Navigating Chemical Space: The Evolving Synergy Between Natural Product Diversity and Purchasable Compound Libraries
This article provides a comprehensive comparative analysis of natural product scaffolds and purchasable compound libraries, two foundational pillars in modern drug discovery.
Navigating Chemical Space: The Evolving Synergy Between Natural Product Diversity and Purchasable Compound Libraries
Abstract
This article provides a comprehensive comparative analysis of natural product scaffolds and purchasable compound libraries, two foundational pillars in modern drug discovery. We explore their unique origins, evolutionary trajectories, and defining structural characteristics. We detail methodological approaches for their effective use in screening campaigns, address common challenges in sourcing and application, and present data-driven comparisons of their scaffold diversity and biological relevance. Synthesizing these perspectives, the article concludes with strategic insights on how the complementary strengths of these sources can be leveraged to navigate chemical space more efficiently, ultimately improving the success rates in identifying novel therapeutic leads.
Chemical Origins: The Distinct Evolution and Defining Traits of Natural Products and Synthetic Libraries
Historical Context and Core Thesis
The journey of natural products (NPs) from ecological specimens to laboratory probes and therapeutics represents one of the most productive narratives in science. For centuries, NPs have served as the primary source of medicines, with their structural complexity and evolutionary pre-validation offering unmatched starting points for drug discovery [1]. The advent of high-throughput screening (HTS) and combinatorial chemistry in the late 20th century promised a more efficient, synthetic path forward [2]. However, the initial focus on easily synthesizable, "flat" aromatic compounds often yielded libraries with limited structural diversity and poor success rates in targeting complex biological interfaces [3].
This guide posits that the enduring impact of NPs lies not in their direct replacement by synthetic libraries, but in the strategic convergence of both approaches. Modern drug discovery is increasingly framed by a critical comparison: the evolutionarily honed, three-dimensional complexity of natural product scaffolds versus the synthetic accessibility, scalability, and tailorability of purchasable compound libraries [4]. The most promising contemporary strategies leverage the biological relevance of NP scaffolds to design innovative, NP-inspired libraries and to guide the intelligent curation of purchasable collections for probing underexplored biological space [5].
Structural Comparison: Natural Product Scaffolds vs. Synthetic Libraries
A chemoinformatic analysis of NPs and synthetic compounds (SCs) over time reveals fundamental and persistent differences in their structural landscapes, which directly influence their performance in biological screening [2].
Table 1: Structural and Physicochemical Comparison: Natural Products vs. Synthetic Compounds [2]
| Property Category | Specific Metric | Trend in Natural Products (Over Time) | Trend in Synthetic Compounds (Over Time) | Direct Comparison (NPs vs. SCs) |
|---|---|---|---|---|
| Molecular Size | Molecular Weight | Consistent increase | Constrained, limited variation | NPs are generally larger |
| Heavy Atom Count | Consistent increase | Constrained, limited variation | NPs have more heavy atoms | |
| Ring Systems | Number of Rings | Gradual increase | Moderate increase | NPs have more rings overall |
| Aromatic Rings | Little change | Clear increase | SCs are more aromatic | |
| Non-Aromatic Rings | Gradual increase | Little change | NPs are richer in saturated, 3D rings | |
| Ring Assemblies | Gradual increase | Moderate increase | NPs have larger, more fused systems | |
| Complexity & Drug-Likeness | Fraction of sp3 Carbons (Fsp3) | Higher and increasing | Lower | NPs are more three-dimensional |
| Synthetic Accessibility Score | Generally higher (more complex) | Lower (more accessible) | NPs are synthetically more challenging | |
| Quantitative Estimate of Drug-likeness (QED) | Varies by source; fungal NPs often high | Often optimized for rules (e.g., Rule of 5) | Fungal NPs show superior QED profiles [6] |
The data shows that NPs have evolved to become larger and more complex, exploring chemical space with greater three-dimensionality [2]. In contrast, SCs, while diversifying, have remained constrained by synthetic practicality and traditional drug-likeness rules, leading to a predominance of planar, aromatic structures [2] [3]. This difference is pivotal: the complex, chiral scaffolds of NPs are uniquely suited to interact with challenging biological targets like protein-protein interfaces, while the more accessible chemical space of SCs offers advantages for rapid optimization and lead development [1].
Performance in Drug Discovery: Key Bioactive Examples
The superior performance of NP-derived and NP-inspired molecules in modulating complex biology is evidenced by numerous clinical and preclinical agents. Their success often lies in engaging targets considered "undruggable" by conventional small molecules.
Table 2: Benchmark Bioactive Natural Products and Derived Agents [1] [7]
| Compound Name | Origin / Class | Primary Molecular Target / Mechanism | Therapeutic Area / Use | Key Advantage Demonstrated |
|---|---|---|---|---|
| TNP-470 | Synthetic analog of fumagillin (fungus) | Covalent inhibitor of Methionine Aminopeptidase 2 (MetAP2) | Antiangiogenic (investigational anticancer) | Target Identification: Enabled discovery of MetAP2's role in angiogenesis [1]. |
| FTY720 (Fingolimod) | Synthetic analog of myriocin (fungus) | Sphingosine 1-phosphate (S1P) receptor modulator (functional agonist) | Multiple Sclerosis (FDA-approved) | Mechanistic Insight: Revealed role of S1P pathway in lymphocyte trafficking [1]. |
| Cyclosporine A | Fungal cyclic peptide | Binds cyclophilin A to inhibit calcineurin (protein-protein interaction stabilizer) | Immunosuppression (organ transplant) | PPI Modulation: Pioneered use of macrocycles to disrupt large protein interfaces [1]. |
| Rapamycin (Sirolimus) | Bacterial macrocycle | Binds FKBP12 to inhibit mTOR (induces protein-protein interaction) | Immunosuppression, anticancer, cardiology | Molecular Glue: Creates a novel composite surface to recruit and inhibit a key kinase [1] [7]. |
| Diazonamide A | Marine ascidian | Binds Ornithine δ-Aminotransferase (OAT), disrupting mitotic spindle | Cytotoxic (anticancer investigational) | Novel Target Discovery: Uncovered a non-canonical role for a metabolic enzyme in cell division [1]. |
| dPNP Inhibitor [5] | Synthetic pseudo-natural product | Inhibits Hedgehog (Hh) signaling pathway (target not fully deconvoluted) | Phenotypic screening hit (anticancer potential) | Scaffold Novelty: Novel chemotype from a designed library uncovered new biology [5]. |
These examples underscore a pattern: NPs and their inspired analogs frequently provide the first chemical tools for new targets or pathways, validating novel therapeutic strategies. Their structural complexity is not an artifact but a functional feature enabling high-affinity, selective binding to complex macromolecular surfaces [7].
The Modern Landscape: Purchasable Compound Libraries
Vendors now offer vast libraries designed to capture diverse chemical space. The choice between a diverse, focused, or NP-inspired library is critical for screening success.
Table 3: Commercial Purchasable Compound Libraries: A Representative Comparison [8] [9] [3]
| Library Type / Vendor Example | Size & Description | Key Design & Filtering Principles | Typical Use Case / Advantage |
|---|---|---|---|
| Large Diverse Libraries(e.g., ChemDiv, Enamine) | 100K – 1.6M+ compounds. Broad chemical space coverage [8] [9]. | Lead-like properties; filtered for PAINS/REOS; optimized solubility; Tanimoto diversity [8] [3]. | Primary HTS against novel targets; maximizing scaffold hit rate for unexplored biology. |
| Focused/Targeted Libraries(e.g., Kinase, GPCR, CNS libraries) | 2,000 – 20,000 compounds. Built around known target classes [9] [10]. | Privileged scaffolds for target family; properties tuned (e.g., BBB penetration for CNS) [10]. | Screening targets with known structural motifs; higher hit rates with smaller library size. |
| Natural Product-Inspired & Derived Libraries(e.g., Selvita/AnalytiCon, 3D-Diversity NP-like) | 1,500 – 26,500 compounds. Contains pure NPs, analogs, or NP-like scaffolds [8] [10]. | High Fsp3, stereogenic centers, macrocycles; based on NP fragments or motifs [8]. | Targeting challenging PPIs and phenotypic assays; accessing bio-relevant, "pre-validated" chemical space. |
| Fragment Libraries(e.g., Selvita SLVer-Bio, Enamine Fragments) | 1,000 – 2,500 compounds. Low molecular weight (<300 Da), high solubility [9] [10]. | "Rule of 3" compliance; 3D-enrichment; designed for structural biology (X-ray co-crystallization) [10]. | Fragment-Based Drug Discovery (FBDD); identifying weak binders for efficient optimization. |
| Specialty Libraries(e.g., Covalent, Macrocyclic, Molecular Glues) | 1,300 – 10,000 compounds. Designed with specific modalities [9] [10]. | Warhead chemistry (covalent); ring topology & linkers (macrocycles); bifunctional design (degraders) [9]. | Addressing "undruggable" targets via covalent inhibition, protein degradation, or stabilizing PPIs. |
The strategic selection from these options allows researchers to align library chemistry with biological question. For novel, challenging targets, NP-inspired or highly diverse 3D-enriched libraries may offer a superior starting point compared to traditional flat, aromatic-focused collections [3] [5].
Convergent Strategies: Designing with Natural Product Wisdom
The most significant modern impact of NPs is their role in inspiring new library design philosophies that blend biological relevance with synthetic innovation.
Table 4: Key Strategies for Designing Natural Product-Inspired Compound Collections [4] [5]
| Strategy | Core Principle | Degree of NP Similarity | Primary Advantage | Example Outcome |
|---|---|---|---|---|
| Biology-Oriented Synthesis (BIOS) | Diversification of actual NP core scaffolds. | High | Retains bioactivity profile while improving synthetic tractability. | New analogs of a known NP with improved properties [4]. |
| Pseudo-Natural Products (PNPs) | De novo combination of distinct NP fragments into novel scaffolds not found in nature. | Low (fragments are NP-derived) | Generates unprecedented chemotypes with high biological relevance. | Novel Hedgehog pathway inhibitor from indole/indanone fragments [5]. |
| Diverse PNP (dPNP) | Combines PNP logic with diversification strategies from Diversity-Oriented Synthesis (DOS). | Variable | Maximizes both scaffold diversity and biological relevance from a common intermediate. | A single divergent intermediate yielding 154 PNPs across 8 classes with multiple bioactivities [5]. |
| Complexity-to-Diversity (CtD) | Uses ring-distortion reactions on NP starting materials to rapidly generate complex, diverse scaffolds. | Moderate to Low | Rapid access to highly complex and novel 3D shapes from available NPs. | Ferroptocide, a ferroptosis inducer, from a complex natural product precursor [4]. |
| Function-Oriented Synthesis (FOS) | Aims to synthesize simpler analogs that retain or improve the function of a complex NP. | Variable (focus on function) | Delivers tractable lead compounds by prioritizing key pharmacophores. | Clinically optimized analogs of potent but complex NPs (e.g., bryostatin analogs) [4]. |
These strategies represent a paradigm shift from simply screening NP extracts to actively engineering chemical space informed by nature's blueprints. The dPNP approach, for instance, directly addresses the thesis by creating libraries that rival the scaffold diversity of purchasable collections but are inherently enriched with NP-derived bio-relevance [5].
Experimental Protocols and Data
This protocol outlines the core reaction for generating spiroindolylindanone scaffolds, a class of dPNPs.
- Reaction Setup: In a flame-dried microwave vial equipped with a stir bar, combine the indole starting material 1a (1.0 equiv), N-formyl saccharin (1.5 equiv, CO surrogate), palladium acetate (5 mol%), XantPhos (10 mol%), and sodium carbonate (2.0 equiv).
- Solvent and Atmosphere: Under an inert atmosphere (N₂ or Ar), add anhydrous N,N-dimethylformamide (DMF) (0.1 M concentration relative to 1a). Seal the vial.
- Reaction Conditions: Heat the mixture to 100°C with vigorous stirring for 16-24 hours. Monitor reaction progress by TLC or LC-MS.
- Work-up: After cooling to room temperature, dilute the reaction mixture with ethyl acetate and wash with water and brine. Dry the organic layer over anhydrous sodium sulfate, filter, and concentrate under reduced pressure.
- Purification: Purify the crude residue by flash column chromatography on silica gel to obtain the dearomatized product A1 (spiroindolylindanone).
This describes the workflow for identifying and characterizing a bioactive dPNP.
- Phenotypic Screening: Treat Shh-Light II cells (a Hedgehog pathway-responsive cell line) with the dPNP library compounds (e.g., 10 µM). Measure pathway activity using a luciferase reporter assay (Gli-responsive firefly luciferase). Identify hits that significantly inhibit luminescence without cytotoxicity.
- Secondary Validation: Perform dose-response curves on hits to determine IC₅₀ values. Assess specificity by testing in unrelated pathway reporter assays.
- Chemical Proteomics for Target ID:
- Probe Synthesis: Synthesize a bifunctional analog of the hit dPNP containing a photoaffinity label (e.g., diazirine) and an affinity handle (e.g., alkyne for biotin conjugation via click chemistry).
- Cell Treatment and Crosslinking: Treat target cells with the photoaffinity probe. Irradiate with UV light (365 nm) to crosslink the probe to interacting proteins.
- Cell Lysis and Enrichment: Lyse cells, perform click chemistry to conjugate biotin to the alkyne handle, and enrich probe-bound proteins using streptavidin beads.
- Analysis: Digest enriched proteins on-bead with trypsin. Analyze resulting peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Identify proteins significantly enriched in probe samples versus vehicle/dummy probe controls.
- Target Validation: Use techniques like cellular thermal shift assay (CETSA) to confirm direct binding, siRNA/gene knockout to show loss of compound sensitivity, and biochemical assays to measure direct inhibition.
Diagram: Hedgehog Signaling Pathway Inhibition by dPNP. The dPNP inhibitor blocks signal transduction at the level of the Smoothened (SMO) protein, preventing the activation of GLI transcription factors and subsequent target gene expression [5].
Diagram: Phenotypic Screening & Target Deconvolution Workflow. This workflow integrates phenotypic screening of designed libraries with modern chemical proteomics to identify novel bioactive chemotypes and their molecular targets [5].
Table 5: Key Research Reagent Solutions for NP and Library Research
| Reagent / Resource | Function / Description | Application in Featured Experiments |
|---|---|---|
| N-Formyl Saccharin [5] | A safe, efficient, and environmentally friendly solid surrogate for carbon monoxide (CO) gas. | Used as a carbonyl source in the palladium-catalyzed dearomatization synthesis of spiroindolylindanone dPNPs [5]. |
| Hantzsch Ester | A dihydropyridine derivative used as a mild, biocompatible reducing agent. | Employed in the diastereoselective reduction of indolenine to indoline during dPNP library diversification [5]. |
| Photoaffinity Probe Kits (e.g., Diazirine-Biotin/Alkyne) | Chemical biology tools containing a photoreactive group and an affinity tag for target identification. | Essential for the chemical proteomics step in deconvoluting the cellular target of a phenotypic dPNP hit [5]. |
| RDKit | An open-source cheminformatics toolkit. | Used for calculating molecular descriptors, generating chemical fingerprints, and assessing diversity in library design and analysis [6]. |
| NPBS Atlas Database [6] | A comprehensive resource linking over 218,000 natural products to their biological sources, taxonomy, and bioactivities. | Critical for selecting NP fragments for PNP design, studying structure-activity relationships, and exploring ecological context in drug discovery. |
| PAINS/REOS Filters | Computational filters to identify compounds with functional groups prone to assay interference or poor reactivity. | A mandatory step in curating high-quality purchasable or in-house screening libraries to reduce false-positive hits [8] [3]. |
The historical context confirms natural products as an irreplaceable foundation of drug discovery. Their enduring impact, however, is now most powerfully felt in their role as guides for the intelligent design of synthetic chemical libraries. The comparison is not a contest of replacement, but a synergy of strengths: the evolutionarily validated, three-dimensional scaffold diversity of nature provides the inspiration and biological relevance, while modern synthetic and computational strategies enable the systematic exploration and optimization of this chemical space. The future of productive discovery lies in continued innovation at this interface—designing purchasable libraries with NP-like character, applying rigorous phenotypic and target-agnostic screens, and leveraging new resources to bridge the natural and synthetic worlds.
The quest for novel therapeutics is fundamentally a search for novel chemical matter. This journey is framed by a central thesis: natural product (NP) scaffolds, honed by evolution for biological interaction, offer unparalleled structural diversity and complexity, while modern purchasable compound libraries, born from synthetic and combinatorial chemistry, offer defined, tractable, and highly optimized chemical matter for target-centric discovery [11] [12]. Historically, drug discovery relied heavily on natural products and their derivatives [3]. However, the late 20th century witnessed the "combinatorial explosion," a paradigm shift where the ability to synthesize vast libraries of compounds rapidly outpaced traditional natural product isolation [13]. This era was initially driven by a philosophy of quantity, generating massive libraries that were often plagued by poor physicochemical properties and a lack of "drug-likeness" [12] [3]. The subsequent evolution has been toward quality and intelligence, integrating principles of medicinal chemistry, advanced filtering, and artificial intelligence (AI) to create today's sophisticated, purchasable libraries [14] [12]. This guide compares the legacy of natural product diversity with the engineered diversity of modern compound libraries, providing researchers with a framework for selecting and utilizing these essential tools within a contemporary, integrated drug discovery workflow.
Comparative Analysis: Natural Product vs. Synthetic Library Scaffolds
The choice between natural product-inspired exploration and synthetic library screening is pivotal. The table below summarizes their core characteristics, strengths, and strategic applications.
Table 1: Comparison of Natural Product and Purchasable Synthetic Compound Libraries
| Aspect | Natural Product (NP)-Based Discovery | Modern Purchasable/Synthetic Compound Libraries |
|---|---|---|
| Core Source & Diversity | Secondary metabolites from microbes, plants, marine organisms. Evolutionary-bred, high scaffold complexity, 3D-character, stereochemical richness [11]. | Designed synthetic molecules from combinatorial and parallel synthesis [13]. Diversity is engineered and can be focused (target-class) or broad. |
| Structural Characteristics | High fraction of sp3 carbons, macrocycles, complex polycyclic systems. Often beyond "Rule of 5" [11]. | Adhere to drug-likeness filters (e.g., Lipinski's Rule of 5, PAINS removal) [12] [3]. Lead-like properties are designed in. |
| Primary Screening Format | Historically: crude extracts, requiring bioassay-guided fractionation [3]. Modern: Pre-fractionated, pure compound libraries [3]. | Discrete, pure compounds in ready-to-screen formats (e.g., DMSO solutions) [9]. |
| Key Advantages | Access to biologically pre-validated, novel chemotypes unmatched by synthetic chemistry. High hit rates for novel mechanisms [11]. | Defined structures, immediate availability, high reproducibility. Amenable to rapid SAR through analogue libraries. Strong IP position for novel synthetic compounds [15] [12]. |
| Major Challenges | Supply, re-supply, and synthetic modification can be difficult. Dereplication is essential to avoid known compounds [11]. | Can be biased toward "flat," aromatic structures. May miss complex, bioactive chemotypes found in NPs [12]. |
| Best Strategic Use | Unlocking novel biology, targeting "undruggable" spaces, and inspiring new scaffold designs for library synthesis [11]. | Target-based HTS, focused screening for target classes (kinases, GPCRs), FBDD, and rapid hit-to-lead campaigns [9] [12]. |
Evolution of Library Design and Synthesis
The transformation of compound libraries from large, undirected collections to intelligent, purpose-built sets is captured in the following workflow.
The combinatorial era began in earnest in the 1990s with techniques like one-bead-one-compound (OBOC) and parallel synthesis on solid support, enabling the rapid production of thousands to millions of peptides and small molecules [13]. Early successes, such as the discovery of the kinase inhibitor Sorafenib from a combinatorial library, proved the concept but were exceptions [12]. The initial focus on quantity often resulted in "fat, flat, and happy" molecules with poor pharmacokinetic potential [3]. This led to a necessary correction, integrating medicinal chemistry principles like Lipinski's Rule of Five and filters to remove Pan-Assay Interference Compounds (PAINS) [12] [3].
The field then matured toward purpose-designed libraries: Diversity-Oriented Synthesis (DOS) to recapture NP-like complexity, Fragment-Based Libraries for efficient hit discovery, and Target-Focused Libraries (e.g., for kinases, GPCRs) [9] [3]. The current frontier is dominated by data and AI. Virtual libraries encompassing billions of make-on-demand compounds (e.g., Enamine's REAL Space) are screened computationally [9] [14]. AI models predict activity, selectivity, and ADMET properties, enabling the design of ultra-focused, high-quality subsets for physical screening, dramatically improving hit rates and compound developability [14] [16].
Experimental Protocols for Library Evaluation and Hit Validation
Selecting a library is only the first step. Rigorous experimental protocols are required to evaluate screening outputs and validate hits. Here, we detail two critical, modern methodologies.
Protocol: AI-Enhanced Virtual Screening (VS) and Lead Optimization (LO) Analysis
Objective: To computationally prioritize compounds from ultra-large virtual libraries before synthesis or purchase. Background: The CARA benchmark study highlights that computational prediction tasks fall into two distinct types: Virtual Screening (VS), with diffuse, diverse compounds, and Lead Optimization (LO), with congeneric series [17]. Models must be evaluated accordingly. Procedure:
- Library Sourcing & Preparation: Access a virtual compound library (e.g., from a vendor like Enamine [9]). Standardize structures and generate molecular descriptors or fingerprints [16].
- Model Selection & Training:
- For VS Tasks: Use models robust to diverse chemical space. The CARA benchmark found that strategies like meta-learning and multi-task learning are particularly effective for VS [17].
- For LO Tasks: Train a quantitative structure-activity relationship (QSAR) model on existing congeneric data for the specific target or a closely related one [17].
- Prediction & Prioritization: Apply the trained model to score the virtual library. Prioritize the top-ranked compounds for purchase or synthesis.
- Experimental Confirmation: Test prioritized compounds in a primary biochemical or cellular assay. Supporting Data: In the CARA benchmark, the performance gap between classical machine learning and deep learning models was less pronounced in LO tasks compared to VS tasks, underscoring the importance of task-specific model selection [17].
Protocol: Cellular Thermal Shift Assay (CETSA) for Target Engagement
Objective: To confirm direct, physiologically relevant target engagement of a hit compound in intact cells or tissues, bridging the gap between biochemical potency and cellular efficacy. Background: A major cause of clinical failure is a lack of target engagement in a physiological setting. CETSA measures drug-induced thermal stabilization of the target protein in cells [14]. Procedure:
- Cell Treatment: Treat live cells with the hit compound at various concentrations or a vehicle control.
- Heat Challenge: Aliquot cell suspensions, heat each aliquot to a gradient of temperatures (e.g., 37°C to 65°C).
- Cell Lysis & Protein Quantification: Lyse heated cells, isolate the soluble protein fraction. Quantify the amount of remaining soluble target protein using a method like Western blot or high-resolution mass spectrometry [14].
- Data Analysis: Plot the fraction of intact protein versus temperature. A rightward shift in the melting curve (ΔTm) for compound-treated samples indicates thermal stabilization and direct target engagement. Supporting Data: A 2024 study applied CETSA to quantify engagement of the target DPP9 in rat tissue, confirming dose- and temperature-dependent stabilization both ex vivo and in vivo, showcasing its translational relevance [14].
Table 2: Benchmarking Data for Compound Activity Prediction Models (CARA Benchmark) [17]
| Task Type | Model/Strategy | Key Performance Metric (Example) | Implication for Library Screening |
|---|---|---|---|
| Virtual Screening (VS) | Meta-Learning | Improved AUC and enrichment in few-shot scenarios. | Effective for selecting hits from large, diverse libraries when prior target data is limited. |
| Virtual Screening (VS) | Multi-Task Learning | Leverages data from related assays to boost performance. | Useful for novel targets with assays in related protein families. |
| Lead Optimization (LO) | Single-Task QSAR | Achieved strong performance with sufficient congeneric data. | The preferred method for optimizing a hit series; accuracy depends on quality of internal SAR data. |
| General Finding | Model Agreement | High agreement between different models' outputs correlates with higher prediction confidence. | Can be used as a reliability filter for selecting compounds from virtual screens. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Modern discovery relies on specialized libraries and reagents. The table below catalogs key solutions for various stages of research.
Table 3: Key Research Reagent Solutions for Compound Library Research
| Reagent Solution | Supplier Example | Core Function & Role in Research |
|---|---|---|
| REAL (Enamine) / Other Make-on-Demand Libraries | Enamine [9] | Provides access to >30 billion virtual compounds for AI/VS, with rapid synthesis of top-ranked hits. Expands accessible chemical space far beyond physical collections. |
| Target-Focused Libraries | Various (e.g., Kinase, GPCR, PPI libraries) [9] | Pre-enriched with "privileged scaffolds" known to interact with specific target classes. Increases hit rates and reduces screening costs for known target families. |
| Fragment Libraries | Enamine, other CROs [9] | Collections of very small, low molecular weight compounds. Used in Fragment-Based Drug Discovery (FBDD) to identify weak binders for efficient optimization into high-affinity leads. |
| Covalent Libraries | Enamine [9] | Libraries designed with reactive warheads (e.g., acrylamides). Crucial for targeting non-catalytic cysteine or other nucleophilic residues, enabling drug discovery for "undruggable" targets. |
| DNA-Encoded Chemical Libraries (DECLs) | Various CROs | Ultra-large libraries (billions+) where each compound is linked to a unique DNA barcode. Allows selection-based screening against purified targets, ideal for identifying binders to challenging targets [13]. |
| Specialized Building Blocks | AstraZeneca SRI Program, WuXi AppTec [15] | High-quality, novel chemical reagents (e.g., sp3-rich fragments, chiral amines) not found in standard catalogs. Used to synthesize proprietary, high-quality compound libraries with improved IP potential and drug-like properties. |
The Integrated Future: AI, Synthesis, and Validation
The future of compound libraries lies in the seamless, iterative integration of design, synthesis, and validation, as shown in the following pathway.
The modern workflow is a closed-loop, Design-Make-Test-Analyze (DMTA) cycle. It starts with AI models designing or screening virtual libraries that dwarf physical collections [14] [16]. High-priority compounds are sourced from make-on-demand platforms [9]. Hits from experimental screening are immediately validated using orthogonal assays, with CETSA providing critical, mechanistic evidence of cellular target engagement [14]. All data feeds back into predictive models, refining the next iteration of design. This loop tightly couples the explorative power of vast chemical spaces (both NP-inspired and synthetic) with the rigorous, mechanistic validation required for translational success.
In conclusion, the rise of the combinatorial era has not made natural products obsolete but has instead provided a powerful, complementary synthetic counterpart. The thesis of scaffold diversity is best addressed by a strategic, non-dogmatic approach: using natural products to explore novel biological and chemical space and employing intelligently designed, purchasable libraries for efficient, target-driven optimization. The researcher's toolkit is now richer than ever, blending the wisdom of evolution with the precision of synthetic and computational chemistry, all guided by stringent experimental validation to build a more efficient and successful path to new medicines.
The global compound library market is experiencing significant and sustained growth, driven by the relentless pursuit of novel therapeutics. Compound libraries, which are curated collections of chemical entities, are indispensable tools for initial hit identification in drug discovery pipelines. The market is propelled by increasing R&D investments, the rising prevalence of chronic diseases demanding new treatments, and advancements in screening technologies such as high-throughput screening (HTS) and artificial intelligence (AI) [18] [19].
Table 1: Global Compound Library Market Size Projections
| Report Source | Base Year/Value | Projected Year/Value | Compound Annual Growth Rate (CAGR) | Key Driver Cited |
|---|---|---|---|---|
| Wiseguy Reports [18] | 2024: USD 4,000 Million | 2035: USD 7,500 Million | 5.9% (2025-2035) | Drug discovery demand, personalized medicine |
| Data Insights Market [19] | 2025: USD 11,500 Million | Forecast to 2033 | 8.2% (2025-2033) | Novel drug discovery, chronic disease prevalence |
| Metrics Trend Insights [20] | 2024: USD 1.56 Billion | 2033: USD 3.25 Billion | 8.9% (2024-2033) | AI integration, high-throughput screening |
Regional analysis consistently identifies North America as the dominant market, attributed to its concentration of major pharmaceutical companies and robust R&D infrastructure [18] [19]. The Asia-Pacific region is projected to be the fastest-growing market, fueled by expanding biotechnology sectors, growing research investments, and government initiatives in countries like China and India [18] [20]. Key market players include Thermo Fisher Scientific, Merck KGaA, Enamine Ltd., ChemBridge Corporation, and WuXi AppTec [18] [21].
A critical supporting industry, the compound management market, which handles the storage, tracking, and distribution of these physical libraries, is growing at an even faster rate (CAGR ~14.5%), highlighting the scaling infrastructure behind drug discovery [22] [21]. This growth is underpinned by a shift toward automation and outsourcing to specialized firms to manage costs and complexity [23].
Comparative Guide: Library Types and Strategic Applications
Selecting the appropriate compound library is a strategic decision that can determine the success of a screening campaign. Libraries differ in their design principles, content, and optimal use cases. The choice hinges on the discovery strategy—whether it is target-based, phenotype-based, or focused on novel scaffold identification [24].
Table 2: Comparison of Major Compound Library Types
| Library Type | Core Characteristics | Primary Applications | Advantages | Considerations |
|---|---|---|---|---|
| Diversity/Small Molecule Libraries | Large collections (10⁵–10⁷ compounds) maximizing structural variety and "drug-likeness" [18] [25]. | Primary high-throughput screening (HTS) for novel hit identification across diverse targets [19]. | Broad coverage of chemical space; high probability of finding hits for unoptimized targets. | Can contain redundant scaffolds; hit potency often requires significant optimization. |
| Fragment Libraries | Small molecules (MW < 300 Da) with high binding efficiency per atom [19]. | Fragment-based drug discovery (FBDD); identifying weak binders to build into high-affinity leads. | Efficient exploration of chemical space; high hit rates; ideal for targeting deep binding pockets. | Requires sensitive biophysical detection methods (e.g., SPR, NMR); leads require synthesis. |
| Target-Focused Libraries | Enriched with compounds known to interact with a specific protein family (e.g., kinases, GPCRs) [24]. | Screening against well-validated target classes; lead optimization. | Higher hit rates for the target family; more advanced starting points for medicinal chemistry. | Limited novelty; less effective for unprecedented target classes. |
| Natural Product & Inspired Libraries | Derived from or inspired by natural products (NPs); characterized by high scaffold complexity [25] [2]. | Discovering novel mechanisms of action; tackling difficult targets; phenotype-based screening. | High biological relevance and structural diversity not found in synthetic libraries [2]. | Supply can be complex; structures may be challenging to synthesize or optimize. |
| DNA-Encoded Libraries (DELs) | Vast libraries (10⁸–10¹⁰ compounds) where each molecule is linked to a DNA barcode for identification [24]. | Ultra-high-throughput screening against purified protein targets. | Unparalleled library size; efficient selection process for protein-binding hits. | Requires specialized DNA chemistry and sequencing; limited to in vitro protein targets. |
| Make-on-Demand & Virtual Libraries | Ultra-large (10⁹–10¹¹ compounds), virtually enumerated from available chemical building blocks and reactions [26]. | AI-driven virtual screening; on-demand synthesis of top-ranked virtual hits. | Access to an almost limitless, synthetically accessible chemical space. | Dependent on the accuracy of docking/scoring algorithms and reaction yields. |
Experimental Analysis: Quantifying Scaffold Diversity
A core thesis in modern drug discovery debates the relative value of natural product scaffold diversity versus the practicality of large, purchasable synthetic libraries [2]. Empirical, cheminformatic analysis provides critical data for this comparison.
Experimental Protocol for Scaffold Diversity Analysis [25]:
- Library Standardization: To ensure a fair comparison, researchers downloaded 11 major purchasable libraries (e.g., ChemBridge, Enamine, Mcule) and a Traditional Chinese Medicine compound database (TCMCD). Molecules were standardized (add hydrogens, remove duplicates), and subsets were created with identical molecular weight distributions to remove size bias.
- Scaffold Generation: Multiple structural decomposition methods were applied:
- Diversity Metrics: Key metrics were calculated for each library:
- Number of Unique Scaffolds: The count of distinct Murcko frameworks or Level 1 scaffolds.
- Scaffold Frequency Distribution: Analysis of how many molecules share the same scaffold (high frequency indicates lower diversity).
- PC50C Value: The percentage of unique scaffolds needed to cover 50% of the molecules in the library. A lower PC50C indicates a more diverse library, as fewer scaffolds dominate the collection [25].
- Visualization: Tree Maps and SAR Maps were used to visually compare the scaffold space and structural relationships across libraries [25].
Table 3: Experimental Scaffold Diversity Metrics for Selected Libraries [25]
| Library / Database | Number of Unique Murcko Frameworks | PC50C Value (Murcko Frameworks) | Key Structural Insight |
|---|---|---|---|
| Traditional Chinese Medicine (TCMCD) | 4,821 | 5.3% | Highest structural complexity but with more conservative, frequently repeating scaffolds (low PC50C) [25] [2]. |
| ChemBridge | 5,385 | 7.1% | High number of unique frameworks, indicating high structural diversity. |
| Mcule | 5,561 | 6.8% | One of the largest libraries with high scaffold diversity. |
| Enamine | 4,743 | 8.5% | Large library size, but with a slightly higher scaffold redundancy than leaders. |
| Average (11 Commercial Libraries) | ~4,900 | ~8.0% | Commercial libraries collectively show broad diversity, but some are dominated by common, synthetically accessible scaffolds. |
Key Finding: While commercial libraries like ChemBridge and Mcule demonstrate high scaffold diversity, the TCMCD natural product library occupies a distinct and more complex region of chemical space. However, its lower PC50C shows its molecules are built upon a set of recurring, evolutionarily conserved core scaffolds [25] [2]. This underscores the thesis that natural products offer privileged, biologically relevant scaffolds, whereas purchasable libraries offer broader, but sometimes less unique, synthetic diversity.
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for Compound Library Screening
| Item / Solution | Function in Library Screening | Application Context |
|---|---|---|
| High-Purity Compound Libraries | The core asset for screening; pre-plated in DMSO in 96-, 384-, or 1536-well plates. | All HTS and virtual screening campaigns. Quality control of purity and solubility is critical to reduce false results [24]. |
| Automated Liquid Handlers & Dispensers | Precisely transfer nanoliter to microliter volumes of compound solutions and assay reagents. | Essential for HTS to ensure speed, accuracy, and reproducibility while minimizing reagent use [22] [23]. |
| Acoustic Dispensers (e.g., Labcyte/Beckman Coulter) | Use sound waves to transfer nanoliter volumes of compound directly from source plates without tips. | Critical for assay miniaturization, reducing compound and reagent consumption, and enabling high-density screening [23]. |
| Biophysical Assay Kits (e.g., FP, TR-FRET, SPR) | Provide validated reagents and protocols to measure binding or enzymatic activity in a homogeneous format. | Target-based biochemical screening for kinases, proteases, epigenetic targets, etc. |
| Live-Cell Staining Kits & Viability Assays | Multi-parameter dyes for cell health, apoptosis, mitochondrial function, and calcium flux. | Phenotypic and target-based screening in cellular models [24]. |
| 3D Cell Culture Matrices & Organoid Media | Support the growth of more physiologically relevant 3D cell models, spheroids, and organoids. | Phenotypic screening in disease models with higher translational relevance [24]. |
| Docking & Cheminformatics Software (e.g., Rosetta, MOE, Schrödinger) | Perform virtual screening of ultra-large libraries by predicting how compounds fit into a protein target's structure. | Prioritizing compounds for purchase and testing from make-on-demand libraries (e.g., Enamine REAL) [26]. |
| Laboratory Information Management System (LIMS) | Software to track compound inventory, location, concentration, and screening data. | Mandatory for managing large library collections, ensuring sample integrity, and data provenance [22] [21]. |
Visualizing Workflows and Structural Evolution
Diagram 1: Integrated Drug Discovery Screening Workflow
Diagram 2: Structural Evolution: Natural vs. Synthetic Chemical Space
Foundational Principles and Philosophical Context
The pursuit of novel bioactive compounds in drug discovery is guided by two fundamentally distinct structural philosophies: evolutionary selection and synthetic design. The former leverages billions of years of natural trial and error, resulting in complex, biologically pre-validated scaffolds like those of digoxin or paclitaxel [27]. The latter applies rational engineering principles to construct designed systems or vast libraries of purchasable compounds, aiming for predictability and control [28] [29]. Framed within a broader thesis on natural product scaffold diversity versus purchasable compound libraries, this contrast is not merely methodological but philosophical, asking whether innovative solutions are best found through nature's exploration or human intention.
Evolution operates as a powerful, blind designer. Through variation, selection, and inheritance, it generates molecules exquisitely tuned to interact with biological targets, often for defense or signaling within ecosystems [30]. This "tinkering" process explores a fitness landscape, yielding privileged scaffolds with proven biological relevance, albeit for non-human purposes. In stark contrast, synthetic design is teleological—it begins with a defined function or problem [29]. Inspired by classical engineering, it employs principles like standardization and abstraction to build biological systems or chemical libraries from conceptual blueprints [28]. This rational approach seeks to avoid the "wastefulness" of random exploration by leveraging prior knowledge and models.
Recent scholarship posits that these philosophies are not opposites but exist on a unified evolutionary design spectrum [28]. All design, including rational engineering, involves iterative cycles of generating variants, testing them, and selecting the best performers—a core algorithm shared with natural evolution. The distinction lies in where intent is applied. In nature, intent is absent; selection acts on random variation. In synthetic biology, intent is applied to the process itself—the engineer designs the rules of variation and selection to steer outcomes toward a goal [28]. This meta-engineering perspective is crucial for fields like synthetic biology, where designed gene circuits must persist in evolving, competitive host environments [31].
This philosophical framework directly informs the practical debate in drug discovery. Should one mine nature's evolutionary library of natural products, or rationally design and screen synthetic libraries? The answer shapes investment, platform development, and the very logic of the search for new therapeutics.
Table 1: Contrasting Foundational Principles
| Aspect | Evolutionary Selection | Synthetic Design |
|---|---|---|
| Core Process | Variation, selection, and inheritance without a pre-defined goal (tinkering) [29] [32]. | Purposeful, iterative design-build-test cycles aimed at a specific function [28]. |
| Source of Innovation | Exploration of fitness landscapes via random mutation and recombination over deep time [30]. | Exploitation of prior knowledge and models; rational planning and directed search [28] [33]. |
| Structural Philosophy | "Retrospective" optimization for ecological function; scaffolds are solutions to historical problems [27] [30]. | "Prospective" construction for a target function; scaffolds are solutions to a defined human problem [29]. |
| Typical Output | Natural product scaffolds (e.g., cardiac glycosides, statin precursors) with high stereochemical and functional group complexity [27]. | Designed systems (e.g., gene circuits) or purchasable compound libraries (e.g., targeted kinase inhibitors) with defined building blocks [34] [31]. |
| Underlying Logic | Teleonomy (appearance of purpose) [29]. | Teleology (application of purpose) [28] [29]. |
Experimental Performance and Quantitative Comparison
Empirical data reveals the distinct strengths, limitations, and trade-offs inherent to each philosophy when applied to biological engineering and drug discovery.
Evolutionary Selection in Action: Natural Product Therapeutics Natural products represent a pre-validated, evolutionarily selected library. Structural analyses demonstrate their sophisticated mechanisms. For instance, digoxin binds to a preformed cavity in the Na+/K+-ATPase, acting as a molecular "doorstop" to lock the enzyme in a non-functional conformation—a form of conformational trapping that is difficult to rationally design [27]. Similarly, the statin pharmacophore (e.g., in simvastatin) mimics the natural substrate HMG-CoA, achieving potent competitive inhibition through perfect molecular mimicry refined by evolution [27]. Estimates indicate that natural products or their direct derivatives constitute approximately 65% of all approved small-molecule drugs, a testament to the functional success of evolutionarily selected scaffolds [27].
Synthetic Design in Action: Engineered Biological Systems The performance of synthetically designed systems is measured by their stability, output, and longevity. A critical challenge is evolutionary instability. Engineered gene circuits consume host resources, creating a metabolic burden that reduces growth rate. Cells with mutations that inactivate the circuit thus outcompete the engineered cells. A 2025 study quantified this: a simple, high-expression gene circuit in E. coli could see its population-level output halve (τ50) in a matter of days during serial passaging [31]. The study evaluated controller designs to extend longevity, finding that post-transcriptional feedback controllers could improve circuit half-life more than threefold compared to open-loop designs [31]. This highlights a key performance conflict: maximizing initial output often hastens evolutionary decline.
Purchasable Compound Libraries: Scale vs. Relevance Synthetic design also manifests in commercially available chemical libraries. Companies like OTAVA offer ultra-large virtual spaces (e.g., 55+ billion compounds) and targeted libraries for specific proteins (e.g., G9a, USP30) [34]. The performance of these libraries depends on the search strategy. A 2025 study targeting SARS-CoV-2 Mpro used active learning to prioritize 19 compounds from an on-demand library for purchase and testing. While three showed weak activity, the hit rate underscored the challenge of navigating vast synthetic spaces to find biologically active molecules [33]. The sheer scale of purchasable space (billions) dwarfs the known natural product space (hundreds of thousands), but the "hit rate" for novel, evolutionarily unprecedented targets may be lower without the guiding hand of biological pre-selection.
Table 2: Experimental Performance Metrics
| Metric | Evolutionarily-Selected Systems (Natural Products) | Synthetically-Designed Systems |
|---|---|---|
| Therapeutic Success Rate | ~65% of approved small-molecule drugs are NP-derived or inspired [27]. | Varies widely; hit rates from HTS of synthetic libraries often <<1%. |
| Typical Structural Complexity | High: multiple stereocenters, complex macrocycles, diverse heteroatoms [27]. | Lower: often built from simpler, more synthetically tractable building blocks. |
| Mechanistic Depth | Diverse: conformational trapping, covalent modification, allosteric modulation [27]. | Often designed for predictable inhibition (e.g., competitive active-site binding). |
| Evolutionary Stability | Extremely high; optimized for persistence in biological environments [30]. | Low to moderate; engineered circuits can degrade in days without stabilization strategies [31]. |
| Design Cycle Time | Millions of years (natural evolution). | Days to months (directed evolution, ML design) [33] [35]. |
| Exploratory Power | Has explored an immense but unknown fraction of biologically-relevant chemical space. | Can theoretically explore vast synthetic space (e.g., >55B compounds) [34], but relevance is uncertain. |
Table 3: Case Study: Longevity of Synthetic Gene Circuits [31]
| Circuit Design Type | Description | Key Performance Metric (τ50: Time to 50% Output Loss) | Relative Improvement vs. Open Loop |
|---|---|---|---|
| Open-Loop (No Control) | Constitutive high expression of reporter protein. | Baseline (~1.5-3 days in serial passage) | 1x (Reference) |
| Transcriptional Feedback | Negative feedback via transcription factor sensing circuit output. | Moderate improvement | ~1.5-2x |
| Post-Transcriptional Feedback | Negative feedback via small RNAs (sRNAs) silencing circuit mRNA. | High improvement | >3x |
| Growth-Rate Coupled Feedback | Controller actuates based on host growth rate signal. | Highest long-term persistence | >3x (best for long τ50) |
Methodological Approaches and Experimental Protocols
The implementation of these philosophies requires specialized methodologies, from harnessing evolutionary dynamics to executing rational design workflows.
Protocol 1: Directed Evolution & Mid-Scale Circuit Evolution This protocol applies evolutionary selection principles in a laboratory context to optimize synthetic designs [36].
- Diversification: Create a library of variants of a genetic part (e.g., promoter), device, or entire circuit. Methods include error-prone PCR (random mutation) or DNA shuffling (homologous recombination).
- Selection/ Screening: Link the desired function (e.g., fluorescence, antibiotic resistance, growth rate) to a selectable phenotype. For circuits, this may involve coupling output to host survival or using fluorescence-activated cell sorting (FACS).
- Amplification: Isolate the genetic material from selected variants and amplify it.
- Iteration: Use the amplified material as the template for the next round of diversification, repeating cycles to accumulate beneficial mutations. "Mid-scale" evolution focuses on evolving multi-gene circuits with non-trivial dynamics rather than single enzymes [36].
Protocol 2: Machine-Learning-Driven Prioritization from On-Demand Libraries This protocol exemplifies a modern synthetic design workflow that navigates ultra-large chemical spaces [33].
- Seed Identification: Start with a known hit or fragment bound to the target protein, determined by crystallography or docking.
- Virtual Library Construction: Use software (e.g., FEgrow) to generate a virtual library by growing or linking fragments around the seed. Alternatively, perform a substructure search in a purchasable database (e.g., Enamine REAL, OTAVA CHEMRIYA) to find compounds matching a core scaffold [34] [33].
- Active Learning Loop: a. Build & Score: A subset of the virtual library is built into the protein binding pocket (using docking or ML/MM optimization) and scored for predicted affinity. b. Model Training: These scores train a machine learning model to predict the performance of unseen compounds. c. Informed Selection: The model selects the next, most promising batch of compounds for evaluation in step (a). d. Iteration: The loop continues, efficiently exploring the chemical space.
- Purchase & Test: The top-prioritized compounds are purchased from an on-demand library and tested in biochemical or cellular assays [33].
Protocol 3: Structural Analysis of Natural Product Mechanisms This protocol reverse-engineers the solutions found by evolutionary selection [27].
- Complex Formation: Co-crystallize or prepare cryo-EM samples of the natural product (e.g., digoxin, paclitaxel) in complex with its protein target (e.g., Na+/K+-ATPase, tubulin).
- Structure Determination: Use X-ray crystallography or cryo-electron microscopy (cryo-EM) to solve the high-resolution structure of the complex.
- Mechanistic Analysis: Analyze the binding interface to identify key interactions (hydrogen bonds, hydrophobic contacts, covalent bonds). Compare with structures of the target alone or with substrates/inhibitors to determine the mechanism (e.g., competitive inhibition, conformational trapping, allosteric modulation).
- Knowledge Application: Use these structural insights to guide the synthetic design of more potent analogs or novel chemotypes that mimic the critical interactions.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents and Materials
| Item | Function/Description | Primary Philosophy Association |
|---|---|---|
| Ultra-Large Virtual Chemical Spaces (e.g., OTAVA CHEMRIYA, Enamine REAL) [34] [33] | Searcheable databases of billions of synthetically feasible compounds for virtual screening and hit expansion. | Synthetic Design |
| Targeted Compound Libraries (e.g., G9a, USP30, Covalent Inhibitor libraries) [34] | Curated sets of compounds designed around specific target classes or mechanisms, enriching screening efforts. | Synthetic Design |
| Directed Evolution Kits (e.g., error-prone PCR kits, DNA shuffling kits) | Commercial reagent suites for creating diverse genetic variant libraries for selection experiments. | Hybrid (Applies Evolution to Design) |
| Protein Language Models & Design Tools (e.g., ESM2, Seq2Fitness, BADASS algorithm) [35] | Machine learning models trained on evolutionary sequence data to predict fitness and design novel, high-performing protein sequences. | Hybrid (Uses Evolutionary Data for Design) |
| Cryo-EM & X-ray Crystallography Platforms | Enable atomic-resolution structure determination of natural product-target complexes, revealing evolutionary solutions [27]. | Evolutionary Selection (Analysis) |
| Fragment Screening Libraries | Small, low-complexity chemical fragments used for initial structural screening to identify weak binding starting points. | Synthetic Design |
| Genetic Controller Parts (e.g., inducible promoters, sRNA systems, kill switches) | Biological parts used to implement feedback control in synthetic gene circuits to enhance evolutionary longevity [31]. | Synthetic Design |
Synthesis and Application in Drug Discovery
The future of biotechnology and drug discovery lies not in choosing one philosophy over the other, but in their strategic integration. The evolutionary design spectrum provides a unifying framework [28]. Natural products offer validated, complex starting points whose innate ecological functions can reveal novel therapeutic targets [30]. For example, understanding a plant toxin's target can identify a vulnerability in a human pathogen or cancer cell. The structural solutions refined by evolution—such as digoxin's conformational trapping—provide blueprints for mechanism-based drug design [27].
Synthetic design, empowered by machine learning and vast purchasable libraries, provides scale, speed, and precision. Active learning can efficiently mine billions of compounds [33], while protein language models can now guide the design of novel proteins by learning from evolutionary data [35]. Furthermore, the principles of synthetic design are essential for overcoming the inherent limitations of evolutionary approaches, such as stabilizing synthetic gene circuits against natural selection by designing intelligent genetic controllers [31].
The most powerful strategy is a convergent approach: using evolutionary wisdom to inspire and validate synthetic efforts. This can involve:
- Biology-Inspired Library Design: Creating "natural product-like" synthetic libraries that capture the scaffold complexity and 3D geometry of evolutionarily selected molecules [34].
- Mechanism-Informed Design: Using atomic-resolution structures of natural product-target complexes to rationally design simplified, more druggable synthetic analogs [27].
- Evolution-Guided Machine Learning: Training AI models on evolutionary sequence and structural data to generate novel designs that respect biological constraints and functional imperatives [35].
In conclusion, the core structural philosophies of evolutionary selection and synthetic design represent complementary modes of inquiry and invention. Evolutionary selection is a master of exploration, uncovering deep solutions within the rugged fitness landscapes of biology. Synthetic design is a master of exploitation, channeling knowledge and intent to solve specific problems. By placing them on a continuum and leveraging the strengths of each, researchers can accelerate the discovery of novel therapeutics and the engineering of robust biological systems.
The quest for novel bioactive molecules in drug discovery hinges on the exploration of diverse chemical landscapes. Two primary sources exist: the evolutionarily refined scaffolds of natural products (NPs) and the vast, synthetically accessible purchasable compound libraries. Within the broader thesis of NP scaffold diversity versus purchasable libraries, this guide provides an objective, data-driven comparison of their performance in populating biologically relevant chemical space.
Natural products are small organic molecules produced by living organisms through evolutionary selection. This process grants them unique chemical diversity, structural complexity (including stereochemistry and medium/large rings), and a proven ability to interact with biological macromolecules [1]. They are considered "privileged scaffolds" with high target affinity and specificity, serving as essential modulators of biomolecular function and a historic source of new drugs [1].
In contrast, purchasable compound libraries are commercially available collections of synthetic small molecules, designed for high-throughput screening (HTS). These libraries, offered by suppliers like ChemDiv, Enamine, Mcule, and ChemBridge, prioritize synthetic accessibility, drug-like physicochemical properties, and broad coverage of abstract "chemical space" [37] [8] [38]. Their design often aims for high scaffold count and lead-like properties.
The table below summarizes the core comparative analysis of these two sources.
Table: Core Comparison: Natural Product Scaffolds vs. Purchasable Compound Libraries
| Comparison Aspect | Natural Product Scaffolds | Purchasable Compound Libraries |
|---|---|---|
| Origin & Design Principle | Evolutionary selection for biological interaction [1]. | Synthetic design for drug-likeness and diversity metrics [8] [38]. |
| Structural Hallmarks | High sp³ character, stereochemical complexity, presence of medium/large rings and macrocycles [1] [39]. | Tends toward planarity (lower Fsp³), simpler stereochemistry, dominated by small rings and flat heterocycles [8]. |
| Chemical Space Coverage | Occupies unique, biologically relevant regions often underexplored by synthetic libraries [1] [39]. | Covers a vast, well-defined region of "lead-like" and "drug-like" space, but can suffer from structural redundancy [8] [38]. |
| Biological Performance | High hit rates against challenging targets (e.g., protein-protein interactions); 19% of new small-molecule drugs (2005-07) were NPs or NP-derived [1]. | Enable high-throughput screening; hit rates can be lower for novel or challenging biological targets. |
| Accessibility & Supply | Often requires isolation, purification, or complex total synthesis; supply can be limited [1]. | Immediately purchasable (millions in stock); reliably supplied in milligram to gram quantities [37] [40] [38]. |
| Typical Library Size | Individual NP libraries are smaller (e.g., ~180,000 in Mcule database) [37]. | Extremely large; vendor catalogs contain 1.6M – over 100M compounds [37] [8]. |
| Advantage | Biological relevance, novelty, and success as drug leads. | Immediate accessibility, scalability, and suitability for HTS campaigns. |
A critical quantitative analysis of scaffold diversity was demonstrated in a 2024 chemoinformatic study of 576 Spleen Tyrosine Kinase (SYK) inhibitors [41]. This research provides a framework for comparing diversity and is summarized below.
Table: Scaffold Diversity Analysis of SYK Inhibitors (2024 Study) [41]
| Analysis Method | Tool/Platform | Key Finding | Interpretation for Library Design |
|---|---|---|---|
| Chemical Space Network | ECFP4/MACCS fingerprints, RDKit, NetworkX [41] | Visualization revealed distinct clusters and outlier molecules. | Purchasable libraries should aim for broad cluster coverage, while NP libraries can provide unique outliers. |
| Scaffold Identification | Bemis-Murcko frameworks [41] | A defined number of unique core scaffolds were identified from the 576 compounds. | Highlights the ratio of compounds-to-scaffolds; a higher ratio indicates better exploration of chemical space around privileged cores. |
| Activity Landscape | Pairwise activity difference mapping [41] | Identified "activity cliffs" (e.g., CHEMBL3415598, CHEMBL4780257)—small structural changes causing large potency jumps. | NP scaffolds, with their complex structure, may be richer sources of activity cliffs, informing targeted library design. |
Experimental Protocols for Diversity Generation
This section details key experimental methodologies for generating diverse chemical libraries from both natural product and synthetic approaches. The protocols highlight the contrasting strategies: complexity-driven diversification of NPs versus scaffold-hopping and property-based design for synthetic libraries.
Protocol 1: Diversifying Natural Products via C-H Functionalization & Ring Expansion
This state-of-the-art protocol, adapted from a 2019 Nature Communications study, enables deep diversification of polycyclic natural products (e.g., steroids) to access underpopulated chemical space featuring medium-sized rings (7-11 members) [39].
1. Principle: A two-phase strategy that first installs new functional handles via site-selective C-H oxidation, then uses these handles for ring expansion reactions. This moves beyond simple peripheral modification to alter the core scaffold itself [39].
2. Materials:
- Starting Material: Polycyclic natural product (e.g., Dehydroepiandrosterone/DHEA, Estrone, Isosteviol).
- Reagents for C-H Oxidation: Electrochemical setup (e.g., graphite electrodes, supporting electrolyte), or chemical oxidants (e.g., Cr or Cu-based catalysts for site-selective oxidation) [39].
- Reagents for Ring Expansion: Dependent on the reaction chosen (e.g., Schmidt reagent (NaN₃, acid) for lactam formation, dimethyl acetylenedicarboxylate (DMAD) for two-carbon expansion, ethyl diazoacetate for homologation) [39].
- General: Anhydrous solvents (CH₂Cl₂, THF, MeCN), standard workup and purification materials (silica gel, TLC plates).
3. Step-by-Step Procedure:
- Step 1: Site-Selective C-H Oxidation. For an allylic C-H bond, employ electrochemical oxidation. Dissolve the steroid substrate (e.g., 1.0 mmol) in a solvent mixture (e.g., CH₃CN/H₂O) with an electrolyte (e.g., LiClO₄). Perform electrolysis in an undivided cell with graphite electrodes at a constant current until complete conversion (monitored by TLC/LCMS). Alternatively, use a metal-catalyzed oxidation for benzylic or other C-H bonds [39].
- Step 2: Functional Group Manipulation. Convert the newly introduced oxygenated functionality (e.g., ketone) to a suitable leaving group or reactive species for expansion. For example, transform a ketone into an oxime via reaction with hydroxylamine hydrochloride.
- Step 3: Ring Expansion Reaction. Subject the functionalized intermediate to the expansion condition. For a Beckmann Rearrangement (forming a lactam), treat the oxime with an acid catalyst (e.g., trifluoroacetic anhydride) in an inert atmosphere. For a two-carbon ring expansion, react a β-keto ester intermediate with DMAD via a formal [2+2] cycloaddition and fragmentation sequence [39].
- Step 4: Purification & Characterization. Isolate the product via standard aqueous workup, followed by purification via flash chromatography. Characterize the novel polycyclic medium-sized ring compound using ¹H/¹³C NMR, HRMS, and, if possible, X-ray crystallography [39].
4. Key Outcome: A library of novel, complex molecules that occupy a unique region of chemical space compared to typical commercial libraries, characterized by increased three-dimensionality and the presence of synthetically challenging medium-sized rings [39].
Protocol 2: Designing & Screening a Focused Purchasable Library
This protocol outlines the standard workflow for leveraging purchasable libraries for hit identification, based on vendor information and standard screening practices [8] [38].
1. Principle: Use computational filters and property-based selection to design a focused subset from a multimillion-compound purchasable catalog for a specific biological assay.
2. Materials:
- Compound Source: Access to a vendor database (e.g., Mcule: ~139M compounds; ChemDiv: >1.6M; Enamine Building Blocks: >1.5M) [37] [8] [40].
- Software: Cheminformatics toolkit (e.g., RDKit, KNIME) for filtering and property calculation.
- Assay Materials: Target-specific biochemical or cellular assay reagents, microplates (96- or 384-well), liquid handler.
3. Step-by-Step Procedure:
- Step 1: Define Selection Criteria. Based on the target (e.g., kinase, GPCR) or phenotype, establish filters:
- Physicochemical: Lead-like (MW <450, clogP <3.5) or drug-like (Lipinski's Rule of 5) properties [38].
- Structural: Remove undesirable functionalities (PAINS, reactive groups) using substructure filters [8].
- Diversity: Apply a clustering algorithm (e.g., Bemis-Murcko scaffold-based) to ensure broad scaffold representation [8].
- Target Focus: If prior knowledge exists, use similarity searching or pharmacophore models to enrich for relevant chemotypes [8].
- Step 2: Generate & Procure Library Subset. Submit the final list of compound identifiers (e.g., Mcule IDs) to the vendor. Compounds are typically delivered as 10 mM DMSO solutions in pre-plated 96- or 384-well microplates, ready for screening [38].
- Step 3: Primary High-Throughput Screening (HTS). Using an automated liquid handler, transfer nanoliter volumes of compounds from the library plates into assay plates. Run the target-specific biochemical or cellular assay. Measure activity (e.g., inhibition, fluorescence) and calculate primary hit criteria (e.g., >50% inhibition/activation at test concentration).
- Step 4: Hit Validation & Progression. Retest primary hits in dose-response to confirm potency (IC₅₀/EC₅₀). Apply medicinal chemistry triage: check for purity (LCMS), chemical stability, and potential assay interference (e.g., aggregation). Purchase or synthesize close analogs from the vendor's building block collection to initiate early Structure-Activity Relationship (SAR) exploration [40].
4. Key Outcome: A list of confirmed hit compounds with associated dose-response data, providing a starting point for lead optimization within a readily accessible and easily scalable chemical series.
Visualizing Chemical Landscapes: Pathways and Workflows
Diagram 1: Strategic Pathways in Chemical Exploration. A decision workflow comparing the complexity-driven NP diversification route with the speed- and scale-oriented purchasable library screening route [1] [39] [8].
Diagram 2: Mapping Scaffolds and Libraries in Chemical Space. A conceptual map showing distinct regions occupied by purchasable libraries and NP scaffolds, connected by analog series networks and highlighting underexplored zones [41] [42] [43].
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table: Key Research Reagents and Solutions for Chemical Landscape Exploration
| Item / Solution | Function in Research | Typical Source / Example |
|---|---|---|
| Natural Product Isolates & Derivatives | Serve as starting points for diversification (Protocol 1) or as reference compounds in screening. | Sigma-Aldrich, Cayman Chemical, Mcule Natural Products Library (~180k compounds) [37]. |
| Purchasable Screening Libraries | Provide immediate, diverse compound sets for primary HTS (Protocol 2). | ChemDiv (DIVERSet), ChemBridge CORE Library, Mcule database subsets (e.g., Kinase Targeting) [37] [8] [38]. |
| Building Block Catalogs | Essential for hit follow-up and SAR expansion via analog synthesis. | Enamine Building Blocks Catalog (~1.6M items), Mcule Building Blocks [37] [40]. |
| Cheminformatics Software Suites | Enable chemical space visualization, descriptor calculation, clustering, and virtual screening. | RDKit (open-source), KNIME, Schrödinger Suite. Used for analyses like in the SYK inhibitor study [41]. |
| C-H Functionalization Reagents/Kits | Facilitate the direct diversification of NP scaffolds at inert positions. | Electrochemical cells, metal catalysts (e.g., Cu, Cr, Pd complexes for site-selective oxidation) [39]. |
| Ring Expansion Reagents | Used to alter core scaffold size and complexity, accessing novel chemotypes. | Schmidt reagents (HN₃), diazo compounds (e.g., ethyl diazoacetate), DMAD [39]. |
| Pre-plated Compound Sets | Accelerate screening by providing ready-to-test compounds in assay-ready formats. | ChemBridge pre-plated libraries (10mM DMSO in 384-well plates) [38]. |
| Structure & Property Databases | Provide reference data for drug-likeness, bioactivity, and scaffold analysis. | ChEMBL, PubChem, vendor-specific property-filtered lists (e.g., CNS-MPO optimized) [8] [38]. |
Synthesis and Strategic Implications for Drug Discovery
The comparative analysis reveals that natural product scaffolds and purchasable libraries are not mutually exclusive but complementary tools. NPs provide evolutionarily validated, complex templates that access high-value, underexplored chemical space, particularly for challenging target classes. Purchasable libraries offer unmatched scale, speed, and accessibility for systematic HTS and rapid SAR generation.
The future of efficient chemical exploration lies in hybrid strategies: using computational "constellation" plots [42] and activity landscape models [41] [43] to guide the design of new libraries. These new libraries should integrate privileged NP frameworks (like medium-sized rings [39]) with the synthetic tractability and property optimization of commercial libraries. As visualized in Diagram 1, the strategic choice between starting from NP complexity or synthetic accessibility depends on the project's specific goals regarding novelty, risk, and timeline. Ultimately, the most effective chemical landscape is one charted with both a map of nature's innovations and a compass of synthetic design.
Strategic Implementation: Methodologies for Screening with Natural and Synthetic Collections
The strategic design and selection of compound libraries are foundational to modern drug discovery. Within the context of a broader thesis on natural product scaffold diversity versus purchasable compound libraries, this guide provides an objective comparison of four principal library taxonomies: Focused, Diverse, Fragment, and Natural Product collections [25] [1]. Each library type embodies a distinct philosophy for navigating chemical space, with direct implications for screening efficiency, hit discovery, and lead development.
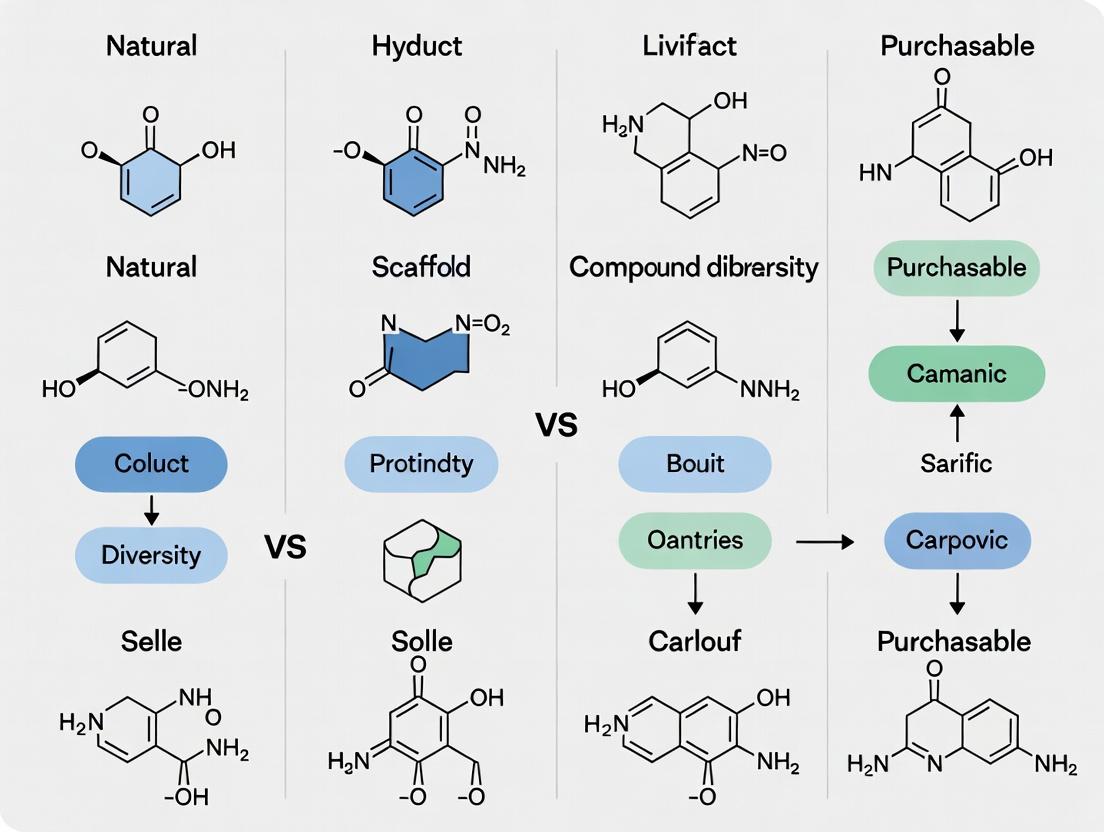
Focused libraries are designed with prior knowledge, targeting specific protein families or pathways to increase the likelihood of identifying hits [33]. Diverse libraries aim for maximal coverage of drug-like chemical space, often built from commercially available building blocks, to serve as general-purpose screening tools [44] [25]. Fragment libraries utilize very small molecules (typically <300 Da) to probe the essential interactions of a target, providing efficient starting points that can be elaborated into leads [45] [46]. Natural Product (NP) and NP-inspired libraries leverage evolutionary-optimized, biologically relevant chemical scaffolds, offering unique structural complexity and a proven track record for yielding novel bioactive compounds [46] [47] [1].
The contemporary convergence of these strategies is evident in approaches like pseudo-natural product (PNP) synthesis, which recombines NP-derived fragments to create novel scaffolds occupying unexplored biologically relevant space [46] [4], and in computational methods that rationally minimize massive NP libraries to focused, high-diversity subsets [47]. The following comparison, supported by recent experimental data, delineates the performance, applications, and ideal use cases for each library taxonomy.
Comparative Performance Analysis of Library Taxonomies
The table below summarizes the core characteristics, typical sources, and performance metrics of the four primary library types, drawing from comparative chemoinformatic and experimental studies.
Table 1: Comparative Overview of Compound Library Taxonomies
| Library Type | Core Design Principle | Typical Size & Source | Key Performance Metrics | Primary Advantages | Common Limitations |
|---|---|---|---|---|---|
| Focused Library | Target- or pathway-informed design; enriched with known pharmacophores. | 1,000 - 50,000 compounds. Derived via virtual screening, on-demand synthesis, or curation from large vendors [33]. | Hit Rate: Highly variable but often increased for the intended target class. Chemical Space: Narrow, focused coverage. | Increased efficiency for specific targets; can leverage extensive prior SAR. | Limited serendipity; bias towards known chemotypes; may miss novel scaffolds. |
| Diverse Library | Maximize coverage of drug-like chemical space; ensure broad scaffold diversity. | 100,000 - 5,000,000+ compounds. Commercially available (e.g., Enamine, Mcule) or via combinatorial synthesis [44] [25]. | Scaffold Diversity: High (e.g., Murcko framework counts). Hit Rate: Generally low (<1%) but provides novel starting points [25]. | General-purpose utility; high probability of finding some hit; explores vast synthetic chemical space. | Very high cost for HTS; high false-positive/negative rates; redundancy. |
| Fragment Library | Small molecules ("rule of three") to probe fundamental binding interactions. | 500 - 5,000 fragments. Often derived from diverse commercial compounds or curated NP collections [45] [46]. | Binding Efficiency: High (LE > 0.3). Hit Rate: Can be high (2-5%) due to efficient sampling of chemical space [45]. | Efficient coverage of chemical space; high ligand efficiency; ideal for structure-based elaboration. | Weak affinity (μM-mM); requires sensitive biophysical detection (SPR, NMR, X-ray). |
| Natural Product (NP) Library | Leverage evolutionarily optimized, biologically pre-validated chemical scaffolds. | Extracts: 1,000 - 100,000+; Pure NPs: 1,000 - 50,000. Isolated from nature or derived from NP databases (e.g., COCONUT) [45] [47]. | Scaffold Complexity/Novelty: High. Hit Rate: Historically high; rational libraries show increased rates (e.g., 22% vs. 11.3% full library) [47]. | High success rate for novel leads; privileged structures for challenging targets (e.g., PPIs) [1]. | Supply, redundancy, rediscovery; complexity can hinder SAR and synthesis. |
Quantitative Comparison: Diversity, Complexity, and Hit Enrichment
Recent studies provide quantitative data for direct comparison of library performance, particularly in scaffold diversity and screening hit rates.
Table 2: Quantitative Performance Comparison from Recent Studies
| Study & Library Type | Key Metric & Result | Experimental Context | Implication for Library Design |
|---|---|---|---|
| Fragment Libraries (Synthetic vs. NP-derived) [45] | Scaffold Count: NP-derived (COCONUT: 2.58M fragments) vs. synthetic (CRAFT: 1,214 fragments). Chemical Space: NP fragments occupy distinct, complementary regions to synthetic fragments. | Chemoinformatic analysis of fragment libraries generated from NP databases (COCONUT, LANaPDB) and a synthetic library (CRAFT). | NP collections are a vast source of unique fragment scaffolds, expanding accessible chemical space for FBDD. |
| Diverse/Purchasable Libraries [25] | Scaffold Diversity (PC50C): Ranged from 1.3% (Mcule) to 4.3% (TCMCD). Lower PC50C indicates greater diversity. Analysis: Commercial libraries (Chembridge, VitasM) showed high diversity. | Analysis of 11 purchasable libraries and TCMCD using Murcko frameworks and Scaffold Trees on standardized subsets. | Library selection for VS should consider scaffold diversity metrics; commercial libraries differ significantly. |
| Focused/Rational NP Library [47] | Hit Rate Enhancement: Anti-P. falciparum hit rate increased from 11.3% (full 1,439-extract library) to 22.0% (50-extract rational library). Library Size Reduction: Achieved 80% scaffold diversity with 50 extracts vs. 109 for random selection. | LC-MS/MS and molecular networking used to create a minimal fungal extract library based on scaffold diversity, tested in phenotypic and target-based assays. | Rational, diversity-focused minimization of NP libraries drastically improves screening efficiency and hit rates. |
| Pseudo-Natural Product (PNP) Library [46] | Chemical Diversity: Intra-subclass similarity high (median 0.75), inter-subclass similarity low (median 0.26). Bioactivity: PNPs exhibited distinct phenotypic profiles from parent NP fragments in Cell Painting. | 244 PNPs synthesized from 4 NP fragments; evaluated via cheminformatics and unbiased Cell Painting assay. | Fragment recombination creates chemically and biologically diverse libraries, accessing new bioactivity. |
| Self-Encoded Library (SEL) [44] | Screening Scale: Single-experiment affinity selection of >500,000 barcode-free compounds. Success: Identified nanomolar binders/inhibitors for carbonic anhydrase IX and FEN1. | Solid-phase combinatorial synthesis and tandem MS decoding enabled massive, tag-free library screening against protein targets. | Next-gen diverse libraries bypass DEL limitations, enabling ultra-large screening without DNA tags. |
Detailed Experimental Protocols for Key Studies
This protocol outlines the chemoinformatic workflow for deriving and comparing fragment libraries, crucial for understanding the unique contributions of NP-derived fragments.
- Source Data Compilation:
- Obtain NP structures from large, curated databases. The study used the Collection of Open Natural Products (COCONUT, >695,133 NPs) and the Latin America Natural Product Database (LANaPDB, 13,578 NPs) [45].
- Obtain synthetic fragment structures from a designed library (e.g., the CRAFT library of 1,214 fragments based on heterocyclic and NP-derived scaffolds) [45].
- Fragment Generation:
- Apply a standardized fragmentation algorithm (e.g., using the RDKit toolkit) to all NP and synthetic compound structures.
- Define fragments according to the "rule of three" (MW ≤ 300, HBD ≤ 3, HBA ≤ 3, cLogP ≤ 3) or similar fragment-like filters.
- Remove duplicates and unwanted small molecules (e.g., salts, solvents) to create non-redundant fragment libraries.
- Cheminformatic Analysis:
- Descriptor Calculation: Compute molecular descriptors (e.g., molecular weight, logP, topological polar surface area, fraction of sp3 carbons) for all fragments.
- Chemical Space Visualization: Use dimensionality reduction techniques like Principal Component Analysis (PCA) or t-distributed Stochastic Neighbor Embedding (t-SNE) on the descriptor sets to map and visualize the distribution of NP-derived versus synthetic fragments.
- Diversity Assessment: Calculate pairwise molecular similarities (e.g., using Tanimoto coefficients on Morgan fingerprints) within and between libraries. Quantify scaffold diversity by generating Murcko frameworks and counting unique scaffolds.
- Data Availability:
- The resulting fragment libraries from this study are made publicly available for download on a GitHub repository.
This experimental protocol describes a method to transform a large, redundant NP extract library into a focused, high-diversity screening set.
- LC-MS/MS Data Acquisition:
- Analyze each extract in the full natural product library (e.g., 1,439 fungal extracts) using untargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS).
- Molecular Networking and Scaffold Definition:
- Process all MS/MS spectra using the Global Natural Products Social Molecular Networking (GNPS) platform.
- Create a "classical molecular network" where nodes represent MS/MS spectra and edges connect spectra with high similarity, grouping molecules with shared fragmentation patterns (and thus, structural scaffolds).
- Define each connected component (molecular family) in the network as a unique "scaffold" for the purpose of library design.
- Rational Library Selection Algorithm:
- Develop a custom script (e.g., in R) to iteratively select extracts.
- Step 1: Identify the extract containing the highest number of unique scaffolds (nodes in the network) and add it to the new "rational library."
- Step 2: For all remaining extracts, count the number of scaffolds they contain that are not already represented in the rational library.
- Step 3: Select the extract with the highest count of new scaffolds and add it to the rational library.
- Step 4: Repeat Steps 2 and 3 until a pre-defined threshold (e.g., 80% or 100% of total scaffolds in the full library) is achieved.
- Validation via Biological Screening:
- Screen both the full library and the rationally minimized library in parallel against relevant biological targets (e.g., the parasite Plasmodium falciparum, the enzyme neuraminidase).
- Compare hit rates (percentage of extracts showing bioactivity) to demonstrate enrichment. The study showed hit rates approximately doubled in the minimized library [47].
This protocol details a novel method for screening ultra-large diverse libraries without DNA encoding.
- Library Synthesis (Solid-Phase Split & Pool):
- Design libraries around drug-like scaffolds (e.g., benzimidazole cores, peptide-like structures, Suzuki cross-coupling products).
- Use solid-phase synthesis on beads. Employ a split-and-pool approach where beads are divided, reacted with a specific building block, then recombined for the next step. This combinatorially generates millions of compounds where each bead carries a single compound.
- Affinity Selection Panning:
- Incubate the pooled bead library with an immobilized, purified target protein (e.g., carbonic anhydrase IX, FEN1).
- Wash away non-binding beads. Elute beads that retain binding to the target protein.
- Hit Decoding by Tandem Mass Spectrometry:
- Cleave compounds from the eluted beads.
- Analyze the mixture via nanoLC-MS/MS.
- Use advanced software tools (SIRIUS and CSI:FingerID) to annotate the MS/MS fragmentation spectra of detected compounds de novo, matching them to the virtual structures in the library's chemical design space. This step eliminates the need for physical DNA barcodes.
- Hit Validation:
- Chemically resynthesize the identified hit compounds as discrete molecules.
- Confirm binding affinity and functional activity using standard biochemical assays (e.g., enzymatic inhibition assays, surface plasmon resonance).
Visualizing Library Design and Screening Workflows
The Continuum of Library Design Strategies
Diagram 1: Relationship between synthetic strategies, library types, and their core properties [46] [4].
Experimental Workflow for Library Evaluation and Hit Identification
Diagram 2: Integrated workflow for screening and evaluating different library types.
The Scientist's Toolkit: Essential Research Reagents & Solutions
The table below catalogs key reagents, software, and databases essential for the design, construction, and screening of the discussed compound libraries.
Table 3: Essential Toolkit for Library-Based Drug Discovery
| Tool Category | Specific Tool / Reagent | Function / Description | Relevant Library Taxonomy |
|---|---|---|---|
| Source Databases | COCONUT [45], LANaPDB [45], Dictionary of Natural Products (DNP) [46] | Public and commercial databases of natural product structures for virtual fragment generation or inspiration. | Natural Product, Fragment, PNP |
| ZINC [25], Enamine REAL [33] | Databases of commercially available/purchasable compounds for virtual screening and library sourcing. | Diverse, Focused | |
| Cheminformatic & AI Software | RDKit [46], Pipeline Pilot [25] | Open-source and commercial toolkits for cheminformatic analysis, descriptor calculation, and scaffold generation. | All |
| FEgrow [33] | Open-source software for growing/optimizing ligands in protein binding pockets, interfacing with active learning. | Focused, Fragment | |
| SIRIUS & CSI:FingerID [44] | Software for de novo structural annotation of compounds from MS/MS spectra, enabling barcode-free screening. | Diverse (SEL), Natural Product | |
| Screening & Assay Platforms | Self-Encoded Library (SEL) Platform [44] | Solid-phase synthesis combined with tandem MS decoding for affinity selection of >500k untagged compounds. | Diverse |
| Cell Painting Assay [46] | High-content, morphological profiling assay for unbiased biological evaluation and mechanism insight. | PNP, Natural Product, Diverse | |
| Classical Molecular Networking (GNPS) [47] | Cloud-based platform for analyzing MS/MS data to group compounds by structural similarity and guide library focusing. | Natural Product | |
| Synthetic & Building Block Sources | Fmoc-Amino Acids, Carboxylic Acids [44] | Building blocks for combinatorial synthesis of peptide-inspired and diverse libraries. | Diverse (SEL), Focused |
| Fragment-sized Natural Products (Quinine, Griseofulvin) [46] | Commercially available complex fragments for the synthesis of pseudo-natural products. | PNP, Fragment |
The selection of an optimal compound library is a foundational decision that predetermines the success or failure of any screening campaign in drug discovery. This choice dictates the accessible chemical space, influences hit rates, and ultimately shapes the profile of resulting lead compounds. The decision is framed within a broader, critical thesis: the inherent scaffold diversity and biological pre-validation of natural products (NPs) offer unique advantages that are often not replicated by commercially available synthetic compound libraries [2]. Historically, NPs have served as the inspiration for a significant proportion of approved small-molecule drugs [2]. However, the rise of high-throughput screening (HTS) in the 1980s created a demand for vast numbers of compounds that NP collections could not initially satisfy, leading to a shift toward synthetic libraries [2].
Modern discovery pipelines now face a triad of screening paradigms, each with distinct library requirements: target-based HTS, phenotypic screening, and virtual screening (VS). This guide provides an objective comparison of library selection strategies for these approaches, underpinned by experimental data and framed by the ongoing research question of how to best harness or mimic the privileged structural diversity of NPs. The resurgence of interest in NPs and NP-inspired libraries stems from analyses showing that while synthetic compounds (SCs) number in the hundreds of millions, they often occupy a more restricted and less biologically relevant region of chemical space compared to NPs, which exhibit greater structural complexity, more chiral centers, and a higher fraction of sp3-hybridized carbons [2].
Library Selection Comparative Analysis
The core properties of a screening library must be aligned with the screening methodology. The following tables provide a quantitative comparison of the key considerations for library selection across the three primary screening paradigms.
Table 1: Comparison of Screening Methodologies and Corresponding Library Requirements
| Screening Paradigm | Primary Goal | Typical Library Size | Key Library Design Principle | Major Consideration |
|---|---|---|---|---|
| Target-Based HTS | Identify ligands modulating a specific protein target. | 100,000 – 4+ million physical compounds [48] [49]. | High purity (>90%), chemical stability, drug-like property filters (e.g., Lipinski’s Rule of 5). | Cost of library acquisition, maintenance, and screening infrastructure can exceed $2 million [48]. |
| Phenotypic Screening | Identify compounds that elicit a desired cellular or organismal phenotype. | 10,000 – 500,000 compounds. | Structural and scaffold diversity to probe multiple mechanisms; inclusion of bioactive tool compounds. | Hit deconvolution (identifying the molecular target) is a major subsequent challenge. |
| Virtual Screening (VS) | Computationally prioritize compounds for experimental testing. | Millions to billions of in silico molecules [50]. | Synthetically accessible (for on-demand libraries), drug-like property filters, diverse chemotypes. | Balance between exploration of vast chemical space and computational feasibility of screening. |
Table 2: Quantitative Performance Metrics of Featured Library Technologies
| Library Technology / Example | Reported Library Size | Key Performance Metric | Experimental Context & Result | Reference |
|---|---|---|---|---|
| Traditional HTS Library (e.g., for 17β-HSD10) | ~350,000 drug-like molecules | Hit identification rate | Screening identified novel, low nanomolar inhibitors of 17β-HSD10 for Alzheimer's/cancer [49]. | [49] |
| DNA-Encoded Library (DEL) | Commonly 10^6 - 10^10 | Affinity selection capability | Standard technology; limited by synthesis complexity and incompatibility with nucleic-acid binding targets [44]. | [44] |
| Self-Encoded Library (SEL) – Barcode-free | Up to 750,000 in a single run | Direct screening & identification of binders | Identified nanomolar binders to carbonic anhydrase IX and FEN1 (a DNA-processing enzyme inaccessible to DELs) [44]. | [44] |
| Ultra-Large Virtual Library (e.g., for docking) | Up to 11+ billion synthesizable molecules | Docking hit rate & novelty | The V-SYNTHES approach enabled discovery of high-affinity, novel chemotypes for GPCR and kinase targets from an 11-billion compound space [50]. | [50] |
Table 3: Structural and Property Analysis: Natural Products vs. Synthetic Compound Libraries
| Property Category | Trend in Natural Products (NPs) Over Time | Trend in Synthetic Compounds (SCs) Over Time | Implication for Library Design |
|---|---|---|---|
| Molecular Size (Weight, Volume) | Marked increase; newer NPs are larger [2]. | Variation within a constrained range (due to synthetic and drug-like rules) [2]. | NP libraries offer access to larger, more complex scaffolds absent from typical SC libraries. |
| Ring Systems | Increasing number of non-aromatic and fused rings (e.g., bridged rings) [2]. | Higher proportion of aromatic rings (e.g., benzene derivatives) [2]. | NP-inspired libraries can enhance 3D shape complexity and saturation, improving odds for difficult targets. |
| Chemical Space | Becomes less concentrated and more diverse over time [2]. | More concentrated within drug-like "rule-based" boundaries [2]. | Supplementing SC libraries with NP-like scaffolds expands the explorable chemical universe for screening. |
| Biological Relevance | Inherently high due to evolutionary selection. | Shows a decline in newer collections [2]. | NPs and pseudo-NP libraries provide biologically pre-validated starting points. |
Experimental Protocols for Key Screening Technologies
Protocol for a High-Throughput Biochemical Screen (Target-Based)
This protocol outlines a standardized HTS campaign as utilized in modern drug discovery, such as the screen that identified 17β-HSD10 inhibitors [49].
1. Assay Development & Validation:
- Objective: Develop a robust, miniaturized biochemical assay measuring target activity (e.g., enzyme inhibition).
- Procedure: Optimize buffer conditions, substrate concentration (at KM), enzyme concentration, and incubation time to achieve a strong signal window. Validate using known inhibitors (positive controls) and DMSO (negative control).
- Quality Control: Calculate the Z'-factor for each assay plate. A Z' > 0.5 indicates an excellent assay suitable for HTS [48]. Implement controls on every plate to monitor edge effects and drift [51].
2. Library Preparation & Reformating:
- Procedure: Thaw source compound plates (e.g., 10 mM DMSO stocks). Using liquid handling robots (e.g., Tecan, Hamilton), perform a dilution series in assay buffer to create intermediate concentration plates [48]. Pin-transfer or acoustically eject (e.g., Acoustic Droplet Ejection) nanoliter volumes of compounds into assay plates (384- or 1536-well format) [51].
3. Automated Screening Run:
- Procedure: Sequentially dispense assay buffer, enzyme, and substrate into assay plates containing compounds. Incubate under controlled conditions. Quench the reaction if necessary. Measure the signal (e.g., fluorescence, absorbance, luminescence) using a plate reader integrated into the robotic system.
4. Primary Data Analysis & Hit Identification:
- Procedure: Normalize raw data using plate-based positive and negative controls. Calculate percent inhibition/activation for each well. Apply statistical methods (e.g., SSMD*) to define a hit threshold, typically >3 standard deviations from the mean of negative controls [51]. Compounds exceeding this threshold are designated "primary hits."
Protocol for Affinity Selection with a Self-Encoded Library (SEL)
This detailed protocol is based on the barcode-free SEL technology that screened 750,000 compounds against FEN1 [44].
1. Library Synthesis (Solid-Phase Split & Pool):
- Objective: Generate a vast, diverse library of small molecules attached to solid-phase beads.
- Procedure: Choose a scaffold (e.g., benzimidazole core). Use a split-and-pool approach: divide beads into separate reaction vessels, couple a different building block (e.g., primary amine) to each pool, then recombine all beads. Repeat for subsequent diversification steps (e.g., heterocyclization with aldehydes). This yields a one-bead-one-compound library where each bead carries ~10^13 copies of a single structure [44].
2. Affinity Selection Panning:
- Objective: Isolate beads carrying compounds that bind to the immobilized target protein.
- Procedure: Incubate the library of beads with the purified, immobilized target (e.g., FEN1) in a suitable binding buffer. Wash extensively to remove non-binding and weakly binding beads. Elute the specifically bound beads using a denaturing buffer (e.g., low pH) or a competitive ligand.
3. Hit Decoding via Tandem Mass Spectrometry (MS/MS):
- Objective: Identify the chemical structure of compounds on selected beads without a DNA barcode.
- Procedure: Cleave the compound from a single selected bead. Analyze via nanoLC-MS/MS. Fragment the molecular ion in the mass spectrometer to generate an MS2 spectrum. Use custom software to compare the experimental MS2 spectrum against a virtual database of predicted spectra for all library members. The software annotates the structure based on the best match, distinguishing between isobaric compounds by their unique fragmentation patterns [44].
4. Hit Validation:
- Procedure: Resynthesize the identified compound as a discrete, pure material. Confirm binding and functional activity (e.g., enzymatic inhibition) using standard biochemical assays (e.g., IC50 determination).
Visualizing Screening Strategies and Library Diversity
Flowchart: Screening Strategy Decision Workflow
Flowchart: Time-Dependent Evolution of Compound Collections
The Scientist's Toolkit: Essential Research Reagent Solutions
Selecting the right tools is critical for executing a successful screening campaign. The following table details key reagents and materials, their function, and application context.
Table 4: Essential Research Reagent Solutions for Screening Campaigns
| Category | Reagent / Material | Function in Screening | Key Consideration / Example |
|---|---|---|---|
| Library Sources | Purchasable Screening Libraries (e.g., ChemBridge, Enamine, Mcule) [25] | Provides physical compounds for HTS and phenotypic screens. | Diversity varies; analysis shows ChemBridge, ChemicalBlock, and Mucle libraries among the most structurally diverse [25]. |
| Natural Product Collections & Databases (e.g., TCMCD) [25] | Provides NPs or NP-inspired compounds with high scaffold diversity. | Traditional Chinese Medicine Compound Database (TCMCD) shows the highest structural complexity among studied libraries [25]. | |
| Virtual/On-Demand Libraries (e.g., ZINC, Enamine REAL) [50] | Source of billions of synthesizable compounds for virtual screening. | Enables ultra-large docking campaigns (e.g., screening 11+ billion compounds) [50]. | |
| Assay Technology | DNA-Encoded Libraries (DELs) | Enables affinity selection of very large (10^6-10^10) encoded libraries. | Limited by water-compatible chemistry and incompatibility with nucleic-acid binding targets [44]. |
| Self-Encoded Libraries (SELs) [44] | Enables barcode-free affinity selection via MS/MS decoding. | Overcomes DEL limitations; used to screen 750k compounds against DNA-binding target FEN1 [44]. | |
| Automation & QC | Automated Liquid Handlers (e.g., Tecan, Hamilton) [48] | Precise, high-throughput dispensing of reagents and compounds. | Essential for miniaturization to 1536-well formats and reducing reagent costs [48] [51]. |
| Acoustic Droplet Ejection (ADE) Systems | Tip-less, non-contact transfer of nanoliter volumes. | Reduces consumable costs and eliminates carryover contamination, alleviating a key HTS bottleneck [51]. | |
| Assay Quality Control Metrics (Z'-factor, SSMD*) | Statistical measures of assay robustness and hit confidence. | Z' > 0.5 indicates a robust assay [48]. SSMD is used for rigorous hit identification in complex phenotypes [51]. | |
| Data Analysis | Pharmacotranscriptomics Platforms | Enables pathway-based screening by measuring genome-wide gene expression changes. | Represents a third screening paradigm alongside target-based and phenotypic approaches [52]. |
| Artificial Intelligence/Machine Learning Platforms | Analyzes HTS data, predicts activity, and designs novel libraries. | AI can design optimized libraries and enable active learning to focus screening efforts [48] [50]. |
The pursuit of novel chemical scaffolds is a fundamental driver in drug discovery. Within this endeavor, two primary reservoirs exist: the vast, evolutionarily refined chemical space of natural products (NPs) and the synthetically curated space of purchasable compound libraries. This guide provides a comparative analysis of the workflows for sourcing and characterizing NPs, framing it within the critical thesis of scaffold diversity. NPs, derived from plants, microbes, and marine organisms, are celebrated for their structural complexity, high fraction of sp3-hybridized carbons, and proven biological relevance, often yielding privileged scaffolds for drug development [53] [5]. In contrast, purchasable libraries offer millions of synthetically accessible, well-defined compounds optimized for drug-like properties but often with lower scaffold diversity and structural complexity [54] [25]. The central challenge lies in effectively navigating the NP workflow—from informed sourcing through rigorous analytical characterization—to access this unique diversity, a process that is inherently more complex than acquiring compounds from a commercial catalog [55].
Comparative Workflows: Natural Products vs. Purchasable Libraries
The journey from source material to an annotated, screening-ready compound differs profoundly between natural and commercial synthetic origins. The table below summarizes the key stages of each workflow.
Table 1: Comparative Workflow for Natural Products and Purchasable Compound Libraries
| Workflow Stage | Natural Product Workflow | Purchasable Compound Library Workflow |
|---|---|---|
| 1. Sourcing & Acquisition | Obtain authenticated biological material (plant, microbial fermentation). Requires voucher specimens, taxonomic verification, and consideration of source variability [55]. | Select vendors (e.g., Enamine, Mcule, ChemBridge). Order based on library composition, purity data, and cost [54] [25]. |
| 2. Preparation & Extraction | Employ extraction (e.g., solvent, SFE, UAE, MAE) to generate a complex crude mixture [56] [57]. | Compounds arrive as purified powders or DMSO solutions. Minimal preparation required; may involve plating or reformatting [58]. |
| 3. Purification & Isolation | Multi-step chromatographic purification (e.g., CPC, Prep-HPLC) is essential to isolate individual compounds from the complex matrix [56]. | Purity is vendor-claimed (typically >90%). Quality control (QC) via LC-MS may be performed upon receipt [58]. |
| 4. Characterization & Annotation | Structural elucidation via NMR, HRMS, IR. Determination of absolute stereochemistry may be required [55] [56]. | Identity confirmed by QC-MS. Full analytical data is typically not provided; structures are vendor-claimed. |
| 5. Library Assembly | Build a screening library through cumulative, labor-intensive isolation efforts. Libraries are smaller (100s-1000s of compounds) but high in scaffold diversity [53]. | Curate a large library (10,000s-100,000s of compounds) from multiple vendors. Focus is on physicochemical property filters and minimizing structural alerts [25] [58]. |
| Key Advantages | Unmatched scaffold diversity, biological pre-validation, and complex, three-dimensional architectures [53] [5]. | Scalability, speed, and cost-efficiency. High synthetic tractability for follow-up chemistry [54] [25]. |
| Major Challenges | Inherent complexity and variability of source material, low abundance of active compounds, resource- and time-intensive process [55]. | Limited scaffold diversity (high redundancy), potential for "flat" aromatic structures, and unknown biological relevance [25]. |
The following diagram illustrates the parallel yet divergent paths of these two critical approaches to populating screening collections.
The Natural Product Workflow: Detailed Protocols and Comparisons
Stage 1: Sourcing and Authentication
Sourcing begins with selecting biologically relevant material. For botanical products, this requires identifying the correct genus, species, and plant part (e.g., root, leaf) used in traditional preparations [55]. A non-negotiable step is the creation of a voucher specimen—a preserved sample deposited in a herbarium for permanent taxonomic verification [55]. For microbial NPs, sourcing involves isolating strains from environmental samples or acquiring them from culture collections, followed by genetic characterization to assess biosynthetic potential [53].
Comparison to Purchasable Libraries: This stage has no direct parallel in the commercial workflow. Vendor selection replaces organism selection, and the "authentication" is replaced by assessing a vendor's reputation and the consistency of their QC data [58].
Stage 2: Extraction & Preparation
The goal is to solubilize bioactive compounds while minimizing degradation. Methods are chosen based on compound polarity and stability.
- Conventional Solvent Extraction: Maceration or Soxhlet extraction using solvents like methanol, ethanol, or dichloromethane. It is simple but can be time-consuming and use large solvent volumes [57].
- Modern Green Extraction Techniques:
- Supercritical Fluid Extraction (SFE): Uses supercritical CO₂. Excellent for thermally labile, non-polar compounds. It is solvent-free and tunable by pressure/temperature [56] [57].
- Ultrasound-Assisted Extraction (UAE): Uses cavitation to disrupt cell walls, enhancing solvent penetration. Faster and more efficient than maceration [57].
- Microwave-Assisted Extraction (MAE): Uses microwave energy to rapidly heat solvents and plant matrices. Highly efficient for specific compound classes [57].
Table 2: Comparison of Key Extraction Techniques for Natural Products
| Technique | Principle | Optimal For | Advantages | Limitations |
|---|---|---|---|---|
| Solvent Maceration | Diffusion of solvent into plant material | Broad range, standard lab prep | Simple, low-cost equipment | Time-consuming, high solvent use, lower efficiency |
| Soxhlet Extraction | Continuous solvent cycling via distillation | Lipophilic compounds | Efficient, good yield | High temperature, not for thermolabile compounds |
| Supercritical Fluid (SFE) | Solvation power of supercritical CO₂ | Non-polar to moderately polar, delicate compounds | Green (no solvent residue), tunable selectivity, fast | High capital cost, poor for very polar compounds |
| Ultrasound-Assisted (UAE) | Cell wall disruption via cavitation | Polar antioxidants, phenolics | Rapid, improved yield, moderate cost | Scale-up challenges, potential for radical degradation |
| Microwave-Assisted (MAE) | Selective heating of moisture/solvent | Essential oils, glycosides | Very fast, high efficiency, low solvent | Optimization needed, risk of overheating target compounds |
Stage 3: Purification & Isolation
This critical stage separates the complex extract into individual compounds.
- Initial Fractionation: Often employs open-column chromatography or Centrifugal Partition Chromatography (CPC). CPC is a liquid-liquid separation technique with high recovery rates and no irreversible adsorption, making it ideal for crude extracts [56].
- Final Purification: Typically achieved via Preparative High-Performance Liquid Chromatography (Prep-HPLC). This scalable technique offers high resolution for isolating pure compounds (≥95% purity) necessary for screening and structure elucidation [56].
Comparison to Purchasable Libraries: For commercial compounds, purification is the vendor's responsibility. Academic labs typically perform QC analyses on a subset of purchased compounds to verify identity and purity (e.g., >80-90%), but do not perform re-purification [58].
Stage 4: Characterization & Annotation
Structural elucidation defines the final "annotation" of the NP.
- High-Resolution Mass Spectrometry (HRMS): Determines the exact molecular formula [56].
- Nuclear Magnetic Resonance (NMR) Spectroscopy: (1D & 2D experiments: ¹H, ¹³C, COSY, HSQC, HMBC) is indispensable for determining planar structure and relative stereochemistry [55] [56].
- Advanced MS/MS and Bioinformatics: Used in untargeted metabolomics to rapidly profile extracts and prioritize novel ions by comparing MS/MS fragmentation patterns against databases [55].
The following diagram details this integrated analytical pipeline.
Comparison to Purchasable Libraries: Characterization of purchased compounds is typically limited to QC-MS for identity and HPLC-UV/ELSD for purity assessment [58]. Full structural elucidation is not performed by the end-user.
Critical Analysis: Scaffold Diversity and Chemical Space
Measuring Scaffold Diversity
Scaffold diversity is quantitatively assessed using cheminformatic tools.
- Murcko Frameworks: Defines the core ring and linker system of a molecule, removing side chains [25].
- Scaffold Tree: Hierarchically deconstructs a molecule to a single ring, assigning levels that show structural relationships [25].
- Analysis: Diversity is measured by the number of unique scaffolds covering a population of compounds (e.g., PC50C, the percentage of scaffolds needed to cover 50% of a library) [25].
Comparative Diversity of NP vs. Purchased Libraries
Data reveals a stark contrast. An analysis of the Natural Products Atlas (microbial NPs) shows high chemical clustering, with 82.6% of compounds grouping into 4,148 scaffold clusters, indicating dense exploration of specific, biologically relevant chemotypes [53]. In contrast, a study of 11 major purchasable libraries found that while vendors like ChemBridge and Mcule offered good diversity, many commercial libraries exhibited high redundancy, with a small number of common scaffolds representing a large fraction of their offerings [25].
Table 3: Scaffold Diversity Metrics: Natural Products vs. Purchasable Libraries
| Metric | Natural Product Libraries (Microbial Focus) | Purchasable Compound Libraries (Selected Vendors) |
|---|---|---|
| Source Data | Natural Products Atlas (v2024_09): 36,454 compounds [53] | Analysis of 11 standardized vendor subsets (e.g., Enamine, Mcule, ChemDiv) [25] |
| # of Unique Murcko Frameworks | Not explicitly stated; high based on cluster analysis. | Ranged from ~5,000 to ~11,000 across different vendor libraries [25]. |
| PC50C (Lower = more diverse) | Not calculated in source, but cluster analysis suggests low PC50C (high diversity per compound). | Varied by vendor. More diverse libraries (e.g., ChemBridge, Mcule) had lower PC50C values [25]. |
| Key Finding on Diversity | Displays "islands of density" – high structural similarity within clusters (e.g., microcystins), but large gaps between cluster types, indicating deep but focused exploration of specific chemotypes [53]. | Scaffold redundancy is common. A small subset of frameworks often accounts for a large percentage of a vendor's library [25]. |
| Structural Complexity | High. Rich in stereocenters and sp3-hybridized carbons [53] [5]. | Generally lower. Often enriched in flat, aromatic structures; lower Fsp³ [25] [58]. |
The Emerging Hybrid: Pseudo-Natural Products (PNPs)
The innovative Pseudo-Natural Product (PNP) strategy directly addresses the thesis of scaffold diversity by merging the strengths of both sources. PNPs are synthetically assembled from distinct NP-derived fragments in combinations not found in nature [5]. For example, a 2024 study generated 154 PNPs from a common indole precursor, creating eight novel classes with high three-dimensionality. Phenotypic screening revealed diverse, unprecedented bioactivities, including Hedgehog signaling inhibition and tubulin modulation [5]. This demonstrates that informed synthetic design based on NP principles can efficiently generate novel, biologically relevant scaffold diversity that complements both traditional NP isolation and large commercial libraries.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Reagents and Materials for Natural Product Workflow
| Item | Function in Workflow | Key Considerations |
|---|---|---|
| Silica Gel (various pore sizes) | Stationary phase for open-column and flash chromatography for initial fractionation. | Different mesh sizes (e.g., 40-63 µm, 63-200 µm) for resolution vs. speed. |
| HPLC-Grade Solvents | Mobile phases for analytical and preparative HPLC (e.g., acetonitrile, methanol, water with modifiers like TFA or formic acid). | Purity is critical for HPLC to prevent column damage and baseline noise. |
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | Solvents for NMR spectroscopy. | Must be anhydrous and of high isotopic purity for accurate spectral acquisition. |
| Sephadex LH-20 | Size-exclusion chromatography gel. Often used for desalting or separating compounds based on molecular size in polar solvents (e.g., methanol). | Excellent for final polish purification of sensitive NPs. |
| Solid Phase Extraction (SPE) Cartridges | Rapid cleanup of crude extracts or fractions to remove salts, pigments, or lipids. | Available in various chemistries (C18, diol, ion-exchange) for selective cleanup. |
| Culture Media Components | For fermentation of microbial NPs (e.g., yeast extract, peptone, specific carbon sources). | Composition dramatically affects the expressed metabolome and NP yield [53]. |
| Reference Standards | Authentic chemical standards for target NPs or marker compounds. | Essential for developing and validating analytical methods (HPLC, GC) and for biological activity comparisons [55]. |
The choice between natural product and purchasable library workflows is not binary but strategic. NPs provide evolutionarily validated, complex scaffolds often inaccessible by commercial synthesis, making them indispensable for probing novel biological mechanisms or targeting "undruggable" sites. The purchasable library workflow offers unmatched efficiency and scale for screening campaigns against well-defined targets.
Recommendations for Researchers:
- For Novelty & Mechanism: Invest in the NP workflow when seeking unprecedented chemotypes or new mechanisms of action. Prioritize source organisms with high biosynthetic potential (e.g., via genomics) [53] and employ modern extraction/chromatography to improve efficiency.
- For Target-Based Screening: Utilize purchasable libraries for their speed and coverage of drug-like chemical space. Employ strict curation filters (e.g., PAINS, property filters) and prioritize vendors with demonstrated scaffold diversity [25] [58].
- Adopt a Hybrid Strategy: Integrate a well-characterized, fractionated NP library alongside a curated commercial library for phenotypic screens. Furthermore, consider adopting or collaborating on Pseudo-Natural Product synthesis to generate focused libraries that bridge the gap between NP-inspired diversity and synthetic feasibility [5].
The future of scaffold discovery lies in intelligently integrating these parallel streams—harnessing the efficiency of synthesis and the inspirational power of nature's chemical innovation.
The pursuit of novel therapeutic agents relies fundamentally on access to chemically diverse small molecules. Within drug discovery, a central thesis contrasts the evolutionarily refined scaffold diversity of natural products (NPs) with the broad, synthetic accessibility of purchasable compound libraries [1] [59]. NPs, honed by nature for biological interaction, exhibit unparalleled structural complexity, three-dimensionality, and success as drug leads, particularly for challenging targets like protein-protein interactions [1] [39]. In contrast, commercially sourced synthetic libraries offer vast numbers of well-characterized, "lead-like" compounds designed for high-throughput screening (HTS) compatibility [3] [8]. The choice of sourcing pathway—whether from individual vendors, digital aggregators, or collaborative consortia—directly impacts a research organization's ability to navigate this chemical space, balancing diversity, quality, cost, and logistical ease to fuel innovation [3] [60].
Comparative Analysis of Purchasing Pathways
The selection of a sourcing model is a strategic decision that influences library quality, cost structure, and research agility. The following table summarizes the core characteristics of the three primary pathways.
Table 1: Comparison of Compound Library Sourcing Pathways
| Feature | Vendors (Direct Manufacturers) | Aggregators (Digital Platforms) | Consortia & Shared Libraries |
|---|---|---|---|
| Core Model | Produce and sell proprietary compound collections [9] [8]. | Curate and list purchasable compounds from multiple vendors under a unified platform [61] [37]. | Facilitate shared access to specialized, often niche, libraries through multi-member collaborations [59]. |
| Chemical Diversity & Focus | Offer both broad diversity libraries and highly targeted sets (e.g., kinases, fragments, covalent inhibitors) [9] [8]. | Provide extremely large virtual databases (e.g., 139M+ compounds) and filtered libraries (e.g., NPs, RNA-binding) [37]. | Often focus on difficult-to-access or ethically sourced chemistry, such as purified natural products or biodiversity-derived extracts [62] [59]. |
| Quality Control | Direct control over synthesis, purification (>90% pure), and analytical validation (LCMS/NMR) [9]. | Variable; depends on the original vendor. Platforms provide filtering tools but may not re-test compounds [61] [37]. | High; governed by consortium research protocols and standards for extraction, characterization, and storage [59]. |
| Primary Advantage | High quality, reliable resupply, and expert design (e.g., AI/ML, focused libraries) [9]. | Unmatched breadth of search, rapid virtual screening capability, and simplified procurement from multiple sources [37]. | Access to unique chemical matter (e.g., novel scaffolds) and shared cost/risk in exploring complex natural product space [62] [59]. |
| Key Challenge | Cost can be high for large, diverse sets; library scope is limited to vendor's own catalog [3]. | Less control over compound quality and availability; physical shipping from disparate global vendors can be complex [61]. | Complex governance, legal agreements (e.g., Nagoya Protocol for biodiversity), and slower access due to collaborative nature [59]. |
| Best For | Organizations prioritizing high-quality, assay-ready compounds with strong vendor support for hit-to-lead [3] [9]. | Virtual screening campaigns and projects requiring the widest possible search of purchasable chemical space [37]. | Academic and industry partnerships aiming to explore high-risk, high-reward chemical space like natural product scaffolds [39] [59]. |
Experimental Focus: Generating Diversity from Natural Product Scaffolds
To objectively compare the potential of natural product-derived libraries versus standard synthetic libraries, experimental data on scaffold diversification is critical. The following protocol outlines a modern, two-phase strategy for diversifying complex natural products into novel polycyclic scaffolds, a process that generates chemical space distinct from commercial collections [39].
Experimental Protocol: C–H Functionalization and Ring Expansion for NP Diversification [39]
- Objective: To generate a library of complex, natural product-like small molecules featuring underrepresented medium-sized rings (7-11 membered) through late-stage diversification of steroid scaffolds.
- Principle: This method combines site-selective C–H bond oxidation to install functional handles, followed by ring expansion reactions. This approach moves beyond simple peripheral modification to alter the core scaffold itself, accessing new skeleta.
- Key Steps & Methodologies:
- Substrate Preparation: Select polycyclic natural product cores (e.g., Dehydroepiandrosterone/DHEA, estrone). Protect reactive functional groups as needed using standard techniques (e.g., silylation, acetylation).
- Site-Selective C–H Oxidation:
- Electrochemical Allylic C–H Oxidation: Use an electrochemical cell with controlled potential to oxidize allylic C–H bonds to enones, as developed by Baran et al. [39].
- Metal-Mediated C–H Oxidation: Apply copper or chromium catalysts to achieve oxidation at specific aliphatic or benzylic C–H sites [39].
- Ring Expansion via Native or Newly Installed C–O Bonds:
- Perform reactions such as the intramolecular Schmidt reaction (using hydrazoic acid) on ketone groups to insert nitrogen and expand rings.
- Employ formal [2+2] cycloaddition-fragmentation with dimethyl acetylenedicarboxylate (DMAD) on β-keto esters to achieve two-carbon ring expansion.
- Execute Beckmann rearrangement of oximes to form medium-sized ring lactams.
- Data & Comparison: Principal component analysis of physicochemical properties (e.g., molecular weight, logP, stereochemical complexity, fraction of sp3 carbons) demonstrates that the resulting library occupies a unique region of chemical space compared to both common commercial synthetic libraries and the parent natural products [39]. This validates the strategy's power to generate purchasable-like libraries with NP-like scaffold diversity.
Diagram: Two-phase strategy for diversifying natural product scaffolds [39].
The Scientist's Toolkit: Essential Reagents & Materials
Building or working with compound libraries, especially those derived from natural products, requires specialized tools and reagents. This table details essential items for the featured diversification experiment and general library management.
Table 2: Research Reagent Solutions for Library Development & Screening
| Item | Function / Application | Sourcing Consideration |
|---|---|---|
| Dimethyl Sulfoxide (DMSO), anhydrous | Universal solvent for dissolving and storing small molecule libraries in HTS; must be of high purity to prevent compound degradation [3] [9]. | Sourced from high-quality chemical vendors; critical for maintaining library integrity over freeze-thaw cycles. |
| Prefractionated Natural Product Extracts | Complex mixtures of NPs used in initial phenotypic or target-based screens to identify bioactive crude fractions [62] [59]. | Sourced from specialized natural product libraries or consortia that ensure taxonomic identification and compliance with biodiversity laws (e.g., Nagoya Protocol) [59]. |
| LC-MS & NMR Analytical Standards | For quality control (QC) of both purchased and synthesized library compounds; verifies purity (>90%) and identity [9]. | Vendors should provide recent QC data. Internal standards are purchased for instrument calibration. |
| Cheminformatics Software (e.g., for PAINS/REOS filtering) | Software to filter virtual or physical libraries for undesirable, promiscuous, or reactive functional groups that cause assay interference [3]. | Commercial packages (e.g., from Schrodinger, OpenEye) or open-source tools are essential for library curation. |
| Building Blocks for Diversification (e.g., Ethyl Diazoacetate, DMAD) | Key reagents for ring expansion and complexity-generating reactions in synthetic diversification campaigns [39]. | Sourced from fine chemical suppliers; stability and safety in handling are paramount. |
| Solid Support & Reagents for Parallel Synthesis | For generating combinatorial libraries via techniques like Diversity-Oriented Synthesis (DOS), inspired by NP scaffolds [1] [62]. | Sourced from manufacturers of combinatorial chemistry supplies. |
Synthesis and Strategic Recommendations
The choice of a sourcing pathway is not mutually exclusive and should align with the research phase and strategic goals. Vendor-sourced libraries are optimal for well-resourced HTS campaigns against established target classes where high-quality, tractable hits are desired [3] [8]. Aggregator platforms are powerful for initial virtual screening across an immense chemical space to identify promising starting points for synthesis or purchase [37]. Consortia and specialized NP libraries are best suited for exploratory research aimed at unprecedented biology or against undrugged targets, where unique scaffold diversity outweighs the need for immediate, large-scale screening [39] [59].
Future success in drug discovery will hinge on a hybrid strategy. This involves leveraging aggregators for breadth, trusted vendors for quality and depth, and consortia for unique scaffold access. Integrating cheminformatics to map the distinct chemical space occupied by NP-derived libraries against commercial collections will allow researchers to make informed, strategic decisions, ultimately bridging the gap between natural product scaffold diversity and purchasable chemical libraries [3] [1] [59].
Diagram: Decision pathway for selecting a compound library sourcing model.
The pursuit of novel therapeutics is fundamentally a quest for novel chemical matter. Within this endeavor, a central thesis has emerged: the structural and scaffold diversity inherent to natural products (NPs) represents a biologically pre-validated and evolutionarily optimized chemical space that is not adequately replicated by traditional synthetic, purchasable compound libraries [63] [64]. While NPs have historically been the source of a significant proportion of all approved drugs, their complexity presents challenges for screening and supply [63]. This has driven the pharmaceutical industry towards large libraries of synthetic compounds (SCs), which, despite their vast numbers, often occupy a more confined and less biologically relevant region of chemical space [2] [1].
This guide examines two advanced strategies designed to bridge this gap: Natural Product-Inspired Libraries and Diversity-Oriented Synthesis (DOS) Libraries. These approaches seek to translate the advantageous properties of NPs—such as structural complexity, three-dimensionality, and proficiency at modulating challenging target classes like protein-protein interactions—into more accessible and screenable compound collections [1] [64]. We objectively compare the performance, output, and application of these strategies against conventional high-throughput screening (HTS) of commercial synthetic libraries, framing the discussion within the critical context of scaffold diversity and its impact on drug discovery outcomes.
Quantitative Comparison: Structural and Performance Landscapes
A time-dependent chemoinformatic analysis reveals fundamental and diverging evolutionary paths for natural products and synthetic compounds. The data below summarizes key structural and property differences that underpin the rationale for NP-inspired strategies [2].
Table 1: Time-Dependent Structural Evolution of Natural Products vs. Synthetic Compounds
| Property Category | Natural Products (NPs) Trend Over Time | Synthetic Compounds (SCs) Trend Over Time | Implication for Library Design |
|---|---|---|---|
| Molecular Size | Consistent increase in MW, volume, and heavy atoms [2]. | Variation within a limited, "drug-like" range (constrained by rules like Lipinski's) [2]. | NP-inspired libraries can access larger, more complex chemical space not covered by standard SC libraries. |
| Ring Systems | Increase in total rings and non-aromatic rings; stable aromatic ring count [2]. | Increase in aromatic rings; high prevalence of 5- and 6-membered rings [2]. | NPs offer more aliphatic and fused ring systems, providing greater three-dimensionality [64]. |
| Complexity & Chirality | Increased sp³-hybridized bridgehead atoms and chiral centers [64]. | Lower sp³ character and fewer chiral centers [64]. | Higher complexity correlates with success in modulating biological macromolecules [63]. |
| Chemical Space | Becoming less concentrated and more diverse over time [2]. | Remains more concentrated, despite vast numbers of compounds [2]. | NP-inspired libraries can populate unique, underexplored regions of chemical space. |
| Biological Relevance | Inherently high due to evolutionary selection [1]. | Shows a decline over time in comparative analysis [2]. | Scaffolds from NPs are "privileged" with pre-validated bioactivity [64]. |
The performance of different library types in drug discovery is further illuminated by market and application data, which reflect their utilization and perceived value in the industry.
Table 2: Market and Application Comparison of Compound Library Types
| Library Type | Estimated Market Share & Growth | Primary Applications | Key Strengths | Notable Examples/Providers |
|---|---|---|---|---|
| Natural Product Libraries | Niche segment within broader market; growth driven by demand for unique diversity [19]. | Phenotypic screening, target ID for novel mechanisms, infectious disease & oncology [65]. | Unparalleled scaffold diversity, biological pre-validation, novel mechanisms of action [63] [1]. | NCI Natural Product Repository, MLSMR [65]. |
| NP-Inspired & DOS Libraries | Growing segment as a hybrid strategy; part of the "Diversity Libraries" type [18]. | Targeting "undruggable" targets (PPIs), fragment-based discovery, lead optimization [1] [64]. | Merges NP-like complexity with synthetic feasibility and library accessibility [66]. | Academic DOS platforms, collaborations (e.g., AstraZeneca-Scripps) [18]. |
| Traditional Small Molecule (HTS) Libraries | Largest market share (e.g., dominant in North America) [18] [19]; expected to grow at a CAGR of ~5.9% [18]. | High-Throughput Screening (HTS), lead generation for well-defined targets [19]. | Vast numbers (10⁶–10⁹ compounds), commercial availability, well-established screening protocols [67]. | Enamine, ChemBridge, WuXi LabNetwork [18]. |
| Fragment Libraries | High-growth segment due to efficiency [19]. | Fragment-Based Drug Discovery (FBDD) [19]. | High ligand efficiency, covers broad chemical space with fewer compounds [19]. | See commercial HTS library providers. |
Experimental Protocols and Methodologies
Protocol for Creating a Prefractionated Natural Product Library
This protocol, based on the NCI's Cancer Moonshot Program for Natural Product Discovery, outlines the creation of a high-quality, screening-ready library [65].
1. Source Collection & Ethics:
- Obtain necessary permits and establish benefit-sharing agreements compliant with the Nagoya Protocol (Access and Benefit-Sharing, ABS) and Convention on Biological Diversity (CBD) [65].
- Collect organism samples (plant, marine, microbial) with detailed voucher specimens, including taxonomy, GPS location, date, and collector metadata [65].
2. Extraction:
- Use accelerated solvent extraction (ASE) or microwave-assisted extraction for efficiency.
- Employ a sequential solvent system (e.g., hexane, dichloromethane, methanol/water) to capture metabolites of varying polarity [65].
- Dry extracts via centrifugal evaporation and store at -20°C.
3. Prefractionation (Critical Step):
- Goal: Separate complex crude extracts into simpler fractions to reduce assay interference and concentrate minor active constituents [65].
- Method: Employ automated reversed-phase High-Performance Liquid Chromatography (HPLC).
- Typical Conditions: Column: C18; Mobile Phase: Water/Acetonitrile gradient (e.g., 5% to 100% ACN over 20 min); Detection: UV at 210 nm & 254 nm.
- Fractionation: Collect time-based fractions (e.g., 12-24 fractions per extract) across the chromatographic run [65].
- Transfer fractions to 384-well plates using liquid handlers, dry, and store.
4. Quality Control & Normalization:
- Use LC-High Resolution Mass Spectrometry (HRMS) to generate a molecular fingerprint for each fraction.
- Employ 1H NMR profiling to assess complexity and identify major components.
- Normalize fractions by weight or by resuspending in DMSO to a standard concentration (e.g., 2 mg/mL or 5 mM estimated) [65].
Protocol for Generating a Natural Product-Inspired Library via Semi-Synthesis
This protocol details the use of an isolated NP as a core scaffold for generating a diverse analogue library [66].
1. Scaffold Selection:
- Choose a "privileged" NP scaffold with demonstrated, interesting bioactivity but suboptimal drug-like properties (e.g., solubility, toxicity) [64]. Examples include oridonin (diterpenoid) or quinine (alkaloid) [64].
- Ensure the scaffold contains multiple, synthetically accessible derivatization points (e.g., hydroxyl, amine, carboxylic acid groups).
2. Synthetic Strategy:
- Employ divergent synthesis from the common NP core.
- Use robust, parallel-compatible reactions (e.g., amide coupling, alkylation, reductive amination, Mitsunobu reactions) to create diversity.
- Focus on modifying peripheral groups to explore Structure-Activity Relationships (SAR) while initially preserving the complex core [66].
3. Library Design & Analysis:
- Design the library to improve target properties: reduce logP, modulate H-bond donors/acceptors, and remove structural alerts (e.g., PAINS) [66].
- Characterize all analogues by LC-MS for purity and identity.
- Analysis: Test the library in the relevant biological assay. Compare the hit rate and potency profile to a matched screening of a traditional synthetic HTS library against the same target.
Protocol for a Diversity-Oriented Synthesis (DOS) Library
DOS aims to generate broad scaffold diversity de novo, mimicking the skeletal variety of NPs but using synthetic chemistry [1].
1. Pathway-Driven Library Design:
- Design starting materials with multiple functional handles.
- Plan to use branching reaction pathways where a common advanced intermediate can be transformed into distinct molecular skeletons using different reagents or conditions [1].
2. Synthesis Execution:
- Implement reactions that rapidly increase complexity, such as cycloadditions, ring-closing metathesis, or multi-component reactions.
- Use a combination of appendage diversity (adding different groups) and skeletal diversity (creating different ring systems) within the same library [1].
- Synthesis is typically performed in parallel format.
3. Screening & Evaluation:
- Screen the DOS library in a phenotypic or target-based assay.
- Key Comparison Metric: Analyze the structural uniqueness and scaffold hit rate of active compounds compared to hits from a million-compound commercial HTS library. A successful DOS library may yield hits with entirely novel chemotypes [1].
Visualization of Strategic Pathways
Diagram 1: Strategic Pathways in Modern Drug Discovery
Diagram 2: NP-Inspired Semi-Synthetic Library Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Library Creation and Screening
| Item / Reagent Solution | Function & Application | Example / Notes |
|---|---|---|
| Solid Phase Extraction (SPE) Cartridges | Initial cleanup and fractionation of crude natural extracts. Separates compounds by polarity [65]. | C18, Diol, or Ion-Exchange SPE stationary phases. |
| HPLC & UPLC Systems | Core tool for analytical profiling and preparative prefractionation of extracts [65]. | Systems with C18 columns, PDA/UV detectors, and fraction collectors. |
| High-Resolution Mass Spectrometer (HRMS) | Critical for dereplication. Provides exact mass for formula determination and database searching (e.g., GNPS) [63] [65]. | LC-QTOF-MS or LC-Orbitrap-MS systems. |
| Nuclear Magnetic Resonance (NMR) Spectrometer | Essential for structural elucidation of pure NPs and complex library members [63]. | High-field (e.g., 500 MHz) for small molecule analysis. |
| Automated Liquid Handlers | Enables high-throughput plating of libraries into 384- or 1536-well plates for screening [65]. | Platforms from Hamilton, Beckman Coulter, or Tecan. |
| Chemical Building Blocks | For DOS and NP-inspired synthesis. Diverse sets of amines, carboxylic acids, boronic acids, and alkyl halides. | Available from Sigma-Aldrich, Enamine, Combi-Blocks. |
| Diversity-Oriented Synthesis Kits | Pre-designed reagent sets for generating skeletal diversity (e.g., via multi-component reactions) [1]. | Custom kits from specialist suppliers (e.g., life science vendors). |
| Assay-Ready Plate Libraries | Pre-plated, normalized compound/fraction libraries for immediate screening [18] [65]. | Offered by the NCI, commercial vendors (e.g., Selleck, MedChemExpress). |
| Virtual Screening Software | To computationally prioritize compounds from ultra-large libraries (e.g., Enamine REAL) before purchase or synthesis [67]. | Molecular docking suites (AutoDock, Glide), machine learning platforms. |
| Global Natural Products Social Molecular Networking (GNPS) | Open-access online platform for sharing and analyzing mass spectrometry data to dereplicate known compounds [63]. | https://gnps.ucsd.edu |
Overcoming Hurdles: Key Challenges and Optimization Strategies for Both Sources
The search for novel therapeutic compounds stands at a crossroads, defined by a critical tension between chemical diversity and practical accessibility. On one side, natural products offer an unparalleled source of structural complexity and biological pre-validation, honed by millions of years of evolution to interact with biological macromolecules [1]. On the other, purchasable synthetic compound libraries provide immediate accessibility, scalability, and consistency, fueling modern high-throughput screening (HTS) campaigns [19]. This guide frames the acute supply chain challenges facing natural product research within this broader thesis: while natural products occupy unique and privileged chemical space critical for probing complex biology and discovering new drug classes [1], their sourcing, re-supply, and scale-up are fraught with bottlenecks that synthetic libraries are engineered to avoid. For researchers and drug development professionals, the strategic choice between these sources is no longer merely scientific but fundamentally logistical and economic, influenced by a global landscape of geopolitical tensions, trade tariffs, and climate risks [68] [69].
Comparative Analysis: Natural Products vs. Synthetic Compound Libraries
Sourcing and Initial Acquisition Bottlenecks
The journey from source to screen is fundamentally different for natural and synthetic compounds. Natural product sourcing is a multi-stage, geographically tethered process, whereas synthetic library acquisition is a commercial transaction.
Table 1: Comparative Sourcing Challenges
| Aspect | Natural Products | Purchasable Synthetic Libraries |
|---|---|---|
| Primary Source | Biological material (plants, marine organisms, microbes) [1]. | Chemical synthesis facilities [19]. |
| Key Bottlenecks | Geopolitical & Trade: Tariffs on imported materials [68]. Environmental: Climate events disrupting wild collection [69]. Ethical/Legal: Access & Benefit Sharing (ABS) agreements, biodiversity permits. Technical: Low yields in source organism. | Supply Chain Concentration: Reliance on specific regional manufacturers (e.g., Asia-Pacific) [20]. Tariffs: On precursor chemicals or final compounds [70]. |
| Lead Time | Months to years (collection, identification, initial extraction). | Days to weeks (commercial purchase and delivery). |
| Scalability of Sourcing | Inherently limited by biomass availability; difficult to scale initial collection. | Highly scalable via combinatorial chemistry [19]. |
| 2025 Risk Profile | High exposure to climate disruptions and trade policy shifts [68] [69]. | High exposure to trade tariffs on chemicals and electronics for screening [70]. |
Experimental Protocol: Initial Sourcing and Authentication of a Natural Product
- Permitting and Collection: Secure prior informed consent and mutually agreed terms under the Nagoya Protocol. Collect voucher specimens of the source organism (e.g., plant, marine sponge) for taxonomic authentication by a specialist [1].
- Material Logistics: Transport biomass under controlled conditions (e.g., frozen, in ethanol) to avoid degradation. Navigate customs declarations, which may be subject to new tariffs on biological materials [68].
- Crude Extract Preparation: Lyophilize and mill the biomass. Perform sequential extraction with solvents of increasing polarity (e.g., hexane, dichloromethane, methanol). Concentrate extracts under reduced pressure.
- Chemical Dereplication: Analyze crude extract via LC-HRMS/MS. Compare spectral data against natural product databases (e.g., Dictionary of Natural Products, NPASS) to identify known compounds and prioritize novel chemistry.
Re-supply and Scale-up Challenges
The transition from a milligram-scale screening hit to gram-scale for preclinical studies represents the most severe bottleneck for natural products, a phase where synthetic libraries have a distinct advantage.
Table 2: Re-supply and Scale-Up Pathways
| Aspect | Natural Products | Purchasable Synthetic Libraries |
|---|---|---|
| Primary Re-supply Strategy | Re-collection, cultivation, or large-scale fermentation. | Re-synthesis via validated chemical route. |
| Key Bottlenecks | Ecological: Over-harvesting threatens source populations. Agricultural: Difficulties in cultivating slow-growing organisms (e.g., trees, marine invertebrates). Fermentation: Non-producer strains, low titers. Supply Chain: Single geographic source vulnerability [71]. | Chemistry: Complex, multi-step syntheses with low yields. Cost: Expensive catalysts or building blocks. Regulatory: Need for cGMP compliance for scale-up. |
| Timeframe | 1-5+ years (cultivation/process development). | 6-18 months (route optimization and kilo-lab synthesis). |
| Cost Trajectory | Very high capital investment for aquaculture/agriculture or bioreactor facilities. | High but predictable chemical costs; economies of scale apply. |
| Resilience | Low; susceptible to disease, weather, and geopolitics [69] [72]. | Moderate; dependent on global chemical supply chains, which are diversifying [71]. |
Experimental Protocol: Scale-Up via Semi-Synthesis This protocol is employed when total synthesis is too complex and natural supply is limited.
- Isolation of Advanced Precursor: From the natural source, isolate a structurally complex intermediate in the highest possible yield (e.g., 10-deacetylbaccatin III for paclitaxel analogs).
- Route Scouting: Develop a short, robust synthetic sequence (1-4 steps) from the precursor to the target molecule and diverse analogs. Focus on convergent, high-yielding reactions.
- Process Chemistry Optimization: Adapt the route for kilo-scale production. This includes solvent switching to safer, cheaper alternatives (e.g., from THF to 2-MeTHF), catalyst loading reduction, and purification via crystallization instead of chromatography.
- Supply Chain Securing: Dual-source or secure long-term contracts for key starting materials and reagents to mitigate tariff and trade disruption risks [69] [71].
The Supporting Infrastructure: Compound Management
Efficient management of physical samples is a critical, often overlooked, component that differentiates these two paradigms. The growth of the compound management market, projected to reach USD 1.9 billion by 2034 [22], is a direct response to the need to handle both synthetic and natural product libraries.
Table 3: Management System Requirements
| Aspect | Natural Product Libraries | Synthetic Compound Libraries |
|---|---|---|
| Storage Complexity | High. Extracts and pure compounds often require -20°C or -80°C storage to prevent degradation of sensitive chemotypes [22]. | Moderate to Low. Most stable small molecules stored at ambient or +4°C in controlled humidity. |
| Inventory Tracking | Critical. Must link sample to precise collection data (location, date, specimen), extraction batch, and chromatographic fraction. | Standardized. Tracks structure, vendor, batch, location, and concentration via barcode/RFID systems [23]. |
| Sample Format | Diverse: crude extracts, prefractionated libraries, pure compounds. Requires different handling protocols. | Uniform: Typically solubilized pure compounds in DMSO in microplates or vials, ideal for automation [23]. |
| Market Driver Fit | Aligns with niche, manual or semi-automated systems due to lower volume and higher variability. | The primary driver for automated, high-throughput systems dominating the market [23] [22]. |
Visualization of Workflows and Strategic Pathways
Comparative Workflows for Drug Discovery
Strategic Integration to Overcome Bottlenecks
The Scientist's Toolkit: Research Reagent Solutions
Navigating the bottlenecks requires specialized tools and services. Below is a table of key solutions for natural product and compound library research.
Table 4: Essential Research Reagents & Solutions
| Item/Service | Function | Relevance to Bottleneck |
|---|---|---|
| Diversity-Oriented Synthesis (DOS) Libraries [1] | Provides synthetic compounds with NP-like complexity and 3D architecture. | Mitigates sourcing risk by creating "synthetic natural products" with reliable supply. |
| Fragment Libraries [19] | Collections of very small molecules (<300 Da) for fragment-based drug discovery. | Enables screening with minimal material, reducing initial scale requirements for rare NPs. |
| AI-Powered Supplier Platforms [68] | Platforms (e.g., Z2Data, Supplier.io) map suppliers and identify alternates. | Addresses sourcing bottlenecks by finding dual sources for key reagents or biomass. |
| Contract Compound Management [23] [22] | Outsourced storage, QC, and distribution of compound libraries. | Reduces capital cost of automated storage systems, crucial for academic NP labs. |
| Specialized Natural Product Databases | Digital libraries of NP structures and spectra (e.g., COCONUT, NPASS). | Accelerates dereplication, preventing wasted effort on known compounds early in the pipeline. |
| Bioprospecting/Cultivation CROs | Contract research organizations specializing in microbial fermentation or plant tissue culture. | Provides a path to scale-up without in-house agricultural or fermentation expertise. |
The supply chain bottlenecks in natural product research—sourcing volatility, re-supply uncertainty, and costly scale-up—present significant but not insurmountable barriers. These challenges starkly highlight the trade-off at the heart of modern drug discovery: privileged, biologically relevant chemical diversity versus supply chain resilience and predictability [69] [1].
The future lies in hybrid strategies that leverage the strengths of both paradigms. This includes:
- Designing next-generation synthetic libraries using diversity-oriented synthesis (DOS) principles inspired by natural product scaffolds [1].
- Employing AI and machine learning not just for virtual screening [20], but to predict cultivation conditions for source organisms or engineer biosynthetic pathways for scale-up.
- Building resilient, diversified sourcing networks for both natural biomass and critical chemical building blocks, informed by real-time geopolitical and climate risk data [68] [71].
For the researcher, the decision matrix is clear. Natural products remain the unmatched source for pioneering new therapeutic modalities and probing intractable biological targets. However, their integration into a viable drug discovery pipeline must now account for the "cost of resilience" [69] from the very outset. By strategically integrating synthetic approaches, advanced logistics planning, and robust compound management, the unique value of natural products can be sustained and harnessed in an era of global supply chain uncertainty.
The pursuit of novel chemical matter in drug discovery is fundamentally shaped by the libraries researchers choose to screen. This exploration is framed by a critical thesis: while natural products (NPs) offer unparalleled scaffold diversity and biological pre-validation, large purchasable compound libraries provide accessibility and synthetic tractability, yet often suffer from limited structural novelty and a higher prevalence of deceptive chemotypes [2]. The structural evolution of synthetic compounds (SCs) has been influenced by NPs, but SCs have not fully evolved toward the complexity and uniqueness of NP space, remaining constrained by synthetic convenience and "drug-like" rules [2]. This divergence creates a significant pitfall. The drive to screen vast, commercially available libraries can inadvertently populate projects with Pan-Assay Interference Compounds (PAINS) and other promiscuous actors, leading to wasted resources, scientific dead-ends, and publication of erroneous results [73] [74]. This guide provides a comparative framework for navigating this landscape, equipping researchers with data and protocols to strategically filter compound collections and prioritize scaffolds with genuine therapeutic potential.
Understanding the Adversary: PAINS and Assay Interference
Defining the Problem
PAINS are defined by a common substructural motif that confers a high probability of generating a positive signal in a broad range of biochemical assays, often through mechanisms unrelated to specific, reversible target modulation [73]. They are a major source of false positives and promiscuous hits that derail projects. It is crucial to distinguish a false positive (a compound that modulates the assay readout, not the target) from a false hit (a compound that acts on the target but is chemically intractable or non-progressible) [73]. PAINS often fall into the latter category. Their interference stems from various mechanisms, including covalent protein reactivity, metal chelation, redox cycling, formation of colloidal aggregates, and fluorescence or absorbance at assay detection wavelengths [73] [75].
Key PAINS Mechanisms and Exemplary Chemotypes
The following table summarizes the primary interference mechanisms, their underlying principles, and representative chemical classes.
Table: Primary Mechanisms of Assay Interference by PAINS and Related Compounds [73] [75]
| Interference Mechanism | Principle of Action | Exemplary Chemotypes / Compounds |
|---|---|---|
| Covalent Protein Reactivity | Irreversible, nonspecific binding to protein nucleophiles (e.g., cysteine thiols). | Quinones, alkylidene barbiturates, rhodanines, enones, isothiazolones. |
| Colloidal Aggregation | Formation of sub-micrometer particles that non-specifically inhibit enzymes. | Miconazole, nicardipine, trifluralin, staurosporine aglycone. |
| Redox Cycling | Generation of reactive oxygen species (ROS) that inhibit protein function. | Phenol-sulphonamides, quinones, catechols, β-lapachone. |
| Metal Chelation | Sequestration of metal cofactors essential for protein or assay function. | Hydroxyphenyl hydrazones, 2-hydroxybenzylamine, catechols. |
| Spectroscopic Interference | Compound fluorescence or absorbance overlaps with assay detection signals. | Daunomycin, riboflavin, compounds with quinoxalin-imidazolium substructures. |
The Limitations of Simple Filtering
A critical caveat is that PAINS filters are not infallible. They were derived from a specific dataset (~100,000 compounds screened in six AlphaScreen assays) [73]. Consequently:
- False Negatives: Filters may not recognize new or variant interference chemotypes (e.g., β-aminoketones, toxoflavins) [73].
- False Positives (Innocent Scaffolds): Privileged scaffolds like indoles or even some FDA-approved drugs (e.g., certain quinones) can be flagged. Automatic exclusion is unwise; a "Fair Trial Strategy" involving experimental validation is essential [76] [75].
- Assay Context-Dependence: Interference is not an absolute property. A compound may be a PAIN in one assay (e.g., fluorescence-based) but not in another (e.g., NMR-based) [73].
Comparative Guide: Purchasable Libraries and Natural Product Collections
Selecting the right screening library is a foundational decision. The following tables compare key vendors and highlight the intrinsic differences between synthetic and natural product-derived spaces.
Table 1: Comparative Analysis of Select Purchasable Compound Libraries [77] (Based on standardized subsets for equitable comparison)
| Library (Vendor) | Filtered Compound Count | Notable Description | Relative Scaffold Diversity |
|---|---|---|---|
| Mcule | ~4.9 million | Large, individual service | High |
| Enamine | ~2.0 million | Lead-like, diverse | Medium |
| ChemBridge | ~1.1 million | Selected, derivatives | High (Ranked Top) |
| VitasM | ~1.5 million | Novel compounds | High (Ranked Top) |
| ChemicalBlock | ~125,000 | Selected, diverse | High (Ranked Top) |
| Maybridge | ~57,000 | Highly diverse | Medium |
| TCMCD (NP-Derived) | ~54,000 | Traditional Chinese Medicine compounds | Highest Structural Complexity |
Table 2: Key Structural & Property Differences: Natural Products vs. Synthetic Compounds [2]
| Property / Feature | Natural Products (NPs) | Synthetic Compounds (SCs) | Implication for Screening |
|---|---|---|---|
| Molecular Size & Complexity | Larger, more rings, more chiral centers. | Smaller, constrained by "drug-like" rules. | NPs explore broader, more complex chemical space. |
| Ring Systems | More non-aromatic, aliphatic, and fused rings. | Dominated by aromatic rings (e.g., benzene). | NP scaffolds are more three-dimensional. |
| Heteroatom Content | Higher oxygen content. | Higher nitrogen content. | Different H-bond donor/acceptor profiles. |
| Biological Relevance | Evolved to interact with biomolecules; higher hit rates. | Biologically relevant scaffolds are designed or discovered. | NPs can reveal novel mechanisms of action. |
| Synthetic Accessibility | Often complex, challenging synthesis. | Designed for tractable, scalable synthesis. | SC hits are generally easier to optimize. |
| PAINS/Interference Risk | Can contain redox-active or polyphenolic PAINS [78]. | Contain classic PAINS from combinatorial chemistry [73]. | Both sources require vigilance; interference mechanisms may differ. |
Next-Generation Library Design and AI-Enhanced Filtering
The field is evolving from simple filters to sophisticated, enumerated libraries and AI-driven tools.
- Reaction-Driven Enumeration: Projects like the Pan-Canadian Chemical Library (PCCL) use novel academic chemistry to generate 148 billion virtual compounds, creating regions of chemical space that do not overlap with commercial libraries like Enamine REAL [79]. This represents a powerful strategy to access unprecedented scaffold diversity.
- AI-Powered Multidimensional Filtering: Tools like druglikeFilter 1.0 move beyond simple substructure matching. They use deep learning to collectively assess drug-likeness across four dimensions: physicochemical rules, toxicity alerts, binding affinity, and synthesizability [80].
- Strategic Curation: Best practice involves filtering for interference, lead-like properties, and target class relevance while actively incorporating natural product-inspired scaffolds and privileged structures to enhance diversity and biological relevance [12].
Essential Experimental Protocols for Hit Triage
Relying solely on computational filters is insufficient. The following experimental workflows are mandatory for validating screening hits and exculpating innocent scaffolds.
Objective: To experimentally distinguish truly promiscuous, interfering compounds ("bad" PAINS) from useful scaffolds unfairly flagged by filters. Workflow:
- In Silico Flagging: Identify hits containing PAINS substructures using public filters.
- Dose-Response Analysis: Confirm potency (IC50/EC50). Many PAINS show shallow or implausibly potent curves.
- Orthogonal Assay: Test activity in a fundamentally different assay technology (e.g., follow a fluorescence polarization hit with an SPR or functional cell-based assay). Loss of activity suggests interference.
- Covalent Reactivity Probe: For electrophile-containing suspects, incubate with a nucleophile like β-mercaptoethanol or glutathione (GSH) prior to assay. Activity loss suggests thiol-reactivity.
- Aggregation Testing: Measure dynamic light scattering (DLS) of the compound in assay buffer. Test for detergent reversal by adding non-ionic detergent (e.g., 0.01% Triton X-100); restored enzyme activity suggests aggregate-based inhibition.
- Advanced NMR: Perform an ALARM NMR or related assay to detect nonspecific protein binding or cysteine reactivity.
Diagram Title: Experimental "Fair Trial" Workflow for PAINS Suspect Triage
Objective: To identify compounds that act via redox cycling or metal chelation. Methods:
- Redox Cycling: Include a reducing agent like dithiothreitol (DTT) in the assay buffer. A significant reduction in the compound's inhibitory activity suggests redox interference. Alternatively, use a horseradish peroxidase/phenol red assay to directly detect H2O2 generation.
- Metal Chelation: For metalloenzyme targets, run the assay in the presence of excess exogenous metal ion (e.g., Zn²⁺, Mg²⁺). Restoration of activity suggests the compound acts by chelating the essential metal cofactor. For non-metallo targets, use a colorimetric chelation assay (e.g., with iron ferrozine).
The Scientist's Toolkit: Key Reagents for Interference Testing
Table: Essential Research Reagents for PAINS and Interference Investigation
| Reagent / Material | Primary Function in Triage | Typical Use Case / Assay |
|---|---|---|
| Non-ionic Detergent (Triton X-100, Tween-20) | Disrupts colloidal aggregates; distinguishes aggregate-based from specific inhibition. | Add at 0.01-0.1% v/v to assay buffer; reversal of inhibition is a positive sign of aggregation [73]. |
| Dithiothreitol (DTT) / β-Mercaptoethanol | Acts as a scavenging nucleophile; detects thiol-reactive covalent agents. Prevents redox cycling. | Pre-incubate compound with 1-5 mM DTT before assay; loss of activity indicates reactivity [75]. |
| Glutathione (GSH) | Biological nucleophile; detects electrophilic compounds that may react in cells. | Similar use to DTT; more biologically relevant [75]. |
| Metal Salts (e.g., ZnCl2, MgCl2) | Competes for chelation; identifies metal-chelating compounds. | Add excess metal ion (100-500 µM) to assay; reduced inhibition suggests chelation [75]. |
| Dynamic Light Scattering (DLS) Instrument | Measures hydrodynamic radius; directly detects compound aggregation in buffer. | Analyze compound at 10-50 µM in assay buffer; particles >100 nm indicate aggregation [75]. |
| ALARM NMR Reagents | Detects nonspecific protein binding and cysteine reactivity. | Specialized NMR-based assay using a labeled protein (e.g., LA protein) [75]. |
The pursuit of novel therapeutics hinges on the quality of the initial chemical libraries screened. This guide objectively compares the structural and physicochemical landscapes of purchasable compound libraries against the rich tapestry of natural product (NP) scaffolds, framing the analysis within the critical thesis of natural product scaffold diversity versus purchasable compound libraries research. While purchasable libraries offer vast, synthetically tractable collections, evidence suggests they underutilize the biologically pre-validated chemical space occupied by metabolites and natural products [81]. For instance, one analysis found a two-fold enrichment of metabolite scaffolds in approved drugs (42%) compared to typical lead libraries (23%), and only 5% of NP scaffold space is shared with current lead compounds [81]. This discrepancy highlights a potential opportunity: applying intelligent drug-likeness and lead-likeness filters can curate purchasable libraries that better capture the desirable complexity and biological relevance of NP space, thereby improving the probability of success in drug discovery campaigns [82] [81].
Methodological Framework for Comparative Analysis
The comparative data presented herein is derived from published studies that employ standardized computational methodologies to ensure an objective comparison [77] [81]. A key approach involves the creation of standardized subsets from large libraries to eliminate bias from differing molecular weight (MW) distributions [77]. In one major study, eleven purchasable libraries and the Traditional Chinese Medicine Compound Database (TCMCD) were processed: molecules were standardized, and an equal number of compounds were randomly selected from identical MW bins (100-700 Da) for each library, resulting in comparable subsets of 41,071 compounds each [77].
Structural and scaffold diversity was then quantified using multiple representations:
- Murcko Frameworks: The union of ring systems and linkers, defining core scaffolds [77].
- Scaffold Tree: A hierarchical decomposition of molecules providing Level 1 (simplified framework) and higher-level scaffolds [77].
- ECFP/FCFP Fingerprints: For assessing molecular diversity and similarity between datasets [81].
Physicochemical property analysis routinely extends beyond Lipinski's Rule of Five to include polar surface area (PSA), solubility (logS), and counts of rotatable bonds and rings [81]. The performance of libraries in virtual screening (VS) is evaluated using metrics like enrichment factor (the increase in hit rate over random selection) and the Tanimoto similarity to known actives [83].
Comparative Analysis of Library Characteristics
Physicochemical Property and Scaffold Diversity
The application of lead-like filters (typically MW < 350-450, LogP < 3-4) shapes the chemical space of libraries. A review of vendor offerings notes it is feasible to select approximately 500,000 lead-like compounds from commercial sources [54]. However, significant differences emerge in scaffold diversity.
Table 1: Scaffold Diversity Analysis of Standardized Compound Libraries [77]
| Library Name | Approx. Total Compounds (Source) | Distinct Murcko Frameworks (in 41k subset) | Notes on Diversity & Character |
|---|---|---|---|
| ChemBridge | ~1.06 million | High | Ranked as more structurally diverse than most purchasable libraries. |
| ChemicalBlock | ~126,000 | High | Selected, diverse library. |
| Mcule | ~4.92 million | High | Largest library in ZINC15; high structural diversity. |
| VitasM | ~1.46 million | High | Novel compounds with high diversity. |
| TCMCD (NP Database) | ~54,000 | Moderate (but Highest Complexity) | Contains the highest structural complexity (e.g., chiral centers, rings); more conservative scaffold set. |
| Enamine | ~1.96 million | Not Specified | Marketed as a lead-like, diverse library. |
| LifeChemicals | ~413,000 | Not Specified | Selected library. |
| Specs | ~212,000 | Not Specified | Selected library. |
Table 2: Physicochemical Profile of Biologically Relevant Datasets [81]
| Dataset | Avg. MW (Da) | Avg. logP | Avg. Rings | Avg. Rotatable Bonds | Key Scaffold Characteristic |
|---|---|---|---|---|---|
| Metabolites | ~265 | Low | Lowest | Moderate | Limited chemical space; high scaffold enrichment in drugs. |
| Natural Products | ~360 | Moderate | Highest | Highest | Vast, complex scaffold space; minimally sampled by leads. |
| Drugs | ~335 | Moderate | Moderate | Moderate | Scaffolds overlap with metabolites (42%) and NPs. |
| Lead Libraries | ~310 | Moderate | Moderate | Moderate | Scaffolds overlap poorly with metabolites (23%) and NPs (5%). |
Performance in Virtual Screening
The ultimate test of a curated library is its performance in identifying active compounds. Studies evaluating library quality using known actives show the impact of filter application.
Table 3: Virtual Screening Performance of a Curated Drug-like Library [83]
| Performance Metric | Result | Experimental Context |
|---|---|---|
| Actives Retrieved | 36% | Percentage of 5,847 external active compounds retrieved when screening a fingerprint-similarity curated library (Tanimoto cutoff = 0.75). |
| Enrichment Factor (EF) | 13 | Fold-increase over random selection in identifying actives, indicating high library quality. |
| Target Family Libraries | Constructed & Evaluated | Specific libraries for target families (e.g., GPCRs, kinases) were also built, demonstrating the focused application of filters. |
Detailed Experimental Protocols
Protocol 1: Constructing & Validating a Focused Screening Library
This protocol is adapted from studies that compiled and tested target-focused libraries [83].
- Source Compound Collection: Begin with a large collection of purchasable, drug-like molecules (e.g., ~20 million from ZINC [83]).
- Define Query Set: Assemble a set of known active molecules for a specific target or target family from databases like ChEMBL.
- Similarity-Based Curation: Perform a fingerprint-based similarity search (e.g., using ECFP4 fingerprints) of the source collection against the query set. Retain compounds above a defined Tanimoto similarity threshold (e.g., 0.75) [83].
- Apply Lead-Likeness Filters: Filter the resulting set using strict lead-like criteria (e.g., MW < 350, clogP < 3, rotatable bonds < 7) to ensure developability [84] [54].
- Apply Secondary Filters: Remove compounds with undesirable chemical functionalities using filters such as REOS (Rapid Elimination of Swill) and PAINS (Pan-Assay Interference Compounds) [8].
- Diversity Selection: Apply a clustering algorithm (e.g., based on Bemis-Murcko scaffolds or fingerprint clustering) to the filtered set and select a diverse subset for the final library [84] [8].
- Validation: Test the library's performance by running a virtual screen against a set of known external actives and decoys. Calculate the enrichment factor (EF) and plot recall rates to quantify library quality [83].
Protocol 2: Analyzing Scaffold Diversity of a Compound Library
This protocol details the comparative analysis of scaffold diversity across different libraries [77].
Data Standardization:
- Download library files from vendor websites or public databases.
- Standardize all molecules using pipeline software (e.g., Pipeline Pilot): fix bad valences, remove inorganics, add hydrogens, neutralize charges, and remove duplicates.
- Create a Standardized Subset: Analyze the MW distribution. For each 100 Da MW bin between 100-700 Da, identify the library with the fewest compounds. Randomly select that same number of compounds from every library in that bin. Combine the selections to create weight-standardized subsets for each library [77].
Scaffold Generation:
- Murcko Frameworks: Generate the Murcko framework for every molecule in the standardized subset using a tool like RDKit or Pipeline Pilot's "Generate Fragments" component [77].
- Scaffold Tree: Generate hierarchical Scaffold Tree decompositions (Level 1, Level 2, etc.) for each molecule using dedicated software (e.g., the
sdfragcommand in MOE) [77].
Diversity Quantification & Visualization:
- Count the number of unique scaffolds (Murcko and Level 1) in each library subset.
- Generate Tree Maps: Visualize the scaffold space by clustering molecules based on fingerprint similarity (e.g., ECFP4). Each rectangle in the map represents a cluster, with size proportional to the number of compounds [77].
- Generate SAR Maps: Visualize the structure-activity relationship landscape by overlaying property data (e.g., predicted activity) onto the scaffold Tree Map.
Table 4: Key Resources for Library Curation and Analysis
| Item Name | Function in Library Design/Analysis | Example/Source |
|---|---|---|
| ZINC Database | Primary public repository for purchasable compound structures and metadata for virtual screening. | https://zinc.docking.org [83] [77] |
| ChEMBL Database | Repository of bioactive molecules with drug-like properties, used as a source of known actives for training or validation. | https://www.ebi.ac.uk/chembl/ [81] |
| Pipeline Pilot | Workflow platform for automating compound standardization, descriptor calculation, and filtering. | Dassault Systèmes BIOVIA [77] |
| RDKit | Open-source cheminformatics toolkit for molecular fingerprinting, scaffold decomposition, and property calculation. | https://www.rdkit.org |
| PAINS/REOS Filters | Rule-based filters to identify and remove compounds with undesirable, promiscuous, or reactive substructures. | Implemented in RDKit or commercial software [8] |
| druglikeFilter 1.0 | An AI-powered tool that collectively evaluates drug-likeness across physicochemical rules, toxicity, affinity, and synthesizability. | https://idrblab.org/drugfilter/ [80] |
| Generative AI (VAE) Workflow | Advanced generative model integrated with active learning to design novel, synthesizable, drug-like molecules for specific targets. | As described in [85] |
| DNA-Encoded Library (DEL) Technology | Ultra-high-throughput screening platform where billions of compounds linked to DNA barcodes are screened in a single tube. | Amgen platform [86] |
Workflow and Strategy Visualization
Workflow for Hierarchical Virtual Screening Library Curation
Hierarchical Curation of a VS Library
Strategic Comparison: NP-Inspired vs. Purchasable Library Design
NP vs Purchasable Library Design Strategy
This comparison guide underscores a central tension in modern library design: the broad synthetic accessibility and IP freedom of purchasable libraries versus the biologically relevant complexity and scaffold diversity of natural products. Data confirms that typical lead libraries capture only a small fraction of NP and metabolite scaffold space, which is disproportionately represented in successful drugs [81]. The strategic application of progressive filters—from basic drug-likeness to advanced, AI-powered multi-parameter assessments—is essential for curating quality from commercial collections [83] [80]. The most promising path forward lies in a hybrid approach: using NP-inspired design principles, such as attention to 3D shape, fraction of sp3 carbons (Fsp3), and privileged scaffold motifs, to inform the filtering and selection of purchasable compounds [82] [8]. Furthermore, emerging technologies like DNA-encoded libraries (DELs) and generative AI active learning workflows are revolutionizing library construction, enabling the direct exploration of vast, novel chemical spaces that deliberately incorporate desired lead-like and NP-like properties [86] [85]. Ultimately, curating for quality demands a nuanced strategy that leverages the strengths of both synthetic and natural chemical space to build libraries primed for discovery.
The declining pipeline of New Chemical Entities (NCEs) is a well-documented crisis in drug discovery. While natural products and their derivatives have historically formed the cornerstone of pharmacotherapy, accounting for approximately one-third of all FDA-approved small molecules [63], traditional bioassay-guided discovery often leads to the frequent rediscovery of known compounds [87]. This bottleneck exists alongside a paradoxical abundance of biosynthetic potential. Genomic sequencing has revealed that a single Streptomyces genome typically encodes 25–50 Biosynthetic Gene Clusters (BGCs), yet under standard laboratory conditions, ~90% of these BGCs remain transcriptionally silent or "cryptic" [88]. This vast reservoir of unexpressed genetic material represents a significant untapped source of novel chemical scaffolds.
The imperative to "reactivate" these silent BGCs is framed within a critical thesis: natural product scaffolds offer unparalleled chemical diversity compared to synthetic, purchasable compound libraries. Analyses demonstrate that natural products and their derived libraries occupy distinct and more complex regions of chemical space, featuring greater stereochemical complexity, a higher number of sp³-hybridized carbons, and more varied ring systems [63]. In contrast, while purchasable libraries offer millions of compounds, their scaffolds can be more conservative and less diverse [77]. Therefore, activating silent BGCs is not merely a technical challenge but a strategic necessity to access this privileged chemical space and discover new leads for overcoming antimicrobial resistance, cancer, and other diseases [87] [89].
Comparison Guide: Strategic Approaches to BGC Reactivation
Activation strategies can be broadly categorized into two paradigms: in situ activation within the native host and heterologous expression in an engineered chassis. The choice of strategy depends on the genetic tractability of the native organism, the size and complexity of the target BGC, and the project's specific goals. The following table provides a high-level comparison of the core strategic families.
Table 1: Strategic Comparison of BGC Reactivation Approaches
| Strategy | Core Principle | Key Advantages | Primary Limitations | Typical Experimental Readout |
|---|---|---|---|---|
| In Situ Activation | Manipulate the native host's physiology or genetics to induce expression. | Preserves native regulatory & maturation context; suitable for large, complex BGCs. | Limited by host tractability; can trigger multiple BGCs complicating analysis. | Comparative metabolomics (LC-MS/NMR) of treated vs. control cultures. |
| Heterologous Expression | Clone and express the BGC in a genetically amenable host (e.g., S. albus, S. coelicolor). | Bypasses host-specific repression; enables focused study of a single BGC. | Technically challenging for very large (>100 kb) BGCs; possible lack of essential host factors. | Detection of target compound(s) in chassis host absent in empty control. |
| One Strain Many Compounds (OSMAC) | Systematic variation of cultivation parameters (media, salinity, aeration). | Simple, low-tech, and high-throughput; can elicit diverse metabolites from one strain. | Empirical and unpredictable; requires extensive screening. | Metabolic profiling under each condition to identify novel peaks. |
| Co-cultivation | Cultivate the target strain with another microbe to simulate ecological competition. | Mimics natural triggers; can activate defense-related BGCs. | Complex and poorly reproducible; mechanism often unknown. | Unique compounds produced only in co-culture, identified via metabolomics. |
The logical relationship and workflow integration of these strategies are visualized in the following decision pathway.
Strategic Decision Pathway for BGC Activation (Max Width: 760px)
Experimental Protocols and Supporting Data
The successful implementation of the strategies outlined above relies on robust, reproducible experimental protocols. Below are detailed methodologies for three foundational approaches.
Protocol 1: OSMAC (One Strain Many Compounds) Screening
- Objective: To induce the expression of silent BGCs by altering the physicochemical cultivation environment of the native producer strain.
- Materials: Target microbial strain, diverse culture media (e.g., ISP2, R2A, Marine Broth, minimal media with varying C/N sources), 250 mL flasks or deep-well plates, incubation shaker, LC-MS system for analysis.
- Procedure:
- Inoculate the target strain from a master stock into 5-10 different liquid media with varying compositions (carbon source, nitrogen source, salinity, pH).
- Incubate under standard conditions (e.g., 28°C, 200 rpm) for an initial growth period (e.g., 3 days).
- For each condition, transfer an aliquot (e.g., 1% v/v) into fresh medium of the same type to establish secondary metabolite production cultures. Incubate further (e.g., 3-7 days).
- Extract metabolites from the whole broth or supernatant using a standardized solvent system (e.g., ethyl acetate or 1:1 butanol:methanol).
- Analyze all extracts using High-Resolution Liquid Chromatography-Mass Spectrometry (HR-LC-MS). Use software (e.g., MZmine, XCMS) to align chromatograms and identify peaks unique to specific culture conditions.
- Supporting Data: A landmark study applying OSMAC to the marine fungus Spicaria elegans under 10 conditions resulted in the isolation of eight previously unknown compounds, including the novel spicochalasin A, demonstrating the power of this simple approach [87].
Protocol 2: Bipartite Co-cultivation for Metabolite Induction
- Objective: To activate silent BGCs by simulating microbial competition or interaction.
- Materials: Target strain and inducing partner strain (often from the same ecological niche), suitable solid and liquid media, permeable membranes or dual-compartment plates (e.g., IVA plates), analytical instruments.
- Procedure:
- Co-culture the two strains on solid agar, allowing physical contact.
- Alternatively, use a compartmentalized system where strains share the gaseous headspace or are separated by a semi-permeable membrane, allowing exchange of diffusible signals but not cells.
- Incubate for an extended period (e.g., 7-14 days).
- Extract metabolites from the entire co-culture plate or from each compartment separately.
- Analyze extracts via HR-LC-MS and compare the metabolic profile to those of each strain cultured in isolation. Look for "cross-talk" peaks present only in the co-culture system.
- Supporting Data: This approach is central to projects like the USDA study on suppressive soil microbiomes, where microbial consortia from soil are co-cultured with nematodes to identify species interactions that induce nematicidal metabolites [90]. It is a key method for mimicking ecological triggers.
Protocol 3: Heterologous Expression via TAR Cloning
- Objective: To clone and express a large, silent BGC in a model heterologous host.
- Materials: Genomic DNA from donor strain, yeast (Saccharomyces cerevisiae) host, TAR cloning vector with targeting "hooks," transformation reagents, selective media, PCR reagents, sequencing services.
- Procedure:
- Design and synthesize linear TAR vector arms with 40-60 bp homology sequences matching the 5' and 3' ends of the target BGC.
- Co-transform the linearized vector and high-molecular-weight genomic DNA from the donor into competent yeast cells.
- Plate on selective media to select for yeast colonies containing the recombined plasmid.
- Screen colonies by PCR to confirm correct capture of the entire BGC.
- Isolate the plasmid from yeast and transform it into an E. coli host for propagation.
- Finally, conjugate or transform the plasmid into the heterologous Streptomyces expression host (e.g., S. albus J1074).
- Culture the exconjugant and analyze extracts for metabolite production compared to the host containing an empty vector.
- Supporting Data: The TAR-based pCAP01 system has been successfully used to clone and express BGCs from marine Streptomyces [88]. The related mCRISTAR platform enabled the activation of the tetarimycin BGC by simultaneously replacing multiple promoters with an efficiency of 68% for six promoters [88].
The experimental workflow from strategy selection to compound identification integrates these protocols, as shown below.
BGC Activation and Metabolite Discovery Workflow (Max Width: 760px)
The Scaffold Diversity Thesis: Natural Products vs. Purchasable Libraries
The driving thesis for exploring silent BGCs is the superior and unique chemical space occupied by natural products. A comparative analysis of scaffold diversity provides quantitative support.
Table 2: Scaffold Diversity Analysis of Select Compound Libraries [77]
| Library Name | Total Compounds (Standardized Subset) | Number of Unique Murcko Frameworks | Scaffold Diversity (Frameworks per 1k Cpds) | Notable Features |
|---|---|---|---|---|
| Traditional Chinese Medicine DB (TCMCD) | 54,138 | 5,412 | ~100.0 | Highest structural complexity; conservative core scaffolds. |
| ChemBridge | 1,064,425 | 38,117 | ~35.8 | High structural diversity; "drug-like" focus. |
| Mcule | 4,876,889 | 112,405 | ~23.0 | Largest library; moderate scaffold frequency. |
| LifeChemicals | 412,788 | 9,856 | ~23.9 | Selected, lead-like compounds. |
| Maybridge | 57,490 | 1,955 | ~34.0 | Highly diverse, historically used in HTS. |
Key Analysis: The data shows that while large commercial libraries (Mcule) contain more absolute unique scaffolds, their density of diversity (frameworks per 1,000 compounds) is lower. The natural product-derived TCMCD library has a significantly higher density, confirming that natural products explore chemical space more efficiently. Importantly, >70% of the scaffolds in commercial libraries are not found in natural products, and vice versa, indicating they are complementary sources [63]. Silent BGCs offer access to the entirely unexplored fraction of natural product space, potentially yielding scaffolds with novel protein-binding properties and bioactivity.
Complementary Chemical Space of Natural vs. Synthetic Compounds (Max Width: 760px)
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful BGC reactivation research requires specialized tools and platforms. The following table details key resources.
Table 3: Essential Toolkit for BGC Reactivation Research
| Tool/Reagent Category | Specific Example(s) | Function in Research |
|---|---|---|
| Bioinformatics Platforms | antiSMASH, PRISM, MIBiG | Predict & annotate BGCs from genomic data; compare to known clusters. |
| Cloning & Engineering Systems | TAR (pCAP01), CRISPR-Cas9 (CATCH/mCRISTAR), Red/ET (ExoCET) | Isolate, clone, and refactor large DNA fragments (>50 kb) for heterologous expression. |
| Heterologous Chassis Strains | Streptomyces albus J1074, S. coelicolor M1146, S. lividans TK24 | Clean genetic backgrounds optimized for expression of secondary metabolite BGCs. |
| Metabolomics & Analytics | High-Resolution LC-MS/MS, GNPS (Global Natural Products Social) | Detect, profile, and dereplicate metabolites; identify novel compounds via molecular networking. |
| Cultivation Tools | Microfluidic chips, 24-well Duetz plates, osmotic membrane chambers | Enable high-throughput OSMAC and co-cultivation screens under controlled conditions. |
| Inducing Agents | Small molecule elicitors (e.g., N-acetylglucosamine), histone deacetylase inhibitors (for fungi) | Chemically induce silent BGCs by perturbing global or specific regulatory pathways. |
The strategic reactivation of silent biosynthetic gene clusters represents a frontier in natural product discovery, directly addressing the critical need for novel chemical scaffolds in drug development. As comparative analyses confirm, the structural diversity offered by natural products remains distinct from and complementary to that of vast purchasable libraries [77] [63]. The experimental strategies outlined—from simple OSMAC variations to sophisticated heterologous expression platforms—provide a robust methodological framework for researchers. Continued advancement in this field, powered by the integration of genomics, synthetic biology, and analytical chemistry, is essential for translating the silent genomic potential of microorganisms into the next generation of therapeutic leads.
The initial screening library is a critical determinant of success in early drug discovery. Its design embodies a core trilemma: maximizing structural diversity to explore chemical space, maintaining a manageable physical size to constrain costs and timelines, and ensuring sufficient screening hits to initiate lead optimization [12]. This challenge is framed by two dominant but philosophically distinct paradigms: libraries derived from natural products (NPs) and libraries of purchasable synthetic compounds [53] [63].
Natural products offer unparalleled scaffold complexity and evolutionary-validated bioactivity, but their libraries are often hampered by structural redundancy, supply challenges, and high screening costs per compound [53] [47]. In contrast, purchasable synthetic libraries, often sourced from aggregators, provide millions of readily available, drug-like molecules but may occupy a narrower, more conservative region of chemical space [12]. This guide objectively compares these approaches and the modern computational and analytical strategies designed to optimize their inherent trade-offs, contextualized within the ongoing research to harness NP-like diversity within more tractable screening paradigms [91].
Comparative Analysis of Screening Library Paradigms
The choice between natural product and purchasable compound libraries involves strategic trade-offs across diversity, cost, and practical logistics. The following tables provide a data-driven comparison.
Table 1: Library Composition & Diversity Profile
| Characteristic | Natural Product (Microbial) Libraries | Purchasable Synthetic Libraries (e.g., Enamine REAL) |
|---|---|---|
| Source & Size | Fungi, bacteria; limited by collection & cultivation. Libraries of hundreds to thousands of extracts [47]. | Commercial synthesis; ultra-large libraries of billions of make-on-demand compounds [33] [67]. |
| Chemical Diversity | High scaffold complexity, stereochemical richness, and macrocyclic structures. Exhibits "islands of diversity" with high intra-cluster similarity [53]. | High count of unique structures, but often clustered in "drug-like" space defined by rules (e.g., Lipinski's). Breadth can be vast but potentially less deep in unique scaffold classes [12]. |
| Redundancy | High structural redundancy; e.g., 82.6% of molecules in a 36,454-compound database fell into similarity clusters [53]. | Can be curated to minimize redundancy, but large libraries contain many similar analogues. |
| Dereplication Need | Critical and challenging; requires LC-MS/MS and molecular networking to identify known compounds early [47] [63]. | Straightforward; compound structures are known from the outset. |
| Typical Format for Screening | Crude or prefractionated extracts, introducing complexity and potential for interference [47]. | Pure, solubilized compounds. |
Table 2: Screening Cost & Efficiency Metrics
| Metric | Natural Product Libraries | Purchasable Synthetic Libraries |
|---|---|---|
| Upfront Library Curation Cost | High: involves sample collection, fermentation, extraction, and chemical characterization [63]. | Low to moderate: purchasing cost per compound, but no synthesis R&D for the end user. |
| Cost per Screening Data Point | High for pure compounds (isolation cost). Lower for extract screening, but hits require costly subsequent isolation [47]. | Relatively low and predictable for physical screening. Computational pre-screening is extremely low cost per compound. |
| Hit Rate | Historically high due to bio-relevant scaffolds. Can be significantly increased by reducing redundancy; e.g., from 2.57% to 8.00% for a target enzyme after rational library pruning [47]. | Typically low (often <0.1%) for random HTS. Can be greatly enriched by virtual screening and AI prioritization [92] [12]. |
| Key Cost-Saving Strategy | Pre-screening LC-MS/MS analysis to create minimal diverse libraries (e.g., 84.9% size reduction) [47]. | Computational virtual screening to prioritize a small subset for purchase and testing (e.g., active learning) [33] [92]. |
| Time from Hit to Lead | Long: due to need for hit deconvolution from extracts, re-isolation, and structure elucidation [63]. | Short: structure is known, and analogues are often readily available for purchase. |
Experimental Protocols for Optimizing Library Efficiency
Protocol 1: Rational Minimization of Natural Product Extract Libraries Using Mass Spectrometry This protocol details a method to drastically reduce library size while preserving chemical diversity and bioactive potential [47].
- Data Acquisition: Analyze all extracts in the library using untargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS).
- Molecular Networking: Process MS/MS data through GNPS (Global Natural Products Social Molecular Networking) to group spectra based on fragmentation similarity, creating nodes representing molecular scaffolds or closely related analogues [47].
- Scaffold-Centric Selection: Use custom algorithms (e.g., in R) to select extracts iteratively. a. First, select the extract contributing the highest number of unique scaffold nodes. b. Iteratively add the extract that adds the most new scaffold nodes not yet represented in the growing subset.
- Stopping Point: Continue until a pre-defined percentage of total scaffold diversity (e.g., 80%, 95%) from the full library is captured. This creates a "minimal rational library."
- Validation: Screen both the full library and the rational library against biological targets. The method demonstrates not only retained bioactivity but increased hit rates due to reduced redundancy [47].
Protocol 2: Active Learning-Driven Prioritization from Ultra-Large Purchasable Libraries This protocol uses the FEgrow software and active learning to efficiently search billion-compound spaces for a given protein target [33].
- Initialization: Start with a protein structure and a known bound ligand or fragment. Define this as the core scaffold.
- Virtual Library Generation: Connect a library of purchasable R-groups and linkers to the core using FEgrow's automated workflow, generating millions of virtual molecules [33].
- Active Learning Cycle: a. Batch Scoring: A small, randomly selected initial batch of virtual compounds is built into the protein binding pocket and scored using a fast docking function (e.g., gnina) [33]. b. Model Training: These scored compounds are used to train a machine learning model (e.g., a Gaussian Process model) to predict scores for the entire unexplored virtual library. c. Informed Selection: The model selects the next batch of compounds, focusing on regions of chemical space predicted to be high-scoring or uncertain. d. Iteration: Steps a-c are repeated. The model improves with each cycle, efficiently converging on high-scoring candidates.
- Purchase & Testing: The top-ranked compounds from the final cycle are purchased from on-demand vendors (e.g., Enamine) and tested experimentally [33].
Protocol 3: AI-Accelerated Virtual Screening of Multi-Billion Compound Libraries This protocol employs the OpenVS platform for large-scale structure-based screening [92].
- Receptor and Library Preparation: Prepare the 3D structure of the target protein. Access a multi-billion compound library in a suitable format (e.g., from ZINC).
- Two-Tier Docking with Active Learning: a. Express Screening (VSX Mode): Use a fast, initial docking pass (RosettaVS VSX) for a large subset of the library. This mode uses a rigid receptor for speed [92]. b. Focused, High-Precision Screening (VSH Mode): A machine learning model, trained on-the-fly from VSX results, actively selects the most promising compounds for high-precision docking with flexible receptor side chains (RosettaVS VSH) [92].
- Ranking and Selection: Compounds are ranked by their predicted binding affinity from the VSH stage. The top-ranked compounds are selected for purchase and experimental validation.
Visualization of Workflows and Strategic Relationships
Diagram 1: Strategic Framework for Balancing Screening Library Trade-offs
Diagram 2: Workflow for Rational Natural Product Library Minimization [47]
Diagram 3: Active Learning Cycle for Screening Ultra-Large Libraries [33]
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Resources for Advanced Library Screening Campaigns
| Tool / Resource | Function in Library Management & Screening | Relevance to Trade-off Balance |
|---|---|---|
| GNPS (Global Natural Products Social Molecular Networking) | Cloud-based platform for analyzing MS/MS data to visualize molecular families and scaffold similarity in complex NP extracts [47]. | Enables rational NP library minimization by quantifying scaffold redundancy and diversity. Directly addresses the size-diversity trade-off. |
| ZINC / Enamine REAL Databases | Public (ZINC) and commercial (Enamine) databases cataloging hundreds of millions to billions of purchasable, make-on-demand compounds for virtual screening [93] [67]. | Provides the raw chemical space for virtual screening. Cost-effective exploration of vast diversity without physical synthesis or storage. |
| FEgrow with Active Learning | Open-source software for growing R-groups on a core scaffold in a protein pocket, integrated with active learning for efficient chemical space search [33]. | Reduces computational cost of screening ultra-large virtual libraries by orders of magnitude, managing the cost-diversity trade-off. |
| OpenVS / RosettaVS Platform | Open-source, AI-accelerated virtual screening platform featuring fast (VSX) and high-precision (VSH) docking modes with active learning [92]. | Enables practical structure-based screening of billion-compound libraries on HPC clusters, balancing accuracy with computational expense. |
| jamdock-suite | Suite of Bash scripts automating a local virtual screening pipeline with AutoDock Vina, from library preparation to result ranking [93]. | Lowers the barrier to entry for computational screening, reducing time and expertise cost for hit identification from purchasable libraries. |
| Generative AI Models (e.g., FREED, DeepFrag) | AI models that generate novel molecular structures conditioned on target protein pockets or desired properties [91]. | Bridges NP and synthetic spaces by proposing novel, NP-inspired scaffolds or optimizing leads, expanding accessible diversity. |
| LC-MS/MS with High Resolution | Analytical instrumentation for the metabolomic profiling of NP extract libraries [47] [63]. | Foundational for characterizing and dereplicating NP libraries, the essential first step in rationalizing them. |
Future Outlook: Integrating Paradigms with AI
The future of managing library trade-offs lies in the convergence of NP inspiration and synthetic accessibility, accelerated by artificial intelligence. AI-driven generative models can now design novel compounds that mimic the complex structural features of natural products while adhering to synthetic feasibility rules [91]. Furthermore, active learning protocols can navigate the combined space of natural product-derived scaffolds and purchasable building blocks, efficiently proposing hybrid molecules [33] [92]. This points toward a hybrid screening paradigm: using minimal, diversity-maximized NP libraries for initial, broad-scope discovery, and leveraging AI-powered virtual screening of vast synthetic spaces for targeted optimization and scaffold hopping. This integrated approach promises a more sustainable balance, leveraging the unique strengths of each paradigm to mitigate their inherent limitations.
Data-Driven Decisions: Comparative Analysis of Scaffold Diversity and Biological Relevance
The search for novel bioactive compounds in drug discovery is fundamentally a pursuit of chemical diversity. This pursuit is framed by a critical comparison between two principal sources: natural products (NPs), shaped by billions of years of evolutionary selection, and purchasable compound libraries, designed and synthesized through modern medicinal and combinatorial chemistry [1]. Natural products are celebrated for their unparalleled structural complexity, diverse stereochemistry, and high success rate as drug leads, partly because they have evolved to interact optimally with biological macromolecules [1] [39]. In contrast, purchasable libraries offer vast numbers of readily accessible, often drug-like compounds, yet they have been criticized for limited structural diversity and an over-reliance on flat, aromatic scaffolds [1] [25].
This guide objectively compares these two strategic resources within the broader thesis that natural products explore a broader and more biologically relevant region of chemical space. We quantify this by analyzing key metrics for scaffold and structural complexity—such as Murcko frameworks, scaffold trees, and network diversity—supported by experimental data from comparative studies and synthetic campaigns [25] [39]. For researchers and drug development professionals, understanding these metrics is not academic; it directly informs library selection for high-throughput or virtual screening, guides the design of targeted libraries, and shapes strategies for hit discovery and lead optimization [94] [25].
Key Diversity Metrics and Comparative Analysis
Quantifying chemical diversity requires moving beyond simple compound counts to analyze the underlying structural frameworks, or scaffolds, that define a library's true coverage of chemical space. Several hierarchical and quantitative methods have been established for this purpose.
- Murcko Frameworks: This method dissects a molecule into rings, linkers, and side chains. The Murcko framework is defined as the union of all ring systems and the linkers that connect them, providing a simplified core structure for classification and diversity analysis [25].
- Scaffold Trees: A more systematic hierarchy proposed by Schuffenhauer et al., where molecules are iteratively deconstructed by pruning peripheral rings according to a set of rules until a single ring remains. Each level of the tree (Level 0 to Level n) represents a different abstraction of the original molecule, with Level n-1 typically corresponding to the Murcko framework. This allows for a nuanced analysis of scaffold relationships and frequency [25].
- Network Diversity Score (NDS): Borrowed from complexity science, this metric evaluates the diversity of topological patterns (e.g., star, tree, small-world) within a combinatorial network. Applied to chemistry, a library's structural complexity can be approximated by the diversity of interconnected substructure motifs it contains. A higher NDS indicates greater informational complexity and less redundancy [95].
A pivotal comparative study analyzed eleven major purchasable libraries (e.g., Mcule, ChemBridge, Enamine) and the Traditional Chinese Medicine Compound Database (TCMCD) as a representative natural product-derived collection [25]. After standardizing for molecular weight, the study used Murcko frameworks and Level 1 scaffolds to assess diversity.
Table 1: Comparative Scaffold Diversity Analysis of Compound Libraries [25]
| Library Name | Type | # Unique Murcko Frameworks | PC50C (Murcko) | PC50C (Level 1) | Notable Structural Feature |
|---|---|---|---|---|---|
| TCMCD | Natural Product-Derived | 4, 921 | 1.6% | 2.9% | Highest structural complexity, conservative scaffolds |
| ChemBridge | Purchasable | 6, 018 | 1.9% | 3.1% | High scaffold diversity |
| Mcule | Purchasable | 5, 887 | 2.0% | 3.3% | High scaffold diversity |
| VitasM | Purchasable | 5, 245 | 2.1% | 3.5% | High scaffold diversity |
| Enamine | Purchasable | 5, 502 | 2.8% | 4.5% | Moderate scaffold diversity |
| ChemDiv | Purchasable | 4, 876 | 3.5% | 5.8% | Lower scaffold diversity |
Note: PC50C is the percentage of scaffolds required to cover 50% of the molecules in a library. A lower PC50C indicates greater scaffold diversity, as fewer unique scaffolds account for half the collection.
The data reveals a key insight: while the TCMCD natural product library possesses the highest measured structural complexity, some top-tier purchasable libraries (ChemBridge, Mcule) can achieve comparable or even higher counts of unique Murcko scaffolds [25]. However, the PC50C metric tells a more nuanced story. Natural product-derived libraries and the most diverse commercial libraries have very low PC50C values, meaning their populations are spread across a wide array of scaffolds without heavy dominance by a few common cores.
Experimental Protocols for Diversity Analysis and Library Synthesis
Protocol for Scaffold Diversity Analysis
The standard workflow for comparative scaffold analysis, as used in the study above, involves [25]:
- Library Standardization: Download SDF files from vendor databases or natural product repositories. Preprocess using cheminformatics pipelines (e.g., Pipeline Pilot, KNIME) to fix valences, remove duplicates, add hydrogens, and filter inorganics. To ensure a fair comparison, create standardized subsets by randomly selecting compounds from each library to match an identical molecular weight distribution (e.g., 100-700 Da in 100 Da bins).
- Scaffold Generation:
- Murcko Frameworks: Use the "Generate Fragments" component in Pipeline Pilot or equivalent functions in RDKit or MOE.
- Scaffold Tree Hierarchies: Generate using the
sdfragcommand in MOE or dedicated scripts implementing the Schuffenhauer rules. Reconstruct the hierarchy from Level 0 (single ring) to Level n (original molecule).
- Diversity Quantification: For each representation (Murcko, each Scaffold Tree level), remove duplicate scaffolds. Generate Cumulative Scaffold Frequency Plots (CSFPs) by sorting scaffolds by frequency and plotting the cumulative percentage of molecules covered. Calculate the PC50C metric from these curves.
- Visualization: Use Tree Map software to visualize the distribution and structural similarity of major scaffolds (e.g., Level 1) within each library, where rectangle size represents scaffold frequency.
Protocol for Natural Product-Inspired Library Synthesis
Recent advances focus on diversifying complex natural product cores into new chemical space. A general two-phase strategy for creating polycyclic scaffolds with under-represented medium-sized rings (7-11 members) involves [39]:
- Core Selection & C–H Functionalization: Select a polycyclic natural product core (e.g., a steroid like dehydroepiandrosterone). Employ site-selective C–H oxidation methods (e.g., electrochemical, copper-mediated, or chromium-mediated oxidation) to install new oxygenated functional groups (alcohols, ketones) at previously inaccessible positions.
- Ring Expansion Diversification: Use the newly introduced or native functional groups as handles for ring expansion reactions.
- Schmidt Reaction: React a ketone with sodium azide/acid to expand a ring by one carbon, forming a lactam.
- Formal [2+2] Cycloaddition-Fragmentation: React a β-keto ester with dimethyl acetylenedicarboxylate (DMAD) to effect a two-carbon ring expansion.
- Acylation/Ring Expansion: Treat a cyclic ketone with a diazoester under Lewis acid catalysis to insert a carbonyl-containing chain.
- Library Production & Characterization: Execute parallel synthesis from multiple natural product starting materials and diversification points. Purify all compounds and characterize by NMR and HRMS. Analyze the resulting library's chemoinformatic properties (3D shape, fraction of sp3 carbons, principal component analysis) to confirm occupancy of unique chemical space compared to standard commercial libraries [39].
Diagram 1: Workflow for NP-inspired library synthesis. This two-phase strategy diversifies natural product cores into underexplored chemical space [39].
Case Studies in Diversity Analysis
Case Study 1: Natural Products as Pathway Probes and Drugs
Natural products often possess the unique structural complexity required to modulate challenging biological targets like protein-protein interfaces. Their scaffold diversity translates directly into diverse and potent bioactivities [1].
- FTY720 (Fingolimod): A synthetic analogue of the fungal metabolite myriocin, approved for multiple sclerosis. It is phosphorylated in vivo to become an agonist of sphingosine-1-phosphate (S1P) receptors. This activity causes lymphocyte sequestration, effectively modulating the immune system through a macromolecular signaling pathway [1].
- TNP-470: A synthetic analogue of fumagillin, it potently inhibits angiogenesis by covalently binding to methionine aminopeptidase 2 (MetAP2), a target validated in endothelial cell proliferation [1].
- Macrocyclic Modulators: Compounds like cyclosporine A, rapamycin, and epothilone B are natural product macrocycles that successfully target protein-protein interactions (calcineurin/cyclophilin, mTOR/FKBP12) or complex binding sites (tubulin dimer interface), areas often intractable for small, flat synthetic molecules [1].
Diagram 2: Simplified FTY720-P signaling pathway. The phosphorylated drug modulates immune cell trafficking via S1P receptor agonism [1].
Case Study 2: Computational Optimization of Purchasable Libraries
To address the diversity limitations of purchasable libraries, computational methods like BonMOLière have been developed [94]. This approach optimizes small to medium-sized libraries (1,000–15,000 compounds) for maximal hit potential against arbitrary targets by:
- Filtering: Starting from millions of "in-stock" compounds in ZINC20, apply stringent filters for drug-likeness, reactivity, and pan-assay interference (PAINS) to create a clean candidate pool.
- Target Prediction: Use a validated 2D similarity model to predict likely protein targets for each compound against a broad ChEMBL-based target set.
- Evolutionary Optimization: Employ a genetic algorithm to select a final compound subset that maximizes a "fitness function" combining broad target coverage, target novelty, and desirable physicochemical properties.
This method reported a calculated +60% to +184% improvement in library "fitness" over random selection, demonstrating that intelligent design can significantly enhance the functional diversity and efficiency of purchasable screening decks [94].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents, Databases, and Software for Diversity Analysis
| Item | Type | Primary Function in Diversity Research | Key Feature / Example |
|---|---|---|---|
| ZINC Database | Public Database | Primary source for purchasable compound structures, vendors, and property data [94] [25]. | Contains over 100 million compounds, with subsets like "in-stock" and "anodyne" (clean) for library building [94]. |
| Pipeline Pilot / MOE / RDKit | Cheminformatics Software | Used for molecular standardization, scaffold generation (Murcko, RECAP), and property calculation [25]. | Pipeline Pilot's "Generate Fragments" component is standard for Murcko framework analysis. |
| Scaffold Tree Algorithm | Computational Method | Hierarchically deconstructs molecules to analyze scaffold relationships and frequency [25]. | Implemented in MOE (sdfrag) or custom scripts; essential for calculating PC50C. |
| Tree Map / SAR Map Software | Visualization Tool | Visualizes the distribution and structural similarity of dominant scaffolds in a library [25]. | Provides an intuitive, spatial map of chemical space coverage. |
| C–H Oxidation Reagents | Chemical Reagents | Enable site-selective functionalization of natural product cores for diversification [39]. | Includes electrochemical set-ups, copper catalysts (e.g., Cu(OTf)₂), and chromium-based oxidants. |
| Ring Expansion Reagents | Chemical Reagents | Transform functionalized cores into novel scaffolds with medium-sized rings [39]. | Includes diazo compounds (e.g., ethyl diazoacetate), azides (for Schmidt reaction), and DMAD. |
Implications for Drug Discovery
The quantitative analysis of scaffold diversity has direct, practical implications:
- Library Selection for Screening: For phenotypic or target-agnostic screens, libraries with low PC50C and high structural complexity (like NP-inspired or top-tier commercial libraries) offer a higher probability of hitting diverse biological targets. For focused kinase or GPCR screens, libraries enriched with relevant privileged scaffolds may be more efficient [25].
- Informing Library Design: The dominance of a few simple scaffolds in many commercial libraries highlights a "synthesis bias." This underscores the value of integrating complex, sp3-rich natural product-derived scaffolds—through either isolation, purification, or inspired synthesis—to access more biologically relevant chemical space [1] [39].
- Future Directions: The convergence of computational optimization (like BonMOLière) and advanced synthetic strategies for NP diversification represents a powerful future path. This hybrid approach aims to create smaller, smarter, and more diverse screening collections that blend the synthetic accessibility of commercial compounds with the structural richness and biological validation of natural products [94] [39].
The systematic exploration of chemical space is a foundational pillar of modern drug discovery. At the heart of this endeavor lies the concept of scaffold diversity—the variety of core ring systems and molecular frameworks within a compound collection. A diverse library increases the probability of identifying novel hits against biologically relevant targets and provides a broader foundation for subsequent medicinal chemistry optimization [12]. This guide provides a direct, data-driven comparison of scaffold diversity between two critical sources of chemical matter: large-scale purchasable commercial libraries and natural product (NP) databases.
This comparison is framed within a critical thesis: while synthetic commercial libraries offer unparalleled accessibility and "drug-like" property tuning, natural products and their derivatives represent a distinct and evolutionarily refined region of chemical space characterized by unparalleled structural complexity and scaffold novelty [96]. The integration of NPs, either directly or through NP-inspired design, is increasingly seen as a strategic imperative to overcome high attrition rates in late-stage development by accessing more biologically relevant chemotypes [11] [12].
Quantitative Scaffold Diversity Rankings
The following tables summarize key scaffold diversity metrics for prominent natural product databases and commercial libraries, based on standardized cheminformatic analyses.
Table 1: Scaffold Diversity of Major Natural Product Databases & Generated Libraries
| Database / Library | Total Compounds | Analyzed Subset or Fragments | Key Scaffold Diversity Metric | Value | Reference / Notes |
|---|---|---|---|---|---|
| COCONUT (Collection of Open NPs) | >695,133 NPs | 2,583,127 fragments | Number of derived fragments | 2.58 M | Fragment library for broad chemical space analysis [45]. |
| Natural Products Atlas (Microbial) | 36,454 compounds | 36,454 compounds | Number of similarity clusters (Dice ≥0.75) | 4,148 | 82.6% of compounds clustered; median cluster size = 3 [53]. |
| LANaPDB (Latin America NP DB) | 13,578 NPs | 74,193 fragments | Number of derived fragments | 74,193 | Focused regional diversity [45]. |
| AI-Generated NP-Like Database | 67,064,204 compounds | 67,064,204 compounds | Expansion over known NPs | ~165x | Generated via ML on NP structures; novel scaffold exploration [97]. |
| SuperNatural Database (Purchasable NPs) | ~50,000 compounds | ~50,000 compounds | Number of NPs identical to drugs | 289 | Focus on commercially available NPs [96]. |
Table 2: Scaffold Diversity of Select Purchasable Commercial Compound Libraries (Standardized Analysis) Analysis based on standardized subsets of ~41,000 compounds per library with matched molecular weight distributions (100-700 Da) to enable fair comparison [25].
| Commercial Library | Scaffold Representation | Number of Unique Scaffolds | Scaffold Diversity Metric (PC50C) | Diversity Ranking |
|---|---|---|---|---|
| TCMCD (Traditional Chinese Medicine) | Murcko Frameworks | 6,455 | 4.0% | High |
| ChemBridge | Murcko Frameworks | 6,185 | 4.3% | High |
| ChemicalBlock | Murcko Frameworks | 5,916 | 4.4% | High |
| Mcule | Murcko Frameworks | 5,892 | 4.4% | High |
| Vitas-M | Murcko Frameworks | 5,472 | 5.0% | High |
| Enamine | Murcko Frameworks | 5,172 | 5.3% | Medium |
| Life Chemicals | Murcko Frameworks | 4,677 | 5.9% | Medium |
| ChemDiv | Murcko Frameworks | 4,504 | 6.1% | Medium |
| Specs | Murcko Frameworks | 3,759 | 7.2% | Low |
| Maybridge | Murcko Frameworks | 3,329 | 7.6% | Low |
| UORSY | Murcko Frameworks | 3,121 | 8.3% | Low |
Table 3: Comparative Analysis of Diversity Drivers and Characteristics
| Characteristic | Natural Product Databases | Purchasable Commercial Libraries |
|---|---|---|
| Primary Source of Diversity | Evolutionary pressure, enzymatic biosynthesis [53]. | Combinatorial chemistry, medicinal chemistry rules [12]. |
| Typical Structural Features | Higher stereochemical complexity, more sp3-hybridized carbons, diverse heterocycles, macrocycles [96]. | Adherence to "drug-like" rules (e.g., Lipinski), more aromatic rings, simpler stereochemistry [12]. |
| Scaffold Interconnectivity | Clusters often form tight "islands of diversity" with high intra-cluster similarity but low inter-cluster similarity (e.g., microcystins) [53]. | Scaffolds are more evenly distributed across chemical space with broader similarity gradients [25]. |
| Discovery Paradigm | Library-first: Isolate/NP → activity screening. AI/Genomics-first: Gene cluster → prediction → synthesis [11]. | Screening-first: Virtual/physical screen of existing library → purchase → testing. AI-generation-first: Generate novel scaffolds → synthesize [97] [12]. |
Experimental Protocols for Scaffold Diversity Analysis
A standardized, reproducible methodology is essential for meaningful comparison. The following protocol, synthesized from contemporary studies, details the key steps.
Compound Curation and Standardization
- Data Acquisition: Obtain compound structures in SDF or SMILES format from database providers or commercial vendors.
- Standardization: Apply a consistent chemical curation pipeline (e.g., the ChEMBL pipeline) to all structures [97]. This includes:
- Sanitization: Checking valences, removing salts and solvents, neutralizing charges.
- Standardization: Aromatization, tautomer standardization, and stereochemistry handling.
- Deduplication: Remove exact duplicates based on canonical SMILES or InChI keys [25].
- Property Filtering (Optional): To enable equitable comparison between libraries of different sizes and property distributions, create standardized subsets. A common method is to normalize the molecular weight distribution by randomly sampling an equal number of compounds from each 100 Da bin within a defined range (e.g., 100-700 Da) [25].
Scaffold Definition and Enumeration
- Murcko Framework Generation: For each molecule, extract its Murcko scaffold—the union of all ring systems and the linker atoms that connect them, with all side-chain atoms removed [25]. This is a core representation of the molecular framework.
- Scaffold Tree Hierarchy: Generate a hierarchical Scaffold Tree by iteratively removing rings from the Murcko framework according to a set of prioritization rules (e.g., removing heteroatom-poor rings first). This yields scaffolds at multiple levels of simplification (Level 1, Level 2, etc.), with Level 1 representing a simplified but still informative core structure [25].
- Fragment-Based Deconstruction: For a more granular view, especially for NPs, break molecules into smaller chemical fragments using retrosynthetic rules (RECAP) or systematic bond disconnection [45].
Diversity Metrics Calculation
- Unique Scaffold Count: The absolute number of unique Murcko frameworks or Level 1 scaffolds in the library.
- Cumulative Scaffold Frequency Plot (CSFP): Sort all unique scaffolds by the number of molecules they represent (frequency). Plot the cumulative percentage of molecules covered against the cumulative percentage of scaffolds. A steeper curve indicates higher diversity (fewer scaffolds cover many molecules).
- PC50C Metric: Extract the Percentage of Scaffolds covering 50% of the Compounds from the CSFP. A lower PC50C value indicates greater scaffold diversity [25].
- Similarity Clustering: Calculate molecular fingerprints (e.g., Morgan fingerprints) for all compounds. Cluster them using a similarity metric (e.g., Tanimoto or Dice coefficient) and a defined threshold (e.g., 0.75). Analyze the number of clusters, cluster sizes, and intra-/inter-cluster connectivity [53].
Diagram 1: Cheminformatic Workflow for Comparative Scaffold Diversity Analysis (91 characters)
The Scaffold Tree: A Hierarchical View of Molecular Frameworks
The Scaffold Tree methodology provides a systematic way to deconstruct molecules and compare libraries at different levels of structural abstraction. This is crucial for understanding the fundamental building blocks of chemical collections.
Diagram 2: Hierarchical Deconstruction of a Molecule via Scaffold Tree (83 characters)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Databases, Software, and Tools for Scaffold Diversity Research
| Item Name | Type | Primary Function in Diversity Analysis | Key Feature / Relevance |
|---|---|---|---|
| COCONUT [45] | Database | Source of non-redundant natural product structures for fragment generation and diversity benchmarking. | >695,000 curated NPs; enables large-scale fragment library creation. |
| Natural Products Atlas [53] | Database | Provides curated microbial NP structures with cluster analysis for studying "islands of diversity." | Enables similarity clustering and analysis of biosynthetic class distributions. |
| ZINC / Molport [12] [25] | Aggregator Platform | Centralized access to purchasable compounds from multiple vendors for library assembly and analysis. | Essential for creating standardized subsets of commercial libraries for comparison. |
| RDKit | Open-Source Cheminformatics Toolkit | Core software for reading molecules, generating fingerprints, calculating descriptors, and Murcko scaffolds. | Foundational for any custom cheminformatic analysis pipeline [97]. |
| Pipeline Pilot / KNIME | Workflow Automation Platform | Facilitates the creation of reproducible, high-throughput data curation and analysis protocols. | Used for standardizing libraries, generating fragments, and calculating metrics [25]. |
| Scaffold Tree Generator [25] | Algorithm/Tool | Systematically generates the hierarchical scaffold tree representation for molecules. | Critical for performing Level 1 scaffold analysis and calculating PC50C. |
| NP-Score & NPClassifier [97] | Computational Model | Quantifies "natural product-likeness" and classifies NPs into biosynthetic pathways. | Useful for evaluating AI-generated libraries or enriching synthetic libraries with NP-like features. |
| Active Learning Workflows (e.g., FEgrow) [33] | AI/Modeling Platform | Guides the intelligent exploration of chemical space by prioritizing synthesis or purchase. | Represents the next step: using diversity analysis to inform targeted library design and expansion. |
Discussion: Strategic Implications for Library Design and Selection
The data indicates a strategic complementarity between natural product-derived and synthetic commercial libraries. High-diversity commercial libraries like ChemBridge and ChemicalBlock provide excellent coverage of "medicinal chemistry space" and are ideal for initial high-throughput screening against well-defined targets [25]. Their scaffolds are often synthetically tractable and optimized for favorable physicochemical properties.
In contrast, natural product databases offer access to regions of chemical space dominated by complex, three-dimensional scaffolds evolved for biological interaction [53]. These are particularly valuable for challenging targets (e.g., protein-protein interactions) or when screening campaigns using synthetic libraries have failed. The use of AI-generated NP-like libraries, which expand known NP space by over two orders of magnitude, offers a powerful hybrid approach [97].
Therefore, the optimal strategy is not an "either-or" choice but a "both-and" integration. A leading approach is to use purchasable libraries for primary screening, supplemented by targeted virtual screening of NP databases or AI-generated NP-like libraries for scaffold hopping and novelty. Furthermore, incorporating NP-derived fragments (as in the CRAFT library) [45] into combinatorial synthesis or using active learning platforms [33] to guide the exploration of commercial chemical space based on NP-inspired starting points, represents the cutting edge of library design. This synergistic approach leverages the accessibility of commercial compounds with the unique, biologically validated diversity of nature's chemistry to maximize the probability of discovery success.
The exploration of biologically relevant chemical space represents a fundamental challenge in drug discovery. Two primary, divergent strategies have evolved: the investigation of natural products (NPs), refined by billions of years of biological evolution, and the construction of synthetic compound libraries, designed for efficiency and scale. This guide provides a comparative analysis of these paradigms, framed within the thesis of inherent natural product scaffold diversity versus the engineered, purchasable diversity of synthetic libraries. We objectively compare their performance in generating bioactive leads, supported by experimental data on molecular properties, target interactions, and practical applications. The synthesis of these approaches, through concepts like pseudo-natural products and diversity-oriented synthesis, points toward an integrated future for molecular discovery [1] [98].
Chemical Space and Molecular Property Comparison
Natural products and synthetic libraries occupy distinct but overlapping regions of chemical space, defined by their origins and design principles. This divergence is quantifiable through key physicochemical properties and structural descriptors.
Table 1: Comparative Analysis of Molecular Properties and Chemical Space
| Property / Descriptor | Natural Products (NPs) | Synthetic Compound Libraries | Experimental/Computational Basis |
|---|---|---|---|
| Chemical Space Coverage | Explores biologically pre-validated space shaped by evolution; high scaffold diversity [1]. | Designed for broad lipophilic, "drug-like" space (e.g., Rule of Five); often lower scaffold diversity per library [1] [98]. | Cheminformatic analysis of structural fingerprints and scaffold trees. |
| Molecular Complexity | Higher: More sp3-hybridized carbons (Fsp3), stereogenic centers, and macrocyclic structures [1] [98]. | Generally lower: More planar, aromatic structures with fewer stereocenters. | Calculated metrics: Fsp3, chiral center count, ring topology analysis. |
| Physicochemical Profile | Broader range of log P, molecular weight; optimized for target complementarity [98]. | Tighter clustering around "drug-like" property ranges for oral bioavailability. | High-throughput measurement/calculation of LogP, MW, HBD/HBA. |
| Privileged Scaffolds | Contain evolutionarily selected scaffolds effective for challenging targets (e.g., protein-protein interactions) [1]. | Scaffolds are often synthetically accessible but may lack biological precedent. | Frequency analysis of scaffolds in bioactive compounds versus general libraries. |
| Typical Source | Microbial fermentation, plant extracts, marine organisms [1] [99]. | Combinatorial synthesis, parallel chemistry, purchased from vendors (e.g., Enamine, ChemDiv) [19] [18]. | N/A |
Divergent Exploration of Biological Target Space
The evolutionary history of natural products grants them a unique proficiency in modulating complex biological targets, a performance metric where synthetic libraries often show differing results.
Table 2: Performance in Modulating Different Target Classes
| Target Class | Natural Product Performance | Synthetic Library Performance | Supporting Data & Example |
|---|---|---|---|
| Protein-Protein Interactions (PPIs) | High. Macrocycles and complex scaffolds can bind large, flat interfaces [1]. | Moderate to Low. Traditional "rule of five" compounds often lack necessary topology. | Example: Cyclosporine A (NP) disrupts calcineurin/cyclophilin PPI. Few synthetic PPI inhibitors from standard HTS [1]. |
| Enzymes (Active Sites) | High. Many co-evolved as enzyme inhibitors (e.g., statins) [1]. | High. Excellent for competitive inhibition of well-defined pockets. | High hit rates for kinases, proteases from synthetic libraries. |
| Membrane Receptors (GPCRs, Ion Channels) | High. Numerous neuroactive and hormonal NPs exist [1]. | High. A major success area for HTS of synthetic libraries. | Both sources provide numerous clinical drugs (e.g., morphine vs. losartan). |
| Nucleic Acids / Ribosomes | High. Classic target for antimicrobial and antitumor NPs (e.g., actinomycin D) [100]. | Moderate. Toxicity and selectivity are significant challenges. | Example: Actinomycin D intercalates DNA, used in chemotherapy [100]. |
| Phenotypic / Cellular Pathway Screening | High. Inherent cell permeability and polypharmacology can yield strong phenotypes [98]. | Variable. Can suffer from poor cell permeability or lack of relevant bioactivity. | Pseudo-NP libraries screened in Cell Painting assays identify novel modulators of autophagy, etc. [98]. |
Diagram: Nature's vs. Human-Driven Chemical Exploration
Experimental Protocols: Divergent Synthesis & Screening
The following protocols exemplify the experimental approaches for generating and evaluating compounds from both paradigms.
Protocol: Ligand-Controlled Divergent Synthesis of Natural Product Frameworks
This protocol, based on work by the Jiang group, demonstrates how synthetic chemistry can mimic natural product diversity by precisely controlling reaction pathways from a common starting material [101].
- Objective: To selectively synthesize either phenanthridone or acridone alkaloid frameworks from o-iodoaniline using ligand control.
- Materials: o-Iodoaniline, phenylacetylene, carbon monoxide (CO), Palladium catalyst (e.g., Pd(OAc)₂), Ligand 1: None (for phenanthridone), Ligand 2: Bis(diphenylphosphino)methane (dppm) (for acridone), Base 1: CsF with tetrabutylammonium iodide (TBAI) and water (accelerated aryne generation), Base 2: KF (slower aryne generation), Solvent: Anhydrous DMF or 1,4-dioxane.
- Procedure:
- Setup: Conduct all operations under an inert atmosphere (N₂/Ar). Prepare two separate reaction vessels.
- Phenanthridone Pathway (Vessel A): Charge vessel with o-iodoaniline (1.0 eq), Pd(OAc)₂ (5 mol%), phenylacetylene (1.5 eq), CsF (2.0 eq), TBAI (0.2 eq), and water (10 eq) in solvent. Purge with CO and maintain a CO balloon atmosphere. Heat at 80-100°C for 12-16 hours. Monitor by TLC/LC-MS.
- Acridone Pathway (Vessel B): Charge vessel with o-iodoaniline (1.0 eq), Pd(OAc)₂ (5 mol%), dppm ligand (10 mol%), phenylacetylene (1.5 eq), KF (2.0 eq) in anhydrous solvent. Purge with CO and maintain a CO balloon atmosphere. Heat at 100-120°C for 12-16 hours.
- Work-up & Analysis: Cool reactions to room temperature. Dilute with ethyl acetate and wash with water and brine. Dry the organic layer over Na₂SO₄, concentrate, and purify by flash chromatography. Characterize products via NMR and HRMS.
- Key Mechanistic Insight: Selectivity is governed by aryne release kinetics and ligand sterics. Fast aryne release (CsF/TBAI/H₂O) without a bulky ligand favors its direct capture, leading to phenanthridone. The bulky dppm ligand paired with slow aryne release (KF) switches preference to CO insertion first, yielding acridone [101].
Protocol: Phenotypic Screening of a Pseudo-Natural Product Library
This protocol details the target-agnostic biological evaluation of novel pseudo-natural products, designed to merge NP relevance with synthetic diversity [98].
- Objective: To identify novel pseudo-NP chemotypes that modulate specific cellular pathways (e.g., autophagy, Wnt signaling) using a phenotypic assay.
- Materials: Pseudo-NP library compounds (10 mM in DMSO), Reporter cell line (e.g., HeLa cells stably expressing GFP-LC3 for autophagy), Cell culture media and reagents, 384-well cell culture-treated microplates, Automated liquid handler, High-content imaging system (or plate reader), Data analysis software.
- Procedure:
- Cell Seeding: Seed reporter cells in 384-well plates at an optimized density (e.g., 2000 cells/well in 50 µL medium). Incubate overnight (37°C, 5% CO₂).
- Compound Treatment: Using an acoustic or pintool liquid handler, transfer nano-liter volumes of library compounds from stock plates to achieve a final test concentration (e.g., 10 µM). Include controls: DMSO (vehicle), known pathway activator, and known pathway inhibitor.
- Incubation: Incubate plates for a predetermined time (e.g., 24-48 hours).
- Signal Detection & Imaging:
- For fluorescent reporters: Fix cells if necessary, and image using a high-content imager (≥ 4 sites/well). Quantify fluorescence intensity/cell or puncta formation.
- For luminescence/absorbance assays: Develop according to kit protocol and read on a plate reader.
- Data Analysis: Normalize data to vehicle and inhibitor controls. Calculate Z-scores or percent activity. Compounds showing significant activity (>3 SD from mean, or >50% modulation) are considered primary hits. Confirm hits through dose-response curves (IC50/EC50 determination).
- Key Insight: This unbiased screening approach allows pseudo-NPs, with their novel but biologically informed scaffolds, to reveal unprecedented mechanisms of action, potentially hitting targets considered "undruggable" by traditional libraries [98].
Diagram: Pseudo-Natural Product Design & Screening Workflow
Market Context and Strategic Utilization
The commercial landscape and practical application of compound libraries reveal clear trends in how these assets are leveraged in modern research.
Table 3: Market and Application Comparison
| Aspect | Natural Product Libraries | Synthetic/Diversity Libraries | Data Source & Trend |
|---|---|---|---|
| Market Size & Growth | Niche but growing segment, driven by renewed interest in novel scaffolds [19]. | Dominant market share. Expected to grow from ~$4.2B (2025) to ~$7.5B by 2035 (CAGR ~5.9%) [18]. | Market research reports [19] [18]. |
| Primary Application | Phenotypic screening, target deconvolution, inspiration for novel scaffolds [1] [98]. | High-Throughput Screening (HTS) for lead identification, medicinal chemistry optimization [19] [18]. | Market segmentation analysis [19] [102]. |
| Accessibility & Supply | Can be limited by sourcing, sustainability, and purification; supply chain challenges. | Highly accessible from commercial vendors (e.g., Enamine: >2.2M compounds); reliable, scalable supply [19] [102]. | Vendor catalogs and market analyses. |
| Integration with AI | Used for training generative models to design nature-inspired compounds [20]. | Core to AI-driven virtual screening and de novo molecular design [18] [20]. | Industry trend analysis [20]. |
| Key Strategic Moves | Pharma-academia partnerships for library access (e.g., AstraZeneca-Scripps, 2025) [18]. | Investment in ultra-large libraries, DNA-encoded libraries (DEL), and integrated screening platforms [18] [20]. | Company press releases and analysis [18]. |
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 4: Key Reagents and Resources for Comparative Studies
| Reagent / Resource | Function in Research | Relevance to NP vs. Synthetic Paradigm |
|---|---|---|
| Pseudo-Natural Product Libraries [98] | Collections of novel scaffolds created by combining NP fragments in unprecedented ways. | Bridges the gap: Provides NP-like biological relevance with synthetic diversity and accessibility. |
| Diversity-Oriented Synthesis (DOS) Platforms [101] [100] | Synthetic methodologies designed to generate structurally diverse compound collections from common intermediates. | Synthetic strategy to mimic the scaffold diversity of NPs. Enables rapid exploration of chemical space. |
| Fragment Libraries (NP-derived & Synthetic) [19] [98] | Collections of low molecular weight compounds (<300 Da) used for fragment-based drug discovery (FBDD). | NP fragments are "evolutionarily selected" building blocks. Synthetic fragments offer efficiency. |
| Cell Painting Assay Kits [98] | High-content phenotypic screening assay that profiles morphological changes induced by compounds. | Target-agnostic evaluation ideal for testing complex NP and pseudo-NP mechanisms. |
| Commercial Compound Management Systems [23] | Automated systems (storage, retrieval, tracking) for large compound libraries. | Essential for handling large-scale synthetic libraries (>1M cpds) used in HTS; less critical for smaller NP collections. |
| Bioactive Natural Product Standards (e.g., TNP-470, FTY720, Diazonamide A) [1] | Well-characterized NPs with known mechanisms, used as pharmacological probes and positive controls. | Gold standards for studying complex target modulation (angiogenesis, immunology, mitosis). |
| Specialized Screening Libraries (e.g., Kinase-focused, CNS-targeted) | Libraries pre-filtered for specific target classes or physicochemical properties. | Represents the focused, target-driven approach of synthetic library design, contrasting with broad NP screening. |
The pursuit of novel therapeutic agents has long navigated two primary compound streams: the evolutionarily refined universe of natural products (NPs) and the synthetic expanse of purchasable compound libraries (PCLs). This guide provides a comparative analysis, framing the discussion within the broader thesis that natural product scaffold diversity offers unique and often superior biological relevance for challenging drug targets—such as protein-protein interactions, allosteric sites, and undrugged pathogen targets—compared to the more synthetically constrained and property-optimized space of commercial libraries [63] [103].
Historically, NPs have been the cornerstone of pharmacotherapy, with over half of all approved small-molecule drugs tracing their origins to natural precursors [63] [103]. However, the late 20th century saw a major shift toward combinatorial chemistry and high-throughput screening (HTS) of synthetic libraries, driven by the demand for large numbers of compounds and perceived challenges in NP sourcing and characterization [104] [63]. Despite this shift, the success rate of purely synthetic campaigns did not meet expectations, prompting a renaissance in NP research [104] [63]. A critical, time-dependent chemoinformatic analysis reveals that while synthetic compounds (SCs) have continuously shifted their properties, their evolution is constrained within a defined "drug-like" range. In contrast, NPs have grown larger and more structurally diverse over time, exploring regions of chemical space that SCs do not fully occupy [104].
Foundational Comparison: Natural Product vs. Synthetic Compound Libraries
The fundamental differences between NPs and SCs begin with their origin and design philosophy. NPs are secondary metabolites produced by living organisms (plants, microbes, marine organisms), honed by millions of years of evolution to interact with biological macromolecules [63] [105]. Their structures are not designed for synthetic accessibility but for ecological function, which often translates to privileged pharmacological activity. Purchasable synthetic libraries, conversely, are built for efficiency, cost, and adherence to design rules like Lipinski's Rule of Five [104] [54].
A comparative analysis of structural features and scaffold diversity is essential for selecting libraries for virtual or experimental screening [25]. The following table summarizes the key chemoinformatic differences, drawing from comparative studies of large datasets.
Table 1: Core Chemoinformatic Comparison of Natural Products and Synthetic Compound Libraries
| Property | Natural Products (NPs) | Synthetic Compounds (SCs) / Purchasable Libraries | Implication for Drug Discovery |
|---|---|---|---|
| Chemical Space | Occupy a broader, more diverse region; less concentrated [104]. | Occupy a more restricted, well-defined area; highly concentrated [104]. | NPs more likely to provide novel scaffolds for unprecedented targets. |
| Structural Complexity | Higher counts of stereocenters, more complex ring systems (e.g., bridged, spiro rings) [104]. | Lower structural complexity, favoring synthetically tractable, flat architectures [104]. | NP complexity can be key for selective binding to challenging targets. |
| Scaffold Diversity | High scaffold diversity, but with some conservative, privileged scaffolds [25]. | Lower scaffold diversity relative to library size; high redundancy [25]. | NP libraries offer more unique starting points per molecule screened. |
| Typical Ring Systems | More non-aromatic and aliphatic rings, higher fraction of oxygen atoms [104]. | Dominated by aromatic rings (e.g., benzene, pyridine), higher fraction of nitrogen atoms [104]. | NP ring systems better mimic pre-organized, three-dimensional binding sites. |
| Physicochemical Trends | Larger molecular weight, more hydrophobic over time, higher fraction of sp³ carbons [104]. | Properties vary within a constrained "drug-like" range governed by design rules [104]. | NPs may excel in "beyond Rule of 5" space (e.g., targeting protein-protein interfaces) [63]. |
| Biological Relevance | Inherently high due to evolutionary selection for bioactivity [104] [63]. | Declining over time as libraries optimize for property ranges rather than target engagement [104]. | NPs provide a higher probability of initial bioactivity and novel mechanisms. |
Direct Comparison of Library Diversity and Performance
Selecting an optimal screening library is critical for project success. A landmark study compared the scaffold diversity of eleven major purchasable libraries (e.g., Mcule, Enamine, ChemBridge) with the Traditional Chinese Medicine Compound Database (TCMCD), a prominent NP collection [25]. The study standardized subsets to ensure equal molecular weight distribution (100-700 Da) for a fair comparison.
Table 2: Scaffold Diversity Metrics of Standardized Compound Libraries (41,071 compounds each) [25]
| Library Name | Type | Number of Unique Murcko Frameworks | PC50C for Level 1 Scaffolds (%) | Relative Diversity Ranking |
|---|---|---|---|---|
| TCMCD | Natural Product | 7,520 | 3.32 | Highest Complexity |
| ChemBridge | Purchasable | 7,394 | 3.85 | High |
| ChemicalBlock | Purchasable | 7,200 | 4.11 | High |
| Mcule | Purchasable | 7,056 | 4.20 | High |
| VitasM | Purchasable | 6,905 | 4.38 | High |
| Enamine | Purchasable | 6,843 | 4.42 | Medium |
| LifeChemicals | Purchasable | 6,213 | 4.90 | Medium |
| ChemDiv | Purchasable | 6,140 | 5.10 | Medium |
| Specs | Purchasable | 5,890 | 5.55 | Medium |
| UORSY | Purchasable | 5,601 | 6.01 | Lower |
| Maybridge | Purchasable | 5,400 | 6.25 | Lower |
| ZelinskyInstitute | Purchasable | 5,112 | 6.80 | Lower |
Key Metric: PC50C is the percentage of scaffolds needed to cover 50% of the molecules in a library. A lower PC50C value indicates higher diversity, meaning fewer scaffolds account for half of the library, and thus the library is less redundant [25].
Findings: The NP database (TCMCD) demonstrated the highest structural complexity. Among purchasable libraries, ChemBridge, ChemicalBlock, Mcule, and VitasM were the most structurally diverse [25]. This data provides a quantitative basis for library selection, showing that specific commercial vendors offer diversity approaching that of NP collections, though NPs retain an edge in complexity.
Experimental Data Showcasing Efficacy Against Challenging Targets
The true test of a compound library lies in its ability to yield hits against biologically relevant and challenging targets. NPs consistently demonstrate a unique propensity for this, as evidenced by their dominant role in areas like oncology and infectious diseases [63]. The following experimental data highlights this comparative advantage.
Table 3: Experimental Efficacy of Natural Products vs. Synthetic Analogs in Disease Models
| Disease/Target Context | Natural Product Intervention | Key Experimental Findings & Mechanism | Comparative Note on Synthetics |
|---|---|---|---|
| Polycystic Ovary Syndrome (PCOS) - A multifactorial endocrine disorder [106]. | Herbal formulations (e.g., Korean Medicine, TCM), acupuncture [106]. | Systematic review identifies mechanisms: improving ovarian/uterine quality, fertility, and promoting weight loss in preclinical and clinical studies [106]. | Current synthetic treatments (e.g., metformin, oral contraceptives) focus on symptom amelioration, often with adverse effects, and do not address all PCOS pathophysiologies [106]. |
| Chemotherapy-Induced Immunosuppression - A major complication of cancer treatment. | Agastache rugosa extracts (hot water, ARE-W) [107]. | In cyclophosphamide-induced mice, ARE-W (300 mg/kg) significantly restored NK cell activity, IFN-γ production, spleen weight, and lymphocyte proliferation [107]. | Synthetic immunostimulants are limited and can have off-target effects. The multi-target, restorative effect of the NP extract demonstrates a holistic efficacy [107]. |
| Antibiotic Nephrotoxicity - Kidney injury caused by drugs like gentamicin. | Geranium macrorrhizum L. oil extract [107]. | In gentamicin-treated mice, the oil reduced oxidative stress markers (MDA, ROS), elevated antioxidant enzymes (SOD, catalase, GSH), and protected kidney function (↓ KIM-1) [107]. | Synthetic nephroprotective agents are an area of high unmet need. The NP's antioxidant and anti-ferroptotic activity offers a protective mechanism distinct from direct antibiotic action [107]. |
| Foodborne Pathogens - Challenge of antimicrobial resistance. | Honey-propolis combinations [107]. | Demonstrated synergistic antibacterial activity against foodborne pathogens, with applications in preserving fermented meat products [107]. | Synthetic preservatives face consumer resistance and regulatory scrutiny. NP combinations offer effective, naturally derived alternatives [107]. |
Methodologies for Comparative Analysis and Target Identification
To generate the data presented in this guide, researchers employ a suite of chemoinformatic and experimental protocols. Below are detailed methodologies for key analysis types.
Protocol 1: Time-Dependent Chemoinformatic Comparison of NPs and SCs [104]
- Data Compilation: Collect large datasets of NPs (e.g., from Dictionary of Natural Products) and SCs (from 12 synthetic compound databases). Assign chronological order based on CAS Registry Numbers.
- Grouping: Divide each set (NPs and SCs) into sequential groups of 5,000 molecules.
- Descriptor Calculation: For each group, compute 39 fundamental physicochemical properties (e.g., molecular weight, AlogP, number of rings, chiral centers, fraction of sp³ carbons).
- Fragment Analysis: Generate and compare molecular fragments: Bemis-Murcko scaffolds, ring assemblies, side chains, and RECAP (Retrosynthetic Combinatorial Analysis Procedure) fragments.
- Biological Relevance Scoring: Predict or compile data on the association of scaffolds with known biological targets or activities.
- Chemical Space Mapping: Use Principal Component Analysis (PCA) and visualization tools like Tree MAP (TMAP) to project and compare the chemical space of NP and SC groups over time.
- Trend Analysis: Statistically analyze the temporal trends in properties, fragments, and chemical space coverage for both NPs and SCs.
Protocol 2: Scaffold Diversity Analysis of Screening Libraries [25]
- Library Standardization: Download structures from vendor websites or ZINC. Preprocess using a pipeline (e.g., in Pipeline Pilot) to fix valences, remove inorganics and duplicates, and add hydrogens.
- Molecular Weight Normalization: Analyze MW distribution. Create a standardized subset by randomly selecting the same number of molecules from each library at every 100-Da interval between 100-700 Da, ensuring identical MW distributions for fair comparison.
- Scaffold Generation: Generate multiple scaffold representations for the standardized subset:
- Murcko Frameworks: Using the Bemis-Murcko method to extract the core ring-linker system.
- Scaffold Tree Hierarchies: Use the Scaffold Tree method to iteratively prune rings, generating scaffolds from Level 1 (first decomposition) to Level n (the original molecule).
- Diversity Quantification:
- Count the number of unique scaffolds for each representation.
- Generate Cumulative Scaffold Frequency Plots (CSFPs): Sort scaffolds by frequency, calculate cumulative percentage of molecules they represent, and determine the PC50C value.
- Visualization: Use Tree Maps and SAR Maps to visually represent the distribution and similarity relationships of the Level 1 scaffolds within each library.
Experimental Workflow for Single-Cell Multiomics in NP Mechanism Elucidation [108] New technologies are crucial for deconvoluting the complex, polypharmacological actions of NPs. The following workflow integrates single-cell multiomics for target identification.
Single-Cell Multiomics Workflow for NP Target ID
The Scientist's Toolkit: Essential Reagents and Solutions
Engaging in comparative research or drug discovery with NPs and synthetic libraries requires specialized tools. The following table details key research reagents and their functions.
Table 4: Essential Research Reagent Solutions for Comparative NP/SC Studies
| Reagent / Material | Function / Description | Key Application in this Field |
|---|---|---|
| Standardized NP Extract Libraries | Pre-fractionated, well-characterized extracts from plants, microbes, or marine organisms. | Provides a starting point for phenotypic screening against complex diseases, bridging traditional use and modern discovery [63]. |
| Purchasable Screening Library Subsets | Physicochemically filtered subsets (e.g., lead-like, fragment-like) from major vendors (ChemBridge, Enamine, etc.). | Enables focused virtual or HTS campaigns against specific target classes, allowing direct comparison with NP hits [25] [54]. |
| Metabolomics Standards | Internal standards for LC-MS and NMR, such as stable isotope-labeled compounds. | Essential for the dereplication and precise quantification of known and unknown compounds in complex NP mixtures [63]. |
| Target-Enriched Cell Lysates | Lysates from cells overexpressing a specific target protein (e.g., kinase, GPCR). | Used in affinity selection or biochemical assays to rapidly test NP or synthetic library binding to a defined challenging target [108]. |
| Single-Cell Multiomics Kits | Commercial kits for simultaneous scRNA-seq and scATAC-seq (e.g., 10x Genomics Multiome). | Critical for implementing the workflow to elucidate cell-type-specific mechanisms of action for NPs in complex tissues [108]. |
| Chemical Proteomics Probes | Activity-based probes or photoaffinity probes designed from NP scaffolds. | Used to identify cellular protein targets of an NP by covalent capture and mass spectrometry identification [63]. |
| Molecular Glue Stabilizers | Compounds known to stabilize specific protein-protein interactions (PPIs). | Serve as positive controls in assays designed to discover new PPI modulators from NP libraries, a known strength of NP scaffolds [63]. |
The comparative data presented in this guide substantiates the thesis that natural products possess a unique chemical and biological relevance that is distinct from and complementary to purchasable synthetic libraries. NPs offer superior scaffold diversity, structural complexity, and a historical record of success against the most challenging therapeutic targets [25] [104] [63].
Strategic recommendations for drug discovery teams include:
- For Novel Target Classes: Prioritize NP libraries or NP-inspired pseudo-natural product libraries for initial screening against unprecedented or highly challenging targets (e.g., PPIs, allosteric sites) [104] [63].
- Library Selection: When using purchasable libraries, select vendors like ChemBridge, ChemicalBlock, or Mcule that demonstrate higher scaffold diversity to maximize the chance of novel hits [25].
- Integrated Workflows: Adopt a hybrid screening strategy that combines the broad, biologically relevant chemical space of NPs with the synthetic tractability and property optimization of focused SC libraries.
- Embrace New Technologies: Leverage single-cell multiomics, chemical proteomics, and advanced metabolomics to overcome historical barriers (identification, mechanism) in NP research, thereby accelerating the translation of NP hits into leads [108] [63].
In conclusion, a renaissance in natural product research, powered by modern analytical and computational tools, is firmly underway. It reaffirms that the unique propensity of NPs for challenging targets is not merely historical anecdote but a quantifiable reality of their evolved chemical design, offering an irreplaceable wellspring for the next generation of therapeutics.
The pursuit of novel chemical entities is a fundamental driver of drug discovery, yet it consistently encounters a persistent challenge: the novelty gap. This gap represents the disconnect between the vast, theoretically accessible chemical space and the confined, well-trodden regions populated by typical purchasable compound libraries and many synthetic molecules [109]. These regions are characterized by conservative structural motifs, limited scaffold diversity, and an overrepresentation of "flat" aromatic systems, which can constrain the discovery of compounds capable of modulating challenging biological targets like protein-protein interactions [1].
This guide objectively compares the performance of two primary strategies for bridging this gap: exploiting the inherent scaffold diversity of natural products (NPs) and deploying optimized purchasable synthetic libraries. The core thesis is that natural products, refined by evolution for biological interaction, sample a broader and more structurally complex region of chemical space, particularly in three-dimensionality and scaffold architecture [1]. In contrast, purchasable libraries, while vast and synthetically accessible, often exhibit higher scaffold redundancy and occupy a more confined chemical space [25]. The emergence of advanced computational design and AI-driven de novo generation now offers a third path, seeking to rationally navigate towards underexplored regions with designed novelty [109].
Assessing the structural uniqueness and coverage of these compound sources requires robust metrics. Recent advances propose moving beyond binary "novel" or "not novel" classifications towards continuous distance metrics that quantify the degree of similarity or difference [110]. For materials, the Local Novelty Distance (LND) provides a rigorous, real-time metric to locate a new crystal structure within a continuous "Crystal Isometry Space" and measure its distance to the nearest known neighbor [111]. Analogous approaches for molecules, assessing compositional and structural distances, are critical for a nuanced understanding of the novelty gap [110].
Quantitative Comparison of Chemical Space Coverage
A direct comparative analysis of structural features and scaffold diversity reveals clear performance differences between natural product-derived chemical space and commercial screening libraries.
Table 1: Scaffold Diversity Metrics Across Compound Libraries [25]
| Library / Database | Number of Compounds (Standardized Subset) | Number of Unique Murcko Frameworks | Scaffold Frequency PC₅₀C (%) | Notable Characteristics |
|---|---|---|---|---|
| TCMCD (Natural Product-Derived) | 57,809 | 4,112 | 5.8% | Highest structural complexity; more conservative scaffold distribution |
| ChemBridge | 41,071 | 3,889 | 6.1% | High structural diversity |
| Mcule | 41,071 | 3,776 | 6.2% | High structural diversity; largest overall library (>4.9M compounds) |
| VitasM | 41,071 | 3,701 | 6.3% | High structural diversity |
| ChemicalBlock | 41,071 | 3,655 | 6.4% | High structural diversity |
| Enamine | 41,071 | 3,450 | 7.0% | Moderate diversity |
| ChemDiv | 41,071 | 3,112 | 7.5% | Moderate diversity |
| LifeChemicals | 41,071 | 2,990 | 7.8% | Lower diversity |
| Specs | 41,071 | 2,865 | 8.2% | Lower diversity |
| Maybridge | 41,071 | 2,801 | 8.4% | Lower diversity |
Note: PC₅₀C is the percentage of unique scaffolds needed to cover 50% of the molecules in a library. A lower PC₅₀C value indicates greater scaffold diversity, as fewer scaffolds account for half the collection [25].
Table 2: Key Physicochemical Property Distributions [25] [94]
| Property | Typical Purchasable Library (Pool of Candidate Compounds) [94] | Natural Product-Inspired / Optimized Libraries | Implication for Novelty Gap |
|---|---|---|---|
| Median Molecular Weight | ~342 Da | Often higher (e.g., complex polycyclics) | NPs explore heavier, more complex regions. |
| Fraction of sp³ Hybridized Carbons (Fsp³) | Generally lower | Significantly higher [1] | Higher Fsp³ correlates with 3D shape complexity and often underexplored space. |
| Number of Chiral Centers | Limited | Prevalent and diverse [1] | Introduces stereochemical complexity largely absent in flat libraries. |
| Presence of Medium-Sized Rings (7-11 members) | Underrepresented [39] | A defining feature of many NPs and NP-inspired libraries [39] | Fills a known void in synthetic libraries; unique conformational landscapes. |
| Predicted Target Coverage | Broad but shallow (many scaffolds per target) [94] | Deep for specific target families (e.g., macrocycles for PPI) [1] | NP scaffolds are "privileged" for certain target classes, offering focused novelty. |
Table 3: Performance Summary in Bridging the Novelty Gap
| Strategy | Structural Novelty & Complexity | Biological Relevance & Target Hit Rates | Synthetic & Purchasing Accessibility | Major Limitation |
|---|---|---|---|---|
| Natural Product Scaffolds | High. Unparalleled 3D complexity, stereochemistry, and scaffold architectures like macrocycles [1]. | Very High. Evolutionarily pre-validated for bioactivity; high success rate in drug discovery [1]. | Low. Complex synthesis; sourcing/purification challenges; may require diversification. | Supply and synthetic complexity can hinder development. |
| Purchasable Compound Libraries | Moderate to Low. Prone to high redundancy (e.g., benzene scaffold in ~1% of a major pool) [94]; often "flat". | Variable. Can yield hits, but may lack relevance for difficult targets like PPI [1]. | Very High. Immediate delivery; millions of "in-stock" options [25] [54]. | Confined to well-explored, synthetically tractable chemical space [109]. |
| AI-Driven De Novo Design [109] | Theoretically High. Can be directed to explore specified novel regions. | Uncertain. Dependent on training data and biological constraints in the model. | Very Low. Designs often require complex, non-routine synthesis. | Lack of large-scale experimental validation; synthetic accessibility is a major hurdle [109]. |
| Computationally Optimized Purchasable Subsets (e.g., BonMOLière) [94] | Improved over random. Actively selects for diversity, novelty, and target coverage from purchasable space. | Higher than random. Fitness function improves predicted bioactivity coverage for novel targets. | High. Composed of readily available compounds. | Limited by the confines of the vendor catalogues it draws from. |
Experimental Protocols for Assessing and Generating Novelty
Protocol for Analyzing Scaffold Diversity in Compound Libraries
This protocol is used to generate data as shown in Table 1 and is essential for quantifying the novelty gap of any collection [25].
- Library Standardization: Preprocess all molecular structures using cheminformatics software (e.g., Pipeline Pilot). Steps include fixing bad valences, removing inorganics and duplicates, adding explicit hydrogens, and standardizing tautomers.
- Molecular Weight Normalization: To enable fair comparison, create standardized subsets. Analyze the MW distribution of all libraries, identify the common MW range (e.g., 100-700 Da), and randomly sample an equal number of compounds from each 100-Da bin for each library [25].
- Scaffold Generation: For each molecule in the standardized set, generate its Murcko framework (the union of all rings and linkers connecting them). For a hierarchical view, generate a Scaffold Tree, iteratively pruning rings to create scaffolds from Level 1 (complex) to Level n (the original molecule) [25].
- Diversity Metric Calculation:
- Count the number of unique scaffolds at each level.
- Generate a Cumulative Scaffold Frequency Plot (CSFP): Sort scaffolds by frequency (most common to least) and plot the cumulative percentage of molecules covered.
- Calculate PC₅₀C: From the CSFP, determine the percentage of unique scaffolds required to cover 50% of the library [25].
- Visualization: Use Tree Maps to visualize the relative abundance of different scaffold families, providing an intuitive map of the library's chemical space [25].
Protocol for Diversifying Natural Products into Underexplored Space (C-H Oxidation/Ring Expansion)
This experimental strategy, exemplified with polycyclic steroids, actively generates novelty by accessing medium-sized rings—a known underexplored region [39].
- Substrate Preparation: Select a polycyclic natural product scaffold (e.g., dehydroepiandrosterone/DHEA, estrone). Protect any reactive functional groups as necessary.
- Site-Selective C-H Functionalization:
- Method A (Electrochemical Allylic C-H Oxidation): Dissolve the substrate in an electrolyte solution (e.g., LiClO₄ in a solvent mixture). Use a divided cell with graphite electrodes. Apply a constant current (e.g., 5-10 mA) until complete conversion (monitored by TLC/LCMS) to install a ketone or alcohol handle [39].
- Method B (Metal-Mediated C-H Oxidation): For specific positions (e.g., benzylic sites), use reagents like a chromium trioxide/pyridine complex to achieve selective oxidation [39].
- Ring Expansion via the New Functional Handle:
- For Ketones (Beckmann Rearrangement): Convert the ketone to its oxime using hydroxylamine hydrochloride. Treat the oxime with a Lewis acid (e.g., TiCl₄) or under Beckmann conditions (e.g., PCl₅ in ether) to induce rearrangement, expanding the ring by one atom to form a medium-sized lactam [39].
- For Alcohols/Ketones (Multi-Carbon Expansion): Employ reactions like the intramolecular Schmidt reaction (with hydrazoic acid) or a formal [2+2] cycloaddition/fragmentation sequence with reagents like dimethyl acetylenedicarboxylate (DMAD) to achieve 2+ carbon ring expansions [39].
- Library Production & Characterization: Apply this two-phase strategy (C-H oxidation followed by ring expansion) divergently to a single natural product to create a library of analogs with varied medium-sized rings (7-11 membered). Characterize all products via NMR, HRMS, and X-ray crystallography. Analyze the library's collective properties (e.g., Fsp³, 3D shape) to confirm its occupation of distinct chemical space [39].
Protocol for Computational Optimization of a Purchasable Screening Library
This protocol, based on the BonMOLière method, creates a high-performance subset from commercially available compounds to maximize potential for discovering novel bioactivity [94].
- Define Source Pool: Start with a large, filtered subset of purchasable compounds (e.g., the "in-stock" and "anodyne" subsets from ZINC20, ensuring compounds are available and non-promiscuous) [94].
- Apply Drug-Likeness Filters: Filter compounds based on physicochemical property rules (e.g., molecular weight ≤ 900 Da, logP ≤ 4, hydrogen bond donors/acceptors within range) to create a Pool of Candidate Compounds (PCC) [94].
- Predict Biological Annotation: Use a validated 2D similarity-based target prediction model against a comprehensive target database (e.g., ChEMBL) to assign predicted protein targets to each compound in the PCC [94].
- Define & Maximize a Fitness Function: Use a genetic algorithm to select the optimal library subset. The fitness function should combine:
- Target Space Coverage: Maximize the number of distinct predicted targets covered by the library.
- Target Novelty: Prioritize compounds with predicted activity against understudied ("novel") targets.
- Chemical Diversity: Incorporate a measure of structural dissimilarity among selected compounds.
- Drug-Likeness Score: Optimize the average quantitative estimate of drug-likeness (QED) score of the set [94].
- Library Assembly & Validation: Select the top-performing compound set from the optimization. The resulting small-to-medium-sized library (e.g., 1,000-15,000 compounds) is predicted to have a significantly higher chance of yielding hits against arbitrary novel targets compared to a randomly selected library of the same size [94].
Visualizing Strategies and Metrics
Workflow for Assessing the Novelty Gap
Title: Strategies to Bridge the Novelty Gap Workflow
Natural Product Chemical Diversification Strategy
Title: NP Diversification via C-H Oxidation and Ring Expansion
Hierarchy of Scaffold Analysis for Novelty Assessment
Title: Scaffold Tree Hierarchy for Diversity Analysis
Computational Optimization of a Screening Library
Title: Workflow for Computational Library Optimization
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Key Reagents, Databases, and Tools for Novelty-Gap Research
| Category | Item / Resource | Function in Research | Relevant Source / Example |
|---|---|---|---|
| Chemical Libraries & Sources | ZINC Database | Primary public aggregator of purchasable compounds from multiple vendors; enables virtual screening and library analysis. | Used as source pool in comparative studies and for optimized library design [25] [94]. |
| Traditional Chinese Medicine Compound Database (TCMCD) | Database of natural product-derived molecules; serves as a benchmark for complex, NP-like chemical space. | Used as a comparator for scaffold diversity and complexity [25]. | |
| Vendor Catalogs (Enamine, ChemBridge, etc.) | Source of physical compounds for high-throughput screening (HTS); diversity varies significantly by vendor. | Analyzed for scaffold diversity and property distributions [25] [54]. | |
| Synthesis & Diversification | C-H Activation Reagents (e.g., Electrochemical cells, CrO₃/pyridine) | Enable late-stage, site-selective functionalization of complex cores (like NPs) to introduce handles for diversification. | Key to the C-H oxidation/ring expansion strategy for accessing novel space [39]. |
| Ring Expansion Reagents (e.g., Hydroxylamine, TiCl₄, DMAD) | Transform functional handles (ketones, alcohols) to expand rings, generating medium-sized rings from NPs. | Critical for synthesizing underexplored chemotypes like medium-sized lactams [39]. | |
| Computational Analysis & Design | Cheminformatics Suites (e.g., Pipeline Pilot, MOE) | Generate molecular descriptors, standardize structures, perform scaffold decomposition (Murcko, Scaffold Tree). | Essential for protocol steps like library standardization and scaffold analysis [25]. |
| Target Prediction Models (2D Similarity-based) | Predict the potential protein targets of compounds based on structural similarity to known actives. | Used to biologically annotate libraries and optimize for target coverage/novelty [94]. | |
| Genetic Algorithm Optimization Software | Iteratively select compound subsets that maximize a multi-parameter fitness function (diversity, novelty, etc.). | Core engine for creating optimized screening libraries like BonMOLière [94]. | |
| AI Generative Models (Chemical Language Models) | De novo design of novel molecular structures conditioned on desired properties. | Emerging tool for exploring beyond confined chemical spaces [109]. | |
| Novelty Assessment Metrics | Scaffold Diversity Metrics (PC₅₀C) | Quantifies how evenly compounds are distributed across scaffolds; lower value indicates higher diversity. | Primary metric for comparing library structural novelty [25]. |
| Continuous Distance Functions (e.g., LND, AMD, Magpie) | Provide a continuous, quantifiable measure of similarity/difference between two compounds or materials. | Overcomes limitations of binary novelty assessments; allows nuanced gap analysis [110] [111]. | |
| Visualization | Tree Map / SAR Map Software | Creates intuitive, space-filling maps of scaffold or compound distributions based on similarity. | Helps visualize the coverage and clustering of chemical space for a given library [25]. |
Conclusion
The comparative analysis reveals natural products and purchasable synthetic libraries not as competitors but as complementary, synergistic pillars of drug discovery. Natural products offer unparalleled structural complexity, evolutionary-validated biological relevance, and unique entry points into challenging target spaces like protein-protein interactions. In contrast, modern purchasable libraries provide vast, drug-like chemical space with excellent synthetic tractability, characterized purity, and defined intellectual property pathways. The future lies in strategic integration: using natural product scaffolds to inspire the design of novel synthetic libraries, enriching screening collections with privileged natural product-derived chemotypes, and employing advanced cheminformatic and AI tools to navigate the combined chemical space intelligently. As the compound library market continues its robust growth, the most successful discovery campaigns will be those that adeptly leverage the unique and evolving strengths of both nature and synthesis to illuminate new paths to therapeutic breakthroughs [citation:1][citation:5][citation:8].
References
- 1. Natural product diversity and its role in chemical biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3110584/]
- 2. Time-Dependent Comparison of the Structural Variations ... [https://www.mdpi.com/1422-0067/25/21/11475]
- 3. Selecting, Acquiring, and Using Small Molecule Libraries for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4687755/]
- 4. Unifying principles for the design and evaluation of natural ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08017c]
- 5. A divergent intermediate strategy yields biologically ... [https://www.nature.com/articles/s41557-024-01458-4]
- 6. NPBS Atlas: a comprehensive data resource for exploring the biological... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01116-y]
- 7. advancements in molecular glues derived from Research ... natural [https://pubmed.ncbi.nlm.nih.gov/40256709/]
- 8. Screening Libraries. [https://www.chemdiv.com/catalog/screening-libraries/]
- 9. Compound Libraries [https://enamine.net/compound-libraries]
- 10. Compound Libraries [https://selvita.com/drug-discovery/small-molecules/expertise-areas/chemistry/compound-libraries]
- 11. Emerging strategies to enhance microbial natural product– ... [https://www.sciencedirect.com/science/article/pii/S0958166925001132]
- 12. The evolution of chemical collections and drug-like diversity [https://www.ddw-online.com/the-evolution-of-chemical-collections-and-drug-like-diversity-36277-202509/]
- 13. Combinatorial Chemistry in Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5645069/]
- 14. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 15. Designing novel building blocks is an overlooked strategy ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644614003894]
- 16. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 17. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 18. Global Compound Libraries Market Research Report [https://www.wiseguyreports.com/reports/compound-libraries-market]
- 19. Compound Libraries Consumer Trends [https://www.datainsightsmarket.com/reports/compound-libraries-1028816]
- 20. Compound Libraries Market CAGR 2026-2033 | Key ... [https://www.linkedin.com/pulse/compound-libraries-market-cagr-2026-2033-key-highlights-s624e/]
- 21. Compound Management Market Size 2024 to 2034 [https://www.towardshealthcare.com/insights/compound-management-market-sizing]
- 22. Compound Management Market Size to Hit USD ... [https://www.precedenceresearch.com/compound-management-market]
- 23. Compound Management Market Size & Industry Report ... [https://www.fortunebusinessinsights.com/compound-management-market-112148]
- 24. From Hits to Leads: Improving Early Drug Discovery With ... [https://www.technologynetworks.com/drug-discovery/blog/from-hits-to-leads-improving-early-drug-discovery-with-better-tools-404996]
- 25. Comparative analyses of structural features and scaffold ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0212-4]
- 26. Ultra-large library screening with an evolutionary algorithm ... [https://www.nature.com/articles/s42004-025-01758-x]
- 27. Natural Product-Derived Drugs: Structural Insights into Their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467776/]
- 28. Engineering is evolution: a perspective on design ... [https://www.nature.com/articles/s41467-024-48000-1]
- 29. Evolutionary tinkering vs. rational engineering in the times of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6087506/]
- 30. Innate functions of natural products: A promising path for ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523423007158]
- 31. Genetic controllers for enhancing the evolutionary ... [https://www.nature.com/articles/s41467-025-63627-4]
- 32. Comparing Technological Development and Biological ... [https://www.sciencedirect.com/science/article/pii/S2405471220300740]
- 33. Active learning driven prioritisation of compounds from on ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00343h]
- 34. OTAVAchemicals, Ltd - synthetic organic compounds for ... [https://www.otavachemicals.com/]
- 35. Designing diverse and high-performance proteins with a large ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013119]
- 36. Synthetic gene circuit evolution: Insights and opportunities ... [https://www.sciencedirect.com/science/article/pii/S2451945624002198]
- 37. mcule database [https://mcule.com/database/]
- 38. Diversity Libraries [https://chembridge.com/diversity-and-pre-plated-libraries/diversity-libraries/]
- 39. A general strategy for diversifying complex natural products ... [https://www.nature.com/articles/s41467-019-11976-2]
- 40. Building Blocks Catalog [https://enamine.net/building-blocks/building-blocks-catalog]
- 41. Exploring chemical space, scaffold diversity, and activity ... [https://pubmed.ncbi.nlm.nih.gov/38690773/]
- 42. Finding Constellations in Chemical Space Through Core ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00510/full]
- 43. Chemical space visual navigation in the era of deep ... [https://www.sciencedirect.com/science/article/pii/S1359644625001059]
- 44. Barcode-free hit discovery from massive libraries enabled ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12559350/]
- 45. Fragment libraries from large synthetic compounds and ... [https://chemrxiv.org/engage/chemrxiv/article-details/67a3b0db81d2151a024b2e66]
- 46. Natural product fragment combination to performance ... [https://www.nature.com/articles/s41467-021-22174-4]
- 47. Rationally minimizing natural product libraries using mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11915828/]
- 48. High-Throughput Screening (HTS): A Simple Guide ... [https://www.chemcopilot.com/blog/hts-material-discovery]
- 49. High-Throughput Screening of Potent Drug-like Molecules ... [https://pubmed.ncbi.nlm.nih.gov/40530895/]
- 50. Computational approaches streamlining drug discovery [https://www.nature.com/articles/s41586-023-05905-z]
- 51. A Researcher's Guide to Optimizing High-throughput ... [https://www.ranomics.com/navigating-the-hurdles-a-researchers-guide-to-optimizing-high-throughput-screens-library-design-and-protein-engineering]
- 52. Advances in high-throughput drug screening based on ... [https://www.sciencedirect.com/science/article/pii/S2090123225006897]
- 53. An Assessment of Chemical Diversity in Microbial Natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12464764/]
- 54. Evolution of commercially available compounds for HTS [https://www.sciencedirect.com/science/article/pii/S1359644618302423]
- 55. Selection and Characterization of Botanical Natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6658353/]
- 56. Advances in Chromatographic Techniques for the ... [https://www.chromatographyonline.com/view/advances-in-chromatographic-techniques-for-the-extraction-and-characterization-of-plant-based-bioactive-compounds]
- 57. A review of modern and conventional extraction techniques and their applications for extracting phytochemicals from plants - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2468227623000443]
- 58. Evaluating and evolving a screening library in academia [https://pmc.ncbi.nlm.nih.gov/articles/PMC8131249/]
- 59. Building Natural Product–Based Libraries for Drug Discovery [https://link.springer.com/article/10.1007/s43450-024-00540-9]
- 60. Strategic Sourcing Guide: 10 Steps to Improve Supply Chain [https://learn.g2.com/strategic-sourcing]
- 61. Marketplace Business Model vs Business... - XB Software Aggregator [https://xbsoftware.com/blog/marketplace-vs-aggregator-business-model/]
- 62. Review Modern methods to produce natural-product libraries [https://www.sciencedirect.com/science/article/abs/pii/S1367593102003381]
- 63. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 64. Natural product derived privileged scaffolds in drug discovery [https://www.sciencedirect.com/science/article/abs/pii/S1367593118301455]
- 65. Creating and screening natural product libraries - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8494140/]
- 66. The use of isolated natural products as scaffolds for ... [https://pubs.rsc.org/en/content/articlelanding/2016/np/c5np00121h]
- 67. Structure-based virtual screening of vast chemical space ... [https://www.sciencedirect.com/science/article/pii/S0959440X24000563]
- 68. The Most Destructive Supply Chain Challenges in 2025 ... [https://www.z2data.com/insights/most-destructive-supply-chain-challenges-2025-how-to-navigate]
- 69. Cost and Resilience: The New Supply Chain Challenge [https://www.bcg.com/publications/2025/cost-resilience-new-supply-chain-challenge]
- 70. Top 10 Hardest-to-Source Products in 2025 [https://supplier.io/resources/blog/top-10-hardest-to-source-products-in-2025-is-your-supply-chain-at-risk]
- 71. The Challenges of SCM and How to Overcome Them in 2025 [https://pscs.jabil.com/what-we-think/blog/the-challenges-of-scm-and-how-to-overcome-them-in-2025.html]
- 72. Supply Chain Challenges in 2025 & How to Overcome Them [https://www.extensiv.com/blog/supply-chain-management/challenges]
- 73. Seven Year Itch: Pan-Assay Interference Compounds ( ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5778390/]
- 74. Problems that Can Occur when Assaying Extracts to Pure ... [https://www.sciencedirect.com/science/article/pii/S0011393X21000230]
- 75. Gains from no real PAINS: Where 'Fair Trial Strategy' stands in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8642439/]
- 76. compounds in early Filtering : is it... promiscuous drug discovery [https://sussexdrugdiscovery.wordpress.com/2016/03/14/filtering-promiscuous-compounds-in-early-drug-discovery-is-it-a-good-idea/]
- 77. Comparative analyses of structural features and scaffold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5400773/]
- 78. Polyphenolic promiscuity , inflammation-coupled selectivity: Whether... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9634583/]
- 79. The Pan-Canadian Chemical Library: A Mechanism to ... [https://www.nature.com/articles/s41597-024-03443-5]
- 80. druglikeFilter 1.0: An AI powered filter for collectively ... [https://www.sciencedirect.com/science/article/pii/S2095177925001157]
- 81. Structural diversity of biologically interesting datasets [https://pmc.ncbi.nlm.nih.gov/articles/PMC3179739/]
- 82. chemical transformations to create lead-like libraries [https://www.sciencedirect.com/science/article/pii/S1359644613003802]
- 83. Development and Testing of Druglike Screening Libraries [https://pmc.ncbi.nlm.nih.gov/articles/PMC6756465/]
- 84. Compound Library Design [https://www.sygnaturediscovery.com/drug-discovery/computer-aided-drug-design/library-design/]
- 85. Optimizing drug design by merging generative AI with a ... [https://www.nature.com/articles/s42004-025-01635-7]
- 86. Driving Drug Design Using DNA Encoded Libraries [https://www.amgen.com/stories/2025/04/driving-drug-design-using-dna-encoded-libraries]
- 87. The Sound of Silence: Activating Silent Biosynthetic Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4557003/]
- 88. Recent Advances in Silent Gene Cluster Activation ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2021.632230/full]
- 89. Natural Products for Drug Discovery in the 21st Century [https://pmc.ncbi.nlm.nih.gov/articles/PMC6032166/]
- 90. Emerging Pests and Pathogens Research: Ithaca, NY [https://www.ars.usda.gov/research/project/?accnNo=440840]
- 91. Bridging chemical space and biological efficacy: advances ... [https://link.springer.com/article/10.1007/s13659-025-00521-y]
- 92. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 93. Protocol for an automated virtual screening pipeline including ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12639438/]
- 94. BonMOLière: Small-Sized Libraries of Readily ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8346018/]
- 95. Using structural diversity to measure the complexity of technologies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6528977/]
- 96. a searchable database of available natural compounds [https://pmc.ncbi.nlm.nih.gov/articles/PMC1347494/]
- 97. 67 million natural product-like compound database ... [https://www.nature.com/articles/s41597-023-02207-x]
- 98. Chemical Evolution of Natural Product Structure - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8895405/]
- 99. Natural product synthesis | Nature Communications [https://www.nature.com/subjects/natural-product-synthesis/ncomms]
- 100. Diversity-Orientated Synthesis and Biological Properties of ... [https://www.mdpi.com/1420-3049/29/1/249]
- 101. BJOC - Recent advances in controllable/divergent synthesis [https://www.beilstein-journals.org/bjoc/articles/21/73]
- 102. Screen Compound Libraries 2025-2033 Trends [https://www.archivemarketresearch.com/reports/screen-compound-libraries-26868]
- 103. Challenges in natural product-based drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10613855/]
- 104. Time-Dependent Comparison of the Structural Variations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11547171/]
- 105. Natural products - Latest research and news [https://www.nature.com/subjects/natural-products]
- 106. Comparative advantage and efficacy of natural products for ... [https://pubmed.ncbi.nlm.nih.gov/40783741/]
- 107. Natural Products in Health Promotion and Disease ... [https://www.mdpi.com/journal/pharmaceuticals/special_issues/5I5AT0PDD0]
- 108. When target identification meets single-cell multiomics [https://www.sciencedirect.com/science/article/pii/S2211383522003732]
- 109. Tackling the issue of confined chemical space with AI ... [https://pubmed.ncbi.nlm.nih.gov/40887314/]
- 110. Continuous Uniqueness and Novelty Metrics for ... [https://arxiv.org/html/2510.12405v1]
- 111. Geographic-style maps with a local novelty distance help ... [https://www.nature.com/articles/s41598-025-10672-0]