its predicted activity would be considered unreliable, mandating empirical testing to validate its effect and potentially expand the model's applicability domain.
From Bench to Bedside: Validating and Comparing Optimized Natural Leads
Within the framework of Structure-Activity Relationship (SAR)-directed optimization of natural product leads, preclinical validation serves as the critical bridge between initial compound identification and clinical trials. This phase aims to comprehensively evaluate the safety and efficacy of lead candidates, providing essential data to refine chemical structures for enhanced therapeutic potential [82]. The process is complex and costly, typically spanning 10-15 years and requiring billions of dollars from discovery to approval [83]. A significant challenge is the poor correlation between traditional animal models and human outcomes, particularly for complex events like Drug-Induced Liver Injury (DILI), which contributes substantially to high attrition rates in later development phases [83]. By integrating robust in vitro and in vivo efficacy assessments into the SAR cycle, researchers can make data-driven decisions to prioritize the most promising compounds for further development.
Integrating Preclinical Validation into the SAR Optimization Workflow
The following diagram illustrates how efficacy assessment feeds iteratively into the SAR-driven optimization of natural product leads.
Figure 1: SAR-Driven Preclinical Validation Workflow. This iterative cycle uses efficacy and toxicity data from in vitro and in vivo assays to inform the chemical optimization of natural product leads. PK/PD: Pharmacokinetics/Pharmacodynamics.
Experimental Protocols for Efficacy Assessment
In Vitro Cellular Models for High-Throughput Efficacy Screening
Principle: Cell-based in vitro models are used for primary efficacy screening and account for nearly half of high-throughput screening (HTS) efforts. They provide insights into toxicity profiles, impacts on signaling pathways, and overall cellular effects within a physiologically relevant environment [83].
Protocol:
- Cell Culture:
- Select appropriate cell lines (e.g., primary cells, immortalized lines, or patient-derived cells) relevant to the disease target.
- Maintain cells in optimized media conditions, ensuring consistent passage number and viability.
- Assay Setup:
- Seed cells in 384-well or 1536-well microplates at a density determined by growth kinetics.
- Allow cells to adhere and recover for 24 hours.
- Compound Treatment:
- Prepare serial dilutions of the natural product analogs in DMSO or assay buffer.
- Treat cells with compounds, ensuring minimal solvent concentration (typically <0.1%).
- Include positive (known inhibitor/agonist) and negative (vehicle-only) controls.
- Incubation and Readout:
- Incubate plates for 48-72 hours at 37°C, 5% CO₂.
- Measure cell viability/proliferation using fluorometric, luminescent, or colorimetric assays (e.g., ATP-based luminescence, resazurin reduction).
- For mechanistic studies, employ multiparameter assays to capture apoptosis, cell cycle arrest, or target engagement simultaneously.
- Data Analysis:
- Normalize data to controls.
- Calculate half-maximal inhibitory/concentrations (IC₅₀/EC₅₀) using non-linear regression models.
Advanced Models: For improved predictivity, move beyond monolayer cultures to advanced systems such as 3D co-cultures, organoids, or organ-on-a-chip models. These systems better replicate tissue-specific mechanical and biochemical characteristics, enhancing predictions of in vivo efficacy and hepatic clearance [83].
Ex Vivo Histoculture Drug Response Assay
Principle: This assay evaluates drug efficacy in a more complex, tissue-like environment, preserving native cell-cell and cell-matrix interactions. It is particularly useful for validating hits from initial HTS in a more physiologically relevant context [84].
Protocol:
- Tissue Preparation:
- Obtain fresh tumor or diseased tissue biopsies (e.g., from Patient-Derived Xenografts/PDXs).
- Under sterile conditions, mince the tissue into ~1 mm³ fragments using a scalpel.
- Histoculture:
- Place tissue fragments onto collagen-coated gel supports in culture dishes.
- Add culture medium supplemented with serum and necessary growth factors.
- Compound Treatment:
- After a 24-hour pre-culture period, add the natural product analogs at desired concentrations.
- Maintain control cultures without drug treatment.
- Incubation and Assessment:
- Culture for 5-7 days, refreshing medium as needed.
- Assess efficacy via metabolic activity dyes (e.g., MTT), histopathological analysis, or measurement of specific biomarkers (e.g., apoptosis markers via immunohistochemistry).
In Vivo Efficacy Assessment in Patient-Derived Xenograft (PDX) Models
Principle: PDX models, established by engrafting human tumor tissues into immunodeficient mice, retain the genomic and phenotypic characteristics of the original patient tumor. They are a gold standard for evaluating in vivo efficacy during later-stage lead optimization [84].
Protocol:
- Model Generation:
- Implant patient-derived tumor fragments subcutaneously or orthotopically into immunocompromised mice (e.g., NSG mice).
- Allow tumors to engraft and grow to a palpable size (~100-150 mm³).
- Study Arm Randomization:
- Randomize mice into treatment and control groups (typically n=5-10 per group).
- Ensure similar average tumor volumes and body weights across groups at the start of dosing.
- Dosing Regimen:
- Administer the lead natural product analog via the intended route (oral, intraperitoneal, intravenous).
- Define dosage levels based on prior in vivo toxicity studies.
- Include a vehicle control group and a positive control group (standard-of-care drug) if available.
- Monitoring and Data Collection:
- Measure tumor dimensions and body weight 2-3 times per week.
- Calculate tumor volume using the formula: V = (length × width²) / 2.
- Monitor animals for any signs of overt toxicity.
- Endpoint Analysis:
- Euthanize animals at the study endpoint.
- Harvest tumors for weight measurement and subsequent biomarker analysis (e.g., genomic, transcriptomic).
- Collect blood for pharmacokinetic analysis and key organs for histopathological toxicity assessment.
Key Performance Metrics:
- Tumor Growth Inhibition (TGI): % TGI = [1 - (ΔT/ΔC)] × 100, where ΔT and ΔC are the mean change in tumor volume for treatment and control groups, respectively.
- Log₁₀ Cell Kill: A metric calculated from the tumor growth delay.
The integrated workflow for preclinical efficacy validation, from cellular models to in vivo studies, is depicted below.
Figure 2: Integrated Preclinical Efficacy Validation Pathway. A sequential approach to validating natural product efficacy, increasing in physiological complexity at each stage. HTS: High-Throughput Screening; PDX: Patient-Derived Xenograft; PK/PD: Pharmacokinetics/Pharmacodynamics.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 1: Essential Reagents and Materials for Preclinical Efficacy Assessment.
| Item/Category | Function & Application | Specific Examples |
|---|---|---|
| Primary Cells & Cell Lines | Provide a biologically relevant system for initial efficacy and toxicity screening. | Primary hepatocytes, immortalized cell lines, iPSC-derived cells, engineered cells with humanized targets [83]. |
| 3D Culture Matrices | Simulate the in vivo extracellular matrix (ECM) for 3D cultures, organoids, and ex vivo histocultures. | Collagen I gels, Matrigel, synthetic hydrogels [84]. |
| High-Throughput Assay Kits | Enable rapid, multiplexed readouts of cell viability, cytotoxicity, apoptosis, and mechanism of action in microplates. | ATP-lite luminescence kits, Caspase-Glo apoptosis assays, multiplexed cytokine panels [83]. |
| Animal Models | Evaluate efficacy and PK/PD relationships in a whole-organism context. | Immunocompromised mice (NSG), Patient-Derived Xenograft (PDX) models, genetically engineered mouse models (GEMMs), humanized mice [84] [83]. |
| Specialized Culture Media | Support the maintenance and function of complex in vitro systems and primary cells. | Spheroid/organoid formation media, defined hepatocyte maintenance media, low-serum assay media. |
Quantitative Data Analysis and Comparison
A critical output of preclinical validation is quantitative data that enables direct comparison of analogs to guide SAR optimization.
Table 2: Quantitative Efficacy and Safety Profile of Natural Product Analogs.
| Compound ID | In Vitro IC₅₀ (μM) | In Vitro HepG2 Cytotoxicity CC₅₀ (μM) | Therapeutic Index (TI) in vitro | Ex Vivo TGI (%) | In Vivo TGI (%) | Maximum Tolerated Dose (mg/kg) |
|---|---|---|---|---|---|---|
| NP-A-01 | 0.15 ± 0.02 | 25.0 ± 2.1 | 166.7 | 75 | 82 | 100 |
| NP-A-02 | 0.08 ± 0.01 | 5.5 ± 0.5 | 68.8 | 65 | 45 | 50 |
| NP-A-03 | 0.25 ± 0.03 | >100 | >400 | 60 | 55 | >200 |
| NP-A-04 | 1.50 ± 0.10 | 15.0 ± 1.2 | 10.0 | 30 | 20 | 100 |
| NP-A-05 | 0.05 ± 0.01 | 2.0 ± 0.2 | 40.0 | 80 | 90 | 25 |
Table 3: Key Pharmacokinetic Parameters of Lead Analogs from Rodent Studies.
| Compound ID | Cmax (μg/mL) | T½ (hours) | AUC₀–t (μg·h/mL) | Clearance (mL/min/kg) | Vd (L/kg) | Oral Bioavailability (%) |
|---|---|---|---|---|---|---|
| NP-A-01 | 4.5 | 6.2 | 35.1 | 25.0 | 1.2 | 65 |
| NP-A-02 | 1.8 | 2.1 | 8.5 | 98.5 | 0.8 | 22 |
| NP-A-03 | 0.9 | 1.5 | 5.2 | 160.0 | 1.5 | 15 |
| NP-A-05 | 12.1 | 3.5 | 42.5 | 20.5 | 0.5 | >80 |
A rigorous, multi-faceted approach to preclinical validation is indispensable for the successful SAR-directed optimization of natural product leads. By strategically employing a suite of in vitro, ex vivo, and in vivo models, researchers can generate high-quality, human-relevant data on both efficacy and safety. This integrated workflow enables the rational selection and refinement of lead compounds with the highest probability of success in clinical trials, thereby de-risking the drug development pipeline and accelerating the translation of natural products into novel therapeutics.
Structure-activity relationship (SAR) studies are a cornerstone of modern medicinal chemistry, providing a systematic framework for understanding how a molecule's structure influences its biological activity [76]. Within drug discovery, SAR analysis is particularly critical for the optimization of natural product (NP) "leads," which, despite their evolved bioactivity and structural complexity, often require modification to achieve sufficient potency, selectivity, and pharmacokinetic properties for therapeutic use [85] [2]. NPs and their derivatives constitute a significant proportion of approved drugs, particularly for cancer and infectious diseases [85] [86]. However, they frequently present challenges such as technical barriers to screening, complex synthesis, and suboptimal drug-like properties [85]. This application note provides a comparative analysis of NP leads and their synthetic analogs, detailing key strategic approaches and offering detailed protocols for SAR-driven optimization. The content is designed to equip researchers with methodologies to harness the unique value of NPs while overcoming their inherent limitations through rational design and synthesis.
Strategic Approaches to Natural Product Optimization
Navigating from a bioactive natural product to a optimized clinical candidate requires a multi-faceted strategy. The chosen approach depends on the complexity of the NP scaffold, the feasibility of its synthesis, and the specific optimization goals (e.g., improving potency, reducing toxicity, or modifying pharmacokinetics).
Table 1: Strategic Approaches for SAR-Driven Optimization of Natural Products
| Strategy | Core Principle | Key Advantages | Inherent Challenges | Representative Applications |
|---|---|---|---|---|
| Diverted Total Synthesis | A target-oriented synthesis is designed with deliberate branch points from common intermediates to generate analog libraries [2]. | Enables access to core structural modifications not accessible via semisynthesis; high scaffold diversity [2]. | Often a lengthy, multi-step process requiring significant synthetic expertise and resources. | Synthesis of migrastatin and pleuromutilin analogs with improved stability and activity [2]. |
| Late-Stage Functionalization (LSF) | Direct, selective functionalization of C-H bonds or other "unfunctionalized" positions in a native NP [16]. | Avoids de novo synthesis; allows rapid SAR exploration and "arming" of NPs for target identification [16]. | Requires chemoselectivity and can yield modest conversions; limited by inherent reactivity of the NP. | Rh(II)-catalyzed C–H amination of eupalmerin acetate for probe synthesis and target ID [16]. |
| Build-Up Library Synthesis | NP structure is divided into a constant core fragment and variable accessory fragments, ligated via a highly efficient reaction [18]. | Rapid generation of large libraries (100s of compounds); minimal purification enables direct in situ biological evaluation [18]. | Ligation chemistry must be high-yielding and clean; requires strategic dissection of the NP. | Hydrazone-based library of MraY inhibitors for antibacterial discovery [18]. |
| Computational & AI-Guided SAR | Machine learning and CADD models are trained to predict the bioactivity of analogs from chemical structure or biosynthetic gene clusters [2]. | Accelerates analog prioritization; provides insights into binding modes and mechanism; de-risks synthesis. | Dependent on quality and quantity of experimental data for training; can be a "black box." | Interpretable AI for predicting bioactivity from NP biosynthetic pathways [2]. |
The following diagram illustrates a synergistic experimental-computational workflow that integrates these strategies into a continuous cycle for NP optimization.
Detailed Experimental Protocols
Protocol: Build-Up Library Synthesis and In Situ Screening for MraY Inhibitors
This protocol describes the construction and evaluation of a hydrazone-based build-up library, adapted from a study optimizing MraY inhibitory natural products like caprazamycin and muraymycin [18]. The method enables the rapid generation of hundreds of analogs without individual purification, allowing for direct biological evaluation.
1. Library Design and Reagent Preparation
- Core Fragment Preparation: Synthesize or acquire the aldehyde-bearing core fragments derived from the NP scaffold. These cores should contain the essential pharmacophore (e.g., the uridine moiety for MraY inhibitors) [18]. Example: Capuramycin aldehyde core.
- Accessory Fragment Preparation: Prepare a diverse collection of 98+ hydrazide fragments. Diversity should encompass:
- Simple acyl hydrazides (e.g., benzoyl-type, phenyl acetyl-type).
- N-acyl aminoacyl hydrazides with varying acyl chain lengths and amino acid side chains (e.g., acetylamino acid, lipid amino acid) [18].
- Stock Solutions: Prepare 10 mM stock solutions of all core and accessory fragments in anhydrous DMSO.
2. In Situ Hydrazone Library Assembly
- Reaction Setup: In a 96-well plate, combine 15.5 µL of a core aldehyde solution (10 mM in DMSO) with 15.5 µL of a hydrazine solution (10 mM in DMSO) to achieve a 1:1 stoichiometry in a total volume of 31 µL. No additional catalysts or reagents are required.
- Incubation: Seal the plate and allow the reaction to proceed at room temperature for 30 minutes.
- Solvent Removal: Place the 96-well plate in a centrifugal concentrator and evaporate the DMSO under vacuum at room temperature overnight.
- Library Reconstitution: Add 30 µL of DMSO to each well to resuspend the reaction products, resulting in a nominal 5 mM library solution for screening. LC-MS analysis of select wells is recommended to confirm >80% conversion for most reactions [18].
3. Direct Biological Evaluation
- Biochemical Assay: Directly use the library solutions from the previous step to evaluate MraY inhibitory activity. Assay conditions will vary by target but typically involve measuring the transfer of a radiolabeled or fluorescent phospho-N-acetylmuramoyl-pentapeptide to the lipid carrier undecaprenyl phosphate [18].
- Antibacterial Susceptibility Testing: Due to the clean nature of the hydrazone formation (by-product is only H₂O), the library solutions can be diluted directly into broth microdilution assays to determine minimum inhibitory concentrations (MICs) against relevant bacterial strains [18].
- Data Analysis: Analyze dose-response curves to determine IC₅₀ and MIC values. Given the nominal concentration, these values are relative but effectively prioritize analogs for full synthesis and confirmation.
Protocol: Late-Stage C–H Amination and Arming of Natural Products
This protocol outlines a method for the site-selective functionalization of natural products at unfunctionalized positions via Rh(II)-catalyzed C–H amination, enabling simultaneous SAR studies and the installation of a handle ("arming") for chemical biology studies [16].
1. Synthesis of Alkynyl Sulfamate Reagent
- Prepare the trichloroethyl-substituted sulfamate nitrene precursor (9) from commercially available trichloromethyl-β-lactone, as described in the literature [16]. The terminal alkyne in this reagent enables subsequent conjugation via click chemistry.
2. Rh(II)-Catalyzed C–H Amination/Aziridination
- Reaction Setup: In a microscale reaction vial, combine:
- Natural product substrate (≤ 1 mg).
- Sulfamate reagent 9 (1.0 equivalent).
- Rh₂(esp)₂ or Rh₂(OAc)₄ catalyst (5 mol%).
- PhI(O₂CtBu)₂ oxidant (1.0 equivalent).
- Anhydrous benzene (to a concentration of ~0.13 M).
- Controlling Chemoselectivity:
- For C–H amination preference, add a Brønsted (e.g., HOAc) or Lewis acid (e.g., In(OTf)₃) additive.
- For alkene aziridination preference, add a base (e.g., K₂CO₃) additive [16].
- Reaction Execution: Add the solid oxidant in one portion to the stirred reaction mixture. React at room temperature until completion (monitor by TLC/LC-MS).
- Work-up: Directly concentrate the reaction mixture. Excellent mass recovery is typical, even with modest conversions.
3. Product Characterization and Application
- Purification & Analysis: Purify the derivatized natural product using preparative TLC or HPLC. The trichloromethyl group provides a distinct isotopic pattern for easy identification by mass spectrometry [16].
- SAR Studies: The introduced amine or aziridine can be deprotected under mild reductive conditions (Zn, acetic acid) to yield the primary amine for direct SAR evaluation.
- Target Identification: The alkyne-functionalized NP can be conjugated to an azide-containing biotin or fluorophore tag via Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) for pull-down or cellular imaging experiments to elucidate the mechanism of action [16].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials essential for executing the SAR strategies discussed in this note.
Table 2: Key Research Reagent Solutions for NP Optimization
| Reagent / Material | Function and Application in NP SAR |
|---|---|
| Aldehyde-Core Fragments | Constant fragments derived from the NP scaffold that retain the essential binding pharmacophore; used in build-up library synthesis for ligation with hydrazine accessories [18]. |
| Diverse Hydrazide Library | Variable accessory fragments that introduce chemical diversity; ligated to core aldehydes to rapidly explore SAR and optimize properties like potency and permeability [18]. |
| Rh(II) Catalysts (e.g., Rh₂(esp)₂) | Catalyze the key C–H amination or aziridation step in late-stage functionalization; different catalysts can influence chemoselectivity between allylic C–H bonds and alkenes [16]. |
| Alkynyl Sulfamate Reagent (9) | A metallonitrenoid precursor used in Rh-catalyzed C–H amination; installs a terminal alkyne handle onto the NP for subsequent "arming" and bio-conjugation [16]. |
| PhI(O₂CtBu)₂ | A stoichiometric oxidant used in Rh-catalyzed C–H amination to generate the active rhodium nitrenoid species from the sulfamate reagent [16]. |
| Computational Software (MOE, KNIME) | Integrated software platforms for SAR/QSAR modeling, molecular docking, and dynamics simulations; used to rationalize experimental SAR and guide the design of new analogs [76]. |
The strategic integration of NPs into the drug discovery pipeline is further visualized by analyzing their unique physicochemical properties compared to purely synthetic molecules, as shown in the following diagram.
The strategic optimization of natural product leads through sophisticated SAR studies remains a powerful avenue for addressing challenging therapeutic targets, including antimicrobial-resistant infections. The methodologies detailed herein—from the rapid, in-situ evaluation of build-up libraries to the selective functionalization of native NP scaffolds—provide a robust toolkit for researchers. By integrating these experimental approaches with computational predictions, scientists can systematically navigate the complex chemical space of natural products. This integrated strategy accelerates the transformation of biologically gifted but therapeutically imperfect natural leads into optimized drug candidates with enhanced potency, improved drug-like properties, and novel mechanisms of action, thereby fully realizing the enduring potential of natural products in modern drug discovery.
Natural products (NPs) have served as a cornerstone in pharmacotherapy, particularly in oncology and infectious diseases, providing unparalleled molecular diversity and structural novelty [9] [87]. Historically, between 1981 and 2010, approximately 79.8% of approved anticancer drugs were natural product-based, underscoring their critical role in therapeutic development [9]. However, these naturally occurring molecules often require optimization to enhance their drug efficacy, improve absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles, and address challenges in chemical accessibility [9]. Structure-Activity Relationship (SAR)-directed optimization represents a systematic approach to this challenge, establishing meaningful correlations between chemical structure modifications and biological activity to transform natural leads into clinically viable drug candidates [9]. This application note presents detailed case studies and methodologies that exemplify successful SAR-driven campaigns in anticancer and antibacterial drug discovery, providing researchers with proven frameworks for lead optimization.
Case Studies in Anticancer Drug Development
Optimization of the Pyrrolidine Scaffold
Pyrrolidine, a saturated five-membered nitrogen-containing heterocycle, is a privileged scaffold prevalent in numerous plant and microbial alkaloids with demonstrated biological activities [88]. Its exploration has yielded significant advances in anticancer agent development.
- Natural Origin and Significance: The pyrrolidine moiety is found in bioactive natural products such as horsfiline, coerulescine, and aegyptolidine A, the latter exhibiting notable anticancer properties [88]. The non-aromatic, chiral nature of the pyrrolidine ring contributes to its diverse pharmacological profile.
- SAR-Driven Optimization Strategies: Research has focused on creating diverse pyrrolidine-based derivatives to establish robust SAR. Key structural classes include:
- Spiro pyrrolidine derivatives: Inspired by natural spirooxindole alkaloids, these compounds are synthesized via 1,3-dipolar cycloaddition reactions. SAR reveals that electron-withdrawing substituents on the arylidine moiety of the spirooxindole core significantly enhance cytotoxic activity against various cancer cell lines [88].
- N-substituted pyrrolidines: The basic nitrogen atom serves as a key modification point. For instance, hydroxycinnamide derivatives featuring an N-2-hydroxy pyrrolidine substituent demonstrated potent activity against P388 murine leukemia cells, with IC₅₀ values reaching 2.6 µM [88].
- Pyrrolidine metal complexes: Schiff base ligands derived from pyrrolidine form stable complexes with metals like Copper(II). These complexes often exhibit enhanced DNA binding and intercalation capabilities, leading to improved apoptotic activity in cancer cells [88].
Table 1: Selected Optimized Pyrrolidine Derivatives and Their Anticancer Profiles
| Derivative Class | Key Structural Modification | Cancer Cell Line / Model | Reported Activity (IC₅₀ or MIC) | Inference from SAR |
|---|---|---|---|---|
| Spirooxindole-pyrrolidine | Electron-withdrawing groups (e.g., -F, -Cl, -NO₂) on arylidine ring | Breast Cancer (MDA-MB-231) | IC₅₀: ~2-5 µM [88] | Electron-withdrawing groups enhance cytotoxic potency. |
| Hydroxycinnamide Derivative | N-2-hydroxy pyrrolidine substitution | P388 Murine Leukemia | IC₅₀: 2.6 µM [88] | Hydrophilic substituents on N can improve activity. |
| Pyrrolidine-thiazolidinone Hybrid | Combination with thiazolidinone core | Pancreatic Cancer (MIA PaCa-2) | IC₅₀: <10 µM [88] | Molecular hybridization is a viable strategy. |
From Harmine to the Selective DYRK1A Inhibitor AnnH75
The natural β-carboline alkaloid harmine is a potent inhibitor of DYRK1A, a kinase target in cancer and neurodegenerative diseases. However, its strong simultaneous inhibition of Monoamine Oxidase A (MAO-A) posed a significant clinical safety risk, necessitating a selective optimization campaign [89].
- Initial Challenge: Potent, dual inhibition of DYRK1A (desired) and MAO-A (undesired) by the natural lead compound, harmine.
- SAR Strategy and Outcome: Through the synthesis and profiling of over 60 harmine analogues, researchers established a critical SAR: small polar substituents at the N-9 position of the β-carboline scaffold selectively abrogated MAO-A inhibition while preserving DYRK1A activity [89]. This strategic modification, combined with optimization at the C-1 position (e.g., methyl or chlorine substitution), led to the development of AnnH75.
- Key Achievement: AnnH75 emerged as a potent DYRK1A inhibitor completely devoid of MAO-A inhibition, successfully separating the desired kinase activity from the undesired neurotoxic potential [89]. Binding mode analysis via X-ray crystallography confirmed the molecular interactions underlying this selective inhibition.
Diagram 1: SAR workflow for harmine to AnnH75 optimization.
Case Studies in Antibacterial Drug Development
Discovery and Optimization of the Oxadiazole Class vs. MRSA
The oxadiazole class of antibiotics represents a successful modern discovery originating from an in silico screen against penicillin-binding protein 2a (PBP2a) of methicillin-resistant Staphylococcus aureus (MRSA) [90].
- Lead Identification: The initial lead compound (1) was identified via structure-based docking of 1.2 million compounds from the ZINC library, followed by phenotypic screening against the ESKAPE panel of bacterial pathogens [90].
- Comprehensive SAR Exploration: A focused SAR investigation on the C-5 position of the 1,2,4-oxadiazole core (Ring A) involved the synthesis and evaluation of 120 derivatives [90]. Key findings included:
- Replacement of the phenol with specific heterocycles like 4-halogen-substituted pyrazoles maintained or improved potency (MIC ≤1 µg/mL).
- The 4-ethynyl derivative (66b) achieved the lowest MIC of 0.25 µg/mL against S. aureus.
- Indolyl, imidazolyl, and substituted pyridinyl rings at the C-5 position were also well-tolerated, confirming the versatility of this region for optimization.
- In Vivo Efficacy and PK Optimization: While exploring Ring A for potency, substitutions on the distal diphenyl ether moiety (Rings C and D) were crucial for optimizing pharmacokinetics. Incorporating a trifluoromethyl or fluoro group at the 4-position of Ring D improved metabolic stability and lowered clearance, leading to compound 75b. This optimized candidate was efficacious in a murine MRSA peritonitis model, exhibited oral bioavailability, and demonstrated a bactericidal mode of action [90].
Table 2: Selected SAR Findings for Oxadiazole Antibiotics (Ring A Modifications) [90]
| Ring A Substituent / Heterocycle | Example Compound | MIC vs. S. aureus ATCC 29213 (µg/mL) | Key Inference |
|---|---|---|---|
| 4-Halogen-Substituted Pyrazole | 60a-c, 61a-b, 62a-c | ≤1 | Heterocyclic bioisosteres of phenol are favorable. |
| Pyrazole with -NH-iPr | 65a-b | 0.5 | Bulky alkylamino groups can enhance potency. |
| Pyrazole with -C≡CH | 66b | 0.25 | Linear, sp-hybridized groups can yield highest potency. |
| Indol-5-yl | 75a-c | ≤1 | Fused heteroaromatic systems are tolerated. |
| 3-F, 5-F Phenol | 71b-c | ≤2 | Additional fluorine atoms on phenol maintain activity. |
Experimental Protocol: SAR-Driven Lead Optimization
This protocol outlines a standard iterative cycle for the SAR-based optimization of a natural product lead.
1. Compound Library Design and Synthesis
- Objective: To generate a focused library of analogues for SAR elucidation.
- Procedure: a. Identify Modification Sites: Based on the natural lead's structure, propose sites for derivatization (e.g., aromatic substitution, side chain elongation, heteroatom replacement, stereochemistry alteration) [9] [89]. b. Plan for Diversity: Design analogues to systematically explore electronic, steric, and lipophilic parameters. This includes incorporating bioisosteric replacements (e.g., a carboxylic acid with a tetrazole) [9]. c. Synthetic Execution: Employ solution-phase or solid-phase synthetic methods, potentially leveraging green chemistry approaches like microwave-assisted synthesis to accelerate library production [88].
2. In Vitro Biological Screening
- Objective: To quantitatively assess the biological activity and selectivity of synthesized analogues.
- Materials:
- Test compounds (as 10 mM DMSO stocks)
- Relevant cancer cell lines (e.g., MCF-7, A549) or bacterial strains (e.g., S. aureus ATCC 29213, MRSA clinical isolates)
- Cell culture media and reagents
- 96-well microtiter plates
- Microplate spectrophotometer or fluorometer
- Procedure for Anticancer Activity (MTS/Proliferation Assay): a. Seed cancer cells in 96-well plates at a density of 5,000 cells/well and incubate for 24 h. b. Treat cells with a dilution series of each test compound (typical range: 0.1 - 100 µM). Include a negative control (DMSO vehicle) and a positive control (e.g., doxorubicin). c. Incubate for 72 h. d. Add MTS reagent and incubate for 1-4 h. e. Measure absorbance at 490 nm. Calculate % cell viability and determine IC₅₀ values using non-linear regression analysis [88].
- Procedure for Antibacterial Activity (Broth Microdilution MIC Assay): a. Prepare Mueller-Hinton broth in a 96-well plate. b. Perform two-fold serial dilutions of test compounds directly in the broth. c. Inoculate each well with a standardized bacterial inoculum of ~5 × 10⁵ CFU/mL. d. Incubate plate at 37°C for 16-20 h. e. The Minimum Inhibitory Concentration (MIC) is the lowest concentration of compound that completely prevents visible growth [90].
3. SAR Data Analysis and Hypothesis Generation
- Objective: To correlate structural changes with biological activity and plan the next cycle of optimization.
- Procedure: a. Compile biological data (IC₅₀, MIC) into a spreadsheet alongside structural descriptors (substituents, logP, etc.). b. Identify structural features that enhance, diminish, or nullify biological activity. c. Develop a hypothesis for the next round of synthesis (e.g., "Increasing hydrophobicity at the para-position improves potency, suggesting this region interacts with a hydrophobic pocket") [91]. d. Use computational chemistry (molecular docking, pharmacophore modeling) to visualize and validate hypotheses, especially if a protein structure is available [9] [89].
Diagram 2: Iterative SAR-driven optimization workflow.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Materials for SAR Studies in Natural Product Optimization
| Reagent / Material | Function / Application | Example in Context |
|---|---|---|
| Bioactive Natural Product Lead | Starting template for derivatization and SAR studies. | Harmine, Voacanga alkaloids, or a simple pyrrolidine-containing natural product [89] [88]. |
| Chemical Building Blocks | For introducing diverse functional groups (e.g., aldehydes, boronic acids, alkyl halides). | Used in coupling reactions (e.g., Suzuki, reductive amination) to create analogue libraries [88] [90]. |
| Cell-Based Assay Kits (e.g., MTS, MTT) | To measure cell viability and proliferation for determining IC₅₀ values of anticancer agents. | Essential for profiling pyrrolidine derivatives against a panel of cancer cell lines [88]. |
| Culture Media for Bacterial Strains | To grow pathogenic bacteria for determining Minimum Inhibitory Concentrations (MICs). | Mueller-Hinton broth for testing oxadiazoles against MRSA and other ESKAPE pathogens [90]. |
| Molecular Modeling Software | For structure-based design, docking, and visualizing ligand-target interactions to guide SAR. | Used to hypothesize the binding mode of harmine analogues to DYRK1A and design selective inhibitors [89]. |
The case studies presented herein demonstrate the profound impact of systematic SAR analysis in advancing natural products from mere leads into viable drug candidates. The optimization of the pyrrolidine scaffold for oncology and the development of oxadiazoles as a novel class of antibiotics against MRSA exemplify how iterative cycles of design, synthesis, and testing can solve critical challenges such as selectivity, potency, and pharmacokinetics. These successful frameworks provide a validated roadmap for researchers aiming to navigate the complex journey of natural product-based drug discovery. As the field evolves, integrating modern techniques like AI-driven design and chemoinformatic analysis with classical SAR principles will further accelerate the discovery of new therapeutic agents to address unmet medical needs in cancer and infectious diseases [87] [92].
Within the context of structure-activity relationship (SAR) directed optimization of natural product leads, the path from initial discovery to a clinically viable therapeutic is a multifaceted challenge. This process requires a meticulous balance between enhancing a compound's specificity for its intended target, minimizing its off-target toxicity, and optimizing its physicochemical properties to ensure adequate pharmacokinetic profiles. Natural products, with their inherent structural complexity and bioactivity, provide excellent starting points for drug discovery. However, their frequent lack of specificity, suboptimal pharmacokinetics, and inherent toxicity necessitate systematic optimization to translate their potential into safe and effective medicines. This Application Note provides detailed protocols and frameworks for the critical evaluation of therapeutic potential, specifically tailored for researchers and drug development professionals working on the SAR-driven optimization of natural product-derived leads.
Establishing a Framework for SAR-Driven Optimization
The optimization of a natural product lead is a deliberate process aimed at addressing specific deficiencies while preserving or enhancing its core biological activity. The strategy is fundamentally guided by SAR studies, which systematically correlate chemical modifications with changes in biological output.
Core Optimization Objectives: The primary goals during SAR-driven optimization can be categorized into three key areas, each with its own set of strategic considerations:
- Enhancing Drug Efficacy: Modifications aimed at improving potency and target specificity. This often involves direct chemical manipulation of functional groups, guided by classic medicinal chemistry principles or modern structure-based design [9].
- Optimizing ADMET Profiles: Efforts to improve Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) characteristics. For natural products, this is crucial as their structural complexity can lead to unfavorable solubility, permeability, and metabolic stability [93] [9].
- Improving Chemical Accessibility: Strategies to overcome limited natural availability or synthetic intractability of the lead compound. This may involve simplifying the core structure or developing more efficient synthetic routes [9].
Chemical Strategies for Optimization: Chemically, these objectives are achieved through a tiered approach, progressing from straightforward derivatization to comprehensive molecular redesign [9]:
- Direct Chemical Manipulation: The most straightforward approach, involving derivatization or substitution of functional groups, alteration of ring systems, and bioisosteric replacement.
- SAR-Directed Optimization: This approach involves the initial establishment of robust SARs through systematic modification, followed by data-driven optimization. The accumulation of chemical and biological data enables more rational and predictive design.
- Pharmacophore-Oriented Molecular Design: The most advanced strategy, where the core structure of the natural product may be significantly altered based on the identified pharmacophore. This approach often leverages modern rational drug design techniques, such as structure-based design and scaffold hopping, to generate novel leads with improved properties and chemical accessibility.
Application Note: Optimizing a Natural Lead Against a Promising Antibacterial Target (MraY)
Background and Objective
Antimicrobial resistance (AMR) necessitates the development of new antibacterial agents with novel mechanisms of action. Phospho-N-acetylmuramoyl-pentapeptide-transferase (MraY) is a promising antibacterial target as it is essential for bacterial cell wall synthesis and is not targeted by existing clinical antibiotics. While several natural products, such as capuramycin and muraymycin, are known MraY inhibitors, their development is often hampered by poor drug-like properties and complex synthetic pathways, which impede traditional SAR studies [18].
Objective: To establish an efficient and comprehensive strategy for the simultaneous SAR exploration and optimization of multiple MraY-inhibitory natural product cores. The goal is to identify analogues with potent and broad-spectrum antibacterial activity against drug-resistant strains, acceptable cytotoxicity, and improved chemical accessibility.
Experimental Protocol: Build-Up Library Synthesis and In Situ Screening
This protocol outlines a method for rapidly generating and evaluating a library of natural product analogues via chemoselective fragment ligation, minimizing the need for lengthy multi-step synthesis and purification of each individual compound [18].
Workflow Overview:
Step-by-Step Procedure:
Library Design and Fragment Preparation:
- Core Aldehydes: Select natural product cores (e.g., from capuramycin, muraymycin) that contain the essential uridine moiety for MraY binding. Synthesize or acquire these cores with a conjugated aldehyde functional group to enhance the stability of the subsequent ligation product [18].
- Accessory Hydrazine Library: Prepare a diverse collection of 98+ hydrazide fragments. Diversity should encompass various chemotypes, including [18]:
- Simple acyl hydrazides (e.g., benzoyl-type, phenyl acetyl-type).
- Alkyl acyl hydrazides.
- N-acyl aminoacyl hydrazides with varying acyl chain lengths and amino acid side chains.
In Situ Hydrazone Library Synthesis:
- In a 96-well plate, combine 10 mM DMSO solutions of each aldehyde core and each hydrazine fragment in a 1:1 stoichiometry. The total reaction volume per well should be 31 µL.
- Incubate the plate at room temperature for 30 minutes to allow for hydrazone formation.
- Remove the DMSO solvent by centrifugal concentration under vacuum at room temperature overnight. This step drives the reaction to completion by removing water, the only by-product.
- Redissolve the resulting residues in 30 µL of DMSO to create a 5 mM library solution of the synthesized hydrazones for biological testing. Assume 100% conversion for concentration calculations in initial evaluations [18].
Primary Biological Evaluation (In Situ):
- MraY Enzymatic Inhibition Assay: Directly use the library solutions from Step 2 to assess MraY inhibitory activity using a established assay (e.g., measuring inhibition of lipid I formation) [18]. Screen at a single high concentration (e.g., 100 µM) to identify active wells.
- Whole-Cell Antibacterial Assay: Due to the clean nature of the hydrazone formation reaction (no cytotoxic reagents or heavy metal catalysts), the same library solutions can be used directly in a cell-based assay. Determine the Minimum Inhibitory Concentration (MIC) against a panel of drug-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE) [18].
Hit Confirmation and Secondary Profiling:
- Re-synthesize and purify the most promising hits (showing potent MraY inhibition and antibacterial activity) on a larger scale for confirmatory testing.
- Perform dose-response curves to determine IC₅₀ (enzyme) and MIC₉₀ (cell) values.
- Evaluate cytotoxicity against mammalian cell lines (e.g., HepG2, Vero) to establish a preliminary selectivity index.
In Vivo Efficacy Assessment:
- Advance the most promising analogue (e.g., Analogue 2 from the referenced study, which demonstrated efficacy in a mouse acute thigh infection model) into in vivo studies [18].
- Utilize a murine model of bacterial infection to evaluate the compound's ability to reduce bacterial load.
Key Research Reagent Solutions
Table 1: Essential research reagents for the build-up library synthesis and screening platform.
| Reagent / Material | Function / Description | Key Consideration |
|---|---|---|
| Aldehyde Core Fragments | Core structures of MraY inhibitors (e.g., from capuramycin) with conjugated aldehyde handle. | Must retain the essential uridine moiety for target binding. The aldehyde should be conjugated for hydrazone stability [18]. |
| Diverse Hydrazine Library | Accessory fragments providing chemical diversity; includes acyl and aminoacyl hydrazides. | A wide variety of steric, electronic, and lipophilic properties is critical for exploring SAR and optimizing bacterial accumulation [18]. |
| Anhydrous DMSO | Solvent for reaction and preparation of assay-ready library solutions. | High purity is essential to prevent side reactions and ensure accurate biological screening. |
| MraY Enzyme & Substrates | For the primary biochemical inhibition assay. | The assay must be robust and sensitive for high-throughput screening of library mixtures [18]. |
| Bacterial Strains | Drug-resistant strains (e.g., MRSA, VRE) for whole-cell activity assessment. | Use clinically relevant strains to ensure translational relevance of the discovered leads [18]. |
Data Analysis and SAR Interpretation
Quantitative Assessment of Key Analogues: The following table summarizes the profile of a lead analogue identified through the described protocol, compared to its parent core and a standard drug [18].
Table 2: Representative data for a lead MraY inhibitor analogue identified via build-up library screening.
| Compound | MraY IC₅₀ (nM) | MIC vs MRSA (µg/mL) | MIC vs VRE (µg/mL) | Cytotoxicity (CC₅₀, µM) | In Vivo Efficacy (Thigh Model) |
|---|---|---|---|---|---|
| Aldehyde Core | ~1000 | >128 | >128 | >64 | Not Active |
| Analogue 2 | 6.2 | 2 | 4 | >64 | Significant Bacterial Load Reduction |
| Moxifloxacin (Control) | N/A | 0.06 | 8 | N/A | Active |
SAR Insights and Decision Points:
- Fragment Ligation is Crucial: The dramatic increase in potency from the aldehyde core to Analogue 2 underscores the essential role of the accessory fragment in MraY binding and inhibition [18].
- Balancing Potency and Accumulation: The specific hydrazine fragment in Analogue 2 was found to optimally balance strong MraY inhibition with the physicochemical properties needed for bacterial cell penetration, a common challenge with this target [18].
- Specificity and Toxicity: The high CC₅₀ value indicates low direct cytotoxicity, suggesting a favorable specificity profile for the MraY target over generic mammalian cell toxicity. This is a critical checkpoint for clinical viability.
Generalizable Protocols for Key Evaluations
Protocol for Cytotoxicity and Selectivity Assessment
A critical step in establishing clinical viability is to determine a compound's therapeutic window.
Procedure:
- Cell Culture: Maintain relevant mammalian cell lines (e.g., HepG2 for liver toxicity, HEK293 for renal, or primary cell lines for greater relevance) in appropriate media.
- Compound Treatment: Seed cells in a 96-well plate and treat with a dilution series of the test compound after 24 hours of attachment. Include a vehicle control (e.g., DMSO) and a positive control (e.g., staurosporine).
- Viability Assay: After 48-72 hours of incubation, measure cell viability using a standardized assay such as the MTT or CellTiter-Glo assay, following the manufacturer's protocol.
- Data Analysis: Calculate the percentage viability relative to the vehicle control and determine the CC₅₀ (concentration that kills 50% of cells) using non-linear regression.
- Selectivity Index (SI) Calculation: Calculate the SI using the formula: SI = CC₅₀ (Mammalian Cells) / MIC₉₀ (Pathogen). An SI > 10 is generally considered a positive indicator for further development [94].
Protocol for Structure-Activity Relationship (SAR) Analysis
Systematic SAR analysis is the engine of lead optimization.
Procedure:
- Design Analogues: Based on the initial hit, design a focused library of analogues that systematically vary one region of the molecule at a time (e.g., the northern hemisphere, core scaffold, southern hemisphere).
- Profile Biological Activity: Test all analogues in the primary biochemical (e.g., MraY IC₅₀) and cellular (e.g., MIC) assays.
- Correlate Structure and Activity: Create a SAR table to visualize how specific structural changes impact potency.
- Identify the Pharmacophore: Use the SAR data to map the essential structural features required for activity. This can be done through molecular modeling or simple schematic diagrams.
- Iterate and Optimize: Use the SAR insights to design a second-generation library focused on optimizing the key parameters (e.g., improving potency against a specific substituent, reducing logP by introducing polar groups, blocking a potential metabolic soft spot).
Table 3: A generalized SAR table template for analyzing natural product derivatives.
| Analogue ID | R¹ Group | R² Group | Core Modification | IC₅₀ (nM) | MIC (µg/mL) | Key SAR Insight |
|---|---|---|---|---|---|---|
| NP-01 | -H (Parent) | -CH₃ | None | 100 | 8.0 | Baseline |
| NP-02 | -Cl | -CH₃ | None | 25 | 2.0 | Halogen at R¹ boosts potency. |
| NP-03 | -OCH₃ | -CH₃ | None | 150 | 16.0 | Methoxy at R¹ is detrimental. |
| NP-04 | -Cl | -C₂H₅ | None | 22 | 1.0 | Small alkyl extension at R² is tolerated. |
| NP-05 | -Cl | -CH₃ | Saturated B-ring | >1000 | >64 | Core B-ring unsaturation is critical. |
The journey from a bioactive natural product to a clinically viable drug candidate hinges on a rigorous, iterative process of evaluation and optimization. The integrated strategy presented here—combining innovative library synthesis like the build-up approach with standardized protocols for assessing specificity, toxicity, and efficacy—provides a robust framework for researchers. By deeply integrating SAR analysis with ADMET profiling early and throughout the optimization cycle, drug development professionals can make data-driven decisions that significantly de-risk the path forward. This disciplined approach maximizes the likelihood of transforming a promising natural lead into a safe, effective, and specific therapeutic agent capable of addressing unmet medical needs.
The structure-activity relationship (SAR) directed optimization of natural products represents a cornerstone of modern drug discovery. Natural products, with their inherent structural complexity and biological relevance, serve as privileged starting points for lead development [85]. However, the path from a natural lead to a optimized drug candidate is fraught with challenges, including the intricate elucidation of SAR and the costly, iterative cycle of synthesis and biological testing. Traditionally, this process has been a major bottleneck, consuming significant time and resources. Recent technological advancements are now fundamentally transforming this landscape. This Application Note details how modern computational and experimental platforms are enhancing validation efficiency at critical stages of natural product optimization. By integrating artificial intelligence (AI), collaborative data tools, and sophisticated molecular modeling, researchers can now accelerate the establishment of robust SAR and make more informed decisions, thereby streamlining the entire lead optimization pipeline.
Technological Approaches and Platforms
The integration of advanced technologies is enabling a more rational and efficient approach to natural product optimization. The table below summarizes the core technological platforms and their specific impacts on validation efficiency.
Table 1: Modern Platforms Enhancing Validation Efficiency in Natural Product SAR
| Platform Category | Key Technologies | Impact on Validation Efficiency |
|---|---|---|
| AI & Machine Learning | Machine Learning (ML), Deep Learning (DL), Bayesian Models [95] [96] | Accelerates prediction of bioactivity and ADMET properties, enabling virtual screening of large virtual libraries prior to synthesis. Reduces cycle times from years to months [96]. |
| Collaborative Informatics | CDD Vault, Interactive Visualization, Collaborative Databases [95] | Provides centralized, secure data management. Enables real-time, multidimensional visualization of complex HTS and SAR data for rapid hypothesis generation and team-based analysis. |
| Advanced Computational Modeling | Molecular Docking, Pharmacophore Modeling, Molecular Dynamics (MD) [6] | Offers structural insights for SAR analysis. Predicts binding modes of analogues, rationalizing activity and guiding the design of more potent and selective derivatives. |
AI and Machine Learning Platforms
Artificial intelligence, particularly machine learning (ML) and deep learning (DL), has emerged as a powerful tool to compress the traditional drug discovery timeline. These platforms learn from large-scale historical screening data, such as that found in public databases (e.g., ChEMBL, PubChem) or proprietary corporate collections, to build predictive models [95]. A systematic review of AI in drug discovery found that ML accounts for approximately 40.9% of AI methods used, with DL at 10.3% [96]. These models are applied to predict the biological activity, selectivity, and key absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters of natural product analogues before they are ever synthesized.
The efficiency gain is substantial. For instance, AI-driven platforms can prospectively enrich screening libraries, potentially doubling experimental hit rates and freeing resources to explore broader chemical space [95]. Insilico Medicine demonstrated this potential by using an AI-driven platform to identify a novel target and advance a drug candidate for idiopathic pulmonary fibrosis into preclinical trials in just 18 months, a process that traditionally takes 4–6 years [96]. This represents a dramatic increase in validation efficiency for early-stage leads.
Collaborative Informatics and Data Mining Platforms
Modern drug discovery is increasingly data-driven and collaborative. Platforms like the Collaborative Drug Discovery (CDD) Vault are designed to manage the immense multidimensional data generated from high-throughput screening (HTS) and SAR campaigns [95]. These platforms integrate data mining and visualization tools that allow researchers to interact with thousands of data points in real-time.
The "Visualization in CDD Vault" module, for example, uses technologies like WebGL and SVG to create dynamic scatterplots and histograms [95]. Researchers can visually identify activity clusters, outliers, and trends within complex datasets by adjusting filters or directly selecting data points on plots. This immediate visual feedback allows for rapid triaging of compounds and the formation of SAR hypotheses, significantly accelerating the decision-making process compared to static data analysis. Furthermore, these platforms support secure, selective data sharing among collaborators and are integrated with modeling tools, creating a seamless iterative workflow from data generation to model building and back to experimental design [95].
Molecular Modeling and Simulation Platforms
Structure-based molecular modeling methods, including molecular docking, pharmacophore modeling, and molecular dynamics (MD) simulations, provide a atomic-level rationale for observed SAR and guide lead optimization [6]. While often used for initial hit identification, their strategic application in the hit-to-lead and lead optimization phases is a key driver of efficiency.
A well-validated molecular docking workflow can predict how different substituents on a natural product scaffold will interact with the target protein. This helps prioritize which analogues to synthesize to improve potency or selectivity. The workflow requires a high-quality protein structure, careful selection of a docking algorithm and scoring function, and rigorous validation against known active and inactive compounds [6]. Pharmacophore modeling can distill the essential steric and electronic features responsible for biological activity, providing a query for virtual screening of novel analogues. By using these in silico tools to filter out low-probability candidates, researchers can focus synthetic efforts on a smaller, higher-value set of compounds, thereby reducing the number of iterative cycles needed to arrive at an optimized lead.
Application Notes & Experimental Protocols
Protocol: AI-Guided Bioactivity Prediction for Natural Product Analogues
This protocol outlines the steps for using machine learning to predict the bioactivity of novel natural product analogues, enabling prioritization for synthesis.
1. Research Reagent Solutions
Table 2: Essential Materials for AI-Guided SAR
| Item | Function |
|---|---|
| Public Bioactivity Database (e.g., ChEMBL, PubChem) | Provides a large, curated source of chemical structures and associated bioactivity data for model training [95]. |
| Chemical Descriptor Software (e.g., RDKit, PaDEL) | Generates numerical representations (e.g., fingerprints, molecular weight, logP) of chemical structures for machine learning algorithms. |
| Machine Learning Library (e.g., scikit-learn, TensorFlow) | Provides algorithms (e.g., Random Forest, Neural Networks) to build predictive models from the chemical descriptor and bioactivity data [96]. |
| Natural Product or Derivative Library | A virtual or in-house collection of natural product scaffolds and proposed analogues for prediction. |
2. Procedure
- Data Curation and Preparation: Assemble a dataset of chemical structures and their corresponding bioactivity (e.g., IC50, Ki) for a specific target. The data should be relevant to the natural product scaffold of interest. Carefully address data quality, including removing duplicates and correcting errors [95].
- Descriptor Calculation and Feature Engineering: Calculate a set of chemical descriptors or fingerprints for every compound in the dataset. This transforms the chemical structure into a numerical vector that the ML algorithm can process.
- Model Training and Validation: Split the dataset into training and test sets. Use the training set to build a predictive model, such as a Bayesian model or a deep neural network [95] [96]. Validate the model's performance on the held-out test set using metrics like AUC-ROC, precision, and recall.
- Prediction and Prioritization: Apply the trained model to predict the bioactivity of a virtual library of unsynthesized natural product analogues. Rank the compounds based on their predicted activity and other desirable properties.
- Experimental Validation and Model Refinement: Synthesize and test the top-ranked candidates. Use the new experimental data to further refine and improve the ML model, creating an iterative, learning workflow [97].
3. Diagram: AI-Guided SAR Workflow
The following diagram illustrates the iterative workflow for AI-guided natural product optimization.
Protocol: Structure-Based SAR Analysis Using Molecular Docking
This protocol describes how to use molecular docking to understand the binding interactions of a natural product and its analogues, guiding the design of improved derivatives.
1. Research Reagent Solutions
Table 3: Essential Materials for Structure-Based SAR
| Item | Function |
|---|---|
| Protein Data Bank (PDB) Structure | Source of a high-resolution 3D structure of the biological target, crucial for docking simulations [6]. |
| Molecular Docking Software (e.g., AutoDock, GOLD, Schrödinger) | Program that computationally predicts how a small molecule (ligand) binds to a protein target [6]. |
| Structure Preparation Tool (e.g., MOE, Maestro) | Software used to prepare the protein and ligand structures for docking (e.g., adding hydrogens, assigning charges). |
| Series of Natural Product Analogues | A set of compounds with known biological activity and varying substituents for in silico SAR analysis. |
2. Procedure
- Target and Ligand Preparation: Obtain a high-resolution crystal structure of the target protein from the PDB. Prepare the protein by adding hydrogen atoms, correcting protonation states, and removing water molecules. Prepare the 3D structures of the natural product lead and its known analogues by energy minimization.
- Workflow Validation (Critical Step): Define the binding site and validate the docking protocol by re-docking a known co-crystallized ligand. The goal is to reproduce the experimental binding pose with a root-mean-square deviation (RMSD) of typically <2.0 Å. This step ensures the reliability of subsequent predictions [6].
- Docking of Analogues: Dock the entire series of natural product analogues into the defined binding site using the validated protocol. Generate multiple poses for each compound.
- Pose Analysis and SAR Interpretation: Analyze the top-ranked poses for each analogue. Identify key protein-ligand interactions (e.g., hydrogen bonds, hydrophobic contacts, pi-stacking) and correlate changes in these interactions with the measured biological activity across the series.
- Design and In Silico Evaluation: Propose new analogues with substitutions predicted to enhance favorable interactions or eliminate unfavorable ones. Dock these proposed compounds to validate the design hypothesis before committing to synthesis.
3. Diagram: Docking Workflow for SAR Analysis
The following diagram outlines the key steps for applying molecular docking to SAR analysis.
The integration of modern technological platforms is undeniably enhancing validation efficiency in the SAR-directed optimization of natural products. AI and ML models allow for the predictive triaging of synthetic targets, collaborative informatics platforms enable rapid, visual data exploration and team science, and advanced molecular modeling provides a rational structural basis for compound design. By adopting these tools, researchers can transition from a largely empirical, trial-and-error approach to a more rational, data-driven paradigm. This shift not only accelerates the lead optimization process but also increases the likelihood of successfully advancing high-quality natural product-derived candidates through the drug development pipeline. The future of natural product research lies in the continued refinement and interdisciplinary application of these powerful technologies.
Conclusion
SAR-directed optimization provides a powerful, systematic framework for transforming natural product leads into clinically viable drug candidates. By integrating foundational chemical principles with advanced methodologies like build-up libraries, computational modeling, and AI-driven analysis, researchers can effectively navigate the complex landscape of multi-parameter optimization. The future of this field lies in further embracing digital transformation through platforms that streamline SAR analysis, enhancing the prediction of ADMET properties, and developing more sophisticated in silico tools to reduce reliance on extensive synthetic cycles. These advances will accelerate the delivery of novel therapeutics from nature's chemical repertoire to address pressing unmet medical needs, particularly against drug-resistant pathogens and complex diseases like cancer.
References
- 1. Structure–activity relationship [https://en.wikipedia.org/wiki/Structure%E2%80%93activity_relationship]
- 2. Advances, opportunities, and challenges in methods for ... [https://pubs.rsc.org/en/content/articlehtml/2024/np/d4np00009a]
- 3. SAR | Structure Activity Relationships [https://www.collaborativedrug.com/cdd-blog/what-is-sar]
- 4. On Exploring Structure Activity Relationships - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4852705/]
- 5. Efficient SAR analysis and reporting for rapid compound ... [https://www.discngine.com/blog/2025/3/14/efficient-sar-analysis-and-reporting-for-rapid-compound-optimizationdiscngine-bayer-success-story]
- 6. Structure-based molecular modeling in SAR analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7979990/]
- 7. Comparison of Quantitative and Qualitative (Q)SAR Models ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6192375/]
- 8. Comparative Study of (Q)SAR Models for Predicting the ... [https://www.sciencedirect.com/science/article/pii/S3050620425000491]
- 9. Strategies for the Optimization of Natural Leads to Anticancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4679534/]
- 10. From Patterns to Pills: How Informatics Is Shaping Medicinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114992/]
- 11. The evolution of chemical collections and drug-like diversity [https://www.ddw-online.com/the-evolution-of-chemical-collections-and-drug-like-diversity-36277-202509/]
- 12. Computational Evaluation and Multi-Criteria Optimization ... [https://www.mdpi.com/1467-3045/47/7/577]
- 13. Structure Activity Relationship (SAR) Analysis [https://www.creative-proteomics.com/services/structure-activity-relationship-sar-analysis.htm]
- 14. Quantitative Structure-Activity Relationship - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/quantitative-structure-activity-relationship]
- 15. Quantitative structure–activity relationship [https://en.wikipedia.org/wiki/Quantitative_structure%E2%80%93activity_relationship]
- 16. Simultaneous structure-activity studies and arming of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4041531/]
- 17. Identification and structure-activity relationship optimization ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425008153]
- 18. Development of a natural product optimization strategy for ... [https://www.nature.com/articles/s41467-024-49484-7]
- 19. Impact of Functional Groups in Organic Chemistry on ... [https://www.reachemchemicals.com/blog/impact-of-functional-groups-in-organic-chemistry-on-pharmaceutical-development/]
- 20. Functional Groups In Organic Chemistry [https://www.masterorganicchemistry.com/2010/10/06/functional-groups-organic-chemistry/]
- 21. Functional groups [https://www.biotechacademy.dk/en/functional-groups/]
- 22. Frequency and Importance of Six Functional Groups that ... [https://www.biotech-asia.org/vol15no3/frequency-and-importance-of-six-functional-groups-that-play-a-role-in-drug-discovery/]
- 23. 6.1: Identifying drugs from functional groups [https://chem.libretexts.org/Bookshelves/Biological_Chemistry/Pharmaceutical_Chemistry_of_Molecular_Therapeutics_(de_Araujo_Saqib_Keillor_and_Gunning)/06%3A_Summary/6.01%3A_Identifying_drugs_from_functional_groups]
- 24. Interdisciplinary analysis of drugs: Structural features and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9108004/]
- 25. New strategies to enhance the efficiency and precision of ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1550158/full]
- 26. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 27. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 28. Design, Quantitative Structure–Activity Relationships and ... [https://www.sciencedirect.com/science/article/pii/S2667022425001458]
- 29. Unraveling the structure–activity relationships of ... [https://www.nature.com/articles/s41598-025-23467-0]
- 30. SAR-Guided Development of Small-Molecule SERCA2a ... [https://pubmed.ncbi.nlm.nih.gov/40702921/]
- 31. Structure–activity relationships of fenarimol analogues with ... [https://chemrxiv.org/engage/chemrxiv/article-details/68137329e561f77ed49a9afb]
- 32. A new computational cross-structure-activity relationship (C ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525005207]
- 33. High-Efficiency Discovery and Structure-Activity ... [https://pubmed.ncbi.nlm.nih.gov/40679143/]
- 34. Enabling AI in chemistry and biologics discovery [https://www.ddw-online.com/enabling-ai-in-chemistry-and-biologics-discovery-37278-202509/]
- 35. Fragment-Based Drug Design: From Then until Now, and ... [https://pubmed.ncbi.nlm.nih.gov/39992814/]
- 36. Fragment-Based Drug Discovery | April 15-16, 2025 [https://www.drugdiscoverychemistry.com/25/fragment-based-drug-discovery]
- 37. Fragment Based Drug Discovery & Lead Generation [https://www.sygnaturediscovery.com/drug-discovery/integrated-drug-discovery/fragment-based-drug-discovery-2/]
- 38. Rationally minimizing natural product libraries using mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11915828/]
- 39. Fragment Library of Natural Products and Compound ... [https://www.mdpi.com/2218-273X/10/11/1518]
- 40. Affinity selection mass spectrometry (AS-MS) for ... [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2025.1562501/full]
- 41. New strategies to enhance the efficiency and precision of drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11850385/]
- 42. Room-temperature X-ray fragment screening with serial ... [https://www.nature.com/articles/s41467-025-64918-6]
- 43. Structure Activity Relationships [https://drugdesign.org/chapters/structure-activity-relationships/]
- 44. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes [https://pmc.ncbi.nlm.nih.gov/articles/PMC7666045/]
- 45. Unlocking potent anti-tuberculosis natural products through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12234934/]
- 46. 1.6: Drug Modifications to Improve Stability [https://chem.libretexts.org/Bookshelves/Biological_Chemistry/An_Introduction_to_Medicinal_Chemistry_and_Molecular_Recognition_(de_Araujo_et_al.)/01%3A_Chapters/1.06%3A_Drug_Modifications_to_Improve_Stability]
- 47. Revolutionizing pharmacology: AI-powered approaches in ... [https://www.sciencedirect.com/science/article/pii/S259009862500020X]
- 48. Target fishing: from “needle in haystack” to “precise ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1673688/full]
- 49. Advances, opportunities, and challenges in methods for interrogating... [https://pubs.rsc.org/en/Content/ArticleLanding/2024/NP/D4NP00009A]
- 50. Multi-Task Learning with Evolutionary Relatedness Metrics... Optimizing [https://pubmed.ncbi.nlm.nih.gov/41018566/]
- 51. Exploring QSAR models for activity -cliff prediction | Journal of... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00708-w]
- 52. Structure-based molecular modeling in SAR analysis and ... [https://www.sciencedirect.com/science/article/pii/S2001037021000696]
- 53. Structure-Activity-Relationships can be directly extracted ... [https://chemrxiv.org/engage/chemrxiv/article-details/68304d0a1a8f9bdab564af6d]
- 54. OECD QSAR Toolbox | OECD [https://www.oecd.org/en/data/tools/oecd-qsar-toolbox.html]
- 55. CCG | MedChem by Design 2025 | Agenda [https://www.chemcomp.com/en/MedChem-2025-Europe.htm]
- 56. AI-Integrated QSAR Modeling for Enhanced Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525248/]
- 57. Machine Learning in Drug Discovery Market 2025 Trends [https://www.towardshealthcare.com/insights/machine-learning-in-drug-discovery-market-sizing]
- 58. Integrating artificial intelligence in drug discovery and early ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11909971/]
- 59. AI meets physics in computational structure-based drug ... [https://www.nature.com/articles/s44386-025-00019-0]
- 60. Designing a Planning AI Agent for Drug Discovery [https://www.receptor.ai/news-and-blogs/from-predictions-to-strategy-designing-a-planning-ai-agent-for-drug-discovery]
- 61. Structure-Activity Relationship (SAR) Optimization of Hits [https://www.bioglyco.com/degl/hit-sar-optimization.html]
- 62. MraY: An Emerging Therapeutic Target in Bacterial ... [https://www.sciencedirect.com/science/article/pii/S0223523425009997]
- 63. 1,2,4-Triazole-based first-in-class non-nucleoside inhibitors of ... [https://pubmed.ncbi.nlm.nih.gov/39975250/]
- 64. Antimicrobial triazinedione inhibitors of the translocase MraY ... [https://pubs.rsc.org/en/content/articlehtml/2025/md/d4md00937a]
- 65. Overcoming Analytical Bottlenecks in Oligonucleotide Drug ... [https://www.genedata.com/resources/learn/details/blog/overcoming-analytical-bottlenecks-in-oligonucleotide-drug-development-with-automation]
- 66. Navigating the Peptide Market: Overcoming Synthesis ... [https://www.linkedin.com/pulse/navigating-peptide-market-overcoming-synthesis-bottlenecks-accelerated-lbhpe]
- 67. Future challenges with DNA-encoded chemical libraries in ... [https://pubmed.ncbi.nlm.nih.gov/31111767/]
- 68. Identification and structure-activity relationship optimization ... [https://pubmed.ncbi.nlm.nih.gov/40803168/]
- 69. Encoding and display technologies for combinatorial ... [https://www.sciencedirect.com/science/article/pii/S2211383524001345]
- 70. Overcoming RNA Bottlenecks - Part 1: How to Ensure ... [https://exothera.world/overcoming-rna-bottlenecks-part-1-how-to-ensure-consistent-formulation-stability/]
- 71. ADMETlab 2.0: an integrated online platform for accurate and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8262709/]
- 72. Which ADMET properties are important for me to predict? [https://optibrium.com/knowledge-base/blog-which-admet-properties-are-important-for-me-to-predict/]
- 73. Leveraging machine learning models in evaluating ADMET ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205928/]
- 74. admetSAR3.0: a comprehensive platform for exploration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11223829/]
- 75. ADMET property prediction via multi-task graph learning ... [https://www.sciencedirect.com/science/article/pii/S2589004223023623]
- 76. Structure-Activity Relationship (SAR) Studies [https://www.oncodesign-services.com/discovery-services/medicinal-chemistry/sar-study/]
- 77. HTS-Oracle: A Retrainable AI Platform for High-Confidence ... [https://pubmed.ncbi.nlm.nih.gov/40777482/]
- 78. Vault Snack #32 – Using the Visualization Data Table for ... [https://www.collaborativedrug.com/cdd-blog/vault-snack-32-using-the-visualization-data-table-for-sar-analyses]
- 79. Computational discovery of natural medicines targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12436275/]
- 80. High Throughput Screening (HTS) [https://www.sygnaturediscovery.com/drug-discovery/bioscience/screening/medium-high-throughput-screening-mts-hts-2/]
- 81. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 82. Preclinical Testing - A Pillar of Drug Development [https://www.lidebiotech.com/blog/purpose-preclinical-testing-pillar-drug-development]
- 83. Advanced In Vitro Models for Preclinical Drug Safety [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763796/]
- 84. Validation of a preclinical model for assessment of drug ... [https://pubmed.ncbi.nlm.nih.gov/26909610/]
- 85. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 86. Cheminformatic comparison of approved drugs from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4607632/]
- 87. Natural products in drug discovery: advances and opportunities [https://pmc.ncbi.nlm.nih.gov/articles/PMC7841765/]
- 88. Structure activity relationship (SAR) and anticancer ... [https://www.sciencedirect.com/science/article/abs/pii/S022352342200856X]
- 89. Structure–Activity Relationships (SAR) of Natural Products [https://www.mdpi.com/journal/molecules/special_issues/Molecules_Structure_Activity_Relationships]
- 90. Structure–Activity Relationship for the Oxadiazole Class of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6863074/]
- 91. Case Studies in SAR Analyses [https://drugdesign.org/chapters/structure-activity-relationships-case-studies/]
- 92. Medicinal chemistry perspectives on anticancer drug design ... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra05472a]
- 93. Natural-product-inspired potent anticancer congeners ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089625003335]
- 94. Unlocking potent anti-tuberculosis natural products through ... [https://link.springer.com/article/10.1007/s13659-025-00529-4]
- 95. Data Mining and Computational Modeling of High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6181121/]
- 96. From Lab to Clinic: How Artificial Intelligence (AI) Is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298131/]
- 97. Advances, opportunities, and challenges in methods for ... [https://www.sciencedirect.com/org/science/article/pii/S0265056824000606]