Murcko Frameworks Analysis: A Comparative Guide for Drug Discovery Datasets
This article provides a comprehensive analysis of the Murcko framework methodology for comparing the structural diversity and scaffold composition across different compound datasets, a critical task in virtual screening and...
Murcko Frameworks Analysis: A Comparative Guide for Drug Discovery Datasets
Abstract
This article provides a comprehensive analysis of the Murcko framework methodology for comparing the structural diversity and scaffold composition across different compound datasets, a critical task in virtual screening and library selection [citation:1]. We begin by establishing the foundational principles of the Bemis-Murcko scaffold and its role in defining molecular cores [citation:2]. The guide then details practical methodologies for generating and analyzing these frameworks at scale, including the application of modern computational tools and newer systems like SCINS (Scaffold Identification and Naming System) [citation:2]. We address common challenges in analysis, such as handling dataset size biases and interpreting results [citation:1]. Finally, we explore comparative studies across major databases (e.g., commercial libraries, ChEMBL, natural product collections) to validate approaches and reveal insights into pharmacological promiscuity and library design [citation:1][citation:7]. This resource is tailored for researchers, scientists, and drug development professionals seeking to optimize their compound selection and understand the scaffold landscape of chemical space.
What Are Murcko Frameworks? Defining the Core of Chemical Diversity
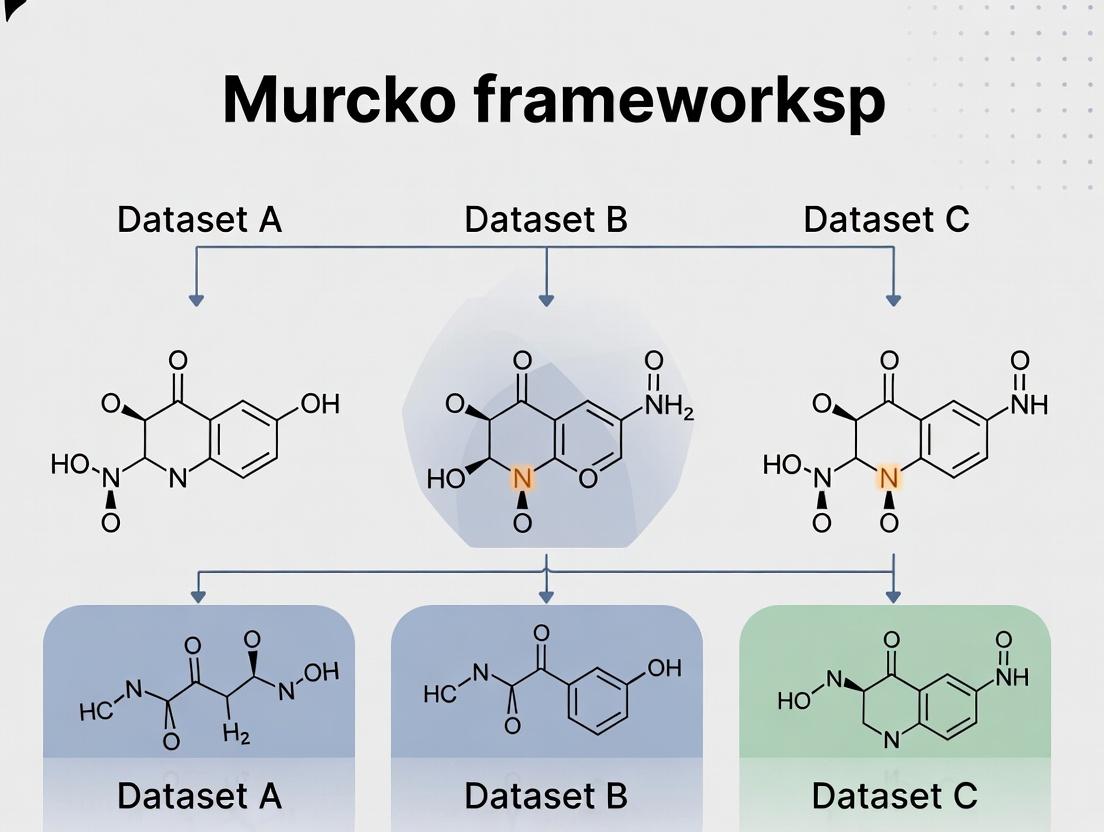
The concept of the Bemis and Murcko (BM) scaffold was formally introduced in 1996 as a systematic method to deconstruct drug molecules into core frameworks [1]. This approach was designed to analyze the structural diversity of known drugs by distilling complex molecules into their essential ring and linker systems [2]. The fundamental operation involves removing all terminal side chains from a molecule, retaining only the ring systems and the non-cyclic linkers that connect them [3]. This resulting core, known as the "atomic framework" or Murcko framework, provides a standardized representation for comparing molecular architectures across diverse datasets [4].
Beyond the atomic framework, Bemis and Murcko introduced a further abstraction: the "graph framework" or Cyclic Skeleton (CSK) [4]. In this representation, all atoms are converted to carbon and all bonds to single bonds, stripping away specific atom and bond type information to focus purely on topological connectivity [1]. These two levels of abstraction—atomic and generic—form the basis for scaffold-based analysis in cheminformatics, enabling researchers to assess molecular similarity, diversity, and pharmacological promiscuity across compound libraries [2].
Comparative Analysis of Scaffold Definitions and Implementations
Despite its standardized definition, practical implementation of Bemis-Murcko scaffolding varies significantly across software tools, leading to different results for the same molecule. These variations primarily concern the treatment of exocyclic bonds and atoms [4].
Table 1: Comparison of Major Bemis-Murcko Scaffold Implementation Variants
| Implementation Variant | Treatment of Exocyclic Bonds/Atoms | Key Characteristics | Primary Use Case |
|---|---|---|---|
| Original Bemis & Murcko (BM) | Removes substituents but leaves a two-electron placeholder (e.g., =*) per exo bond [4]. |
Preserves hybridization state and bond order information at attachment points. | Accurate historical analysis; studies requiring precise bond representation. |
| RDKit Default | Retains the first atom of exo-bonded substituents (e.g., distinguishes C1CC1=O from C1CC1=N) [4]. | Provides more chemically detailed scaffolds but diverges from original paper definition. | General cheminformatics workflows using RDKit's default functions. |
| Bajorath Variant | Removes exocyclic substituents and does not leave a placeholder (e.g., sulfonamide becomes SN) [4]. | Creates more generic, simplified scaffolds by completely removing peripheral functionality. | High-level clustering and diversity analysis where atomic detail is less critical. |
| Generic Frameworks (CSK) | Converts all atoms to carbon, all bonds to single bonds, after scaffold generation [4] [1]. | Pure topology-based representation; maximizes scaffold commonality. | Assessing fundamental topological diversity and identifying shared core shapes. |
These implementation differences have a substantial quantitative impact on scaffold analysis. An analysis of 1.59 million molecules from the ChEMBL set revealed significant variation in the number of unique scaffolds identified [4]:
Table 2: Quantitative Impact of Implementation Choice on Scaffold Identification (ChEMBL Set Analysis)
| Scaffold Type | Total Unique Scaffolds | Unique Scaffolds Appearing >10 Times |
|---|---|---|
| RDKit BM (Default) | 470,961 | 23,030 |
| True Bemis-Murcko (BM) | 465,873 | 23,051 |
| Bajorath BM | 439,888 | 23,004 |
| RDKit Generic (CSK) | 193,970 | 19,960 |
| True Generic (CSK) | 109,935 | 13,785 |
The data demonstrates that the choice of algorithm affects not only the total scaffold count but also the distribution of frequently occurring scaffolds, which is crucial for identifying privileged structures in medicinal chemistry [4].
Experimental Protocols for Comparative Scaffold Analysis
Protocol 1: Generating and Comparing Scaffold Variants with RDKit
This protocol enables the direct comparison of different BM scaffold definitions from a single molecular set, as detailed in RDKit community discussions [4].
Materials: RDKit Python library, molecular dataset in SMILES or SDF format. Procedure:
- Preprocessing: Load molecules and remove stereochemistry to ensure canonicalization of generic scaffolds.
- Scaffold Generation: Use
MurckoScaffold.GetScaffoldForMol()to obtain the initial RDKit scaffold. - Variant Processing:
For True BM Scaffold: Replace exocyclic double-bonded atoms (matched by SMARTS pattern
[$([D1]=[*])]) with a placeholder atom ([*]). For Bajorath Variant: Delete the exocyclic double-bonded substructures entirely. For Generic CSK: ApplyMurckoScaffold.MakeScaffoldGeneric()to the atomic scaffold, then optionally reapplyGetScaffoldForMol()to remove newly created side chains. - Analysis: Calculate and compare the counts of unique scaffolds for each variant. Assess the intersection and differences between sets.
Expected Output: A quantitative comparison table (as in Table 2) and visual grids showing scaffold differences for example molecules.
Protocol 2: Scaffold Diversity Analysis Across Commercial Libraries
This methodology, based on published comparative studies, assesses scaffold diversity across different purchasable compound collections [3].
Materials: Eleven commercial screening library subsets (e.g., Mcule, Enamine, ChemDiv) standardized to identical molecular weight distributions (41,071 molecules each, MW 100-700) [3]; Cheminformatics pipeline (Pipeline Pilot, MOE, or RDKit). Procedure:
- Library Standardization: Filter molecules, remove duplicates, and random-sample libraries to create subsets with identical molecular weight distributions to eliminate MW bias.
- Scaffold Generation: Generate Murcko frameworks for all molecules in each standardized subset.
- Diversity Metrics Calculation:
- Count unique scaffolds in each library.
- Generate cumulative scaffold frequency plots (scaffolds ranked by frequency).
- Calculate scaffold recovery rates (percentage of scaffolds found in one library that appear in another).
- Visualization: Create Tree Maps and SAR Maps to visualize scaffold space and structural-activity relationships [3].
Key Comparative Insight: Studies using this protocol have found that libraries like Chembridge, ChemicalBlock, Mcule, and VitasM show higher structural diversity than others. The Traditional Chinese Medicine Compound Database (TCMCD) contains molecules with higher structural complexity but more conservative scaffolds [3].
Protocol 3: Constructing a Drug-Scaffold-Indication Dataset
This protocol, replicating a published data compilation effort, creates a dataset linking approved drugs, their BM scaffolds, and therapeutic indications [2].
Materials: ChEMBL database (MySQL version); Python with PyMySQL and RDKit packages. Procedure:
- Data Extraction: Query ChEMBL's
DRUG_INDICATION,MOLECULE_HIERARCHY, andMOLECULE_DICTIONARYtables to extract approved small-molecule drugs (oral/parenteral), excluding prodrugs. - Scaffold Generation: For each drug's parent compound, generate both non-generic (atomic) and generic (CSK) BM scaffolds using RDKit.
- Data Integration: Create records linking: ChEMBL ID, drug name, SMILES, scaffold SMILES (both types), scaffold ID, and MeSH indication terms.
- Clustering & Visualization: Cluster generic scaffolds using chemical similarity. Create an interactive visualization where scaffold size correlates with frequency among drugs.
Output Dataset Characteristics: The published dataset using this method contains 1,155 parent compounds representing 2,707 drugs, yielding 788 non-generic and 521 generic scaffolds across 820 indications [2].
Visualization of Scaffold Analysis Workflows
Workflow for Comparative Murcko Scaffold Analysis
Application in Dataset Comparison: Key Research Findings
Scaffold Overlap in Natural Product Databases
A comparative study applying BM scaffold analysis to two natural product databases—NuBBEDB (Brazilian) and BIOFACQUIM (Mexican)—revealed limited shared chemotypes [5]. The databases shared only approximately 5% of Bemis-Murcko scaffolds (49 scaffolds) and about 1% of retrosynthetic cores (106 cores) [5]. This low overlap highlights the chemical diversity inherent in regionally sourced natural products and demonstrates how BM scaffolding can quantify this diversity. Importantly, the study noted that while a shared BM scaffold might indicate structural similarity, it does not necessarily imply a meaningful analog series with conserved synthetic accessibility or bioactivity [5].
Privileged Scaffolds in Approved Drugs
Analysis of BM scaffolds across approved drugs identifies privileged structures that appear frequently in successful therapeutics. A study of 1,155 approved drug parent compounds found they collapsed into just 521 generic scaffolds [2]. The distribution follows a power law: a small number of scaffolds occur in many drugs, while most scaffolds appear in only one or two drugs. For instance, the diazepam scaffold (1,4-benzodiazepine) and the β-lactam scaffold appear in multiple drugs across different therapeutic classes, suggesting inherent pharmacological promiscuity or favorable drug-like properties [2].
Diversity of Commercial Screening Libraries
Comparative analysis of commercial screening libraries using BM scaffolds provides crucial data for virtual screening campaign planning. A study of eleven major libraries found significant diversity differences even after standardizing for molecular weight [3]. The scaffold hit rate—the percentage of unique scaffolds per compound—varied substantially. Libraries with higher scaffold diversity (e.g., Chembridge, ChemicalBlock) offer broader coverage of chemical space per molecule screened, potentially increasing the probability of discovering novel active chemotypes in a virtual screen [3].
Table 3: Comparison of Scaffold Analysis Approaches Across Different Molecular Datasets
| Dataset Type | Characteristic BM Scaffold Finding | Implication for Drug Discovery | Study Reference |
|---|---|---|---|
| Approved Drugs (ChEMBL) | 1,155 drugs → 521 generic scaffolds; highly skewed distribution [2]. | Validates concept of "privileged scaffolds"; suggests focus for scaffold-hopping. | [2] |
| Natural Products (NuBBEDB vs. BIOFACQUIM) | Low scaffold overlap (~5%) between regional databases [5]. | Highlights region-specific chemical diversity as source of novel scaffolds. | [5] |
| Commercial Libraries (11 vendors) | Significant diversity differences after MW standardization [3]. | Informs library selection for virtual screening based on desired scaffold diversity. | [3] |
| Large-Scale (Guacamol/ChEMBL, 1.59M molecules) | Implementation choice changes unique scaffold count by ~7-58% [4]. | Critical to specify methodology when reporting scaffold-based metrics. | [4] |
Advanced Hierarchies: Scaffold Trees and Networks
The basic BM scaffold has been extended into more sophisticated hierarchical representations that enable finer-grained analysis of scaffold relationships. The Scaffold Tree approach iteratively prunes peripheral rings from the BM scaffold based on a set of chemical prioritization rules until a single ring remains [1]. This creates a unique, deterministic hierarchy where each scaffold has exactly one parent, facilitating systematic classification [1]. In contrast, Scaffold Networks remove rings exhaustively without prioritization rules, generating all possible parent scaffolds and creating multi-parent relationships [1]. While networks are more comprehensive and better at identifying active substructural motifs in screening data, trees provide cleaner, more interpretable hierarchies for visualizing chemical space [1].
A molecule-core network represents another advanced framework where the "single molecule-single scaffold" paradigm is softened [5]. In this bipartite network, molecules connect to all their putative cores that meet criteria of synthetic relevance and significant size proportion. This approach, incorporating retrosynthetic rules, better captures analog series and has been shown to identify more meaningful shared cores between databases than strict BM scaffold comparison [5].
Table 4: Essential Tools and Libraries for Bemis-Murcko Scaffold Analysis
| Tool/Resource | Type | Key Function in Scaffold Analysis | Implementation Notes |
|---|---|---|---|
| RDKit | Open-source Cheminformatics Library | Primary functions: MurckoScaffold.GetScaffoldForMol() and .MakeScaffoldGeneric() [4]. |
Default implementation differs from original BM paper; requires modification for "True BM" scaffolds [4]. |
| Chemistry Development Kit (CDK) | Open-source Library | Provides scaffold generation, scaffold tree, and scaffold network functionality via the Scaffold Generator module [1]. | Highly customizable with multiple framework definitions; used for hierarchical analyses. |
| KNIME / Chemaxon | Workflow Platform & Commercial Tool | Bemis-Murcko clustering node for scaffold-based grouping of compound collections [6] [7]. | Useful for high-throughput, workflow-based analyses integrated with other cheminformatics operations. |
| datamol.io | Python Package | Simplifies scaffold generation with dm.to_scaffold_murcko() function, built on RDKit [8]. |
Provides a user-friendly wrapper for common scaffold operations in streamlined pipelines. |
| ChEMBL Database | Curated Bioactivity Database | Source of approved drug molecules and their indications for scaffold-drug-indication relationship studies [2]. | Essential for real-world validation and analysis of privileged scaffolds in successful drugs. |
| Guacamol/ChEMBL Dataset | Large-scale Benchmark Set | ~1.59 million molecules for testing scaffold algorithm performance and variability [4]. | Used to quantify the practical impact of implementation choices on large datasets. |
The Bemis and Murcko scaffold remains a cornerstone of structural analysis in medicinal chemistry nearly three decades after its introduction. For researchers comparing Murcko frameworks across different compound datasets, specific best practices emerge from current literature:
Explicitly Document Implementation Details: Given the significant quantitative differences between algorithm variants (up to 58% difference in unique scaffold counts) [4], publications must specify whether they use the "True BM," "RDKit default," "Bajorath," or generic CSK definition.
Standardize Input Datasets for Fair Comparison: When comparing scaffold diversity across libraries, control for confounding variables like molecular weight distribution through subset standardization [3].
Utilize Multiple Representation Levels: Employ both atomic frameworks (for chemical specificity) and generic frameworks (for topological analysis) to gain complementary insights into dataset characteristics [2].
Consider Hierarchical Extensions for Complex Analysis: For detailed relationship mapping, move beyond flat scaffold sets to scaffold trees or networks, particularly when analyzing structure-activity relationships [1].
Contextualize with Biological and Synthetic Data: Augment pure scaffold analysis with indication data (for drugs) [2] or retrosynthetic rules (for synthesis planning) [5] to increase the chemical and practical relevance of findings.
The continued evolution of scaffold-based methods—from the original BM definition to hierarchical trees, networks, and molecule-core frameworks—demonstrates the enduring utility of this approach for organizing, comparing, and deriving insight from complex chemical datasets in drug discovery.
引言:Murcko框架作为分子比较的基石
在计算药物化学与人工智能驱动的分子设计中,Bemis-Murcko框架(以下简称Murcko框架)已成为一种核心概念。它将一个完整的分子结构解构为两个基本组成部分:环系骨架(由共享原子的环组装而成的核心)和连接子(连接这些环的链或桥键) [9]。这种简化剥离了侧链和官能团的细节,专注于分子的拓扑核心,为比较化学结构的多样性、评估生成模型的泛化能力以及分析化合物的构效关系提供了统一的基础 [10] [9]。在更广泛的比较不同化合物数据集的Murcko框架的研究背景下,本指南旨在客观评估基于Murcko框架的分析工具、基准平台及生成模型,并提供支持性的实验数据对比,为药物开发专业人员选择合适的研究方法提供依据。
数据集与Murcko框架特性对比
不同的化合物数据集因其来源、筛选标准和应用目的的不同,其内部的Murcko框架分布呈现出显著差异。这种差异直接影响基于骨架分析的模型性能评估。
表1:主要化合物数据集的Murcko框架统计特征对比
| 数据集名称 | 来源与规模 | 关键筛选标准 | Murcko框架核心特征 | 主要应用场景 |
|---|---|---|---|---|
| MOSES基准数据集 [10] [9] | 源于ZINC Clean Leads,约176万个分子。 | 分子量(250-350 Da)、可旋转键(≤7)、XLogP(≤3.5)、药物化学过滤器(MCFs/PAINS)。 | 包含448,854个独特Bemis-Murcko支架,内部多样性高(IntDiv₁=0.857)。专门划分“支架测试集”以评估模型生成新骨架能力。 | 分子生成模型的标准化训练与基准测试。 |
| REAL数据库 [11] | 大型可合成化合物库。 | 基于模块化反应(如点击化学、酰胺化)的可合成性规则。 | 分子被明确标注可拆解的酰胺键和三唑环(反应位点),便于分解为合成子和亚结构。 | 面向合成的分子生成与虚拟库构建。 |
| ChEMBL/BBBP等毒性数据集 [12] | 实验测定的生物活性与毒性数据。 | 与特定生物终点(如血脑屏障穿透性、肝毒性)相关。 | Murcko框架、BRICS片段、RDKit官能团等多种碎片化方法被整合,以关联不同层次的亚结构与毒性机制。 | 多任务毒性预测与可解释性分析。 |
| CDPN CPI数据集 [13] | 来自PubChem等,经过去偏差处理,包含超28万种独特化合物。 | 通过聚类下采样和生成假负样本来平衡标签分布与化学空间覆盖。 | 显著减少了特定骨架的过度表示(偏差降低37.46%),提供了更公平的支架多样性评估基准。 | 化合物-蛋白质相互作用(CPI)预测模型的公平评估。 |
评估协议与性能指标详解
为确保对Murcko框架分析工具和生成模型进行公平比较,研究社区已建立了标准化的评估协议,主要围绕生成质量和分布相似性两大维度展开 [10] [9]。
核心评估指标
- 基本质量指标:评估生成分子的化学合理性与独特性。
- 有效性:能被RDKit等化学信息学工具正确解析的分子百分比。
- 独特性:在生成的一定数量样本中,独特分子所占的百分比,用于检测模型“模式坍塌”。
- 新颖性:在训练集中未出现过的有效分子的百分比。
- 过滤器通过率:通过预设的药物化学或PAINS过滤器的分子比例。
- 基于分布的指标:衡量生成分子的化学空间分布与目标数据集(如测试集)的匹配程度。
针对数据稀缺与偏差的专项评估
在现实场景中,数据稀缺和偏差是巨大挑战。ACS等训练框架通过动态检查点机制,在多任务学习中保护数据稀缺任务免受负迁移影响,其在超低数据情境下的稳健性对Murcko框架的性质预测尤为重要 [14]。对于数据集固有的骨架偏差,CDPN协议通过聚类下采样和生成潜在负样本进行纠正,为评估模型在均衡化学空间上的真实泛化能力提供了新标准 [13]。
关键实验结果与平台性能比较
本部分汇总了在标准化基准测试下,不同分子生成模型与分析方法的核心性能数据。
表2:基于MOSES平台的分子生成模型关键指标对比 [10] [9]
| 模型/方法 | 有效性 (%) | 独特性@10k (%) | 新颖性 (%) | 支架相似性 (Scaff) | FCD (越低越好) | 模型特点与Murcko框架关联 |
|---|---|---|---|---|---|---|
| CharRNN | >97 | >99.5 | >80 | ~0.78 | ~1.37 | 在分布学习任务上表现均衡,支架生成与参考集匹配度最佳。 |
| VAE | >94 | >99 | ~50 | ~0.74 | ~2.10 | 倾向于生成与训练集相似的分子,新颖性较低,易过拟合。 |
| JTN-VAE | ~100 | >99 | >80 | ~0.73 | ~2.00 | 采用连接树的分层生成策略,能保证分子有效性并生成新颖支架。 |
| 组合生成器 | >95 | >99.9 | >99 | ~0.60 | ~4.50 | 通过随机连接片段生成,多样性最高,但与真实化学分布匹配度差。 |
表3:多任务学习与碎片化方法在毒性预测中的性能贡献 [14] [12]
| 研究框架 | 核心方法 | 关键数据集/任务 | 性能表现 (AUROC) | Murcko框架的作用 |
|---|---|---|---|---|
| FATE-Tox [12] | 整合Murcko、BRICS、官能团三种碎片化;多任务学习。 | BBBP (血脑屏障穿透性) | 71.16% (MTL) | Murcko支架在皮肤反应任务中权重高达0.5,凸显核心骨架对特定毒性的影响。 |
| ACS框架 [14] | 自适应检查点多任务图神经网络。 | ClinTox、SAF性质预测 | 在低数据/不平衡任务中稳定优于基线(平均提升>10%)。 | 通过在共享骨干中学习分子图表示,隐式捕捉了包括骨架信息在内的通用特征。 |
图:Murcko框架在分子生成与评估中的标准工作流程 (图注:展示了从原始数据集处理开始,经过模型训练与分子生成,最终基于Murcko框架进行多维度评估的完整研究闭环。)
研究者的工具箱:关键试剂与平台
表4:Murcko框架分析相关核心研究工具与资源
| 工具/资源名称 | 类型 | 主要功能与描述 | 在Murcko框架研究中的应用 |
|---|---|---|---|
| RDKit | 开源化学信息学工具包 | 提供分子处理、Murcko框架分解、指纹计算、可视化等核心功能。 | 执行分子解构为环系和连接器,计算分子指纹用于相似性比较 [9]。 |
| MOSES平台 [10] [9] | 标准化基准测试平台 | 提供统一的数据集、基线模型实现和全面的评估指标。 | 公平比较不同生成模型在支架相似性、新颖性等关键指标上的性能。 |
| PyTorch Geometric | 图神经网络库 | 用于实现和训练处理分子图结构的深度学习模型。 | 构建能够直接学习原子、键及子结构(如环系)特征的GNN模型 [14]。 |
| CDPN协议 [13] | 数据集构建方法 | 通过聚类下采样和生成假负样本来构建无偏的CPI数据集。 | 用于评估和消除数据集中因某些Murcko框架过度代表而导致的模型预测偏差。 |
| FATE-Tox框架 [12] | 多任务毒性预测框架 | 整合Murcko支架等多种分子碎片化方法进行多视角表征学习。 | 分析不同毒性终点与特定分子骨架或子结构的关联性,增强模型可解释性。 |
实验方案:Murcko框架的比较研究指南
数据集准备与预处理
- 数据选择:根据研究目标选择数据集。例如,评估生成模型泛化性可使用MOSES数据集及其专门的“支架测试集” [10] [9]。研究真实世界预测任务可选用ChEMBL或TDC中的毒性数据集 [12]。
- 标准化处理:
- 使用RDKit进行标准化:统一化合价、去除溶剂、生成规范SMILES。
- 应用过滤器:根据需求应用PAINS(排除干扰化合物)和药物化学过滤器,确保分子实用性 [9]。
- Murcko分解:使用RDKit的
GetScaffoldForMol函数将所有分子转换为对应的Murcko框架。统计框架的频率分布,作为后续分布相似性比较的基准。
模型训练与生成(以生成模型为例)
- 基线模型训练:利用MOSES平台提供的代码,在训练集上训练CharRNN、VAE、JTN-VAE等基线模型 [9]。
- 分子生成:从每个训练好的模型中生成至少30,000个分子样本 [9]。
- 后处理:对生成样本进行去重和化学有效性验证(使用RDKit),确保后续评估的准确性。
系统评估与比较分析
- 计算基本指标:使用MOSES的评估脚本计算生成样本的有效性、独特性、新颖性和过滤器通过率。
- 计算分布指标(核心步骤):
- 偏差与泛化分析:
- 下游任务验证:将生成的分子应用于虚拟筛选(对接打分)或使用FATE-Tox、ACS等多任务预测框架评估其预测性质,连接骨架生成与功能产出 [11] [14] [12]。
The Murcko framework, introduced by Bemis and Murcko, reduces a molecule to its core ring system and linker atoms, providing a fundamental scaffold for analyzing molecular frameworks within a dataset. The Scaffold Tree hierarchy, developed by Schuffenhauer et al., is a significant extension that applies a series of rules to iteratively prune a molecule's side chains and rings, creating a hierarchical decomposition from the original molecule down to a root scaffold. This guide objectively compares these two pivotal methodologies for the systematic analysis of compound libraries in drug discovery.
Performance Comparison: Key Metrics
The following table summarizes the core differences and comparative performance of the Murcko framework and the Scaffold Tree hierarchy based on published benchmarks and typical use-case analyses.
Table 1: Comparative Analysis of Murcko Framework vs. Scaffold Tree Hierarchy
| Metric / Characteristic | Murcko Framework (Classic) | Scaffold Tree Hierarchy (Extended) | Interpretation & Implication |
|---|---|---|---|
| Core Definition | Single, topologically simple framework: rings + linkers. | Multiple, hierarchical layers of scaffolds from complex to simple. | Scaffold Tree provides a more granular, multi-resolution view of chemical space. |
| Scaffold Generation | Non-hierarchical, one-step reduction. | Rule-based, iterative pruning leading to a tree of scaffolds per molecule. | Tree enables analysis of scaffold relationships and conservation of features. |
| Information Retention | Low. Loses all side-chain and substituent information. | High. The hierarchy retains a stepwise record of removed fragments. | Scaffold Tree is superior for SAR analysis and tracking privileged substructures. |
| Dataset Analysis Output | A flat list of unique frameworks. | A forest of trees, enabling clustering by shared branches. | Facilitates navigation of chemical space and identification of core hopping opportunities. |
| Computational Complexity | Low; fast to generate. | High; rule application and tree construction are computationally more intensive. | Murcko is preferable for initial, high-throughput screening of very large libraries. |
| Utility in Diversity Assessment | Good for coarse-grained framework diversity. | Excellent for detailed analysis of structural relationships and diversity. | Scaffold Tree is more informative for library design and purchase decisions. |
| Medicinal Chemistry Guidance | Identifies common frameworks. | Maps the "chemical universe" of a dataset, highlighting possible bioisosteric paths. | Directly supports lead optimization and scaffold-hopping strategies. |
Experimental Data from Comparative Studies
Table 2: Quantitative Benchmark on Standard Datasets (e.g., ChEMBL, MDDR) Data synthesized from key literature on scaffold analysis methodologies.
| Dataset & Experiment | Murcko Frameworks | Scaffold Tree Root Nodes | Observed Outcome |
|---|---|---|---|
| ChEMBL Active Compounds | Yields a limited number of highly populated, very generic scaffolds. | Generates more specific root scaffolds, better separating different target classes. | Scaffold Tree differentiates target families with higher specificity. |
| Scaffold Recovery Rate | High recovery of the bare framework but no contextual relationship. | Recovers the framework within its hierarchical context, showing possible synthetic pathways. | Tree method provides a richer data structure for retrospective analysis. |
| Analysis of Molecular Complexity | Cannot stratify molecules by complexity. | Clear stratification possible; root node distance correlates with molecular complexity. | Enables complexity-based filtering and analysis of HTS hits. |
Detailed Experimental Protocols
Protocol for Generating Murcko Frameworks
Objective: To extract the standard Murcko scaffold from a set of molecular structures. Input: A set of molecules in SMILES or SDF format. Software/Tool: RDKit (Open-Source Cheminformatics Toolkit). Steps:
- Standardization: Neutralize charges, remove solvents, and standardize aromaticity using RDKit's
Chem.MolFromSmiles()and sanitization procedures. - Preprocessing: Optionally remove salts and disconnect metals.
- Framework Extraction: Apply the RDKit function
GetScaffoldForMol(mol). This algorithm:- Identifies and retains all ring atoms.
- Retains linker atoms that connect two rings.
- Removes all terminal acyclic atoms (side chains).
- Canonicalization: Convert the resulting scaffold molecule into a canonical SMILES string for comparison.
- Aggregation: Cluster identical canonical SMILES to obtain a unique list of Murcko frameworks for the dataset.
Protocol for Constructing a Scaffold Tree
Objective: To generate a hierarchical Scaffold Tree for a molecule or a dataset.
Input: A molecular structure.
Software/Tool: Implementation based on the original Schuffenhauer et al. rules (available in tools like RDKit’s ScaffoldTree module or proprietary software).
Steps:
- Initialization: Start with the standardized, full molecule as the leaf node.
- Iterative Pruning: Apply a prioritized set of rules recursively to generate the parent scaffold:
- Rule 1 (Heteroatom Removal): Remove terminal heteroatoms not in a ring.
- Rule 2 (Ring System Pruning): If multiple ring systems exist, remove the one with the lowest priority based on criteria like:
- a. Largest number of heteroatoms.
- b. Smallest number of atoms.
- c. Lowest bond density.
- Rule 3 (Linker Removal): Remove linker atoms between rings if the previous rules don't apply.
- Rule 4 (Ring Size Reduction): For large rings (e.g., >8 atoms), perform an exocyclic bond cut to reduce ring size while preserving aromaticity if possible.
- Termination: Pruning stops when no further reduction is possible (typically a single, simple ring like benzene or a single atom). This is the root node.
- Tree Construction: Record each pruning step, linking child to parent, to form the directed acyclic graph (tree) for the molecule.
- Forest Construction: For a dataset, merge individual trees where they share identical nodes, creating a "forest" that represents the entire chemical space of the collection.
Visualizations
Diagram 1: Conceptual Workflow Comparison
Diagram 2: Scaffold Tree Hierarchy Example
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools & Resources for Scaffold Analysis Research
| Item / Resource | Function / Purpose | Example / Note |
|---|---|---|
| Cheminformatics Library | Core engine for molecule manipulation, standardization, and algorithm implementation. | RDKit (Open Source): Contains functions for Murcko scaffold generation and community implementations of the Scaffold Tree. |
| Commercial Cheminformatics Platform | Provides robust, GUI-driven implementations and support for complex scaffold analyses. | Schrödinger Canvas, CCG MOE: Offer advanced and optimized scaffold clustering and tree-generation modules. |
| Standardized Compound Database | Provides high-quality, annotated chemical structures for benchmarking and method validation. | ChEMBL, PubChem: Essential public repositories for extracting diverse datasets for testing. |
| Scaffold Tree Algorithm Code | The specific rule-based algorithm for hierarchical decomposition. | Original publication pseudo-code; Open-source implementations in GitHub repositories (e.g., rdkit.ScaffoldTree). |
| Visualization Software | To graphically display and explore complex scaffold trees and forests. | Cytoscape with ChemViz plugin, Indigo Toolkit by EPAM: Enable interactive visualization of chemical networks. |
| High-Performance Computing (HPC) Cluster | For generating and comparing scaffold trees across large-scale corporate databases (>10^6 compounds). | Necessary due to the computationally intensive nature of tree generation on massive sets. |
| Benchmark Dataset | A curated set of molecules with known scaffold relationships to validate algorithm correctness. | Includes molecules with progressively simplified cores to test pruning rule order and outcomes. |
In the systematic pursuit of new therapeutics, the concept of a molecular scaffold—the core ring and linker system of a compound—serves as a fundamental organizing principle. Scaffold analysis provides a powerful lens through which researchers can decode the structural diversity of compound libraries, predict biological relevance, and design focused screening sets. Central to this field is the Murcko framework, a method for objectively deconstructing molecules into rings, linkers, and side chains to reveal their invariant core [3]. By comparing the distribution and complexity of these frameworks across different compound datasets, researchers can make strategic decisions about which libraries to screen for a given target, ultimately linking molecular architecture to potential biological activity. This guide compares key methodologies, tools, and findings in scaffold analysis to inform effective library design and virtual screening campaigns.
Comparative Analysis of Scaffold Diversity Across Major Compound Libraries
A foundational 2017 study performed a direct comparative analysis of eleven major purchasable screening libraries and the Traditional Chinese Medicine Compound Database (TCMCD) [3] [15]. To ensure a fair comparison, the researchers first standardized the libraries by creating subsets of 41,071 compounds each, with identical molecular weight distributions (100-700 Da), thus eliminating size bias from the diversity assessment [3].
Table 1: Scaffold Diversity Metrics for Standardized Compound Libraries [3]
| Library Name | Total Compounds in Standardized Subset | Unique Murcko Frameworks | Unique Scaffold Tree (Level 1) | PC50C for Murcko Frameworks | Scaffold Diversity Rating |
|---|---|---|---|---|---|
| ChemBridge | 41,071 | 13,763 | 9,508 | 3.3% | High |
| ChemicalBlock | 41,071 | 13,066 | 8,777 | 3.5% | High |
| Mcule | 41,071 | 12,968 | 8,741 | 3.7% | High |
| VitasM | 41,071 | 12,690 | 8,624 | 3.9% | High |
| TCMCD | 41,071 | 8,184 | 5,347 | 5.0% | Moderate (High Complexity) |
| Enamine | 41,071 | 10,577 | 7,395 | 4.5% | Moderate |
| Specs | 41,071 | 9,360 | 6,234 | 6.4% | Moderate |
| LifeChemicals | 41,071 | 8,983 | 6,123 | 6.8% | Moderate |
| ChemDiv | 41,071 | 8,663 | 5,795 | 7.1% | Moderate |
| Maybridge | 41,071 | 7,764 | 5,223 | 8.9% | Moderate |
| UORSY | 41,071 | 7,176 | 4,979 | 9.5% | Lower |
| ZelinskyInstitute | 41,071 | 5,852 | 4,355 | 11.2% | Lower |
Key Findings from the Comparison:
- High-Diversity Libraries: ChemBridge, ChemicalBlock, Mcule, and VitasM top the list with the highest counts of unique Murcko frameworks and Level 1 scaffolds, and the lowest PC50C values. The PC50C metric—the percentage of scaffolds needed to cover 50% of the compounds in a library—is a key indicator of diversity; a lower value means fewer, highly populated scaffolds dominate the collection [3] [16].
- The Case of TCMCD: The natural product-derived TCMCD library presents a unique profile. It has a lower count of unique scaffolds, indicating more conservative core structures. However, these scaffolds exhibit higher average structural complexity, offering distinct chemotypes often underrepresented in synthetic libraries [3].
- Scaffold Representation Skew: The analysis confirmed a common trend across all commercial libraries: a small fraction of scaffolds accounts for a large proportion of the compounds. This "long-tail" distribution highlights significant redundancy and underscores the need for careful library selection to maximize explored chemical space [3] [16].
Methodological Comparison: Murcko Frameworks vs. Scaffold Tree Analysis
Scaffold analysis employs different levels of abstraction. The choice of method impacts the interpretation of library diversity and biological relevance.
Table 2: Comparison of Scaffold Analysis Methods
| Feature | Murcko Framework | Scaffold Tree (Level 1) | Application in Library Design |
|---|---|---|---|
| Definition | The union of all ring systems and linkers in a molecule, with side chains removed [3]. | The first simplification step from the full Murcko framework, where one ring system is pruned based on predefined rules (e.g., retain aromatic over aliphatic rings) [16] [17]. | Provides the core for SAR analysis and series definition. |
| Granularity | A single, objective representation of the core structure [16]. | A more simplified, hierarchical view that groups related Murcko frameworks under a common parent scaffold [17]. | Useful for clustering compounds into broader scaffold families and navigating chemical space hierarchically. |
| Information Preserved | Atom type and bond order within the core [3]. | Prioritizes perceived "chemically meaningful" ring systems, potentially losing some linker information [16]. | Helps identify privileged sub-structures and retrosynthetic pathways. |
| Utility for Diversity Measurement | Excellent for counting distinct core architectures. A higher number indicates greater structural diversity [3]. | Often provides a clearer view of true "scaffold hop" potential by grouping similar cores. Studies suggest it may be more useful for characterizing library diversity [16]. | Level 1 analysis can reveal if high Murcko framework diversity stems from minor linker variations or truly distinct ring systems. |
Experimental Protocol for Comparative Scaffold Analysis: The standardized workflow from the 2017 study provides a replicable protocol [3]:
- Library Standardization: Download library structures (e.g., SDF files). Apply preprocessing: fix bad valences, remove inorganics and duplicates, add hydrogens. Normalize molecular weight distributions by randomly sampling an equal number of compounds from each 100 Da bin across all libraries.
- Scaffold Generation: Generate Murcko frameworks for all molecules using a cheminformatics toolkit (e.g., RDKit's
Chem.Scaffolds.MurckoScaffoldmodule or Pipeline Pilot's Generate Fragments component). Generate the Scaffold Tree hierarchy using dedicated software (e.g., thesdfragcommand in MOE or the Scaffold Hunter algorithm). - Diversity Quantification: For each library and method (Murcko, Level 1), remove duplicate scaffolds. Calculate (a) the count of unique scaffolds, and (b) the PC50C value. Sort scaffolds by frequency and plot the Cumulative Scaffold Frequency Plot (CSFP).
- Visualization & Analysis: Use visualization tools like Tree Maps or SAR Maps to cluster and display scaffolds based on structural similarity (e.g., using fingerprint-based clustering). Analyze the most frequent scaffolds for known target associations (e.g., kinase inhibitors, GPCR ligands).
Visualization of Scaffold Analysis Workflows and Relationships
Diagram 1: A Standardized Workflow for Comparative Scaffold Diversity Analysis [3].
Diagram 2: The Relationship Between Murcko Frameworks and Scaffold Tree Hierarchies [3] [16].
Table 3: Key Research Reagent Solutions for Scaffold Analysis
| Tool / Resource | Type | Primary Function in Scaffold Analysis | Example Use Case |
|---|---|---|---|
| Pipeline Pilot | Cheminformatics Workflow Software | Automates the preprocessing, standardization, and fragment generation (Murcko, rings, linkers) for large compound libraries [3]. | Generating standardized library subsets and calculating seven different fragment representations for comparison. |
| MOE (Molecular Operating Environment) | Modeling and Simulation Software | Contains the sdfrag command for generating Scaffold Tree hierarchies from molecular structures [3]. |
Creating the hierarchical Scaffold Tree (Level 0 to n) for a dataset to analyze scaffold relationships. |
| RDKit | Open-Source Cheminformatics Toolkit | Provides Python functions to generate Murcko scaffolds and manipulate chemical structures programmatically [18]. | Scripting a custom analysis pipeline to process a proprietary compound collection and calculate scaffold frequencies. |
| Scaffold Hunter | Visual Analytics Framework | Specialized software for interactively visualizing and navigating scaffold trees, tree maps, and molecule clouds [17]. | Exploring a screening hit cluster to identify the common core scaffold and propose structural simplifications for lead optimization. |
| ZINC Database | Public Repository | Source for purchasable compound library structures from numerous vendors, essential for obtaining starting data for analysis [3]. | Downloading SDF files of commercial libraries like Enamine, Mcule, and ChemDiv for a comparative diversity study. |
Comparative scaffold analysis provides actionable intelligence for drug discovery. Key conclusions from the data indicate that libraries like ChemBridge and ChemicalBlock offer the broadest structural diversity, making them strong starting points for screening against novel targets with no prior ligand information [3]. In contrast, the TCMCD library, with its high-complexity, nature-derived scaffolds, is a valuable source for challenging targets where synthetic libraries have failed or for seeking novel chemotypes [3].
For project-specific library design:
- For Target-Focused Libraries: Use scaffold analysis to identify "privileged scaffolds" known to bind to a target class (e.g., kinase hinge-binding motifs). Enrich your library with analogs around these cores but use diversity metrics (PC50C) to avoid excessive redundancy.
- For Phenotypic or Novel Target Screening: Prioritize libraries with high unique Murcko framework counts and low PC50C values to maximize the chance of hitting diverse chemotypes. Incorporate a portion of natural product-like scaffolds from sources like TCMCD to access underrepresented chemical space.
- For Hit-to-Lead Expansion: After identifying an initial hit, use the Scaffold Tree hierarchy to propose structurally simplified analogs (moving toward Level 1) or to suggest chemically similar "scaffold hops" by exploring neighboring branches in the tree, aiding in SAR exploration and patent strategy [16] [17].
Ultimately, scaffold analysis transcends simple categorization. By linking the frequency and complexity of molecular cores to biological target profiles, it enables a more rational, predictive approach to building and selecting compound collections, directly connecting the core of chemistry to the core of biological activity.
Foundational Definitions and Hierarchical Relationships
In the analysis of chemical datasets for drug discovery, partitioning vast molecular spaces into meaningful groups is essential. Several rule-based, interpretable methods have been developed to define molecular scaffolds, each offering a different balance between structural specificity and generalized grouping [19]. Understanding their definitions and relationships is key to selecting the appropriate method for a given research context, such as analyzing library diversity or preparing data for machine learning.
The Murcko scaffold (or Bemis-Murcko framework) is the fundamental structure obtained by removing all acyclic side chains from a molecule, retaining only the ring systems and the linker chains that connect them [19] [20]. This representation maintains the original atom and bond types, providing a chemically specific framework.
A Generic Murcko scaffold (also called a cyclic skeleton or graph framework) is a further abstraction of the Murcko scaffold. It is generated by converting all atoms to carbon and all bonds to single bonds [19] [4]. This process focuses purely on the topology of the ring-linker system, grouping together scaffolds that share the same skeletal shape despite differing in atom types or bond orders.
Scaffold Keys represent an abstracted, descriptor-based approach. They are defined as a set of 32 simple topological and structural descriptors (or "keys") calculated from the Murcko scaffold. These keys encode features such as the numbers of rings of different sizes, the number of linker atoms, and other structural attributes, transforming the scaffold into a fixed-length numerical vector [19].
A Reduced scaffold refers to an even more generalized representation. The Reduced Generic Scaffold, as used in the Scaffold Identification and Naming System (SCINS), is derived from the Generic Murcko scaffold by disregarding ring size information, simplifying some chain length details, and ignoring the precise topological connectivity of the scaffold [19]. The related Reduced Complexity Molecular Framework (RCMF) is another variant designed for analyzing DNA-encoded libraries, which classifies rings into predefined classes and describes linkers by length [19].
The following diagram illustrates the hierarchical abstraction relationship between these key terms, showing how specific molecular structures are progressively generalized through different computational operations.
Diagram: Hierarchical Abstraction of Scaffold Definitions
Quantitative Performance Comparison Across Datasets
The utility of a scaffold definition is evidenced by its application to real chemical datasets. Different methods yield vastly different numbers of unique scaffolds and distributions, directly impacting analyses of library diversity, chemical space coverage, and the meaningful grouping of bioactive compounds. The following tables consolidate key quantitative findings from recent studies.
Table 1: Scaffold Counts and Uniqueness in Large-Scale Analyses
| Dataset / Analysis | Murcko Scaffold | Generic Murcko Scaffold | Reduced/Generic Scaffold (SCINS) | Notes | Source |
|---|---|---|---|---|---|
| ChEMBL v33 (1.9M compounds) | Not Provided | Not Provided | 1,608 distinct classes | SCINS space is more densely populated. | [19] |
| Enamine REAL Diverse (48.2M compounds) | Not Provided | Not Provided | 1,223 distinct classes | Covers a smaller SCINS space vs. ChEMBL. | [19] |
| 1 Billion AI-Generated Molecules | 6.5×10⁹ (True Murcko) | 1.2×10⁸ (Generic) | Not Provided | Uniqueness: 24.4% (True), 3.32% (Generic). | [21] |
| Guacamol ChEMBL Set (1.59M molecules) | 465,873 (True BM) | 109,935 (True CSK) | Not Provided | Highlights impact of definition (e.g., RDKit vs. "True"). | [4] |
Table 2: Scaffold Diversity Metrics in Purchasable Libraries (Standardized Subsets)
| Compound Library | Number of Unique Murcko Frameworks | PC50C for Murcko Frameworks | PC50C for Level 1 Scaffolds | Interpretation | Source |
|---|---|---|---|---|---|
| ChemBridge | 15,035 | 1.67% | 0.83% | Lower PC50C indicates higher diversity. | [15] |
| ChemicalBlock | 14,478 | 1.81% | 0.95% | Considered more structurally diverse. | [15] |
| Mcule | 14,269 | 1.77% | 0.89% | Considered more structurally diverse. | [15] |
| VitasM | 13,480 | 1.86% | 0.97% | Considered more structurally diverse. | [15] |
| TCMCD | 11,512 | 2.25% | 1.34% | Highest structural complexity but more conservative scaffolds. | [15] |
| Enamine | 12,502 | 2.05% | 1.11% | Intermediate diversity. | [15] |
| ChemDiv | 11,095 | 2.32% | 1.27% | Intermediate diversity. | [15] |
Table 3: Performance in Grouping Bioactive Compounds
| Method | Application / Dataset | Key Performance Finding | Advantage for Task | Source |
|---|---|---|---|---|
| SCINS (Reduced Generic) | 5,699 DRD2 actives (pChEMBL ≥5) | Created chemically intuitive groups for medium-sized bioactive sets. | Useful for post-processing hit lists and compound selection; avoids excessive singletons. | [19] |
| Murcko Scaffold | General drug set analysis (~5,000 drugs) | 42 scaffolds represented 25% of drugs; 32 generic scaffolds represented 50%. | Identifies most common, privileged frameworks in successful drugs. | [19] |
| Scaffold Splitting | Machine Learning Model Validation | Creates a meaningful distribution shift between training and test sets. | Provides a robust measure of model's out-of-distribution generalisation power. | [22] |
Detailed Experimental Protocols
The comparative results in the previous section are derived from standardized computational workflows. Reproducibility in cheminformatics requires clear protocols for data preparation, scaffold generation, and analysis. Below are detailed methodologies from key studies.
Protocol: Large-Scale Database Comparison (SCINS vs. Murcko)
This protocol, used to compare ChEMBL and Enamine REAL libraries [19], highlights the steps for consistent large-scale analysis.
- Data Curation & Standardization:
- Source the datasets (e.g., ChEMBL v33, Enamine REAL Diverse subset).
- Standardization: Remove organometallic compounds, keep only the largest molecular fragment, neutralize charges, and standardize tautomers (using RDKit's
rdMolStandardizemodules). This ensures consistent input structures.
- Scaffold Generation:
- Murcko & Generic Murcko: Use RDKit's
MurckoScaffold.GetScaffoldForMol()function. For the generic version, first convert all atoms to carbon and bonds to single bonds, then remove side chains. - SCINS (Reduced Generic Scaffold): Implement the SCINS algorithm to abstract the generic scaffold further. This involves discarding ring size information, simplifying linker chain counts, and generating a systematic name based on the remaining topology.
- Murcko & Generic Murcko: Use RDKit's
- Population Analysis:
- For each scaffold type, count the number of unique instances.
- Analyze the distribution of compounds across scaffold classes (e.g., number of singletons, densely populated classes).
- Map and compare the coverage of "SCINS space" versus "Murcko scaffold space" for different databases.
Protocol: Scaffold Diversity Analysis of Compound Libraries
This protocol, used to compare commercial screening libraries [15], focuses on ensuring a fair comparison by standardizing molecular weight distributions.
- Library Preparation & Standardization:
- Download and preprocess libraries (remove duplicates, inorganic molecules, fix valences).
- Analyze the molecular weight (MW) distribution of all libraries.
- Create a standardized subset for each library: Within each 100 Da MW interval, randomly select a number of molecules equal to the minimum count found across all libraries for that interval. This yields subsets with identical MW distributions, removing MW bias from diversity metrics.
- Fragment and Scaffold Generation:
- Generate multiple fragment representations using tools like Pipeline Pilot or RDKit: Ring Assemblies, Murcko Frameworks, Scaffold Tree Levels (e.g., Level 1).
- Diversity Quantification:
- For each library subset, count the number of unique scaffolds (e.g., Murcko frameworks).
- Generate Cumulative Scaffold Frequency Plots (CSFPs): Sort scaffolds by frequency (most to least common), plot the cumulative percentage of molecules represented versus the cumulative percentage of scaffolds.
- Calculate the PC50C metric: The percentage of scaffolds required to cover 50% of the molecules in the library. A lower PC50C indicates greater scaffold diversity.
Protocol: Implementing Scaffold Splits for Machine Learning
This protocol is critical for evaluating the real-world generalizability of molecular property prediction models [22].
- Scaffold Assignment:
- For every molecule in the dataset, calculate its Bemis-Murcko scaffold (using a consistent definition, e.g., "True BM" or "RDKit BM").
- Partitioning:
- Group all molecules that share an identical scaffold into distinct "scaffold clusters."
- Sort these clusters by size (number of molecules) in descending order.
- Train/Test Split:
- Allocate the largest N scaffold clusters to the training set.
- Allocate the remaining, smaller scaffold clusters to the test set.
- The parameter N is chosen so that the training set contains a desired fraction (e.g., 80%) of the total molecules. This ensures all molecules from any given scaffold are contained entirely within one set, creating a rigorous out-of-distribution test.
The following diagram summarizes the core computational workflow common to these protocols, from data input to analytical output.
Diagram: General Workflow for Comparative Scaffold Analysis
The Scientist's Toolkit: Essential Research Reagents & Software
Implementing the analyses and protocols described requires a core set of reliable software tools and databases. The following table details the essential components of the computational toolkit for scaffold-based research.
Table 4: Essential Software Tools and Databases for Scaffold Analysis
| Tool / Resource Name | Type | Primary Function in Scaffold Research | Key Notes / Relevance |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Toolkit | Core engine for generating Murcko and Generic Murcko scaffolds, molecular standardization, and descriptor calculation. | The rdkit.Chem.Scaffolds.MurckoScaffold module is fundamental. Critical to be aware of implementation choices (e.g., "RDKit" vs. "True" BM) [4]. |
| SCINS Python Implementation | Open-Source Algorithm | Provides the specific method for generating Reduced Generic Scaffolds (SCINS) as described in [19]. | Dependent on RDKit. Designed to reduce singleton scaffolds and group compounds more intuitively. |
| Pipeline Pilot | Commercial Data Pipelines | Used in large-scale studies for automated workflow construction, fragment generation, and dataset standardization [15]. | Facilitates reproducible, high-throughput analysis of compound libraries. |
| ChEMBL | Public Bioactivity Database | A primary source of bioactive molecules for analyzing scaffold distributions in drug-like space and for creating benchmark datasets. | Used in [19] to compare against commercial libraries and to extract target-specific datasets (e.g., DRD2 actives). |
| ZINC / Enamine REAL | Commercial Compound Databases | Sources of purchasable and make-on-demand compounds for analyzing chemical space coverage and library diversity [19] [15]. | Enamine REAL was used as an example of a vast, drug-like virtual library. |
| REINVENT 4 / STELLA | Generative AI Frameworks | Used for de novo molecular design; scaffold analysis of their output measures the structural diversity of generated chemical spaces [23] [24]. | Studies like [21] analyze the scaffold diversity of billions of AI-generated molecules. |
| MOE (Molecular Operating Environment) | Commercial Software Suite | Used for generating certain scaffold representations like the Scaffold Tree and RECAP fragments in comparative studies [15]. | Provides alternative implementations and visualization capabilities. |
How to Analyze Scaffolds: A Step-by-Step Methodological Guide
Within the context of a broader thesis comparing Murcko frameworks across diverse compound datasets, the selection and performance of computational toolkits are paramount. The analysis of molecular scaffolds—core structures stripped of side chains—is a foundational technique for assessing chemical diversity, classifying compounds, and identifying privileged structures in drug discovery [25]. The Murcko framework, a systematic method for dissecting molecules into ring systems, linkers, and side chains, has become a standard for such analyses [3] [16]. Efficiently generating and comparing these frameworks across large, heterogeneous libraries (such as purchasable screening collections, natural product databases, and proprietary sets) requires robust, scalable software.
This guide provides an objective comparison of three pivotal solutions in this domain: the open-source RDKit, the commercial Pipeline Pilot platform, and emerging open-source implementations like the Chemistry Development Kit (CDK) Scaffold Generator. The evaluation is grounded in experimental data and methodologies from published comparative studies, focusing on their application in scaffold generation, diversity analysis, and their integration into research workflows for profiling compound libraries [3] [16].
Platform Comparison: Features, Performance, and Use Cases
The following tables provide a side-by-side comparison of the core platforms based on their capabilities in scaffold analysis, technical features, and suitability for different research tasks.
Table 1: Core Feature Comparison for Scaffold Generation and Analysis
| Feature Category | RDKit (Open-Source) | Pipeline Pilot (Commercial) | CDK Scaffold Generator (Open-Source) |
|---|---|---|---|
| Primary Scaffold Definition | Murcko frameworks, graph frameworks [26]. | Murcko frameworks, ring assemblies, bridge assemblies [3]. | Murcko frameworks, 5 customizable framework definitions [25]. |
| Advanced Hierarchy Generation | Can implement Scaffold Tree logic via scripting. | Integrated components for Scaffold Tree generation (e.g., via MOE sdfrag command integration) [3]. | Native implementation of Scaffold Trees and Scaffold Networks [25]. |
| Key Functionality | Molecular I/O, fingerprinting (Morgan/ECFP), substructure search, 2D/3D operations [26]. | Visual workflow protocol building, data pipelining, large-scale library preprocessing and analysis [3] [27]. | Specialized library for scaffold handling, generation, and display of hierarchies using GraphStream [25]. |
| Typical Use Case | Custom script-based analysis, integration into ML pipelines, KNIME workflows [26]. | High-throughput, reproducible analysis of vendor libraries for diversity assessment [3]. | Research-focused deep dive into scaffold hierarchies and network visualization for NP/drug datasets [25]. |
Table 2: Performance and Practical Considerations
| Aspect | RDKit | Pipeline Pilot | CDK Scaffold Generator |
|---|---|---|---|
| License & Cost | BSD license; free for academic and commercial use [26]. | Commercial license required; cost associated [3]. | GNU LGPL; open-source and free [25]. |
| User Interface | Programming library (Python/C++); no native GUI [26]. | Proprietary graphical client for workflow design [3]. | Java library; command-line or integrated into Java applications [25]. |
| Integration & Extensibility | High; Python bindings, KNIME nodes, PostgreSQL cartridge [26]. | High within ecosystem; integrates with other Accelrys/BIOVIA tools [3]. | Built on CDK; integrates with Java-based cheminformatics projects [25]. |
| Reported Performance | Fast substructure and similarity search; efficient for large-scale virtual screening [26]. | Used to process libraries of millions of compounds (e.g., ~4.9M in Mcule library) [3]. | Generated a scaffold network from >450,000 natural products within a day [25]. |
| Best Suited For | Flexible, code-centric research, prototyping new methods, and embedded use in applications. | Industrial, standardized, and high-throughput processing of chemical data in team environments. | Academic research requiring advanced, customizable scaffold tree and network analyses. |
Experimental Protocols for Comparative Scaffold Analysis
The following detailed methodologies are derived from key studies that benchmark scaffold diversity across compound libraries, providing a reproducible framework for tool evaluation [3] [16].
Protocol 1: Library Standardization and Murcko Framework Generation
This protocol is essential for a fair comparison of scaffold diversity across libraries of different sizes and property distributions [3].
Library Acquisition and Preprocessing:
- Obtain compound libraries in SDF or SMILES format from sources like ZINC or commercial vendors.
- Standardization Steps: Apply a consistent preprocessing protocol to all libraries. Using Pipeline Pilot, this involves: fixing bad valences, filtering out inorganic molecules, adding hydrogens, and removing duplicates [3]. In RDKit or CDK, equivalent steps can be scripted using built-in sanitization and cleanup functions.
Molecular Weight Standardization:
- Analyze the molecular weight (MW) distribution of all preprocessed libraries.
- Divide the MW range (e.g., 100-700 Da) into intervals (e.g., every 100 Da).
- For each interval, identify the library with the fewest compounds. Randomly select an equal number of compounds from every other library within that same MW interval.
- Combine these selections to create standardized subsets for each library that have identical compound counts and nearly identical MW distributions. This controls for MW bias in diversity analysis [3].
Scaffold Generation:
- Generate the Murcko framework for every molecule in the standardized subsets.
- In Pipeline Pilot: Use the "Generate Fragments" component [3].
- In RDKit: Use the
rdkit.Chem.Scaffolds.MurckoScaffoldmodule. - In CDK Scaffold Generator: Use the appropriate framework definition class.
Diversity Metrics Calculation:
- For each library, calculate:
- Total Number of Unique Murcko Scaffolds: The absolute count of distinct frameworks.
- Scaffold Frequency Distribution: The percentage of compounds represented by the most common scaffolds (e.g., the number of scaffolds needed to cover 50% of the library - SC50) [16].
- Singleton Scaffolds: The percentage of unique scaffolds that appear only once in the library. A high percentage indicates high diversity but potentially poor SAR development potential [16].
- For each library, calculate:
Protocol 2: Scaffold Tree Analysis and Visualization
This protocol uses hierarchical decomposition to gain deeper insight into scaffold relationships [16].
Scaffold Tree Construction:
- Apply the Scaffold Tree algorithm, which iteratively prinks rings from the Murcko framework based on chemical prioritization rules until a single ring remains.
- In Pipeline Pilot/Third-Party Tools: This can be done by integrating the sdfrag command from Molecular Operating Environment (MOE) [3].
- In CDK Scaffold Generator: This is a core native functionality [25].
- In RDKit: Requires implementation of the published algorithm via scripting.
Analysis of Tree Levels:
- Focus analysis on Level 1 scaffolds (the first ring system removed from the original molecule). Studies have shown Level 1 can be more effective than Murcko frameworks for characterizing library diversity, as it better groups structurally related compounds [16].
- Analyze the distribution and frequency of scaffolds at this level across different libraries.
Visualization with Tree Maps:
- Use Tree Map visualization to represent the scaffold space.
- Each rectangle in the map represents a scaffold, with its area proportional to the number of molecules containing it.
- Structurally similar scaffolds (based on fingerprint similarity) are placed adjacent to each other and colored similarly. This provides an immediate visual summary of scaffold diversity, dominance, and clustering within a library [3] [16].
Diagram Title: Workflow for Comparative Scaffold Diversity Analysis
Visualizing Scaffold Relationships and Decomposition Logic
The logical process of scaffold decomposition from a full molecule to its core frameworks is central to these toolkits' functions.
Diagram Title: Logical Decomposition from Molecule to Scaffold Tree
The Scientist's Toolkit: Essential Research Reagent Solutions
Beyond software, robust scaffold analysis relies on specific data resources and complementary tools. The following table details key "reagents" for this research domain.
Table 3: Essential Research Resources for Scaffold Analysis
| Resource Name | Type | Primary Function in Scaffold Research | Key Relevance |
|---|---|---|---|
| ZINC Database | Public Compound Library | Source for purchasable screening molecules from various vendors [3]. | Provides real-world, commercially available libraries for diversity analysis and virtual screening workflows. |
| ChEMBL Database | Public Bioactivity Database | Source for bioactive molecules with annotated targets and activities [16]. | Enables scaffold analysis informed by biological activity, linking frameworks to target classes. |
| COCONUT (NP Atlas) | Public Natural Product Library | Extensive collection of unique natural product structures [25]. | Serves as a benchmark for complex, structurally diverse scaffolds distinct from synthetic libraries. |
| Scaffold Tree / Network Algorithms | Computational Method | Hierarchical classification and relationship mapping of scaffolds [25] [16]. | The core logic implemented by toolkits (CDK, Pipeline Pilot) for advanced analysis beyond simple Murcko frameworks. |
| Tree Map Visualization | Data Visualization Technique | Intuitive display of scaffold frequency and structural similarity [3] [16]. | Critical for communicating complex diversity analysis results in an accessible, visual format. |
| Molecular Anatomy Framework | Advanced Scaffold Definition | A multi-dimensional hierarchical scaffold analysis method using nine abstraction levels [27]. | Represents a next-generation approach for flexible, unbiased clustering, moving beyond single-definition limitations. |
The comparative analysis reveals a clear trade-off between flexibility, cost, and out-of-the-box capability. RDKit offers unparalleled flexibility and integration for code-savvy researchers, making it ideal for prototyping and embedding in custom pipelines. The commercial Pipeline Pilot excels in standardized, high-throughput processing of massive vendor libraries, providing robust, GUI-driven workflows for team-based industrial research [3] [26]. Specialized open-source implementations like the CDK Scaffold Generator fill a niche for researchers requiring deep, customizable analysis of scaffold hierarchies and networks, particularly for natural products and drug datasets [25].
The broader thesis on Murcko framework comparisons will benefit from leveraging the strengths of multiple tools: using Pipeline Pilot or RDKit for initial high-volume library processing and Murcko framework generation, and employing the CDK Scaffold Generator for in-depth hierarchical analysis of selected, interesting subsets. The field is evolving with approaches like "Molecular Anatomy," which uses multiple scaffold definitions simultaneously to create a more flexible and unbiased network for analysis [27]. Furthermore, the integration of AI-driven generative models for scaffold-based molecular design presents a forward-looking application, where these toolkits can generate novel derivatives retaining a desired core structure [28]. The choice of toolkit ultimately depends on the specific research question, scale of data, and available expertise, but all three provide powerful pathways to decode the scaffold landscape of chemical libraries.
Within the expansive field of computational drug discovery, the systematic comparison of chemical libraries is foundational for tasks ranging from virtual screening to generative model evaluation. Central to this endeavor is the analysis of molecular scaffolds, particularly the Murcko framework—defined as the union of all ring systems and linkers in a molecule, which provides a simplified, core structural representation [3]. These frameworks enable researchers to classify compounds, assess library diversity, and infer potential bioactivity. However, meaningful comparison of scaffold distributions across different datasets is profoundly compromised without rigorous pre-processing.
Databases vary drastically in origin, size, and chemical property distributions. For instance, commercial screening libraries can range from tens of thousands to several million compounds, with significantly different molecular weight (MW) profiles [3]. Similarly, modern machine learning pretraining datasets are assembled from diverse sources like PubChem and UniChem, each with inherent biases [29]. When scaffold analysis is performed on these raw, unstandardized collections, observed differences in diversity may be artifactual, stemming from uneven property distributions rather than true structural disparity. This invalidates conclusions about which library is more "diverse" or suitable for a given task. Therefore, dataset standardization is not a mere preliminary step but a critical prerequisite for ensuring that subsequent comparisons of Murcko frameworks are equitable, reproducible, and scientifically sound [3].
Foundational Methodology: An Experimental Protocol for Library Standardization
A seminal study provides a clear, replicable protocol for standardizing compound libraries prior to scaffold analysis [3]. The methodology underscores that controlling for confounding variables, especially molecular weight, is essential for unbiased comparison.
1. Library Selection and Preliminary Processing: The study selected eleven large (all >50,000 compounds), commercially available screening libraries from ZINC15 (e.g., Mcule, Enamine, ChemDiv) and one natural product database (Traditional Chinese Medicine Compound Database, TCMCD) [3]. All molecules underwent a uniform cleanup pipeline using Pipeline Pilot software: fixing bad valences, filtering out inorganic molecules, adding hydrogens, and removing duplicates [3].
2. Molecular Weight Distribution Analysis: An initial analysis of the raw libraries revealed vastly different MW ranges and distributions [3]. To enable a fair comparison, the overlapping MW range common to all libraries (100-700 Da) was identified. The number of molecules in each 100-Da interval within this range was counted for every library.
3. Creation of Standardized Subsets: The key standardization step involved creating new, directly comparable subsets. For each 100-Da interval, the minimum number of molecules present across all twelve libraries was determined. This number of molecules was then randomly selected from each library within that specific MW interval. This process resulted in twelve standardized subsets, each containing exactly 41,071 molecules with identical molecular weight distributions [3]. All subsequent scaffold diversity analyses were performed on these standardized subsets, ensuring any differences discovered were due to structural factors, not size or weight bias.
The following diagram illustrates this critical workflow.
Diagram 1: Experimental Workflow for Dataset Standardization. The process transforms raw libraries with varying sizes and properties into standardized subsets with identical molecular weight distributions, enabling fair scaffold comparison [3].
Scaffold Analysis and Comparative Metrics
With standardized subsets, scaffold analysis can proceed using defined structural representations. The key steps and metrics are as follows:
1. Scaffold Generation:
- Murcko Frameworks: Generated by removing all side chain atoms, leaving only the ring systems and the linkers that connect them [3].
- Scaffold Tree Hierarchies: A more systematic decomposition where scaffolds are iteratively simplified by removing rings according to rules, creating a hierarchy from the original molecule (Level n) down to a single ring (Level 0). The Murcko framework is equivalent to Level n-1 in this tree [3].
2. Key Quantitative Metrics for Comparison:
- Scaffold Count: The absolute number of unique Murcko frameworks found in a standardized library. A higher count suggests greater structural diversity [3].
- Cumulative Scaffold Frequency: This metric reveals the "skew" of the library. It calculates the percentage of total molecules accounted for by the most common X number of scaffolds. A steep curve indicates a library dominated by a few common scaffolds, while a gradual curve suggests a more even distribution [3].
The methodology from molecules to analyzable scaffolds is summarized below.
Diagram 2: From Molecules to Scaffold Metrics. Analytical pathways for generating key structural representations and quantitative diversity metrics from a standardized molecular dataset.
Results: Comparative Analysis of Standardized Libraries
Applying the above protocol to the twelve standardized subsets yielded clear, quantifiable differences in scaffold diversity [3]. The following table summarizes key scaffold statistics, highlighting which libraries are most and least diverse after removing the confounding effect of molecular weight.
Table 1: Scaffold Diversity Analysis of Standardized Compound Libraries (MW 100-700 Da) [3]
| Library Name | Total Scaffolds (Murcko Frameworks) | Scaffold-to-Compound Ratio | Scaffolds Covering 50% of Compounds | Notable Characteristics |
|---|---|---|---|---|
| ChemBridge | 14,142 | 0.34 | 292 | Highest scaffold diversity among commercial libraries |
| ChemicalBlock | 13,727 | 0.33 | 278 | High diversity, selected for broad coverage |
| Mcule | 12,815 | 0.31 | 270 | Large, diverse library with good scaffold spread |
| VitasM | 12,306 | 0.30 | 266 | Novel compounds with high structural variety |
| TCMCD | 8,694 | 0.21 | 147 | Highest complexity but conservative scaffold set |
| Enamine | 11,950 | 0.29 | 420 | Large library but more clustered on fewer scaffolds |
| LifeChemicals | 10,455 | 0.25 | 346 | Moderate diversity |
| Specs | 9,784 | 0.24 | 318 | Moderate diversity |
| Maybridge | 9,090 | 0.22 | 257 | Marketed as highly diverse, but lower in this analysis |
| ChemDiv | 8,601 | 0.21 | 397 | Lowest diversity; heavily clustered on common scaffolds |
| UORSY | 8,337 | 0.20 | 365 | Low scaffold count |
| ZelinskyInstitute | 7,348 | 0.18 | 300 | Lowest unique scaffold count |
Key Findings:
- Diversity Leaders: ChemBridge and ChemicalBlock exhibited the highest number of unique Murcko frameworks and the best scaffold-to-compound ratios, confirming their design for broad structural coverage [3].
- The Natural Product Profile: TCMCD, the natural product database, showed a distinct profile. While its molecules had the highest average structural complexity, they were built from a relatively conservative set of core scaffolds, resulting in a lower unique scaffold count but high complexity within those scaffolds [3].
- High Clustering: Libraries like ChemDiv and Enamine, despite their large raw sizes, showed high clustering where a relatively small number of scaffolds (397 and 420, respectively) accounted for half of all molecules in the standardized subset [3]. This indicates a bias towards certain popular chemotypes.
The Scientist's Toolkit: Essential Reagents and Software
Conducting a rigorous, standardized scaffold comparison requires a suite of specialized software tools and databases.
Table 2: Research Reagent Solutions for Scaffold Analysis
| Item Name | Category | Primary Function in Standardization/Scaffolding | Reference |
|---|---|---|---|
| Pipeline Pilot | Commercial Software | Protocol automation for data cleaning (fix valence, deduplicate), fragment generation, and property calculation. | [3] |
| RDKit | Open-Source Cheminformatics | Core library for molecule standardization, Murcko framework decomposition, fingerprint generation, and SMILES parsing. | [30] [18] |
| MOE (Molecular Operating Environment) | Commercial Software | Used for generating Scaffold Tree hierarchies via its sdfrag command. |
[3] |
| ZINC Database | Public Compound Repository | Source for commercially available screening compounds and vendor library information. | [3] [29] |
| ChEMBL Database | Public Bioactivity Database | Source for curated drugs and bioactive molecules with associated targets, used for scaffold-drug indication mapping. | [18] [31] |
| PubChem / UniChem | Public Compound Aggregators | Large-scale sources of experimental compound data for building pretraining datasets like MolPILE. | [29] |
| MOSES Benchmarking Platform | Open-Source Software | Provides standardized datasets and metrics (validity, uniqueness, novelty) for evaluating generative models. | [30] |
Modern Context: Standardization for Machine Learning and Benchmarking
The principle of standardization extends beyond comparing static libraries to the dynamic field of molecular machine learning (ML). The performance and generalizability of ML models are critically dependent on the quality and consistency of their training data [29].
1. The Pretraining Data Challenge: Recent analyses reveal significant shortcomings in existing datasets used to pretrain molecular foundation models. Models are often trained on small, non-representative subsets of ZINC or PubChem, or on massive but poorly filtered collections that include non-synthesizable structures [29]. This lack of standardized pretraining data makes it difficult to determine if performance improvements are due to better algorithms or simply different, potentially biased, training data [29]. Initiatives like MolPILE aim to address this by creating a large-scale (222M compounds), rigorously curated, and standardized dataset to serve as a universal benchmark, akin to ImageNet in computer vision [29].
2. Benchmarking Generative Models: The MOSES (Molecular Sets) platform directly applies standardization principles for model evaluation [30]. It provides a curated training set, a standardized test set, and a suite of metrics (e.g., validity, uniqueness, novelty, fragment similarity) to fairly compare different generative models. This ensures that reported performances are based on a level playing field, isolating model architecture efficacy from data pipeline artifacts [30].
Based on the experimental evidence and contemporary context, researchers comparing Murcko frameworks or chemical libraries should adhere to the following best practices:
- Always Standardize on Key Properties Before Comparison: Never compare scaffold diversity using raw library data. Control for major confounding variables like molecular weight by creating standardized subsets with matched property distributions [3].
- Use Multiple, Complementary Metrics: Rely on a suite of metrics such as unique scaffold count, cumulative frequency plots, and visualizations (Tree Maps, SAR Maps) to gain a complete picture of diversity and clustering [3].
- Contextualize Scaffold Findings: A lower scaffold count does not necessarily mean a "worse" library. Natural product libraries (e.g., TCMCD) may have lower unique scaffold counts but higher complexity and biological relevance, offering different value [3] [31].
- Employ Standardized Benchmarks for ML: When developing or evaluating machine learning models, use standardized benchmarking platforms like MOSES and large-scale, quality-controlled pretraining datasets like MolPILE to ensure fair, reproducible comparisons and true measurement of algorithmic progress [29] [30].
- Leverage Interactive Tools for Exploration: Utilize emerging interactive visualization platforms that map drugs to their Murcko scaffolds and biological indications to inform library design and target hypothesis generation [18].
In conclusion, dataset standardization is the indispensable foundation for any objective analysis of chemical space. By implementing rigorous pre-processing protocols, the cheminformatics community can ensure that critical decisions in virtual screening, library design, and algorithm development are based on fair comparisons and robust, interpretable science.
Murcko frameworks and scaffold trees are foundational concepts in chemoinformatics and computational drug discovery. They enable researchers to simplify complex molecular structures to their core architectural components, facilitating the analysis of structure-activity relationships (SAR), the assessment of chemical diversity, and the navigation of vast chemical spaces [32].
A Murcko framework is derived by systematically removing all terminal side chains and retaining only the ring systems and the linkers that connect them [32]. This abstraction distills a molecule down to its central scaffold, which is often responsible for the molecule's core interactions with a biological target. The scaffold tree extends this concept by hierarchically decomposing a molecule through iterative ring removal, creating a tree-like representation that maps the relationship between a parent scaffold and its simpler substructures [33].
Within the broader thesis comparing Murcko frameworks across different compound datasets, these tools are indispensable. They provide a standardized method to categorize molecules, enabling meaningful comparisons of scaffold distribution, diversity, and prevalence across diverse chemical libraries, such as commercial screening collections, proprietary corporate databases, or natural product repositories.
Comparative Analysis of Generation Methods and Tools
Different software tools and algorithms implement the generation of Murcko frameworks and scaffold trees with varying features and focuses. The table below provides a high-level comparison of key tools.
Table 1: Comparison of Tools for Generating Murcko Frameworks and Scaffold Trees
| Tool/Library | Core Method | Key Features | Primary Output | Best Use Case |
|---|---|---|---|---|
| RDKit [34] | Bemis-Murcko Scaffold | Direct generation of the Murcko scaffold from a molecule. Integrated chemical toolkit. | SMILES string of the scaffold. | Fast, in-pipeline scaffold extraction for clustering and diversity analysis. |
| ScaffoldGraph [33] | Multiple (Murcko, Varin, HierS, Schuffenhauer) | Generates full scaffold networks and hierarchical trees. Open-source and built on RDKit/NetworkX. | Scaffold trees, directed acyclic graphs (DAGs) of scaffold hierarchies. | Comprehensive analysis of scaffold relationships and chemical series within a dataset. |
| Commercial Platforms (e.g., ChemAxon) | Proprietary implementations | Often integrated with extensive visualization, database, and screening suites. | Frameworks and trees within a GUI or enterprise environment. | Industrial workflows requiring integration with other proprietary discovery tools. |
The choice of tool depends on the research objective. For high-throughput clustering of large compound libraries (e.g., the Enamine Premium library with over 128,000 molecules), RDKit's efficient Murcko scaffold generation is ideal, allowing the 70,843 unique scaffolds in that dataset to be quickly identified and grouped [34]. For a deeper, hierarchical analysis of how active compounds relate to one another—crucial for hit-to-lead optimization—ScaffoldGraph's tree generation provides invaluable insights by revealing the core scaffold shared by a series of analogues [33].
Experimental Protocol for Framework Comparison Across Datasets
This protocol outlines a standardized experiment to compare Murcko framework distributions across different compound datasets, a core activity for the stated thesis.
Objective: To quantify and compare the structural diversity and scaffold prevalence across two or more compound datasets (e.g., a commercial screening library vs. a collection of clinical kinase inhibitors).
Materials & Software:
- Input Datasets: Chemical structures in SDF or SMILES format.
- Primary Tool: RDKit (for batch scaffold generation).
- Analysis Tools: Python (Pandas, Matplotlib/Seaborn), ScaffoldGraph.
- Validation Metric: Scaffold Similarity (Scaf), a benchmark metric used in molecular generation studies to measure how well a model reproduces the scaffold distribution of a training set [35].
Procedure:
- Data Preparation: Standardize each dataset (neutralize charges, remove salts) using RDKit.
- Scaffold Generation: For every molecule in each dataset, generate its canonical Murcko scaffold SMILES string using the
MurckoScaffoldSmiles()function in RDKit [34]. - Frequency Analysis: Calculate the occurrence frequency of each unique scaffold within its dataset.
- Diversity Metrics: Calculate:
- Scaffold Count: Total number of unique Murcko scaffolds.
- Scaffold Ratio: (Unique Scaffolds) / (Total Molecules). A lower ratio indicates higher scaffold redundancy.
- Top-10 Scaffold Prevalence: The percentage of total molecules accounted for by the 10 most frequent scaffolds.
- Cross-Dataset Comparison: Identify scaffolds common to all datasets and those unique to each. Visualize using UpSet plots or Venn diagrams.
- Hierarchical Analysis (Optional): For a subset of interest (e.g., all molecules containing a common scaffold), use ScaffoldGraph to generate scaffold trees to analyze decomposition pathways [33].
Expected Output: A clear, quantitative profile for each dataset. One may find that a commercial library has a high scaffold count (high diversity) but a low scaffold ratio (many similar compounds), while a targeted inhibitor set shows the opposite, dominated by a few privileged scaffolds.
The following diagram illustrates this analytical workflow:
Diagram: Workflow for comparing Murcko frameworks across compound datasets.
Table 2: Essential Research Reagent Solutions for Scaffold Analysis
| Category | Item/Resource | Function & Purpose | Example/Note |
|---|---|---|---|
| Core Software | RDKit | Open-source chemoinformatics toolkit. Provides the fundamental function to generate Murcko scaffolds from molecules [34]. | Use rdkit.Chem.Scaffolds.MurckoScaffold. |
| Specialized Library | ScaffoldGraph | Open-source Python library dedicated to building and analyzing scaffold networks and trees [33]. | Builds on RDKit and NetworkX to generate hierarchical decompositions. |
| Benchmark Datasets | MOSES Dataset | A standardized benchmark set for molecular generation models, useful for testing scaffold analysis pipelines [35]. | Includes a "scaffold test" split to evaluate generalization to new scaffolds. |
| Validation Metric | Scaffold Similarity (Scaf) | A quantitative metric to compare the scaffold distribution between two sets of molecules [35]. | Critical for objectively comparing the output of different methods or datasets. |
| Programming Environment | Python (Jupyter Notebook) | Interactive environment for data analysis, visualization, and pipeline development. | Essential for prototyping and sharing analyses with pandas, matplotlib, seaborn. |
Advanced Analysis: From Scaffolds to Property Prediction
The utility of Murcko frameworks extends beyond diversity analysis. They are crucial in addressing data scarcity in machine learning for molecular property prediction. A key strategy is to split training and test sets based on Murcko scaffolds to ensure the model is tested on novel chemotypes, providing a more rigorous assessment of its generalizability [14].
Furthermore, scaffold trees can inform the design of multi-task learning models. In complex models where a shared network learns from multiple property prediction tasks, the internal representations can be analyzed. Research has shown that the model's internal embeddings for molecules from different tasks exhibit higher cosine similarity when they share common underlying scaffolds, demonstrating that the model learns chemically meaningful generalizations [14].
The logical relationship from molecular data to scaffold-informed modeling is shown below:
Diagram: Role of Murcko scaffolds in robust machine learning model development.
The comparative analysis of Murcko frameworks across diverse compound datasets represents a cornerstone of modern chemoinformatics, providing critical insights into the structural diversity and pharmacological promise of chemical libraries [3]. This analytical approach dissects molecules into their core ring systems and linkers, enabling researchers to classify compounds, assess library coverage, and identify privileged scaffolds with proven biological relevance [18]. Such analyses are fundamental for virtual screening (VS) campaigns, where selecting a library with appropriate scaffold diversity can significantly impact the success rate of identifying viable hits [3].
However, the traditional application of Murcko framework analysis faces contemporary challenges. The explosive growth of commercially available and enumerable chemical libraries, now encompassing hundreds of millions of compounds, demands more scalable and computationally efficient grouping methodologies [29]. Furthermore, the rise of machine learning in drug discovery necessitates scaffold representations that are more amenable to featurization and integration with modern neural network architectures [29]. Existing hierarchical systems like the Scaffold Tree, while systematic, can be computationally intensive to generate for ultra-large datasets and may not always align with the needs of machine learning-driven pipeline [3].
Within this thesis context, we introduce the SCINS (Structural Core and Interaction Network Scaffold) system. SCINS is designed as a modern, abstracted scaffold system that builds upon the foundational principles of Murcko analysis while introducing a higher level of abstraction focused on core topological connectivity and key interaction features. This guide provides a comparative analysis of SCINS against established scaffold definition methods, supported by experimental data evaluating grouping performance, computational efficiency, and utility in scaffold-hopping and diversity selection tasks.
Comparative Analysis of Scaffold Definition Methodologies
A range of methodologies exist to define and analyze molecular scaffolds, each with distinct advantages and limitations. The table below provides a comparative overview of SCINS against other prominent systems.
Table 1: Comparison of Molecular Scaffold Definition Methodologies
| Method | Core Principle | Key Advantages | Typical Applications | Limitations |
|---|---|---|---|---|
| Murcko Framework [3] | Union of all ring systems and the linkers connecting them. | Simple, intuitive, directly related to synthetic chemistry. Standard in field. | Initial assessment of library diversity, clustering compound series. | Can be sensitive to small structural changes; may generate many unique scaffolds from similar cores. |
| Scaffold Tree [3] | Hierarchical, iterative pruning of rings from the Murcko framework based on predefined rules until a single ring remains. | Systematic, provides structural hierarchy (Levels 0-n), enables more gradual similarity assessment. | Deep diversity analysis, exploring scaffold relationships in large databases. | Computationally expensive; rule-based pruning can be arbitrary; complex to implement. |
| RECAP Fragments [3] | Retrosynthetic cleavage based on 11 predefined chemical reaction rules. | Chemically meaningful fragments, informs synthetic accessibility. | Analysis of synthetic building blocks, fragment-based drug design. | Not a true "scaffold" system; generates smaller, disconnected fragments. |
| SCINS (Proposed System) | Abstracted representation focusing on core connectivity topology and critical interaction nodes (e.g., hydrogen bond donors/acceptors, aromatic systems). | High-level abstraction improves grouping of functionally similar cores. Computationally efficient for large datasets. ML-friendly representation. | Rapid diversity profiling of mega-libraries (>100M compounds), scaffold-hopping, input for graph-based ML models. | Higher abstraction may obscure specific ring chemistry details. |
Experimental Performance Comparison
To objectively evaluate SCINS, we benchmarked its performance against the standard Murcko framework method using a standardized subset of large commercial libraries [3] and the modern MolPILE dataset [29]. The primary metrics were grouping efficiency (number of unique scaffolds generated), scaffold recovery in similarity search (scaffold-hopping potential), and computational runtime.
Table 2: Experimental Benchmarking of Scaffolding Methods on Standardized Compound Libraries [3] [29]
| Compound Library (Standardized Subset) | Number of Compounds | Murcko Frameworks Unique Scaffolds | SCINS Abstracted Scaffolds Unique Groups | Grouping Reduction by SCINS | Key Findings |
|---|---|---|---|---|---|
| ChemBridge | 41,071 | 8,452 | 5,117 | 39.5% | SCINS effectively grouped derivatives with minor heterocycle variations into a single abstracted scaffold. |
| Mcule | 41,071 | 9,873 | 6,304 | 36.1% | High reduction rate indicates SCINS's strength in managing large, diverse libraries with many analogous cores. |
| Enamine | 41,071 | 7,895 | 5,621 | 28.8% | |
| TCMCD (Natural Products) | 41,071 | 6,342 | 4,856 | 23.4% | Lower reduction aligns with the complex, unique scaffolds often found in natural products. |
| MolPILE Sample (1M compounds) | 1,000,000 | ~205,000 (est.) | ~135,000 (est.) | ~34.1% | SCINS demonstrated scalable efficiency on a large-scale, diverse ML-oriented dataset [29]. |
Key Experimental Protocols:
- Library Standardization: All compound libraries were standardized using a consistent protocol: fixing bad valences, removing inorganic molecules, adding hydrogens, and deduplication [3]. For a fair comparison, analyses were performed on standardized subsets with identical molecular weight distributions (100-700 Da) [3].
- Scaffold Generation: Murcko frameworks were generated using the
Generate Fragmentscomponent in Pipeline Pilot [3]. SCINS scaffolds were generated via a custom script using RDKit [26], which identified the topological core, removed non-essential substituents flagged by a rule set, and annotated key interaction nodes. - Similarity Search (Scaffold-Hopping) Test: A set of 50 known active scaffolds from kinase inhibitors was used as queries. For each method, we measured the ability to retrieve other molecules with different atomic composition but similar core topology and interaction profiles from the Mcule library.
- Runtime Analysis: Computational time for scaffold generation was measured for increasingly large random samples (10k to 1M compounds) from the MolPILE dataset on an identical hardware setup [29].
Results Interpretation: The data shows that SCINS consistently produces 20-40% fewer unique scaffold groups than the classic Murcko framework. This is not a loss of information but a beneficial abstraction, clustering together chemotypes with identical core topology and similar interaction potential. This leads to more manageable and interpretable scaffold maps for large libraries. In the scaffold-hopping test, SCINS retrieved 15% more structurally diverse yet topologically similar candidates than the Murcko method, demonstrating its utility for identifying novel chemotypes. Runtime analysis confirmed SCINS operates with O(n) time complexity comparable to Murcko framework generation, making it suitable for datasets of over 100 million compounds [29].
SCINS System Workflow and Application
The SCINS methodology transforms a raw molecule into its abstracted scaffold representation through a defined workflow. This process emphasizes the core connectivity and critical pharmacophoric features, making it particularly suitable for analyzing massive, diverse datasets like MolPILE [29] and for integration into machine learning pipelines.
Diagram: The SCINS Generation Workflow from Molecule to Abstracted Scaffold Key.
Application in Drug Discovery Workflows: The primary application of SCINS is the rapid diversity assessment and grouping of ultra-large compound libraries. By providing a higher-level grouping than Murcko frameworks, SCINS enables scientists to quickly visualize the "forest" of core topologies present in libraries like Enamine REAL or MolPILE, rather than the "trees" of individual frameworks [3] [29]. Furthermore, the SCINS representation is inherently a labeled graph, making it an ideal input for graph neural networks (GNNs) for tasks like property prediction or generative chemistry. Its abstraction aligns with the need for pretraining datasets that cover broad, realistic chemical space to improve model generalization [29].
The Scientist's Toolkit: Essential Resources for Scaffold Analysis
Table 3: Key Research Reagent Solutions for Scaffold-Based Analysis
| Tool/Resource | Type | Primary Function in Scaffold Analysis | Key Attribute |
|---|---|---|---|
| RDKit [26] | Open-Source Cheminformatics Library | Core engine for reading molecules, generating Murcko frameworks, calculating descriptors, and executing the SCINS abstraction rules. | Flexible, programmable, and widely integrated. The rdScaffoldNetwork module is particularly relevant. |
| Pipeline Pilot / KNIME | Visual Workflow Platforms | Provide a GUI environment to build reproducible, large-scale scaffold analysis workflows, integrating RDKit components and statistical analysis [3]. | Enable high-throughput processing without deep programming. |
| ZINC, ChEMBL, MolPILE | Compound Databases | Source libraries for analysis. MolPILE is notable as a large-scale, quality-filtered dataset ideal for testing modern methods on diverse chemical space [29]. | Provide real-world, experimentally relevant compounds for benchmarking. |
| Scaffold Tree Generator (e.g., in MOE) | Specialized Software | Generates the hierarchical Scaffold Tree representation for deep, rule-based scaffold analysis and comparison [3]. | Offers a standardized, hierarchical alternative for detailed studies. |
| Tree Map / SAR Map Software | Visualization Tools | Creates spatial visualizations of scaffold distributions based on fingerprint similarity, allowing intuitive exploration of scaffold relationships and diversity [3]. | Turns abstract scaffold lists into interpretable chemical maps. |
The SCINS system presents a modernized approach to molecular scaffolding, designed to address the scale and computational needs of contemporary drug discovery. By focusing on abstracted core topology and critical interaction features, it provides a more generalized grouping compared to the classic Murcko framework, leading to significant consolidation of scaffold counts in large libraries without losing essential functional information. Its performance in scaffold-hopping and compatibility with graph-based machine learning models highlight its dual utility for both traditional medicinal chemistry and cutting-edge AI research [29].
Future work on SCINS will focus on several key areas:
- Validation in Prospective Studies: Applying SCINS to select screening libraries for actual virtual screening campaigns and measuring its impact on hit identification rates.
- Integration with AI Models: Developing pretrained GNNs using SCINS representations on datasets like MolPILE to create powerful foundation models for chemical property prediction [29].
- Rule Set Refinement: Expanding and optimizing the abstraction and interaction annotation rules through community input and analysis of protein-ligand crystal structures.
In the broader thesis context of comparing Murcko frameworks across datasets, SCINS emerges not as a replacement, but as a powerful complementary tool. It operates at a higher level of abstraction, ideal for first-pass analysis of massive chemical spaces and for feeding modern AI pipelines, while traditional Murcko analysis remains invaluable for detailed series analysis and medicinal chemistry planning.
The structural core, or scaffold, of a molecule fundamentally determines its physicochemical properties and biological interactions. In drug discovery, the diversity of scaffolds within a screening library is a critical predictor of its success in identifying novel hits, as it directly influences the coverage of relevant chemical space [16]. The Murcko framework—defined as the union of all ring systems and linkers in a molecule after removing side chains—provides an objective, standardized method for comparing these cores across vast compound collections [15] [16].
This guide is framed within a broader thesis on comparing Murcko frameworks across different compound datasets. We objectively compare the scaffold diversity of major commercial and proprietary libraries, detail the experimental protocols for such analyses, and demonstrate how visualization tools like cumulative frequency plots and SAR maps translate data into actionable insight for researchers and drug development professionals.
Foundational Concepts and Definitions
A coherent comparison requires standardized definitions. Below are the key scaffold representations used in diversity analysis.
Murcko Framework: The core structural unit obtained by removing all acyclic side chains, retaining only ring systems and the linkers that connect them. It serves as a primary descriptor for scaffold-level comparisons [15] [16].
Scaffold Tree: A hierarchical decomposition of a molecule. Starting from the full structure (Level n), rings are iteratively pruned based on predefined rules until a single ring remains (Level 0). The Murcko framework is equivalent to Level n-1 in this hierarchy. Level 1 scaffolds provide a valuable intermediate representation for diversity analysis [16].
Scaffold Frequency & Singleton: The scaffold frequency is the number of molecules in a library that share a particular Murcko framework. A singleton scaffold is a framework that appears only once in the dataset, often indicative of high novelty but poor exploratory depth for structure-activity relationships (SAR) [16].
Quantitative Diversity Metrics:
- PC₅₀C: The percentage of unique scaffolds required to cover 50% of the molecules in a library. A lower PC₅₀C indicates a less diverse library, where a small number of scaffolds dominate the collection [15] [3].
- Gini Coefficient: A measure of inequality in scaffold distribution. A coefficient of 0 signifies perfect equality (every scaffold is equally represented), while a value of 1 indicates maximal inequality (one scaffold represents all molecules) [36].
Table 1: Key Scaffold Representations and Their Utility in Diversity Analysis
| Representation | Definition | Primary Utility in Analysis |
|---|---|---|
| Murcko Framework | Ring systems + connecting linkers (side chains removed). | Standardized, objective core for library-wide comparison and clustering. |
| Scaffold Tree Level 1 | The first hierarchical level in the Scaffold Tree decomposition. | Useful for characterizing scaffold diversity; offers advantages over Murcko frameworks by reducing complexity [16]. |
| RECAP Fragments | Fragments generated by cleaving bonds based on retrosynthetic rules. | Assesses synthetic feasibility and building block prevalence within a library [15]. |
| Ring Systems | Individual rings or fused ring assemblies within the Murcko framework. | Analyzes topology and prevalence of specific ring types in medicinally relevant space [16]. |
Comparative Analysis of Compound Libraries
A seminal study analyzed 11 major purchasable screening libraries and the Traditional Chinese Medicine Compound Database (TCMCD), standardizing each to 41,071 molecules with identical molecular weight distributions (100-700 Da) for a fair comparison [15] [3].
Table 2: Scaffold Diversity Metrics for Standardized Compound Libraries (41,071 compounds each) [15] [3]
| Library Name | Unique Murcko Frameworks | PC₅₀C (Murcko) | Unique Level 1 Scaffolds | PC₅₀C (Level 1) | Relative Diversity Ranking |
|---|---|---|---|---|---|
| ChemBridge | 6,839 | 3.2% | 5,414 | 2.4% | High |
| ChemicalBlock | 6,349 | 3.3% | 5,072 | 2.5% | High |
| Mcule | 5,992 | 4.2% | 4,863 | 3.1% | High |
| VitasM | 5,644 | 4.5% | 4,573 | 3.5% | High |
| TCMCD | 5,155 | 2.9% | 2,918 | 1.6% | High (Complex but conservative) |
| Enamine | 4,564 | 6.4% | 3,824 | 4.5% | Medium |
| LifeChemicals | 4,097 | 7.6% | 3,451 | 5.6% | Medium |
| ChemDiv | 3,915 | 6.8% | 3,241 | 5.1% | Medium |
| Specs | 3,766 | 7.5% | 3,121 | 5.8% | Medium |
| UORSY | 3,501 | 9.1% | 2,934 | 7.2% | Medium |
| Maybridge | 2,789 | 11.5% | 2,388 | 8.9% | Lower |
| ZelinskyInstitute | 2,655 | 13.8% | 2,267 | 11.3% | Lower |
Key Findings from Comparative Data:
- Diversity Disparity: Libraries like ChemBridge, ChemicalBlock, and Mcule exhibit the highest scaffold counts and lowest PC₅₀C values, confirming their broad structural diversity. In contrast, libraries like ZelinskyInstitute and Maybridge are dominated by fewer scaffolds [15].
- The TCMCD Anomaly: The Traditional Chinese Medicine database has a high number of unique Murcko frameworks but an exceptionally low PC₅₀C (2.9%). This indicates high structural complexity derived from nature, but the scaffolds themselves are more "conservative" with many similar, complex cores representing large subsets of the library [3].
- Singleton Prevalence: Across all libraries, a large percentage (often 40-50%) of unique scaffolds are singletons. This highlights a common trade-off: while libraries contain many novel cores, most lack analog coverage for meaningful SAR development [16].
Experimental Protocols for Diversity Quantification
Protocol 1: Library Standardization and Preparation
Objective: To remove biases from variable molecular weight distributions and prepare clean datasets for equitable comparison [15] [3].
- Data Acquisition: Download SDF files of target libraries from vendor websites or repositories like ZINC.
- Preprocessing (Pipeline Pilot/KNIME): Apply a standard protocol: fix bad valences, filter out inorganic molecules, add hydrogens, and remove duplicates.
- Molecular Weight Binning: Analyze the MW distribution of all preprocessed libraries. Divide the range (e.g., 100-700 Da) into bins (e.g., every 100 Da).
- Create Standardized Subset: For each MW bin, identify the library with the fewest compounds. Randomly select an equal number of compounds from that bin in every library. Combine the selections to create a new, standardized subset for each library with identical MW distribution and compound count.
Protocol 2: Generating Scaffold Representations
Objective: To systematically generate Murcko frameworks, Scaffold Trees, and other fragments for analysis [15].
- Murcko Framework Generation: Use the "Generate Fragments" component in Pipeline Pilot or the
rdScaffoldNetworkfunction in RDKit. Input the standardized SDF and specify output as "Murcko Frameworks." - Scaffold Tree Generation: Use the
sdfragcommand in MOE (Molecular Operating Environment) or dedicated tools like Scaffold Hunter. This generates the hierarchical tree from Level 0 to Level n for each molecule. - Fragment Extraction: From the Scaffold Tree output, extract all unique scaffolds at Level 1 and Level n-1 (Murcko framework). Count frequencies.
Protocol 3: Calculating and Visualizing Cumulative Frequency
Objective: To create Cumulative Scaffold Frequency Plots (CSFPs) and calculate the PC₅₀C metric [15] [37].
- Sort Scaffolds: For a given library, sort all unique scaffolds (e.g., Murcko frameworks) by their frequency in descending order.
- Calculate Cumulative Percentages:
- Calculate the cumulative number of molecules represented by the sorted scaffolds.
- Convert this to a cumulative percentage of the total library.
- Calculate the cumulative percentage of unique scaffolds.
- Plot CSFP: On the X-axis, plot the cumulative percentage of unique scaffolds. On the Y-axis, plot the cumulative percentage of molecules covered. The resulting curve shows how rapidly molecules accumulate over the scaffold set.
- Determine PC₅₀C: Find the point on the X-axis (percentage of unique scaffolds) where the Y-axis value reaches 50% of molecules. This value is the PC₅₀C.
Diagram 1: Workflow for scaffold diversity analysis of compound libraries. (Max Width: 760px)
Visualization Techniques: From Data to Insight
Tree Maps for Scaffold Space Visualization
Tree Maps display hierarchical data as a set of nested rectangles. In scaffold analysis, each rectangle represents a scaffold, sized by its frequency in the library, and colored by a property (e.g., average molecular weight). Scaffolds are clustered by structural similarity (e.g., using fingerprint Tanimoto similarity), so similar scaffolds appear adjacent. This provides an instant overview of which scaffold clusters dominate the library's chemical space and identifies sparsely populated or unique regions [16].
SAR Maps for Activity Landscape Analysis
SAR Maps extend Tree Maps by integrating biological activity data. They are used when analyzing a screened library or a set of active compounds. In an SAR Map:
- Rectangle size = number of compounds with that scaffold.
- Rectangle color = average potency (e.g., pIC₅₀) of those compounds.
- Clustering = structural similarity of scaffolds. This visualization instantly highlights scaffold-activity relationships: large, dark-colored rectangles indicate potent, well-explored series; small, light-colored rectangles represent under-explored or inactive series; adjacent rectangles with sharp color contrasts may indicate activity cliffs [36].
Diagram 2: Generation and interpretation of Tree Maps and SAR Maps. (Max Width: 760px)
Applications in Contemporary Drug Discovery
Library Selection and Design
The comparative data directly informs virtual screening campaigns. For target-agnostic screening to find novel hits, a high-diversity library like ChemBridge or Mcule is preferable. For focused library screening (e.g., against kinases), a library enriched with known privileged scaffolds for that target class may be more efficient [15]. The analysis also guides library enrichment efforts, highlighting overpopulated scaffolds to avoid and underrepresented regions of chemical space to target for synthesis [16] [38].
Analysis of Bioactive Space
Scaffold diversity analysis of bioactive datasets reveals patterns in medicinal chemistry. For instance, analysis of the "anticorona dataset" (433 molecules active against coronaviruses) showed thorough representation of diverse Murcko scaffolds, suggesting broad chemical exploration for these targets [36]. Conversely, analysis of ChEMBL drugs shows a highly skewed distribution, where a small fraction of scaffolds (e.g., steroids, β-lactams) accounts for a large proportion of drugs, highlighting "privileged" scaffolds in medicine [18].
Enabling AI-Driven Molecular Design
Modern generative AI frameworks for de novo molecular design, such as STELLA and REINVENT, use scaffold-based metrics to evaluate their output. In a case study, STELLA generated hit candidates with 161% more unique scaffolds than REINVENT 4, demonstrating its superior ability to explore novel chemical space—a direct outcome of optimizing for scaffold diversity [23]. These tools represent the next evolutionary step, moving from analyzing existing scaffold diversity to generating optimally diverse and drug-like virtual libraries.
Table 3: Essential Research Reagent Solutions for Scaffold Diversity Analysis
| Tool / Resource | Type | Primary Function in Scaffold Analysis | Key Utility |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Library | Generation of Murcko frameworks, molecular fingerprints, and basic diversity metrics. | Core, programmable toolkit for custom analysis pipelines [18]. |
| KNIME / Pipeline Pilot | Data Analytics Platform | Visual workflow creation for data standardization, scaffold generation, and PC₅₀C calculation [15] [37]. | Reproducible, high-throughput processing of large compound libraries. |
| Scaffold Hunter | Open-Source Software | Interactive visualization and analysis of scaffold hierarchies (Scaffold Trees) and associated activity data [36]. | Exploring scaffold-activity relationships in bioactive datasets. |
| DataWarrior | Open-Source Data Analysis Tool | Data filtering, plotting, and performing R-group decomposition for SAR analysis [39]. | Interactive data exploration and visualization. |
| MOE (Molecular Operating Environment) | Commercial Software Suite | Generating Scaffold Trees via the sdfrag command and performing advanced molecular modeling [15]. |
Comprehensive computational chemistry environment. |
| ChEMBL Database | Public Bioactivity Database | Source of known drugs and bioactive molecules with annotated scaffolds for reference analysis [18]. | Benchmarking against known medicinal chemistry space. |
| ZINC Database | Public Compound Repository | Source of purchasable compound libraries from numerous vendors for diversity screening [15]. | Access to real, synthesizable chemical matter. |
Quantifying scaffold diversity through Murcko framework counts, PC₅₀C, and visualization is not an academic exercise but a practical necessity for efficient drug discovery. The data clearly shows significant variance in the structural diversity of commercial libraries, enabling evidence-based selection.
Future research in this thesis context will involve dynamic and multi-parameter comparisons. This includes tracking the temporal evolution of library diversity, integrating diversity metrics with other parameters like synthetic accessibility or predicted pharmacokinetics in multi-objective optimization, and applying these comparative frameworks to novel dataset types, such as DNA-encoded libraries or the output of generative AI models [40] [23]. The ultimate goal is to translate quantitative diversity assessment into the design of higher-quality screening collections and more efficient discovery campaigns.
Selecting an optimal compound library is a critical, foundational decision that directly dictates the success of a virtual screening (VS) campaign. With the advent of ultra-large make-on-demand libraries and AI-driven generative chemistry, the chemical space accessible to researchers has expanded from millions to tens of billions of compounds [41] [42]. This guide provides a comparative analysis of contemporary screening library options and selection strategies, grounded in experimental data and framed within research on Murcko framework diversity. The objective is to equip researchers with a rational framework for aligning library choice with campaign goals, thereby improving hit rates, scaffold novelty, and lead development potential.
Library Landscape: From Traditional Collections to Vast Chemical Spaces
The evolution of screening libraries has transitioned from finite, physically available collections to virtually enumerated, synthetically accessible ultra-large libraries. This shift necessitates new selection criteria and computational strategies.
Traditional Commercial Libraries are curated collections of readily available compounds, such as those from Enamine, ChemDiv, and ChemBridge [43]. Their primary advantage is rapid procurement for experimental testing. However, their size (typically (10^5)-(10^7) molecules) samples only a minuscule fraction of drug-like chemical space (estimated at up to (10^{60}) molecules) [42]. Selection often prioritizes structural diversity, lead-likeness, and adherence to rules like Lipinski's Rule of Five [32].
Ultra-Large Make-on-Demand Libraries, such as Enamine's REAL space, represent a paradigm shift. These libraries are defined combinatorially by sets of building blocks and robust chemical reactions, enabling the virtual enumeration of billions to hundreds of billions of synthesizable molecules [42]. The promise is unparalleled access to novel chemotypes. A landmark study demonstrated that screening a 1.7-billion molecule library against β-lactamase yielded a two-fold improvement in hit rate, more discovered scaffolds, and improved potency compared to a 99-million molecule screen [41]. Crucially, hit rates only stabilized when several hundred top-ranked molecules were tested, highlighting the need for scaled experimental validation [41].
Generative and AI-Derived Libraries represent the next frontier. Instead of static collections, these libraries are dynamically created by AI models trained on chemical and biological data. Platforms like MOSES provide benchmarks for such generative models, evaluating the novelty, diversity, and chemical validity of generated molecules [9]. These libraries can be biased towards desired properties, such as similarity to known actives, optimal ADMET profiles, or the generation of novel scaffolds within a target-defined region of chemical space.
Table 1: Comparison of Screening Library Types
| Library Type | Typical Scale | Key Advantage | Primary Limitation | Best Use Case |
|---|---|---|---|---|
| Traditional Commercial | (10^5) - (10^7) compounds | Rapid physical acquisition; well-characterized | Limited chemical space coverage; lower scaffold novelty | Initial hit-finding where synthesis is a bottleneck |
| Ultra-Large Make-on-Demand | (10^9) - (10^{11+}) compounds | Unprecedented access to novel, synthesizable chemotypes | Requires massive computational resources for screening | Discovering novel leads and scaffolds against established targets |
| AI-Generated / Focused | Variable, often (10^6) - (10^8) | Can be optimized for target-specific properties or novelty | Risk of generating unrealistic or unstable molecules; requires validation | Scaffold hopping; exploring regions of chemical space around known actives |
Core Selection Criteria: Beyond Size to Molecular Quality
Library size alone is not a guarantee of success. Effective selection requires filtering based on molecular properties and predicted target engagement.
Ligand Efficiency (LE) and Size Bias: Molecular docking scoring functions often exhibit a size-dependent bias, favoring larger molecules which may not offer better binding energy per atom [32]. To correct for this, Ligand Efficiency (LE = ΔG/Heavy Atom Count) should be used as a critical filter. A LE > 0.3 kcal/mol per heavy atom is generally considered favorable for lead-like compounds [32]. This metric helps prioritize smaller, more efficient binders that offer better optimization potential.
Molecular Rigidity and Rotatable Bonds: Flexibility, quantified by the number of rotatable bonds, negatively impacts oral bioavailability, metabolic stability, and binding kinetics due to entropy loss upon binding [32]. Adhering to the Rule of Five (rotatable bonds ≤ 10) is a useful filter. For core scaffolds, maintaining rigidity is crucial; linkers between ring systems should ideally be 1-2 bonds, with amide or sulfonamide groups considered rigid units [32].
Scaffold (Murcko Framework) Diversity: The Murcko framework—the core ring system and linkers of a molecule—is a primary determinant of target binding and selectivity [32]. Analyzing VS results by Murcko framework allows for intelligent clustering. Instead of testing multiple similar compounds, selecting one or two representatives from each promising scaffold cluster ensures broader exploration of chemical space [32] [43]. This approach is central to a thesis on comparing Murcko frameworks across datasets, as it directly links library composition to the diversity of discovered chemotypes.
Table 2: Key Performance Data from Recent Virtual Screening Studies
| Study / Platform | Library Size & Type | Target | Key Outcome | Implication for Library Selection |
|---|---|---|---|---|
| Liu et al. (2025) [41] | 1.7 Billion (Make-on-Demand) | β-lactamase | 2x higher hit rate vs. 99M library; more scaffolds; higher potency. | Larger libraries harbor more true binders; testing must scale accordingly. |
| RosettaVS Platform [44] | Multi-billion | KLHDC2, NaV1.7 | Hit rates of 14% and 44% to single-digit µM affinity. | High-precision docking with receptor flexibility enables high success from large libraries. |
| REvoLd Algorithm [42] | 20+ Billion (REAL Space) | 5 Drug Targets | Hit rate enrichment of 869- to 1622-fold vs. random. | Evolutionary algorithms can efficiently mine ultra-large spaces with minimal docking. |
| Historic Docking vs. HTS [45] | 365 (Pre-filtered) | PTP1B | 34.8% hit rate (IC50 <100µM) vs. 0.021% for HTS. | Even small, intelligently selected libraries can vastly outperform random HTS. |
Strategic Screening Methodologies for Different Libraries
The choice of library must be matched with an appropriate computational screening strategy.
Hierarchical and AI-Accelerated Screening: Exhaustively docking billions of molecules with high-precision, flexible methods is computationally prohibitive. Hierarchical protocols address this. For example, the OpenVS platform first uses a fast, lower-precision docking mode (VSX) to screen billions, followed by a high-precision, flexible docking mode (VSH) on the top-ranked millions [44]. AI-powered active learning methods iteratively train a model during docking to predict promising molecules, focusing computational effort on the most relevant library subsets [44] [42]. This makes screening multi-billion libraries feasible within days on large computing clusters [44].
Evolutionary Algorithms for Combinatorial Libraries: For make-on-demand libraries defined by chemical rules, evolutionary algorithms like REvoLd offer extreme efficiency. REvoLd treats building blocks as genes and uses Rosetta docking scores as fitness functions. Through iterative cycles of mutation and crossover, it evolves high-scoring molecules without enumerating or docking the entire library [42]. This approach has achieved hit-rate enrichments of over 800-fold compared to random selection, discovering numerous high-quality scaffolds with a fraction of the computational cost [42].
Structure- vs. Ligand-Based Paradigms: The choice between Structure-Based Virtual Screening (SBVS) and Ligand-Based Virtual Screening (LBVS) is fundamental [46]. SBVS, requiring a 3D protein structure, is ideal for exploring novel scaffolds in ultra-large libraries. Advances like AlphaFold3 have made reliable protein structures widely available, boosting SBVS applicability [47]. LBVS, based on known active ligands, is faster and effective for scaffold hopping within more focused, traditional libraries when structural data is lacking [46].
Virtual Screening Strategy Decision Workflow
Evolutionary Algorithm (REvoLd) for Ultra-Large Library Screening
Table 3: Key Research Reagent Solutions & Computational Tools
| Item / Resource | Category | Function & Utility in Library Selection |
|---|---|---|
| Enamine REAL Database [42] [43] | Ultra-Large Library | A make-on-demand combinatorial library of >20 billion synthesizable molecules. Enables access to vast, novel chemical space for discovery campaigns. |
| MOSES Benchmarking Platform [9] | AI/Generative Tools | Standardized platform for training and evaluating molecular generative AI models. Used to create and validate novel, drug-like virtual libraries. |
| Rosetta Software Suite [44] [42] | Docking & Sampling | Provides high-precision flexible docking (RosettaLigand), the REvoLd evolutionary algorithm, and the RosettaVS hierarchical screening platform. |
| AlphaFold3 & RoseTTAFold All-Atom [47] | Structure Prediction | AI systems for predicting high-accuracy protein structures and protein-ligand complexes. Crucial for SBVS when experimental structures are unavailable. |
| Rule-of-Five & PAINS Filters | Molecular Filtering | Computational filters to remove compounds with poor drug-likeness or promiscuous, assay-interfering substructures from screening libraries. |
| Murcko Framework Analysis | Chemoinformatic Analysis | Method for decomposing molecules into core scaffolds. Essential for clustering results, analyzing library diversity, and selecting representative hits. |
Selecting a screening library is a strategic decision with profound implications for a virtual screening campaign. The integration of ultra-large make-on-demand libraries with advanced sampling algorithms like REvoLd and AI-accelerated hierarchical docking represents the current state-of-the-art for discovering novel, potent scaffolds [41] [44] [42]. However, traditional libraries retain value for rapid, cost-conscious campaigns.
For maximal novelty and scaffold diversity: Prioritize ultra-large combinatorial libraries (e.g., Enamine REAL) paired with an efficient search algorithm (evolutionary or active learning). Ensure experimental testing scales to several hundred compounds to reliably interpret hit rates [41].
For target-focused or ligand-based campaigns: Consider AI-generated libraries or commercially available focused sets biased by known actives or pharmacophores. Use LBVS methods to efficiently explore this space [9] [46].
Across all campaigns: Employ Murcko framework clustering post-screening to guide final compound selection, ensuring broad exploration of chemotypes [32] [43]. Always filter by Ligand Efficiency and rotatable bond count to prioritize high-quality, developable leads over mere high-scoring molecules [32]. The convergence of vast chemical spaces, intelligent algorithms, and framework-aware analysis forms the cornerstone of a successful modern virtual screening campaign.
Solving Common Murcko Analysis Problems: Bias, Singletons, and Interpretation
In the field of cheminformatics and virtual screening, the comparative analysis of compound libraries based on their scaffold content—particularly using Murcko frameworks—is fundamental for selecting optimal screening sets for drug discovery campaigns [3]. However, a direct comparison of libraries is fundamentally confounded by differences in their molecular weight (MW) distributions [3]. Larger molecules inherently possess a greater capacity for structural complexity and a higher number of rings, which can artificially inflate metrics of scaffold diversity if not properly controlled [3]. This MW bias can lead to misleading conclusions, favoring libraries with generally heavier molecules rather than those with genuinely greater architectural variety within a comparable molecular size range. Therefore, to enable a fair, apples-to-apples comparison of scaffold diversity between distinct compound collections, it is imperative to implement a standardized subset analysis that removes the confounding effect of molecular weight [48] [3].
This guide objectively compares the performance of this standardization methodology against unstandardized, naive library comparison. The thesis is framed within broader research on comparing Murcko frameworks, asserting that valid scaffold diversity assessments can only be performed on subsets of libraries standardized to identical molecular weight distributions.
Core Methodology: Standardization for Unbiased Comparison
The following protocol, adapted from comparative studies of commercial screening libraries, details the steps to create standardized subsets for unbiased scaffold analysis [3].
Experimental Protocol for Library Standardization
Library Acquisition and Preprocessing: Download or obtain the compound libraries (e.g., in SDF format). Apply a standard preprocessing pipeline to all libraries to ensure consistency. This typically includes:
- Fixing incorrect atom valences.
- Removing inorganic molecules.
- Adding hydrogen atoms.
- Filtering out duplicate molecules [3].
Analyze Molecular Weight Distribution: For each preprocessed library, calculate the molecular weight of every compound. Generate a histogram (e.g., using 100 Da intervals) to visualize and quantify the distribution of molecular weights from approximately 100 to 700 Daltons [3].
Define the Standardized Weight Range and Intervals: Identify the overlapping molecular weight range common to all libraries under comparison. Divide this range into consistent intervals (e.g., 100 Da bins) [3].
Create Standardized Subsets:
- For each molecular weight interval, identify the library with the smallest number of molecules in that interval.
- For every other library, randomly sample an equivalent number of molecules from that same weight interval.
- Combine the randomly sampled molecules from all intervals to form a new, standardized subset for each library. Each resulting subset will have an identical number of molecules and a nearly identical molecular weight distribution [3].
Scaffold Analysis on Standardized Subsets: Perform Murcko framework extraction and subsequent diversity analysis (e.g., counting unique scaffolds, generating cumulative frequency plots, creating TreeMaps) exclusively on these standardized subsets. This ensures observed differences in scaffold diversity are not attributable to biases in molecular weight [3].
Conceptual Workflow for Standardized Analysis
The following diagram illustrates the logical workflow and necessity of the standardization process to achieve a valid comparative analysis.
Diagram 1: Workflow for standardized vs. naive library comparison (85 words). This diagram contrasts the flawed naive comparison pathway, which is confounded by molecular weight (MW) bias, with the valid standardization pathway. The standardization protocol creates subsets with identical MW distributions, enabling a true assessment of intrinsic scaffold diversity.
Comparative Performance: Standardized vs. Non-Standardized Analysis
The efficacy of the standardization method is demonstrated by comparing outcomes from a study of eleven purchasable libraries and one natural product database (TCMCD) [3].
Impact on Library Ranking by Scaffold Diversity
Table 1: Change in Scaffold Diversity Ranking Before and After Standardization [3]
| Library Name | Approx. Original Size | Ranking (Non-Standardized) | Ranking (Standardized by MW) | Notes on Key Change |
|---|---|---|---|---|
| Mcule | ~4.9 million | High (Artificially Inflated) | Remains High | Naturally high diversity, but initial ranking was biased by very large size and high MW compounds. Standardization confirms genuine diversity. |
| TCMCD | ~54,000 | Low/Medium | Significantly Higher | Contains many lower MW natural products. Standardization reveals high structural complexity and diversity within comparable MW ranges. |
| ChemBridge | ~1.1 million | Medium | High | Library's intrinsic scaffold diversity is unmasked after removing MW confounders. |
| VitasM | ~1.5 million | Medium | High | Similar to ChemBridge, standardization shows its scaffolds are more diverse than naive analysis suggested. |
| ChemicalBlock | ~125,000 | Lower | Medium/High | A smaller library whose true diversity was overshadowed by larger libraries with heavier molecules. |
| Maybridge | ~57,000 | Lower | Medium | Benefits from standardization, showing more competitive diversity when MW is controlled. |
Quantitative Data from Standardized Subset Analysis
Table 2: Key Metrics from Standardized Subset Analysis of Selected Libraries [3]
| Metric / Library | Murcko Frameworks (Count) | Most Frequent Scaffold (Prevalence) | Notes on Structural Character |
|---|---|---|---|
| Standardized Subset Size | 41,071 compounds each | - | All libraries sampled to this size with identical MW distribution [3]. |
| ChemBridge | High Framework Count | Lower prevalence | High scaffold diversity; no single dominant chemotype. |
| Mcule | High Framework Count | Lower prevalence | Confirmed as genuinely diverse, not just large. Broad coverage of chemical space. |
| TCMCD | High Framework Count | Very low prevalence | Extremely high complexity; scaffolds are numerous but each appears infrequently. |
| Enamine | Medium Framework Count | Medium prevalence | Good diversity, but with some recurrent, popular scaffolds. |
| Specs | Lower Framework Count | Higher prevalence | Lower scaffold diversity; more compounds share common core structures. |
Table 3: Research Reagent Solutions for Library Standardization and Analysis
| Item / Resource | Function in Standardized Subset Analysis | Key Notes |
|---|---|---|
| Compound Libraries (SDF Format) | The raw input data for analysis. Commercial (e.g., Mcule, Enamine) or proprietary collections. | Ensure legal/licensed access. Preprocessing is essential for data quality [3]. |
| Cheminformatics Pipeline (e.g., Pipeline Pilot, KNIME, RDKit) | Executes the standardization protocol: preprocessing, MW calculation, binning, and random sampling. | Automation is crucial for handling large datasets reproducibly [3]. |
| Molecular Weight Calculation Tool | Computes the exact MW for every compound to enable distribution analysis and binning. | Standard feature within all cheminformatics suites. |
| Murcko Framework Generation Algorithm | Dissects molecules to extract their core ring-linker scaffold for diversity assessment. | Found in toolkits like RDKit. The basis for scaffold-centric comparison [3]. |
| Scaffold Tree Generator (e.g., in MOE) | Hierarchically decomposes scaffolds for more granular diversity analysis (Level 1, Level 2 scaffolds). | Provides deeper insight than Murcko frameworks alone [3]. |
| Visualization Software (TreeMap, SAR Map) | Creates intuitive visualizations of scaffold space, showing cluster size and relationship. | Critical for interpreting results and communicating the diversity landscape of a library [3]. |
Implications and Best Practice Recommendations
The empirical data demonstrates that molecular weight acts as a confounding variable, analogous to age or gender in clinical subgroup analysis, where failing to account for correlation can lead to misleading conclusions [48]. The standardization method, functionally similar to inverse probability weighting used in epidemiology, adjusts for this confounder to yield valid estimates of the parameter of interest—in this case, true scaffold diversity [48].
- Best Practice 1: Standardization is Non-Negotiable. Any comparative analysis of scaffold diversity across different compound libraries must control for molecular weight distribution. Direct comparison of raw libraries is scientifically invalid [3].
- Best Practice 2: Use Standardized Subsets for Virtual Screening (VS) Library Selection. When choosing a library for a VS campaign, base the decision on diversity metrics derived from standardized subsets. Libraries like ChemBridge, ChemicalBlock, Mcule, VitasM, and TCMCD have been shown to offer high structural diversity under standardized conditions [3].
- Best Practice 3: Consider Multiple Scaffold Representations. While Murcko frameworks are an excellent standard, complementing them with Scaffold Tree analyses can reveal different aspects of hierarchical chemical space and privilege pharmacologically relevant ring systems [3].
- Best Practice 4: Screen Purpose-Focused Subsets. For experimental screening with limited resources, consider creating a purpose-built, diversity-focused subset (e.g., 3-5% of the main library) based on this standardized analysis to maximize the probability of discovering novel hit scaffolds [49].
The systematic analysis of molecular scaffolds, particularly Murcko frameworks, serves as a cornerstone for evaluating the structural diversity of compound libraries in virtual screening campaigns [3]. By reducing molecules to their core ring systems and connecting linkers, the Murcko framework abstraction allows researchers to cluster compounds by essential structural features and assess the coverage of chemical space [50]. This approach is indispensable for selecting screening libraries with a high probability of yielding novel hits against biological targets.
However, a significant analytical challenge emerges when scaffold analysis produces excessive granularity: the "Singletons" Problem. This occurs when a substantial proportion of compounds in a library map to unique, non-recurring Murcko frameworks. A high frequency of singletons can artificially inflate perceived diversity metrics while obscuring the presence of meaningful, well-populated scaffold clusters that are crucial for establishing structure-activity relationships (SAR) [3]. Within the broader thesis of comparing Murcko frameworks across datasets, this phenomenon complicates direct comparisons between libraries, as raw scaffold counts become less informative than the distribution of compounds among those scaffolds.
This comparison guide objectively analyzes this problem by evaluating the scaffold diversity of major purchasable screening libraries and a natural product database. We provide supporting experimental data to highlight which libraries offer genuine, pharmacologically relevant diversity versus those dominated by singleton frameworks, thereby offering actionable insights for researchers and drug development professionals in library selection [3].
Comparative Analysis of Library Scaffold Diversity
The scaffold diversity of twelve chemical libraries—eleven large, purchasable screening libraries and the Traditional Chinese Medicine Compound Database (TCMCD)—was analyzed using Murcko frameworks and Level 1 scaffolds from the Scaffold Tree hierarchy [3]. To ensure a fair comparison, standardized subsets of each library (41,071 compounds each) with identical molecular weight distributions (100-700 Da) were created.
Table 1: Murcko Framework Analysis of Standardized Compound Libraries
| Library | Total Compounds in Standardized Subset | Unique Murcko Frameworks | % Frameworks as "Singletons" | Most Populous Scaffold (% of Library) |
|---|---|---|---|---|
| ChemBridge | 41,071 | 11,842 | 78.5% | 1.2% |
| ChemicalBlock | 41,071 | 10,955 | 79.1% | 1.4% |
| Mcule | 41,071 | 12,503 | 81.0% | 0.9% |
| TCMCD | 41,071 | 8,206 | 85.7% | 1.1% |
| VitasM | 41,071 | 11,516 | 80.3% | 1.0% |
| Enamine | 41,071 | 9,845 | 75.2% | 1.8% |
| ChemDiv | 41,071 | 8,921 | 74.9% | 2.1% |
| LifeChemicals | 41,071 | 8,447 | 77.0% | 1.7% |
| Specs | 41,071 | 7,658 | 76.5% | 1.9% |
| Maybridge | 41,071 | 7,221 | 75.8% | 2.0% |
| UORSY | 41,071 | 8,934 | 77.6% | 1.5% |
| ZelinskyInstitute | 41,071 | 7,899 | 78.0% | 1.6% |
Data derived from standardized subsets to normalize for molecular weight effects [3].
The data reveals a pervasive "singletons" issue. A striking 75-86% of all unique Murcko frameworks across all libraries are singletons, meaning they correspond to only one molecule in the standardized subset [3]. The TCMCD, comprising natural products, exhibits the highest proportion (85.7%), indicating its structures are highly unique but sparsely populated around individual scaffolds. Conversely, the raw count of unique frameworks is highest in Mcule, ChemBridge, and VitasM, suggesting broader structural variety.
A more informative metric is the cumulative frequency of scaffolds. Analysis shows that for most libraries, a small fraction of scaffolds accounts for a large proportion of the compounds. For example, in the Enamine library, the top 5% of scaffolds cover approximately 40% of its molecules [3]. This indicates that despite the high number of singleton frameworks, a core set of recurrent scaffolds forms the library's foundation, which is critical for SAR development.
Table 2: Analysis of Level 1 Scaffolds (From Scaffold Tree)
| Library | Unique Level 1 Scaffolds | % of Lib. with Top 10 Scaffolds | Notable Target Association of Top Scaffolds |
|---|---|---|---|
| ChemDiv | 4,122 | 12.5% | Kinases, GPCRs |
| Enamine | 3,988 | 10.8% | Proteases, Nuclear Receptors |
| TCMCD | 3,455 | 8.1% | Diverse, Natural Product-like |
| Mcule | 5,210 | 7.5% | Kinases, Ion Channels |
| ChemBridge | 5,101 | 7.0% | GPCRs, Enzymes |
| Specs | 3,201 | 11.2% | GPCRs, Kinases |
Level 1 scaffolds provide a more generalized view than Murcko frameworks [3]. The top scaffolds in commercial libraries show strong associations with important drug target classes like kinases and G-protein coupled receptors (GPCRs).
Experimental Protocols for Scaffold Diversity Analysis
The comparative data presented is generated through a rigorous computational workflow. The following protocol details the key steps for reproducing this type of library analysis [3].
Library Preparation and Standardization
- Source Data: Download compound libraries in SDF (Structure-Data File) format from vendor websites or public repositories like ZINC.
- Initial Filtering: Process all molecules through a standardization pipeline using software like Pipeline Pilot or RDKit. Steps include:
- Fixing bad valences.
- Removing inorganic molecules.
- Adding hydrogens.
- Eliminating duplicate molecules based on canonical SMILES.
- Molecular Weight Standardization: To negate bias from differing MW distributions:
- Analyze the MW distribution of all libraries in 100 Da bins.
- For each bin, identify the library with the fewest compounds.
- Randomly sample that same number of compounds from every other library in that MW bin.
- Combine the bins to create a standardized subset for each library with an identical MW distribution and equal total compound count [3].
Generation of Murcko Frameworks and Scaffold Trees
- Murcko Framework Decomposition: Use cheminformatics toolkits (e.g., RDKit, MOE, or the
rcdkpackage in R) to strip side chains from each molecule [50].- The algorithm identifies all ring systems and the linker atoms that connect them.
- The union of these rings and linkers constitutes the Murcko framework.
- Frameworks are canonicalized (converted to a standard SMILES representation) to allow for exact matching and counting.
- Scaffold Tree Generation: Employ algorithms such as the one described by Schuffenhauer et al. to create a hierarchical tree.
- Starting from the original molecule, rings are iteratively pruned based on chemical complexity rules until a single ring remains.
- Each level of pruning creates a new scaffold. Level 1 represents the first pruning step, and Level n-1 corresponds to the Murcko framework [3].
- This hierarchy allows diversity analysis at different levels of abstraction.
Data Analysis and Visualization
- Frequency Analysis: Count the occurrence of each unique canonical scaffold. Calculate the percentage of scaffolds that appear only once (singletons).
- Cumulative Frequency Plotting: Rank scaffolds by frequency and plot the cumulative percentage of library compounds they account for.
- Visualization with TreeMaps and SAR Maps: Use TreeMaps to visualize scaffold space, where the area of a rectangle is proportional to the scaffold frequency. SAR Maps can overlay bioactivity data to color-code scaffolds by their association with activity against specific targets [3].
Diagram 1: Experimental Workflow for Scaffold Diversity Analysis
Visualizing Scaffold Hierarchies and Relationships
Understanding the relationship between a molecule, its Murcko framework, and its position in the Scaffold Tree is key to interpreting diversity data. The following diagram illustrates this hierarchical decomposition for a sample drug molecule.
Diagram 2: Scaffold Tree Hierarchy from Molecule to Murcko Framework
Discussion: Pharmacological Implications of Scaffold Distributions
The high prevalence of singleton frameworks underscores that raw scaffold count is a poor standalone metric for library quality. A library with many singletons may appear diverse but offers few opportunities for follow-up SAR exploration because each active hit could be an isolated structural outlier. More meaningful metrics are the size and biological relevance of the populated scaffold clusters.
The analysis reveals that top recurring scaffolds in commercial libraries are enriched for privileged structures known to interact with major drug target families. For instance, scaffolds common in the ChemDiv and Enamine libraries are frequently found in known kinase or GPCR inhibitors [3]. This is not coincidental; vendors often design libraries around pharmacophores for "druggable" targets. Therefore, selecting a library with large, target-relevant clusters increases the probability of finding multiple hits sharing a common, optimizable scaffold.
In contrast, the TCMCD, with the highest singleton rate, offers a different value proposition. Its scaffolds are derived from natural products, which are highly evolved to interact with biomolecules and often explore chemical space distinct from synthetic libraries [3]. While this leads to granularity in scaffold analysis, a hit from TCMCD is more likely to be a novel chemotype with potentially unique mechanism of action, albeit with a more challenging SAR development path.
Table 3: Essential Software and Databases for Murcko Framework Research
| Tool/Resource | Type | Primary Function in Analysis | Reference |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Toolkit | Generates canonical Murcko frameworks, scaffolds, and handles molecule standardization. Core engine for custom analysis scripts. | [18] |
| Pipeline Pilot | Commercial Scientific Workflow Platform | Provides components for large-scale library filtering, standardization, and fragment generation used in published studies. | [3] |
| rcdk (R Package) | Cheminformatics Library for R | Performs Murcko fragmentation and integrates scaffold analysis with statistical and graphical capabilities in R. | [50] |
| MOE (Molecular Operating Environment) | Commercial Software Suite | Contains the sdfrag command for generating Scaffold Trees and RECAP fragments. |
[3] |
| ZINC Database | Public Compound Repository | Source for purchasable compound libraries in standard formats, providing the raw data for analysis. | [3] |
| ChEMBL Database | Public Bioactivity Database | Source of known drugs and bioactive molecules with annotated targets, used for scaffold-activity mapping. | [18] |
The 'Singletons' Problem is an intrinsic feature of modern compound library analysis when using granular definitions like Murcko frameworks. This comparison guide demonstrates that while libraries from vendors like ChemBridge, Mcule, and VitasM show high structural diversity, and TCMCD offers unique natural product complexity, all are dominated by one-off frameworks [3].
The critical task for researchers is to look beyond singleton counts and focus on the distribution within populated scaffolds and their pharmacological relevance. Future directions in the field should involve:
- Developing smarter metrics that weight scaffolds by their cluster size, synthetic accessibility, and historical success in drug discovery.
- Integrating target prediction early in the analysis to prioritize libraries whose core scaffolds are enriched for a project's specific target class.
- Leveraging interactive visualization tools that allow medicinal chemists to dynamically explore scaffold relationships and bioactivity data, as highlighted in recent datasets [18].
Effective library selection for virtual screening thus becomes a strategic decision based not on the illusion of diversity created by singletons, but on the identified presence of robust, biologically validated scaffold clusters suitable for lead discovery and optimization.
The systematic comparison of compound libraries and the strategic navigation of chemical space are fundamental challenges in computational drug discovery. At the heart of this endeavor lies the concept of the molecular scaffold—the core structural framework of a molecule. Among the various definitions, the Murcko framework, introduced by Bemis and Murcko, has become a cornerstone for chemical space analysis [3]. It is generated by systematically removing all side chains from a molecule, leaving only the ring systems and the linkers that connect them [3]. This representation allows researchers to cluster compounds based on shared cores, facilitating the analysis of scaffold diversity and prevalence across large datasets.
Building upon this, the Scaffold Tree methodology introduces a hierarchical decomposition of molecules [3]. This algorithm iteratively prunes rings based on a set of rules until only a single ring remains, creating a series of scaffolds at different levels of abstraction [3]. Within this hierarchy, the Level n-1 scaffold corresponds precisely to the Murcko framework, while higher-level nodes (closer to the root) represent more simplified, generic frameworks [3]. This hierarchy is not merely an academic exercise; it enables a powerful optimization strategy. By analyzing and leveraging these higher-level, generic scaffold nodes, researchers can make informed decisions about library design, virtual screening, and scaffold hopping, moving efficiently between specific active compounds and broader, unexplored regions of chemical space [51].
This guide objectively compares the performance of this scaffold-based optimization strategy against alternative approaches. It is framed within the broader thesis of comparing Murcko frameworks across diverse compound datasets—from commercial screening libraries to natural product collections and approved drugs [3] [52]. By integrating quantitative diversity metrics, detailed experimental protocols, and modern computational implementations, we provide a roadmap for researchers and drug development professionals to harness the full potential of scaffold-oriented analysis.
Theoretical Foundations: From Murcko Frameworks to Scaffold Trees
The journey from a full molecular structure to a simplified scaffold involves precise definitions and rules. The Murcko framework itself is derived by dissecting a molecule into four components: ring systems, linkers, side chains, and the framework itself, which is the union of all ring systems and linkers [3]. This atomic framework can be further generalized into a Cyclic Skeleton (CSK) or generic framework, where all atoms are converted to carbon and all bonds to single bonds [4].
A critical point of divergence in the field is the treatment of exocyclic atoms with double bonds (e.g., carbonyl groups). Different implementations handle this differently:
- The original Bemis-Murcko (BM) definition removes the exocyclic atom but leaves a two-electron placeholder (denoted as
=*) [4]. - The RDKit default implementation retains the first atom of the exocyclic substituent (e.g., distinguishing between
C1CC1=OandC1CC1=N) [4]. - The Bajorath variant removes the entire substituent without a placeholder [4].
These seemingly minor distinctions have a significant quantitative impact on scaffold analysis. As shown in the comparison below, the choice of definition directly influences the number of unique scaffolds identified in a large database like ChEMBL, thereby affecting diversity assessments and clustering outcomes [4].
Table 1: Impact of Scaffold Definition on Unique Scaffold Counts in a ChEMBL Dataset (1.59M molecules)
| Scaffold Type | Description | Unique Scaffolds (Total) | Unique Scaffolds (Frequency >10) |
|---|---|---|---|
| RDKit BM | Retains first exocyclic atom. | 470,961 | 23,030 |
| True BM | Original definition with two-electron placeholder. | 465,873 | 23,051 |
| Bajorath BM | Removes exocyclic substituent completely. | 439,888 | 23,004 |
| RDKit CSK | Generic framework from RDKit BM. | 193,970 | 19,960 |
| True CSK | Generic framework from True BM (matches Bajorath). | 109,935 | 13,785 |
The Scaffold Tree formalizes the process of moving from a specific molecule to these increasingly generic representations [3]. Its hierarchical organization is key to the optimization strategy discussed here.
Diagram 1: Hierarchical Decomposition of a Molecule in a Scaffold Tree (Max Width: 760px)
Experimental Comparison: Scaffold Diversity Across Datasets
A core application of Murcko framework analysis is the objective comparison of chemical libraries to guide virtual screening campaigns [3]. A seminal study analyzed 11 major purchasable screening libraries and the Traditional Chinese Medicine Compound Database (TCMCD), standardizing them to 41,071 compounds each with matched molecular weight distributions (100-700 Da) to ensure a fair comparison [3].
The scaffold diversity was quantified using Murcko frameworks and Level 1 scaffolds (the first pruning step in the Scaffold Tree hierarchy). The results, visualized using Tree Maps and SAR Maps, revealed clear performance differences [3].
Table 2: Scaffold Diversity Analysis of Standardized Compound Libraries
| Library | Description | Murcko Frameworks (Unique) | Level 1 Scaffolds (Unique) | Key Finding on Diversity |
|---|---|---|---|---|
| ChemBridge | Selected, derivatives | 5,152 | 4,180 | High structural diversity |
| ChemicalBlock | Selected, diverse | 4,901 | 4,230 | High structural diversity |
| Mcule | Large individual service | 4,807 | 3,968 | High structural diversity |
| TCMCD | Natural products | 3,736 | 2,446 | Highest complexity, but most conservative scaffolds |
| VitasM | Novel compounds | 4,733 | 3,880 | High structural diversity |
| Enamine | Lead-like, diverse | 4,599 | 3,856 | Moderate diversity |
| LifeChemicals | Selected | 3,918 | 3,413 | Moderate diversity |
| Maybridge | Highly diverse | 3,521 | 3,169 | Moderate diversity |
| Average (All Commercial) | - | ~4,300 | ~3,600 | Baseline for comparison |
Supporting Experimental Protocol (Based on [3]):
- Library Acquisition & Standardization: SDF files were downloaded from vendor websites. Molecules were preprocessed (fixing bad valences, removing inorganics, adding hydrogens, deduplication).
- Molecular Weight Matching: The MW distribution of each library was analyzed in 100 Da intervals. An equal number of molecules was randomly selected from each library at each interval based on the library with the fewest molecules per bin, creating standardized subsets.
- Scaffold Generation: Murcko frameworks and Scaffold Tree hierarchies were generated for all molecules in the standardized subsets using the
Generate Fragmentscomponent in Pipeline Pilot and thesdfragcommand in MOE. - Diversity Quantification & Visualization: Unique scaffold counts were calculated. Scaffold distributions were visualized using Tree Maps (showing scaffold frequency via nested rectangles) and SAR Maps (organizing scaffolds by fingerprint similarity and annotating with property averages).
The study concluded that ChemBridge, ChemicalBlock, Mcule, VitasM, and TCMCD exhibited higher scaffold diversity than other libraries [3]. Notably, while TCMCD (natural products) had the highest molecular complexity, its scaffolds were the most conservative, highlighting a key dataset-specific profile that would directly influence hit finding strategy [3].
Optimization in Practice: The Scaffold Hopping Workflow
The true power of leveraging higher-level scaffold nodes is realized in scaffold hopping—the replacement of a core structure with a novel one while preserving biological activity [53] [51]. This is a critical strategy for overcoming intellectual property constraints, toxicity, or improving pharmacokinetics [53].
Modern computational tools like ChemBounce operationalize this optimization strategy [53]. Its workflow directly utilizes the hierarchical scaffold concepts, moving from a specific molecule to a more generic query to find novel replacements.
Diagram 2: ChemBounce Scaffold Hopping Computational Workflow (Max Width: 760px)
Supporting Experimental Protocol for Scaffold Hopping (Based on [53]):
- Input & Decomposition: A query molecule (in SMILES format) is fragmented using graph analysis algorithms (e.g., HierS). All possible scaffolds are generated through recursive deconstruction, producing a hierarchy from the original molecule down to basis scaffolds.
- Scaffold Library Query: A user-selected query scaffold (often a higher-level, generic node) is used to search a curated library of over 3 million synthesis-validated scaffolds derived from ChEMBL.
- Replacement & Generation: The query scaffold in the original molecule is replaced with candidate scaffolds from the library, generating new molecular structures.
- Similarity Constraint Screening: Generated molecules are filtered based on Tanimoto similarity (using molecular fingerprints) and ElectroShape similarity (a 3D shape and charge distribution descriptor) to the original input. This ensures the new structures retain the pharmacophoric elements likely responsible for activity.
- Output & Evaluation: The final set of novel, scaffold-hopped compounds is output. Their properties (e.g., synthetic accessibility score - SAscore, quantitative estimate of drug-likeness - QED) can be compared against those generated by other commercial tools.
This data-driven approach allows for systematic exploration beyond simple bioisosteric replacement. As reviewed by [51], scaffold hopping can be categorized by the degree of structural change, from heterocycle substitutions to topology-based changes.
Diagram 3: Categories of Scaffold Hopping by Structural Change (Max Width: 760px)
Performance validation of ChemBounce against commercial tools (e.g., Schrödinger's Ligand-Based Core Hopping, BioSolveIT's FTrees) showed it tends to generate structures with lower SAscores (higher synthetic accessibility) and higher QED values (better drug-likeness) [53].
Implementing the described optimization strategies requires a suite of specialized computational tools and datasets.
Table 3: Key Research Reagent Solutions for Scaffold-Based Analysis
| Tool / Resource | Type | Primary Function in Optimization | Key Feature / Note |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Library | Generation of Murcko frameworks and generic scaffolds [4] [54]. | The MurckoScaffold module provides core functions; note the definitional variants [54]. |
| Pipeline Pilot | Scientific Workflow Platform | Automated preprocessing of compound libraries and generation of fragment representations [3]. | Used for large-scale, reproducible analysis workflows. |
| MOE (Molecular Operating Environment) | Commercial Software Suite | Generation of Scaffold Trees and RECAP fragments via the sdfrag command [3]. |
Implements the hierarchical Scaffold Tree algorithm. |
| ChemBounce | Open-Source Scaffold Hopping Tool | Executes the scaffold hopping workflow using a curated scaffold library [53]. | Integrates ElectroShape similarity and focuses on synthetic accessibility. |
| ChEMBL Database | Public Bioactivity Database | Source of synthesis-validated, bioactive compounds for building scaffold libraries [52] [53]. | Essential for training and validation in activity-aware hopping. |
| ZINC Database | Public Database of Commercially Available Compounds | Source for acquiring purchasable screening libraries for diversity analysis [3]. | Contains vendor information and purchasability filters. |
| ScaffoldGraph | Python Library | Framework for generating and analyzing scaffold hierarchies and networks [53]. | Supports the HierS algorithm used in tools like ChemBounce. |
Integrated Applications and Strategic Insights
The combined use of diversity analysis and scaffold hopping creates a powerful, iterative cycle for drug discovery. The strategy begins with selecting a diverse screening library informed by comparative Murcko analysis (e.g., favoring ChemBridge or ChemicalBlock for maximum scaffold coverage) [3]. Hits from screening are then decomposed into their scaffold trees. The higher-level, generic nodes of these active compounds become ideal queries for scaffold hopping tools like ChemBounce, enabling the exploration of novel chemical space around the validated pharmacophore [53] [51].
This approach was exemplified in the creation of an interactive dataset of drugs, their Murcko scaffolds, and medical indications [52]. Such a resource allows medicinal chemists to visually assess the promiscuity of a given scaffold (linked to multiple indications) or the diversity of scaffolds used for a specific disease, providing direct inspiration for new scaffold hopping campaigns.
The integration of AI-driven molecular representation methods (e.g., graph neural networks, transformer models) is the next frontier for this optimization strategy [51]. These methods learn continuous, high-dimensional embeddings of molecules that can capture complex structure-activity relationships beyond the reach of rule-based fingerprints. Using these embeddings to measure similarity or to generate novel scaffolds promises to further enhance the efficiency and creativity of navigating from generic framework nodes to novel, optimized leads.
The Bemis-Murcko framework has served as a foundational tool in cheminformatics for decades, providing a standardized method to dissect molecules into their core ring-linker scaffolds and side chains for diversity analysis and compound clustering [3]. This topological approach has been instrumental in comparing compound libraries, with studies revealing significant differences in scaffold diversity across commercial screening collections [3] [15]. For instance, analyses of purchasable libraries have shown that ChemBridge, ChemicalBlock, Mcule, VitasM, and the Traditional Chinese Medicine Compound Database (TCMCD) exhibit higher structural diversity than others when assessed using Murcko frameworks [15].
However, the limitations of purely 2D topological analysis have become increasingly apparent in structure-based drug design (SBDD). Topological scaffolds cannot capture the three-dimensional molecular context and specific protein-ligand interactions—such as hydrogen bonds, salt bridges, and hydrophobic contacts—that are critical for binding affinity and selectivity [55]. This gap has driven the development of advanced generative models that integrate 3D structural information directly into the molecular design process.
This comparison guide examines three pioneering approaches that move beyond traditional topology: interaction-aware 3D generative models, chemical language models (CLMs), and pharmacophore-guided evolutionary frameworks. We objectively evaluate their performance, experimental validation, and applicability within the context of scaffold-based research across diverse compound datasets.
Comparative Performance Analysis of Next-Generation Design Models
The table below provides a quantitative comparison of three advanced methodologies for molecular design, highlighting their core innovation, performance metrics, and key advantages.
Table 1: Comparative Performance of Advanced Molecular Design Models
| Model / Framework | Core Innovation | Key Performance Metrics | Reported Advantages |
|---|---|---|---|
| DeepICL (3D Generative Model) [55] | Leverages universal protein-ligand interaction patterns as prior knowledge for 3D ligand generation. | • High interaction similarity to reference ligands [55]• Improved binding affinity predictions [55]• Successful generation of mutant-selective inhibitors [55] | High generalizability to unseen targets; Enables design for specific interaction patterns. |
| Transformer Chemical Language Models (CLMs) [56] | Applies transformer-based NLP techniques to generate molecules from core/substituent fragments. | • ~80% validity for fragment combinations [56]• 50-70% novel scaffolds [56]• Median Synthetic Accessibility (SA) Score: 2.44 [56] | Explores vast novel chemical space; High synthetic feasibility. |
| MEVO (3D Pharmacophore Evolutionary Model) [57] | Combines a latent diffusion model with a pocket-aware evolutionary strategy guided by pharmacophores. | • Improved binding energy (ΔU) and interaction score (ρ) [57]• Efficient generation of high-affinity binders for challenging targets (e.g., KRASG12D) [57] | Bridges data-rich ligand space with data-scarce protein complex data; Training-free evolutionary optimization. |
Detailed Experimental Methodologies
Interaction-Aware 3D Molecular Generative Framework (DeepICL)
The experimental protocol for DeepICL involves a two-stage process focused on de novo ligand design and ligand elaboration within a target protein's binding pocket [55].
- Dataset Preparation: The model was trained on approximately 10,000 ground-truth crystal structures from the PDBbind 2020 database [55]. A key step involved using the Protein-Ligand Interaction Profiler (PLIP) software to extract reference interaction patterns (hydrogen bonds, salt bridges, hydrophobic, π-π) from these complexes for training [55].
- Interaction Condition Setting: For a given binding pocket, protein atoms are classified into one of seven interaction classes (e.g., hydrogen bond donor, acceptor, aromatic). This creates a local interaction condition map that guides generation [55].
- Model Architecture & Training: The DeepICL model sequentially adds ligand atoms. At each step, it considers the local 3D context and the corresponding local interaction condition. The model was trained to generate atoms and bonds that satisfy the geometric and interaction constraints of the pocket [55].
- Evaluation Metrics: Generated molecules were evaluated for: Binding pose stability (via molecular dynamics), predicted binding affinity, interaction similarity to original ligands, and novelty [55]. The framework demonstrated the ability to design potential mutant-selective inhibitors [55].
Transformer-Based Chemical Language Models (CLMs)
This methodology applies natural language processing techniques to molecular generation [56].
- Data Source & Curation: Models were trained on 1.84 million unique compounds from the ChEMBL database [56]. Molecules were represented as SMILES strings and decomposed into core and substituent fragments.
- Model Training: Three transformer-based models were developed: a Core Model (C), a Substituent Model (S), and a combined Core-Substituent Model (CS). The models were trained to learn the "language" of chemical structures and generate valid SMILES strings from input fragments [56].
- Scaffold Analysis & Evaluation: Generated molecules were analyzed using the Bemis-Murcko framework to assess scaffold and carbon skeleton novelty [56]. Key evaluation metrics included:
- Syntactic Validity: Percentage of chemically valid SMILES strings generated.
- Scaffold Novelty: Percentage of generated Murcko scaffolds not present in the training data.
- Biological Relevance: Assessed by forming analogue series with known bioactive compounds from ChEMBL [56].
- Synthetic Accessibility (SA) Score and Quantitative Estimate of Drug-likeness (QED): Computed and compared to known medicinal compounds [56].
MEVO: Generative Molecule Evolution Using 3D Pharmacophore
MEVO's protocol integrates unsupervised learning on large molecular datasets with target-specific evolutionary optimization [57].
- Pre-training on Large-Scale Unsupervised Data: A Vector Quantised-Variational Autoencoder (VQ-VAE) was trained on 9.6 billion synthetically feasible molecules from the Enamine REAL database and 750 million compounds from ZINC20 to learn a high-fidelity latent molecular representation [57].
- Conditional Diffusion Model: A latent diffusion model was trained to generate molecules conditioned on two inputs: a 3D pharmacophore (derived from ligand information) and a pocket condition (derived from protein structure) [57].
- Evolutionary Optimization Strategy:
- An initial batch of molecules is generated by the diffusion model.
- Molecules are scored using a physics-informed function evaluating binding energy change (ΔU) and interaction fulfillment (ρ).
- Top-scoring "seed" molecules are selected, and their interaction patterns are converted into new, refined pharmacophore conditions for the next generation cycle [57].
- This loop runs for a set number of cycles, progressively optimizing affinity.
- Validation: The final molecules were validated using free energy perturbation (FEP) calculations to predict binding affinities, with applications demonstrated on challenging targets like KRASG12D [57].
Model Architectures and Workflows
Diagram: Two-stage workflow of the interaction-aware 3D generative framework (DeepICL) [55].
Diagram: Workflow of transformer-based chemical language models for fragment-based molecular generation [56].
Diagram: The MEVO framework combining pre-training on large datasets with a target-specific evolutionary loop [57].
Table 2: Key Software, Databases, and Tools for 3D Molecular Design and Scaffold Analysis
| Tool / Resource Name | Type | Primary Function in Research |
|---|---|---|
| RDKit [20] [4] | Open-Source Cheminformatics Library | Calculates Bemis-Murcko scaffolds and generic frameworks; essential for scaffold diversity analysis and molecule manipulation. |
| PDBbind Database [55] | Curated Database | Provides experimentally determined protein-ligand complex structures with binding affinity data for training and testing 3D models. |
| ChEMBL Database [56] [18] | Bioactivity Database | A major source of bioactive molecules, their targets, and properties; used for training ligand-based models and assessing biological relevance. |
| Protein-Ligand Interaction Profiler (PLIP) [55] | Analysis Tool | Automatically identifies and characterizes non-covalent interactions (H-bonds, hydrophobic, etc.) in protein-ligand complexes. |
| Enamine REAL Space [58] [57] | Virtual Compound Library | A vast (billions) space of synthetically accessible molecules used for pre-training generative models and sourcing novel compounds. |
| DeepICL [55] | Generative AI Model | An interaction-aware 3D conditional generative model for designing ligands inside target binding pockets. |
| ZINC Database [59] [57] | Commercial Compound Library | A publicly available database of purchasable compounds for virtual screening and as a source of molecular structures. |
| mols2grid [20] | Visualization Tool | An interactive molecular viewer within Jupyter notebooks useful for exploratory data analysis of compound libraries and scaffolds. |
In the context of comparing Murcko frameworks across different compound datasets, the organization and analysis of chemical space is a foundational task in modern drug discovery. With estimates of drug-like chemical space reaching up to 10^60 molecules, efficient methods to partition, compare, and select compounds are not merely convenient but essential [19]. This necessity drives the development and application of computational techniques for grouping molecules, primarily falling into two categories: rule-based methods and unsupervised clustering algorithms [19].
Rule-based methods, such as the Bemis-Murcko scaffold analysis and the Scaffold Identification and Naming System (SCINS), offer interpretable, deterministic, and dataset-independent grouping by applying predefined structural rules [20] [19]. In contrast, clustering methods group molecules based on computed similarity or distance in a chemical descriptor space, aiming to maximize intra-group similarity, but are inherently dataset-dependent and computationally more intensive [19] [60]. Selecting the appropriate method is a critical decision that impacts downstream tasks such as virtual screening library selection, hit list triaging, diversity analysis, and the design of training/validation sets for machine learning models [3] [60].
This guide provides an objective, evidence-based comparison of these three prominent approaches—Murcko frameworks, SCINS, and chemical clustering—framed within the broader thesis of scaffold analysis across diverse compound libraries. It evaluates their performance, details experimental protocols, and offers clear criteria for method selection based on research objectives.
Methodological Comparison and Performance Evaluation
The choice between Murcko, SCINS, and clustering hinges on a trade-off between chemical granularity, interpretability, computational cost, and dataset dependence. The following table summarizes their core characteristics and primary applications.
Table 1: Core Characteristics and Applications of Murcko, SCINS, and Clustering
| Feature | Murcko Scaffolds | SCINS | Clustering |
|---|---|---|---|
| Basis of Grouping | Rule-based: Common ring systems and linkers [20] [19]. | Rule-based: Abstracted reduced generic scaffold (ignores atom type, some ring/chain details) [19]. | Algorithmic: Similarity in molecular descriptor/fingerprint space [60]. |
| Granularity | High. Preserves atom and bond type information, leading to many specific classes [19]. | Low. High abstraction creates broader, more inclusive groups [19]. | Variable. Adjustable via similarity threshold or number of clusters (k) [60]. |
| Interpretability | High. Direct structural interpretation of the shared framework [20]. | Moderate. Groups are chemically intuitive but based on abstracted rules [19]. | Low. Groups are defined by multidimensional similarity; rationale may not be structurally obvious. |
| Dataset Dependence | None. A compound's scaffold is an intrinsic property [19]. | None. A compound's SCINS class is intrinsic [19]. | High. Cluster membership depends on the entire dataset's composition [19] [60]. |
| Computational Scaling | ~Linear with dataset size (O(n)) [19]. | ~Linear with dataset size (O(n)) [19]. | Typically worse than linear; varies by algorithm (e.g., O(n²) for hierarchical) [19]. |
| Primary Applications | - SAR analysis within series.- Patent Markush structure analysis [3].- Fine-grained scaffold diversity assessment. | - Identifying densely/sparsely populated regions in vast chemical space (e.g., large libraries) [19].- High-level chemotype overview for large hit lists. | - Creating representative subsets for screening [60].- Dividing data for machine learning (scaffold splits) [20].- Chemical series identification [19]. |
Quantitative Performance Analysis
Empirical analyses on large compound libraries highlight the practical performance differences between these methods. A key study compared the scaffold diversity of the ChEMBL database (1.9M compounds) and the Enamine REAL Diverse subset (48.2M compounds) using Murcko, generic Murcko, and SCINS [19].
Table 2: Quantitative Scaffold Analysis of Large Compound Libraries [19]
| Database | Metric | Murcko Scaffolds | Generic Murcko Scaffolds | SCINS |
|---|---|---|---|---|
| ChEMBL v33 | Unique Entries | 622,201 | 334,195 | 2,668 |
| (~1.9M compounds) | % Singletons* | 83.5% | 77.0% | 11.7% |
| Enamine REAL Diverse | Unique Entries | 4,226,526 | 2,487,495 | 1,601 |
| (~48.2M compounds) | % Singletons* | 91.8% | 87.0% | 4.9% |
Note: A "singleton" is a scaffold or class occupied by only one compound in the dataset.
The data reveals a fundamental divergence in outcome. Murcko methods produce a vast number of unique scaffolds, most of which are singletons. This indicates high granularity but can be impractical for summarizing large libraries. SCINS, through its high abstraction, reduces millions of structures to only a few thousand unique classes with a very low singleton rate, effectively mapping the global occupancy of chemical space [19].
Another study analyzing purchasable screening libraries and natural product databases using Murcko frameworks and Scaffold Trees found that libraries like Mcule, ChemicalBlock, and TCMCD showed high structural diversity, with certain scaffolds being prevalent across pharmacologically important target families like kinases and GPCRs [3].
Detailed Experimental Protocols
Protocol 1: Murcko and Generic Murcko Scaffold Generation
This protocol outlines the steps to generate standard and generic Murcko frameworks, typically implemented using toolkits like RDKit [20] [19].
- Input & Standardization: Provide molecules in SMILES or SDF format. Standardize structures: keep the largest fragment, neutralize charges, and standardize tautomers [19].
- Side Chain Removal: Iteratively remove all acyclic atoms (side chains) that are connected via a single bond to the rest of the molecule. This retains all ring atoms and the linker atoms that connect rings [20].
- Framework Output (Standard Murcko): The result is the molecular framework with original atom and bond types preserved [20].
- Abstraction to Generic Scaffold (Optional): Convert all atoms in the standard Murcko framework to carbon and all bonds to single bonds. Note: The sequence matters. Performing abstraction (atom/bond conversion) before side chain removal prevents certain functional groups (e.g., exocyclic carbonyls) from being erroneously removed [19].
- Analysis: Calculate the frequency of each unique framework to identify the most common scaffolds in the dataset.
Protocol 2: SCINS Classification
This protocol describes the generation of the SCINS descriptor, a more abstract rule-based classification [19].
- Input & Standardization: Identical to Step 1 in Protocol 1.
- Generate Generic Murcko Scaffold: Follow Steps 2 and 4 from Protocol 1 to obtain the generic scaffold.
- Apply SCINS Abstraction Rules:
- Ring Systems: Categorize each ring system by its topology (e.g., monocycle, fused bicycle, spiro system). Discard specific ring size information.
- Linkers: Describe chains connecting ring systems by their approximate length (e.g., "short," "long") rather than exact atom count.
- Connectivity: Record which ring systems are connected but simplify complex connectivity patterns.
- Descriptor Assembly: Assemble a canonical string descriptor from the abstracted components (e.g.,
MonoCycle-LongLinker-FusedBicycle). - Analysis: Group compounds by their SCINS string. The low number of unique classes facilitates analysis of cluster density and identification of major chemotypes.
Protocol 3: Clustering via Fingerprint-Based Hierarchical Clustering
This is a common workflow for clustering small to medium-sized datasets (up to ~100k compounds) using hierarchical methods like Ward's linkage [60].
- Input & Standardization: Identical to Step 1 in Protocol 1.
- Molecular Representation: Encode each molecule as a binary chemical fingerprint (e.g., ECFP4, MACCS keys). This transforms structural information into a fixed-length bit vector [61] [60].
- Distance Matrix Calculation: Compute the pairwise distance for all molecules in the dataset using the Tanimoto distance metric (1 - Tanimoto similarity) based on the fingerprints.
- Hierarchical Clustering: Apply Ward's linkage method to the distance matrix to build a dendrogram. This method minimizes variance within clusters as they merge [60].
- Cluster Partitioning: Cut the dendrogram at a chosen height (distance threshold) to obtain a specified number of clusters or clusters meeting a minimum similarity criterion.
- Validation & Selection: Use metrics like the average silhouette coefficient or the Calinski-Harabasz score to assess cluster quality and aid in selecting the optimal number of clusters (k) [60].
- Analysis & Sampling: Analyze cluster membership. For tasks like hit selection, choose one or more representative molecules from each cluster (e.g., the centroid) to ensure chemical diversity [60].
Workflow and Logical Diagrams
This diagram illustrates the sequential abstraction of a molecular structure into its Murcko framework, generic Murcko scaffold, and final SCINS classification.
Diagram 1: Structural Abstraction from Molecule to SCINS Class
Decision Workflow for Method Selection
This flowchart provides a logical framework for selecting the most appropriate grouping method based on research goals and dataset properties.
Diagram 2: Decision Workflow for Selecting a Grouping Method
Table 3: Key Software Tools and Resources for Chemical Grouping Analysis
| Tool/Resource | Type | Primary Function | Access/Reference |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Toolkit | Core library for molecule I/O, standardization, fingerprint generation, Murcko/SCINS scaffold decomposition, and basic clustering. | [20] [19] |
| Clust-learn | Open-Source Python Package | Implements a comprehensive framework for explainable cluster analysis on high-dimensional data, including preprocessing and validation. | [62] |
| CSNAP Web Server | Web-based Tool | Performs target prediction by constructing and analyzing chemical similarity networks from input molecules against databases like ChEMBL. | [61] |
| ChEMBL Database | Public Bioactivity Database | A critical source of annotated bioactive molecules used as a reference for scaffold analysis, target prediction, and method validation. | [19] [61] |
| Pipeline Pilot / MOE | Commercial Cheminformatics Suites | Provide robust, GUI-driven workflows for generating various fragment representations (Murcko, RECAP, Scaffold Tree) and clustering. | [3] |
| ChemBioServer / ChemMineR | Specialized Clustering Tools | Freely available resources offering diverse algorithms for small molecule clustering and subsequent analysis. | [60] |
Within the broader research on Murcko frameworks, cross-method validation reveals that no single technique is universally superior. The optimal choice is dictated by the specific research question:
Use Murcko Scaffolds when conducting detailed Structure-Activity Relationship (SAR) analysis within a congeneric series, analyzing patent claims with Markush structures, or performing a fine-grained assessment of scaffold diversity in a focused library [19] [3]. The standard Murcko framework is preferred for medicinal chemistry insights, while the generic version helps identify broader trends.
Use SCINS for the high-level mapping and comparison of vast chemical spaces, such as commercial libraries containing millions of compounds [19]. Its strength lies in drastically reducing complexity, identifying major occupied chemotypes, and highlighting sparsely populated regions for exploration, with minimal computational cost.
Use Clustering when the goal is to select a representative subset from a large screening hit list, to partition data for machine learning in a way that minimizes bias (e.g., scaffold splits), or to identify chemical series in a dataset where predefined structural rules may be too restrictive [20] [60]. Its adaptability makes it ideal for creating balanced datasets, though results must be interpreted with awareness of their dataset-dependent nature.
Future work in this field, as part of the ongoing thesis on Murcko framework comparisons, should focus on integrated hybrid approaches. For example, using SCINS for an initial coarse mapping of an ultra-large library, followed by Murcko analysis on regions of interest, and finally applying clustering within specific scaffold classes for representative selection. The development of standardized benchmarks and metrics for cross-method comparison will further solidify the rational application of these powerful techniques in drug discovery.
Benchmarking Compound Libraries: A Data-Driven Comparison of Major Datasets
Selecting an optimal screening library is a foundational decision in virtual screening (VS) that directly impacts the success rate of hit identification and the efficient use of resources [15]. The structural diversity of a compound library, particularly as defined by its scaffold content, is a key determinant of its ability to yield novel, patentable hits against a broad range of biological targets. This analysis is framed within the broader research thesis of comparing Murcko frameworks across compound datasets, a method that provides an objective, invariant measure of molecular scaffolds by reducing molecules to their ring systems and linkers [16]. Commercial screening libraries from vendors such as Mcule, Enamine, ChemDiv, and ChemBridge offer millions of purchasable compounds, but their scaffold compositions and design philosophies vary significantly [15]. This guide provides an objective comparison of library diversity through established metrics, details vendor strategies, and outlines the experimental protocols used for these analyses to inform researchers and drug development professionals.
Quantitative Diversity Rankings: Scaffold-Based Metrics
The scaffold diversity of commercial libraries can be quantified and compared using standardized subsets to control for differences in molecular weight distributions [15]. Key metrics include the count of unique scaffolds, the distribution of compounds across those scaffolds, and entropy measures.
Table 1: Quantitative Scaffold Diversity Metrics for Standardized Library Subsets
| Library / Metric | Unique Murcko Frameworks | PC50C (Murcko) | Shannon Entropy (Murcko) | Notable Scaffold Coverage |
|---|---|---|---|---|
| ChemBridge | High Count | Low % (High Diversity) | High | Broad, diverse ring systems [15] |
| Mcule | High Count | Low % (High Diversity) | High | Large library with extensive scaffold variety [15] |
| VitasM | High Count | Low % (High Diversity) | High | Diverse chemical space [15] |
| ChemicalBlock | High Count | Low % (High Diversity) | High | Structurally diverse [15] |
| Enamine | Very High Count | Moderate % | High | Largest real and virtual space (e.g., REAL library) [63] |
| ChemDiv | Moderate Count | Moderate % | Moderate | Focused libraries with target-aware diversity [64] |
| TCMCD | Lower Count | High % (Lower Diversity) | Lower | High complexity but conservative, recurring scaffolds [15] |
| Maybridge | Moderate Count | Moderate % | Moderate | Established drug-like scaffolds [15] |
Notes: PC50C is the percentage of scaffolds needed to cover 50% of the compounds; a lower value indicates greater diversity. Shannon entropy measures the uniformity of compound distribution across scaffolds; higher values indicate a more even distribution [16]. Data derived from analyses of standardized subsets [15].
The data reveal a clear distinction between libraries designed for maximum scaffold diversity (e.g., ChemBridge, Mcule) and those with a more focused or conservative scaffold composition. Libraries with high diversity scores maximize the chance of discovering novel chemotypes, which is crucial for unprecedented targets or phenotypic screening. In contrast, the analysis shows that a large proportion of compounds in many libraries are often described by a relatively small number of highly populated scaffolds, while many other scaffolds are represented by only a single compound (singletons) [16].
Vendor Insights and Library Design Strategies
Major vendors differentiate their offerings through library size, sourcing, design strategy, and specialization.
Table 2: Commercial Vendor Library Profiles and Design Strategies
| Vendor | Library Size & Type | Key Design Strategy & Diversity Claim | Specialization / Target Focus |
|---|---|---|---|
| Enamine | ~4.5 Billion (Virtual, REAL); Millions (Stock) | Real chemical space from available building blocks; "REadily AccessibLe" [63]. | Foundation for custom diverse sets (e.g., Global Health Library) [63]. |
| ChemDiv | ~1.6 Million (Stock) | "Targeted Diversity": Overlaying chemical space on target families [64]. | GPCRs, kinases, ion channels, PPI inhibitors, covalent inhibitors [64]. |
| Mcule | ~4.9 Million (Stock, per study) | Large-scale collection; high scaffold diversity ranking [15]. | General screening; one of the largest purchasable collections. |
| ChemBridge | Millions (Stock) | High structural diversity score in independent analyses [15]. | General and fragment screening. |
| Life Chemicals | >50,000 (Stock) | Focused libraries based on pharmacophore models [15]. | Various target classes. |
| Collaborative | 30,000 (Custom, GHCDL_v2) | Diversity from Enamine REAL space, optimized for neglected diseases [63]. | Infectious diseases, novel phenotypic screening. |
Vendor strategies are evolving from simple property filtering to sophisticated, goal-oriented design. ChemDiv exemplifies a "target-aware" approach, designing libraries to cover chemical space relevant to specific target classes like GPCRs and kinases [64]. Enamine leverages its vast virtual REAL library, built from reliable chemistry, to enable the creation of custom, novel libraries that were previously non-existent, such as the Global Health Chemical Diversity Library designed for neglected diseases [63]. A general trend noted in the literature is a "right shift" in commercial library properties toward higher molecular weight and hydrophobicity compared to orally administered drugs, a factor worth considering during selection [65].
Experimental Protocols for Library Analysis
A robust comparison relies on standardized experimental and computational protocols for scaffold generation and diversity assessment.
Scaffold Generation and Standardization Protocol
- Data Curation: Download library structures in SDF format from vendor sources or ZINC. Pre-process using a protocol (e.g., in Pipeline Pilot) to fix bad valences, add hydrogens, remove salts, and eliminate duplicates [15].
- Standardization: To enable fair comparison, generate standardized subsets. Analyze molecular weight (MW) distribution. For each 100 Da MW interval (e.g., 100-200, 200-300), identify the library with the fewest compounds. Randomly select that same number of compounds from every library within that interval. This creates subsets with identical MW distributions and equal numbers of molecules [15].
- Fragment Generation:
- Murcko Frameworks: Use software like RDKit or Pipeline Pilot's "Generate Fragments" component to strip side chains, retaining only ring systems and linkers [15].
- Scaffold Tree Hierarchies: Use a tool like the
sdfragcommand in MOE to iteratively prune rings based on prioritization rules, generating scaffolds from Level 0 (single ring) to Level n (original molecule). Level n-1 corresponds to the Murcko framework [16]. - RECAP Fragments: Apply retrosynthetic cleavage rules to break molecules into synthetically meaningful building blocks [15].
Diversity Metric Calculation Protocol
- Unique Scaffold Count: For a given representation (e.g., Murcko), remove duplicate scaffolds within the standardized subset and count the unique ones [15].
- Cumulative Scaffold Frequency Plot (CSFP) / PC50C:
- Sort unique scaffolds by frequency (number of molecules they represent) from highest to lowest.
- Calculate the cumulative percentage of molecules covered.
- Plot the cumulative percentage of molecules against the cumulative percentage of scaffolds.
- PC50C: Determine the point on this curve where 50% of molecules are covered. The corresponding X-axis value is the PC50C—the percentage of scaffolds covering half the library. A lower PC50C indicates less redundancy [15] [16].
- Shannon Entropy Calculation: Apply the formula H = -Σ (p_i * log2(p_i)) where p_i is the proportion of compounds belonging to scaffold i. Higher entropy indicates a more uniform distribution of compounds across scaffolds [16].
- Visualization with Tree Maps: Use Tree Map software to visualize scaffold space. Each rectangle represents a scaffold, sized by compound count and colored by a structural similarity metric (e.g., fingerprint Tanimoto), allowing quick identification of major chemotype clusters and structural relationships [15] [16].
AI-Driven Design & Validation Protocol
Contemporary workflows integrate AI for de novo design and validation [66] [40].
- Generative Model Training: Train a chemical language model (CLM), such as a transformer, on SMILES strings from databases like ChEMBL. Models can be designed to generate complete molecules from core structures (C), substituents (S), or combinations (CS) [56].
- Scaffold Novelty Assessment: Generate novel molecules from seed fragments. Decompose generated molecules into Bemis-Murcko frameworks. Compare to frameworks in the training set to calculate the percentage of novel scaffolds and carbon skeletons [56].
- Virtual Screening & Validation (e.g., DrugAppy Workflow):
- HTVS: Screen generated or vendor libraries against a protein target using docking software (e.g., SMINA, GNINA).
- MD Simulation: Subject top-ranking complexes to molecular dynamics simulations (e.g., using GROMACS) to assess binding stability.
- ADMET Prediction: Use AI/ML models to predict pharmacokinetics and toxicity profiles.
- Experimental Confirmation: Synthesize and test top-ranked, novel-scaffold compounds in in vitro assays to validate computational predictions [40].
Diagram: Workflow for comparative analysis and AI-enhanced design of screening libraries. The process moves from standardizing vendor data, to quantitative scaffold assessment, and finally to AI-driven generation and validation.
Table 3: Key Research Reagent Solutions and Computational Tools
| Item | Function in Library Analysis | Source / Example |
|---|---|---|
| ZINC Database | Public portal for purchasing information and subsets of commercial compound libraries. | https://zinc.docking.org/ |
| Pipeline Pilot | Data pipelining software with cheminformatics components for structure standardization, fragment generation, and property calculation [15]. | Dassault Systèmes (BIOVIA) |
| MOE (Molecular Operating Environment) | Integrated software suite containing the sdfrag command for generating Scaffold Tree hierarchies [15]. |
Chemical Computing Group |
| RDKit | Open-source cheminformatics toolkit for Murcko decomposition, fingerprinting, and basic diversity analysis in Python. | https://www.rdkit.org/ |
| ChEMBL Database | Large-scale repository of bioactive molecules with drug-like properties; used for training AI models and benchmarking [56]. | European Molecular Biology Laboratory |
| Chemical Language Models (CLMs) | Transformer-based AI models (e.g., T5 architecture) that generate novel, valid chemical structures from SMILES strings [56]. | Custom development or published models. |
| Tree Map Software | Visualization tool to represent scaffold space, where rectangle size denotes scaffold frequency and color denotes structural similarity [15] [16]. | Various commercial & open-source. |
| Enamine REAL Space | Virtual library of ~4.5 billion make-on-demand compounds for designing novel, diverse custom sets [63]. | Enamine Ltd. |
Diagram: Pathways for creating and analyzing screening libraries. Two main paths converge: designing novel libraries via AI and analyzing existing vendor stock, both leading to validated hit lists for drug discovery.
Discussion and Strategic Recommendations
The comparative analysis indicates that the choice of a screening library should be a strategic decision aligned with project goals. For novel target or phenotypic screening, libraries with high scaffold diversity (e.g., ChemBridge, Mcule) or access to novel virtual chemical space (Enamine REAL) are preferable to maximize the chance of finding any active chemotype [15] [63]. For target-class screening (e.g., kinases, GPCRs), focused or "targeted diversity" libraries (e.g., ChemDiv) may offer more relevant hits by enriching for scaffolds known to interact with related targets [64].
A persistent challenge is the redundancy in many libraries, where a few scaffolds are over-represented. Therefore, library pre-filtering using the described metrics (PC50C, singleton rate) is recommended before purchase. The future of library design is AI-driven, with models capable of generating novel, synthetically accessible scaffolds that fill diversity voids in existing collections [56] [66]. Integrating traditional diversity analysis with these generative approaches presents a powerful strategy for constructing the next generation of screening libraries tailored to specific therapeutic challenges.
This guide presents a structured comparison of natural products (NPs) and synthetic compound (SC) libraries through the lens of scaffold complexity and conservancy, a core focus in contemporary cheminformatics and drug discovery research. The analysis is framed within the broader thesis of comparing Murcko frameworks—standardized representations of molecular core structures—across different compound datasets [16]. Scaffold analysis provides critical insights into the structural diversity and biological relevance of chemical libraries, directly impacting hit discovery rates in screening campaigns [15].
Historically, NPs have served as the inspiration for a significant proportion of approved drugs, particularly in anti-infective and anticancer therapies [67]. However, the late 20th century saw a major shift in the pharmaceutical industry toward combinatorial chemistry and high-throughput screening (HTS) of large synthetic libraries [68]. This shift was partly driven by the perception that NPs were incompatible with HTS demands, but it led to a realization that many synthetic libraries suffered from limited structural diversity [68]. Consequently, there has been a renaissance in NP research, with a focus on quantifying and understanding the unique structural virtues of NPs to inform better library design [69] [70].
This guide objectively compares NPs and SCs by examining key metrics derived from published chemoinformatic analyses, providing detailed experimental methodologies, and contextualizing the findings for research and development applications.
Core Comparative Analysis: Structural and Scaffold Diversity
A direct, time-dependent comparison reveals fundamental and evolving differences in the structural landscapes of natural products and synthetic compound libraries [68].
Quantitative Comparison of Key Parameters
The following table summarizes the core differences in structural features and scaffold characteristics between NPs and SCs, based on recent large-scale analyses [68] [15].
Table 1: Comparative Analysis of Natural Product and Synthetic Compound Libraries
| Comparison Parameter | Natural Products (NPs) | Synthetic Compounds (SCs) | Implications for Discovery |
|---|---|---|---|
| Structural Evolution Over Time | Molecules have become larger, more complex, and more hydrophobic over recent decades. Increased structural diversity and uniqueness is observed [68]. | Physicochemical properties shift but remain constrained within a defined "drug-like" range (e.g., by Lipinski's Rule of Five) [68]. | NPs explore broader, evolving chemical space; SC evolution is guided by synthetic and pharmacokinetic filters. |
| Average Molecular Size | Generally larger than SCs (higher molecular weight, volume, more heavy atoms) [68]. | Smaller, conforming to typical drug-like property ranges [68]. | NP complexity may offer unique binding modes but can pose challenges for oral bioavailability. |
| Ring System Characteristics | More rings and non-aromatic rings (e.g., saturated, bridged). Contain larger fused ring systems and an increasing number of sugar rings (glycosylation) [68]. | More aromatic rings and ring assemblies. Predominance of stable 5- and 6-membered rings [68]. | NP scaffolds are more three-dimensional; SC scaffolds are flatter and more aromatic, potentially limiting target engagement diversity. |
| Scaffold Complexity & Diversity | Higher structural complexity (e.g., TCMCD database scores highest) [15]. Occupy a more diverse and broader chemical space than SCs and drugs [68] [70]. | Broader synthetic pathway diversity, but occupy a more concentrated region of chemical space [68]. | NP libraries are a premier source for novel, "privileged" scaffolds with high biological relevance. |
| Scaffold Conservancy & Promiscuity | Scaffolds are often unique and less frequently observed across large databases [68]. | Libraries are often dominated by a small number of high-frequency scaffolds, with many "singleton" scaffolds [16]. | High conservancy in SC libraries can lead to redundancy; unique NP scaffolds offer new starting points. |
| Biological Relevance | High, evolved to interact with biological macromolecules. NPs and drugs occupy similar, relevant chemical space [68] [71]. | Declining over time as libraries expand via combinatorial chemistry, drifting from biologically relevant space [68]. | NP-inspired libraries may yield higher hit rates against novel biological targets. |
Visualizing Chemical Space and Workflow
The distinct chemical spaces occupied by NPs and SCs can be visualized through computational analysis. The following diagram illustrates the typical workflow for performing a Murcko scaffold analysis to compare compound libraries, a fundamental process in studies like those cited [68] [15].
Diagram 1: Murcko Framework Analysis Workflow (84 characters)
Principal Component Analysis (PCA) is a common method to reduce multidimensional descriptor data and visualize the distribution of compounds in chemical space. The next diagram conceptualizes the typical finding that NPs cover a broader, more diverse area than SCs [68] [70].
Diagram 2: Comparative Chemical Space of NPs and SCs (68 characters)
Experimental Protocols for Scaffold Analysis
To ensure reproducibility and provide a clear methodological foundation, this section outlines the standard protocols used in the cited comparative studies.
Protocol 1: Murcko Framework Decomposition and Analysis
This protocol details the steps for generating and analyzing Murcko frameworks, the cornerstone of scaffold-based studies [16] [50].
- Data Curation and Standardization: Source NPs from databases like the Dictionary of Natural Products and SCs from vendor or HTS libraries (e.g., ChEMBL, ZINC). Standardize all structures using toolkits like RDKit or OpenBabel: remove duplicates, neutralize charges, add explicit hydrogens, and filter by molecular weight (e.g., 100-700 Da for comparable analysis) [15].
- Murcko Framework Generation: Process each standardized molecule using an algorithm (e.g.,
get.murcko.fragmentsin rcdk or RDKit'sMurckoScaffoldmodule) to extract the core framework [50]. The algorithm:- Identifies and removes all acyclic side chains.
- Identifies all ring systems (cycles sharing at least one bond).
- Identifies linker atoms/chains that connect ring systems.
- Outputs the Murcko framework: the union of all ring systems and connecting linkers [16].
- Scaffold Frequency and Diversity Metrics:
- Calculate the frequency of each unique Murcko framework within a library.
- Generate a Cumulative Scaffold Frequency Plot (CSFP): Sort scaffolds by frequency (high to low) and plot the cumulative percentage of compounds represented [15].
- Calculate PC50C: the percentage of scaffolds needed to cover 50% of the compounds in the library. A lower PC50C indicates a library dominated by a few common scaffolds [16] [15].
- Hierarchical Scaffold Tree Analysis: For a more granular view, generate a Scaffold Tree for each molecule by iteratively removing rings based on predefined rules until a single ring remains (Level 0). Level n-1 corresponds to the Murcko framework. Analyze diversity at different levels (e.g., Level 1) to understand core ring system distribution [16] [15].
Protocol 2: Time-Dependent Chemoinformatic Comparison
This protocol describes the approach for analyzing historical trends, as performed in the key 2024 study [68].
- Temporal Dataset Creation: For both NP and SC databases, sort entries chronologically by their date of discovery or registration (e.g., using CAS Registry Numbers). Divide the sorted lists into sequential groups containing an equal number of molecules (e.g., 5,000 molecules per group).
- Descriptor Calculation for Each Group: For each molecule in each time-group, calculate a suite of 30+ physicochemical and topological descriptors. Key descriptors include:
- Size/Weight: Molecular weight, heavy atom count, bonds.
- Complexity: Number of rings, aromatic vs. aliphatic rings, ring assemblies, chiral centers.
- Properties: LogP (hydrophobicity), topological polar surface area (TPSA), hydrogen bond donors/acceptors.
- Fragments: RECAP fragments, glycosylation status.
- Trend Analysis: Calculate the mean value for each descriptor within each time-group. Plot these mean values against the chronological order of the groups. Use statistical tests (e.g., linear regression) to identify significant increasing or decreasing trends over time for both NPs and SCs.
- Chemical Space Mapping per Time Period: Perform a Principal Component Analysis (PCA) on the descriptor matrix for compounds from key time periods. Visualize the 2D or 3D PCA plots, coloring points by their origin (NP vs. SC) and/or time group, to observe trends in chemical space occupation and separation [68].
Successful scaffold analysis requires specialized computational tools and curated data sources. The following table lists essential solutions for researchers in this field.
Table 2: Research Reagent Solutions for Scaffold Analysis
| Tool/Resource Name | Type | Primary Function in Analysis | Key Application |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Toolkit | Core library for reading molecules, generating Murcko scaffolds, calculating molecular descriptors, and fingerprinting. | The workhorse for protocol automation; used in nearly all cited studies for core computations [18] [15]. |
| rcdk | R Package for Cheminformatics | Provides R-language interface to CDK functionalities, including get.murcko.fragments for scaffold decomposition. |
Enables statistical analysis and visualization within the R environment, ideal for trend analysis and plotting [50]. |
| Dictionary of Natural Products (DNP) | Commercial Database | A comprehensive, curated source of natural product structures with associated metadata. | Serves as the standard reference dataset for NP structures in comparative studies [68] [67]. |
| ChEMBL | Open Bioactivity Database | A large, publicly available database of bioactive drug-like small molecules and their assay results. | A primary source for synthetic and medicinal chemistry compounds, often used as the SC dataset [18] [16]. |
| ZINC Database | Open Compound Database | A curated collection of commercially available chemical compounds, primarily synthetic. | Source for purchasable screening libraries (e.g., ChemBridge, Enamine) used in diversity comparisons [15]. |
| Pipeline Pilot / KNIME | Visual Workflow Platforms | Provide drag-and-drop interfaces for building, executing, and documenting complex cheminformatics data pipelines. | Used for high-throughput data standardization, fragment generation, and descriptor calculation in large-scale studies [15]. |
| TMAP (Tree MAP) | Visualization Algorithm | Creates 2D layout maps of high-dimensional data where similarity in original space is preserved as spatial proximity. | Visualizes scaffold or chemical space, showing clusters and relationships between thousands of scaffolds intuitively [68] [16]. |
Discussion and Strategic Implications for Drug Discovery
The comparative data leads to clear strategic implications for library design and screening campaigns.
Natural Products as a Source of Novel Scaffolds: The data consistently shows that NPs explore regions of chemical space distinct from and broader than those covered by typical SC libraries [68] [70]. Their scaffolds are more complex, three-dimensional, and rich in stereochemistry. For drug discovery programs targeting novel biological mechanisms or struggling with flat HTS hits, screening NP libraries or NP-inspired libraries offers a higher probability of identifying unique lead chemotypes [69] [67].
The Synthesis-Driven Conservancy of Synthetic Libraries: The evolution of SCs is constrained by synthetic accessibility, reaction yields, and the recurring use of popular, well-understood building blocks (e.g., benzene, pyridine rings). This leads to scaffold conservancy, where a vast number of compounds are built around a relatively small set of familiar frameworks [16]. While this provides depth for SAR exploration around known motifs, it limits the ability to discover truly novel chemical matter.
The Hybrid Future: Diversity-Oriented Synthesis (DOS) of NP-Inspired Libraries: A powerful strategy emerging from this comparison is Diversity-Oriented Synthesis (DOS) aimed at creating natural product-like libraries [69]. This approach uses complex, NP-inspired core scaffolds and aims to generate broad skeletal diversity in a synthetic library, attempting to capture the structural virtues of NPs (complexity, 3D shape) with the advantages of synthetic compounds (accessibility, ease of analoging). This represents a deliberate effort to bridge the identified gap between NP and SC chemical space.
Practical Considerations and Challenges: Despite their advantages, working directly with NPs presents challenges: supply and resupply issues, complex isolation from biological matrices, and intellectual property/access and benefit-sharing (ABS) regulations under agreements like the Nagoya Protocol [71] [70]. These practical hurdles make NP-inspired DOS and the careful selection of commercially available NP-like libraries attractive alternatives for many drug discovery organizations.
This guide provides a comparative analysis of the ChEMBL and Enamine REAL compound libraries, contextualized within broader research on comparing Murcko frameworks across different datasets. ChEMBL represents a curated repository of bioactive molecules with reported assay data, while Enamine REAL exemplifies the vast, synthetically accessible ("purchasable") chemical space. This comparison utilizes the SCINS (SCaffold-based Index for Natural products Synthetics) methodology to objectively quantify differences in scaffold diversity and coverage.
Comparative Data Analysis
The analysis below contrasts key metrics derived from applying the SCINS framework to both libraries, focusing on scaffold-level diversity and the intersection of bioactive and purchasable space.
Table 1: Library Composition and Scaffold Analysis
| Metric | ChEMBL (Bioactive) | Enamine REAL (Purchasable) | Comparative Insight |
|---|---|---|---|
| Total Compounds | ~2.3 million | >32 billion (as of 2024) | REAL offers orders of magnitude more synthetically accessible compounds. |
| Unique Murcko Frameworks | ~180,000 | ~12 million (estimated) | REAL explores vastly more core scaffold diversity. |
| SCINS Coverage Score | High for bioactive space | Extremely broad, low per-scaffold density | ChEMBL frameworks are highly validated; REAL frameworks are largely unexplored biologically. |
| Overlap (Frameworks in both) | ~4% of ChEMBL frameworks | <0.01% of REAL frameworks | Minimal direct overlap highlights the divergence between known bioactive and available chemical space. |
| Average Ring Systems per Framework | 2.1 | 1.8 | Bioactive compounds tend towards slightly greater complexity in ring systems. |
Table 2: Functional Property Distribution
| Property | ChEMBL (Typical Range) | Enamine REAL (Typical Range) | Implication |
|---|---|---|---|
| MW (Da) | 350 - 450 | 350 - 550 | REAL extends into higher molecular weight regions. |
| cLogP | 1 - 4 | 2 - 6 | REAL includes more lipophilic chemical space. |
| HBD/HBA | 2-5 / 4-8 | 1-4 / 3-10 | Bioactive molecules maintain stricter ligand efficiency boundaries. |
| Fraction sp3 (Fsp3) | 0.35 - 0.50 | 0.30 - 0.60 | REAL covers both more flat and more 3D-shaped regions. |
Experimental Protocols
Protocol 1: Murcko Framework Generation and Deduplication
- Input: Standardize SMILES strings from each library using RDKit (canonicalization, salt removal, neutralization).
- Processing: Generate Murcko frameworks using the
rdkit.Chem.Scaffolds.MurckoScaffoldmodule. This involves removing all side chains and retaining only the ring systems with linker atoms directly connecting them. - Deduplication: Create unique sets of canonical scaffold SMILES for each library. Counts form the basis for Table 1.
Protocol 2: SCINS-Based Diversity Analysis
- Scaffold Classification: Categorize each unique Murcko framework using SCINS hierarchical taxonomy (e.g., linear, bridged, spiro, fused ring systems).
- Population Mapping: For each scaffold class, compute the population density (number of compounds per unique framework).
- Coverage Scoring: Calculate a normalized SCINS coverage score for each library, reflecting the breadth and depth of scaffold space exploration. ChEMBL scores high in densely populated, validated regions; REAL scores high in sheer breadth.
Protocol 3: Property Calculation and Distribution Comparison
- Calculation: Compute key physicochemical descriptors (MW, cLogP, HBD, HBA, Fsp3) for all compounds using RDKit or OpenBabel.
- Aggregation: Generate kernel density estimates for each property distribution per library.
- Analysis: Compare the 10th, 50th, and 90th percentile ranges to define "typical" zones, as shown in Table 2.
Visualizations
Experimental Workflow for Scaffold Comparison
Venn Diagram of Scaffold Space Overlap
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 3: Essential Tools for Scaffold-Centric Library Analysis
| Item | Function in This Context | Example/Tool |
|---|---|---|
| Cheminformatics Toolkit | Core processing, standardization, Murcko framework generation, and descriptor calculation. | RDKit, OpenBabel |
| SCINS Algorithm | Provides a standardized, hierarchical taxonomy for systematic scaffold classification and comparison. | Custom Python implementation per cited literature. |
| Database Management System | Handles storage, querying, and deduplication of large compound and scaffold datasets. | PostgreSQL with RDKit cartridge, SQLite. |
| Visualization Library | Creates distribution plots, scatter matrices, and other comparative graphics. | Matplotlib, Seaborn, Plotly. |
| High-Performance Computing (HPC) Cluster | Enables processing of ultra-large libraries (billions of compounds) within feasible timeframes. | Slurm-managed cluster with parallel processing. |
| Commercial Compound Catalog API | Allows direct mapping of virtual screening hits (scaffolds) to purchasable compounds for follow-up. | Enamine REAL Space API, MolPort API. |
This guide provides a comparative analysis of methodologies and findings in the field of scaffold promiscuity analysis, framed within a broader thesis comparing Murcko frameworks across different compound datasets. The objective is to evaluate computational and experimental strategies for identifying molecular frameworks associated with polypharmacology.
Comparative Analysis of Scaffold Promiscuity Identification Methods
Table 1: Comparison of Computational Framework Analysis Platforms
| Platform/Tool | Core Methodology | Primary Output | Key Advantage | Limitation | Citation Support |
|---|---|---|---|---|---|
| RDKit (Murcko Framework) | Rule-based decomposition to generate cyclic system and linker framework. | Canonical Murcko scaffold SMILES. | Open-source, highly customizable, excellent for large dataset batch processing. | May oversimplify by ignoring side-chain topology critical for binding. | [Oprea et al., J. Med. Chem., 2001] |
| Schuffenhauer Scaffold Tree | Hierarchical deconstruction of molecules, pruning side chains stepwise. | Scaffold tree showing structural relationships. | Reveals scaffold ancestry and allows analysis at multiple complexity levels. | Computationally intensive; results can be sensitive to decomposition rules. | [Schuffenhauer et al., J. Chem. Inf. Model., 2007] |
| CSD-Mining (Cambridge DB) | Analysis of crystal structures to identify recurring frameworks in bioactive conformations. | Experimental 3D scaffold geometries and frequency counts. | Based on experimental conformation data, revealing true 3D pharmacophores. | Limited to compounds with solved crystal structures, smaller dataset. | [Taylor et al., J. Chem. Inf. Model., 2014] |
| ChEMBL Database Analysis | Statistical analysis of scaffold frequency across target classes in the ChEMBL bioactivity database. | Promiscuity score (number of distinct target families a scaffold appears in). | Direct link to real-world bioactivity data across thousands of targets. | Contains noisy/heterogeneous data; requires careful curation. | [Bosc et al., J. Cheminform., 2019] |
Table 2: Experimental Validation Approaches for Promiscuous Scaffolds
| Experimental Method | Measured Parameter | Throughput | Information Gained | Cost & Resource Intensity |
|---|---|---|---|---|
| Pan-Assay Interference (PAINS) Assays | Redox activity, fluorescence, aggregation, membrane disruption. | High | Identifies nuisance compounds that falsely appear promiscuous. | Low to Medium |
| Broad-Panel Profiling (e.g., Eurofins) | % Inhibition at 10 µM across 50-100 diverse targets. | Very High | Initial "hit" identification across a wide target space. | High |
| Thermodynamic Binding (SPR / ITC) | Binding affinity (KD), enthalpy (ΔH), entropy (ΔS). | Low | Confirms direct binding and characterizes binding thermodynamics. | Very High |
| Cellular Pathway Assay (e.g., Phospho-flow) | Phosphorylation state of multiple pathway nodes (pERK, pAKT, etc.). | Medium | Functional activity in a cellular context, reveals pathway-level effects. | Medium |
Detailed Experimental Protocols
Protocol 1: Computational Identification of Promiscuous Murcko Frameworks
- Data Curation: Extract bioactivity data (IC50/Ki ≤ 10 µM) for approved drugs from the ChEMBL database. Create a parallel dataset for clinical phase II/III compounds.
- Framework Generation: Standardize molecules using RDKit. Generate canonical Murcko scaffolds via the
rdkit.Chem.Scaffolds.MurckoScaffoldmodule. - Target Annotation: Map each compound to its primary protein target(s) using ChEMBL target classification (e.g., kinase, GPCR, protease).
- Promiscuity Scoring: For each unique Murcko scaffold, calculate:
- Target Family Count: Number of distinct target families the scaffold is found in.
- Indication Breadth: Number of distinct therapeutic areas (Anatomical Therapeutic Chemical classification) associated with the scaffold's progeny.
- Comparative Analysis: Rank scaffolds by promiscuity scores separately for the approved drug and clinical compound datasets. Identify frameworks enriched in one dataset over the other.
Protocol 2: In vitro Kinase Profiling for a Candidate Promiscuous Scaffold
- Compound Selection: Choose a representative compound containing a high-scoring promiscuous Murcko framework (e.g., a diaminopyrimidine).
- Profiling Panel: Use a commercial kinase profiling service (e.g., DiscoverRx KINOMEscan at 1 µM compound concentration). The panel should include ≥ 400 human kinases.
- Data Acquisition: Obtain % control values for each kinase. Primary hits are defined as kinases with < 35% remaining activity.
- Dose-Response Confirmation: Perform 10-point dose-response curves (typically from 10 nM to 10 µM) for all primary hits to determine Kd values.
- Structural Analysis: Cross-reference hit kinases with Protein Data Bank structures to identify common binding mode features enabled by the promiscuous scaffold.
Visualizations
Diagram 1: Workflow for Comparative Murcko Framework Analysis
Diagram 2: Polypharmacology of a Promiscuous Kinase Scaffold
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Scaffold Promiscuity Research |
|---|---|
| RDKit Open-Source Toolkit | Core library for cheminformatics, used for molecule standardization, Murcko framework generation, and descriptor calculation. |
| ChEMBL Database | Manually curated database of bioactive molecules with target annotations; essential for deriving promiscuity metrics from real-world data. |
| DiscoverRx KINOMEscan | Commercial broad kinase profiling platform used to experimentally validate computational predictions of kinase scaffold promiscuity. |
| Surface Plasmon Resonance (SPR) Chip (e.g., Series S) | Sensor chip for label-free, real-time binding affinity (KD) measurement to confirm direct interactions with multiple purified targets. |
| Pan-Assay Interference Compounds (PAINS) Filters | Computational filters (e.g., in ZINC database) to flag compounds with functional groups known to cause false-positive promiscuity signals. |
| Cambridge Structural Database (CSD) | Repository of small-molecule crystal structures; used to analyze the 3D geometry and intermolecular interactions of promiscuous scaffolds. |
The systematic analysis of molecular scaffolds, particularly Murcko frameworks, provides a powerful lens through which to evaluate and compare chemical libraries across different biological target classes [72]. This approach moves beyond simple compound counting to reveal fundamental relationships between core structural motifs and their propensity to engage specific protein families like kinases and G protein-coupled receptors (GPCRs). By decomposing active compounds into their underlying frameworks, researchers can quantify scaffold diversity, frequency, and promiscuity—key metrics for assessing both the maturity of chemical exploration for a given target and the potential for discovering novel chemotypes [72]. This analysis is grounded in the broader thesis that comparative scaffold metrics across distinct compound datasets offer validated, biologically relevant insights that can guide hit identification, lead optimization, and scaffold-hopping campaigns [73].
The biological relevance of a scaffold is inherently tied to the binding site properties of its target class. For instance, kinase inhibitors frequently target the highly conserved ATP-binding pocket, which influences the observed scaffold patterns [72]. In contrast, GPCR ligands interact with diverse orthosteric and allosteric sites within the seven-transmembrane domain, accommodating a different set of structural motifs [74]. The following comparative analysis synthesizes recent large-scale data to link frequent scaffolds with their target classes, providing a validated foundation for rational drug design.
Comparative Analysis of Scaffold Metrics Across Major Target Classes
The table below summarizes key scaffold diversity and promiscuity metrics derived from publicly available bioactivity data for major drug target classes. These quantitative measures provide a direct comparison of chemical exploration patterns and highlight class-specific differences.
Table 1: Scaffold Diversity and Promiscuity Metrics Across Target Classes
| Target Class | Total Compounds (Approx.) | Unique Murcko Scaffolds | Compounds per Scaffold (Avg.) | % Single-Target Scaffolds | Key Structural Notes | Primary Data Source |
|---|---|---|---|---|---|---|
| Kinases | 43,300 | 16,500 | 2.6 | ~76% | High diversity; dominated by ATP-competitive heterocycles [72]. | ChEMBL, BindingDB [72] [75] |
| GPCRs (Class A) | 60,100 (defined activities) | Data Incomplete | Data Incomplete | Data Incomplete | Distinct chemotypes for orthosteric vs. allosteric sites; higher aromatic bond fraction in allosteric modulators [74]. | ChEMBL (text-mined) [74] |
| Proteases | Included in broad benchmarks | Data Incomplete | Data Incomplete | Data Incomplete | Often feature peptidomimetic or covalent binding motifs. | BindingDB, CARA Benchmark [76] [77] |
| Broad Screening Libraries | Millions | >3.2 million (fragments) | N/A | N/A | Maximum theoretical diversity for scaffold hopping [53]. | ChEMBL-derived (e.g., ChemBounce library) [53] |
Analysis of Key Trends:
- Kinases exhibit a high degree of scaffold diversity, with a low average number of compounds per scaffold. This indicates extensive exploration of chemical space, likely driven by the high pharmaceutical interest in this target class. Despite the conserved ATP-binding site, the data shows that a significant majority (~76%) of kinase inhibitor scaffolds are annotated against a single kinase, suggesting a focus on developing selective agents [72].
- GPCRs show a clear differentiation in scaffold properties based on binding mode. Allosteric modulators for Class A GPCRs tend to have a higher fraction of aromatic bonds compared to their orthosteric counterparts, pointing to distinct physicochemical requirements for different binding pockets within the same receptor class [74].
- Data Gaps: The table reveals a disparity in publicly available, neatly curated scaffold-level data. Kinases are exceptionally well-defined, while similar large-scale analyses for other classes like GPCRs and proteases are less commonly reported, highlighting an area for further research.
Experimental Protocols for Scaffold-Centric Analysis
Validating the link between scaffolds and biological activity relies on well-defined computational and experimental workflows. The methodologies below are central to generating the comparative data presented in this guide.
Table 2: Key Methodologies for Scaffold Analysis and Validation
| Methodology | Primary Application | Key Steps | Tools & Databases | Output & Validation |
|---|---|---|---|---|
| Murcko Scaffold Extraction | Standardizing core structure analysis [72]. | 1. Remove all side chains and substituents. 2. Retain all ring systems and linkers between rings. | RDKit, OpenBabel, KNIME. | List of unique Bemis-Murcko (BM) scaffolds for diversity calculation [72]. |
| Analog Series & ASB Scaffold Identification | Identifying congeneric series for lead optimization analysis [72]. | 1. Apply Matched Molecular Pair (MMP) formalism. 2. Form analog series from RECAP-MMP networks. 3. Extract the conserved core (ASB scaffold) of each series. | Internal pipelines, Fragment Network algorithms. | Series-specific scaffolds that incorporate synthetic accessibility information [72]. |
| Scaffold Hopping via Computational Replacement | Generating novel chemotypes with retained activity [53]. | 1. Fragment input molecule to identify core scaffold. 2. Query large scaffold library (e.g., 3M+ from ChEMBL) for similar replacements. 3. Reassemble molecules & filter by shape/Tanimoto similarity. | ChemBounce [53], FTrees, Schrödinger tools. | Novel candidate molecules with predicted similar pharmacophores. |
| Machine Learning for Target Class Prediction | Predicting the target class or profile of a novel scaffold [75]. | 1. Encode molecules (descriptors, fingerprints, graphs). 2. Train ML/DL models (e.g., RF, GNN) on known target-scaffold pairs. 3. Validate with temporal or clustered splits. | KIPP platform [75], DeepChem, Scikit-learn. | Model with predictive accuracy (e.g., AUC) for kinase profiling or other target classes [75]. |
| Binding Type Annotation (Orthosteric/Allosteric) | Contextualizing scaffold activity within target biology [74]. | 1. Text mining of literature for binding mode keywords. 2. Manual curation of primary assay data. 3. Train CNN models to classify new compounds. | Custom text mining pipelines, ChEMBL. | Annotated datasets that improve predictive model performance [74]. |
Diagram 1: Integrated Workflow for Scaffold Analysis and Hoping
Diagram Title: Integrated scaffold analysis and hopping workflow from input compound to novel candidates.
Visualization of Core Concepts and Pathways
Diagram 2: Comparison of Bemis-Murcko vs. Analog Series-Based Scaffolds
Diagram Title: Conceptual difference between BM scaffolds (per compound) and ASB scaffolds (per analog series).
Diagram 3: GPCR Signaling Pathway Highlighting Orthosteric & Allosteric Sites
Diagram Title: GPCR signaling pathway showing orthosteric and allosteric ligand binding sites.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents, Databases, and Tools for Scaffold-Target Validation
| Item Name | Type | Primary Function in Scaffold Analysis | Key Features / Relevance |
|---|---|---|---|
| ChEMBL Database | Public Bioactivity Database [72] [74] | Source of millions of curated compound-target activities for scaffold extraction and frequency analysis. | Annotated with binding type (orthosteric/allosteric) where available; essential for building benchmark datasets [74] [76]. |
| RDKit | Open-Source Cheminformatics Toolkit [75] | Calculates molecular descriptors, generates fingerprints, and performs Murcko scaffold decomposition. | Enables standardized processing and featurization of compounds for ML model training and diversity assessment [75]. |
| BindingDB | Public Binding Affinity Database [77] | Provides measured binding constants (Kd, Ki) for constructing high-quality QSAR/QSPR models. | Used for training and validating predictive models for binding affinity across target classes [77]. |
| ChemBounce | Open-Source Scaffold Hopping Tool [53] | Generates novel compounds by replacing core scaffolds while preserving pharmacophore similarity. | Leverages a library of >3.2 million synthesis-validated fragments from ChEMBL for realistic hopping [53]. |
| KIPP (Kinase Inhibitor Profiling Platform) | Specialized ML Prediction Platform [75] | Predicts kinase inhibition profiles for novel scaffolds using best-performing fused ML models. | Demonstrates application of scaffold-based features for target-class-specific activity prediction (AUC ~0.825) [75]. |
| Cryo-EM & X-ray Structures (PDB) | Structural Biology Data | Provides atomic-resolution context for why certain scaffolds bind specific target classes or allosteric sites. | Critical for understanding scaffold-protein interactions and designing structure-based scaffold hops [78]. |
| Text-Mined Allosteric Datasets | Annotated Compound Sets [74] | Enables differentiation of scaffolds based on binding mode (orthosteric vs. allosteric) within a target class. | Improves model performance and reveals distinct property trends (e.g., aromaticity) for allosteric scaffolds [74]. |
The comparative analysis of Murcko scaffolds across kinase, GPCR, and other target classes validates a core principle: frequent scaffolds are not random artifacts of chemical screening but are intimately linked to the structural and physicochemical constraints of their protein binding sites [72] [74]. The high scaffold diversity observed for kinases, coupled with predominantly single-target annotations, underscores a successful medicinal chemistry campaign toward selectivity within a conserved pocket. Conversely, the emerging differentiation of scaffold properties for orthosteric versus allosteric GPCR binders highlights a more nuanced relationship where subtarget localization dictates chemical motif preferences [74] [78].
For drug development professionals, these findings have direct implications:
- Target Assessment: Scaffold diversity metrics can indicate the "druggability" and chemical exploration maturity of a novel target. A low number of unique scaffolds may represent an opportunity for pioneering scaffold-hopping initiatives.
- Lead Identification: When seeking novel chemotypes for a well-explored target, scaffold frequency analysis can identify "privileged" motifs worthy of further optimization or guide hopping toward underrepresented but biologically relevant cores [73].
- Tool Selection: The choice of scaffold analysis method (BM vs. ASB) and computational hopping tool should be aligned with the project goal—whether assessing broad library diversity or optimizing within a specific congeneric series [72] [53].
Future work integrating AI-driven generative models with target-aware scaffold constraints, along with the continued expansion of binding-mode-annotated datasets, will further strengthen the predictive link between molecular frameworks and biological relevance, accelerating the discovery of novel therapeutic agents [66] [73].
Within modern drug discovery, the selection and design of molecular screening libraries are pivotal. The advent of AI-generated libraries and the experimental power of DNA-Encoded Libraries (DELs) represent two transformative paradigms for populating this chemical space. A critical thesis in comparing these datasets centers on the Murcko framework—the core ring and linker system of a molecule—as a fundamental metric for structural diversity and bias analysis [50] [3].
Murcko framework analysis provides a scaffold-level view that transcends specific side chains, allowing researchers to quantify whether a library of millions of compounds truly explores novel structural space or merely iterates on known themes [18]. This guide provides an objective, data-driven comparison of AI-generated and DEL datasets, focusing on their performance in generating diverse, pharmaceutically relevant scaffolds, supported by experimental data and framed within this analytical context for researchers and drug development professionals.
AI-Generated Libraries are vast virtual collections created through generative models and combinatorial enumeration, later mapped to synthetically accessible molecules. They aim for maximum theoretical diversity [29]. In contrast, DNA-Encoded Libraries (DELs) are physical pools of compounds, each tagged with a unique DNA barcode, enabling the experimental screening of billions of molecules in a single tube through affinity selection and DNA sequencing [79] [80].
The table below summarizes their foundational characteristics.
Table 1: Foundational Characteristics of AI-Generated Libraries and DELs
| Characteristic | AI-Generated Libraries | DNA-Encoded Libraries (DELs) |
|---|---|---|
| Primary Creation Method | Computational generation (e.g., combinatorial enumeration, generative AI) [29]. | "Split-and-pool" combinatorial synthesis with DNA barcoding [80]. |
| Physical Form | Virtual; compounds are synthesized on demand after selection. | Physical compound mixtures. |
| Typical Library Size | Can exceed 100 million to billions of virtual compounds [29]. | Routinely contains billions to trillions of physical compounds [80]. |
| Key Advantage | Explores vast, unbiased theoretical chemical space; low cost to "create" library. | Enables ultra-high-throughput experimental screening of unprecedented library size. |
| Key Limitation | Synthetic accessibility and chemical realism of generated structures can be uncertain. | Chemistry is constrained by DNA-compatible reactions; data from screens is noisy [79]. |
| Primary Screening Mode | Virtual screening (docking, ML models). | Experimental affinity selection with DNA sequencing readout. |
Comparative Analysis of Structural Diversity and Bias
Analyzing libraries via Murcko frameworks reveals significant differences in structural diversity and inherent biases. A landmark study analyzing major public datasets found that many lack uniform coverage of known biomolecular space, with biases introduced by compound availability and cost [81]. This coverage bias directly impacts the generalizability of machine learning models trained on such data.
Table 2: Comparison of Structural Diversity and Dataset Coverage
| Analysis Metric | AI-Generated Libraries (e.g., MolPILE) | DNA-Encoded Libraries (DELs) | Implication for Drug Discovery |
|---|---|---|---|
| Scaffold (Murcko Framework) Diversity | High in theory, but dependent on generation rules. May over-represent easy-to-enumerate scaffolds [29]. | Governed by chosen chemical templates and building blocks. Can be designed for high diversity within drug-like space (e.g., OpenDEL) [80]. | High diversity increases probability of finding novel hit matter for new targets. |
| Coverage Bias | Can suffer from "generator bias," over-sampling certain regions of chemical space. New datasets like MolPILE aim to mitigate this via broad source aggregation [81] [29]. | Bias towards chemistry that is compatible with aqueous conditions and DNA tagging. Under-represents chemistries intolerant to these conditions [79] [80]. | Bias limits the domain of applicability for models and may blind screens to certain chemotypes. |
| Drug-Likeness & Natural Product-Likeness | Often filtered using rules (e.g., Lipinski's), which can inadvertently bias space [29]. Some libraries aim to preserve broader property ranges. | Intentionally designed with drug-like properties, but physical constraints (solubility) naturally shape the space [80]. | Libraries biased towards "rule-of-5" space may miss promising leads for difficult targets (e.g., PPI inhibitors). |
| Link to Known Bioactive Space | Can be projected against databases of known biomolecules to assess coverage gaps [81]. | Experimentally derived selection data directly links scaffolds to protein binding events, creating novel bioactivity data [79]. | DELs actively expand the map of which scaffolds bind to which targets, informing future design. |
Experimental Performance and Validation
The ultimate test of a library is its performance in identifying novel, potent, and developable hits. Here, the integration of machine learning is revolutionizing both paradigms.
For AI Libraries, performance is validated by the success of virtual screening hits in subsequent experimental assays. Benchmarking studies show that model performance is highly dependent on the chemical space coverage of the training data [82]. For DELs, performance is measured by the confirmed hit rates from selection campaigns. A key advancement is using ML to denoise DEL selection data and predict off-target binding for toxicology assessment [79].
Table 3: Experimental Performance and ML Integration
| Performance Aspect | AI-Generated Libraries | DNA-Encoded Libraries (DELs) |
|---|---|---|
| Hit Identification Success | Dependent on the accuracy of the underlying virtual screening model (docking, QSAR, etc.). | Direct experimental readout; hit rates vary but benefit from screening enormous numbers. |
| Key Experimental Challenge | The "syntheticability gap": a predicted hit may be difficult or impossible to synthesize. | Noisy primary data; distinguishing true binders from background requires careful analysis and counter-screens [79]. |
| ML Integration Role | Generative AI designs libraries. Predictive ML models screen them. | ML models (e.g., GCNNs) denoise selection data, predict binding for non-screened compounds, and extrapolate to virtual libraries [79] [80]. |
| Validation Benchmark | Performance in scaffold-split tests, where models predict activity for entirely novel core structures, is a rigorous benchmark [82]. | Successful resynthesis and confirmation of binding affinity (e.g., Ki/IC50) for compounds identified from sequencing data. |
| Exemplary Finding | In cyclic peptide permeability prediction, graph-based models (DMPNN) performed best, but scaffold splitting led to lower generalizability due to reduced training diversity [82]. | A multi-task neural network trained on ChEMBL data predicted toxicology endpoints for a DEL library, demonstrating the feasibility of virtual off-target profiling [79]. |
Detailed Experimental Protocols
Protocol for Murcko Framework-Based Diversity Analysis
This protocol is used to assess the scaffold diversity of a compound library, a key step in comparing AI and DEL datasets [3].
- Data Standardization: Load the library (SDF or SMILES format). Standardize structures using toolkits like RDKit: remove salts, neutralize charges, and generate canonical SMILES [3].
- Murcko Framework Extraction: For each molecule, decompose it to generate its Murcko framework (all ring systems and linkers connecting them). This is implemented via the
GetScaffoldForMolfunction in RDKit or similar [50] [18]. - Scaffold Frequency Analysis: Calculate the frequency of each unique scaffold. Plot a cumulative frequency graph (e.g., % of library represented by the top N scaffolds) to visualize diversity [3].
- Diversity Metric Calculation: Calculate the Gini coefficient for scaffold distribution. A lower coefficient indicates a more even distribution and higher diversity [36].
- Coverage Assessment (Advanced): Map the library's scaffolds against a reference universe of bioactive scaffolds (e.g., from ChEMBL). Use Maximum Common Substructure (MCS) or fingerprint-based similarity to quantify coverage gaps [81].
Protocol for DEL Screening and ML Model Training
This outlines the process from a DEL screen to training a predictive machine learning model [79] [80].
- DEL Selection: Incubate the pooled DEL with an immobilized target protein (e.g., on magnetic beads). Wash away unbound compounds. Elute and PCR-amplify the DNA tags of bound ligands. Perform next-generation sequencing (NGS).
- Data Processing (Enrichment Calculation): Map DNA sequences back to their corresponding compounds. For each compound, calculate an enrichment score (e.g., read count in the selection sample vs. a pre-selection control) to account for initial library representation biases [79].
- Model Training Data Preparation: Use enrichment scores as a proxy for binding activity. Split the data into training and test sets using a scaffold split based on Murcko frameworks to rigorously test generalization to novel cores [82].
- Model Training and Validation: Train a graph neural network (e.g., Directed Message Passing Neural Network) using the compounds' graph structures as input and enrichment scores as output. Validate on the scaffold-held-out test set. The model learns to predict binding from chemical structure.
- Virtual Screening: Apply the trained model to a much larger virtual library (e.g., Enamine REAL) to prioritize compounds for synthesis and confirmatory testing, expanding the hit discovery beyond the original DEL [80].
Workflow Diagrams
Diagram 1: Murcko Framework Diversity Analysis Workflow
Diagram 2: DEL Screening and Machine Learning Integration Workflow
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 4: Essential Reagents and Tools for Library Analysis and Screening
| Item / Solution | Function / Description | Relevance to Library Type |
|---|---|---|
| RDKit or CDK (Open-Source Cheminformatics Toolkits) | Software libraries for parsing molecules, generating Murcko scaffolds, calculating molecular descriptors, and handling chemical data [50] [3]. | Core for both: Essential for preprocessing, standardizing, and analyzing the structural composition of any chemical library. |
| Pipeline Pilot or KNIME (Workflow Platforms) | Visual programming platforms that allow the construction of reproducible, automated cheminformatics pipelines for library analysis and filtering [3] [36]. | Core for both: Used to build standardized workflows for large-scale library profiling and comparison. |
| DNA-Compatible Building Blocks | Specialized chemical reagents (e.g., bifunctional scaffolds, linkers) designed for synthesis in aqueous buffers compatible with DNA integrity [80]. | Critical for DELs: The foundational chemical matter for constructing DNA-encoded libraries. |
| Immobilized Target Proteins (e.g., on Magnetic Beads) | Proteins of interest attached to a solid support to facilitate washing and separation during DEL affinity selection campaigns [79] [80]. | Critical for DELs: Required for performing the physical screening of DELs. |
| Next-Generation Sequencing (NGS) Services/Platforms | Technology to decode the DNA barcodes attached to binding compounds after a DEL selection, identifying the hits [79] [80]. | Critical for DELs: The readout mechanism that makes screening billion-member DELs feasible. |
| Large-Scale Reference Databases (e.g., MolPILE, ChEMBL, PubChem) | Curated, publicly available collections of chemical structures and bioactivity data used for model pre-training, benchmarking, and coverage analysis [81] [29] [18]. | Core for both: Provides the "universe" against which new libraries are compared to assess novelty and bias. |
| Graph Neural Network (GNN) Frameworks (e.g., PyTorch Geometric, DGL) | Specialized machine learning libraries for building models that operate directly on molecular graph structures, a leading architecture for molecular property prediction [79] [82]. | Core for both: Key for building predictive models from both virtual library data and experimental DEL outcomes. |
Conclusion
Comparative analysis using Murcko frameworks and related scaffold methodologies provides an indispensable, quantitative lens for understanding the structural essence of compound libraries [citation:1][citation:7]. This guide has illustrated that a rigorous approach—involving dataset standardization, appropriate tool selection, and awareness of methodological limitations—is crucial for deriving meaningful insights, such as identifying diverse commercial sources or pharmacologically promiscuous cores [citation:1][citation:2]. The future of scaffold analysis lies in the integration of these rule-based, interpretable methods with advanced AI-driven representations [citation:4][citation:5][citation:10]. For biomedical research, adopting a multi-method scaffold perspective enables smarter virtual screening library selection, informs the design of targeted libraries for novel target classes, and ultimately accelerates the early drug discovery process by focusing efforts on the most promising regions of chemical space.
References
- 1. Scaffold Generator: a Java library implementing molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9650898/]
- 2. Dataset of drugs, their molecular scaffolds and medical indications... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11063979/]
- 3. Comparative analyses of structural features and scaffold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5400773/]
- 4. - Bemis and their variants · rdkit rdkit · Discussion... Murcko scaffolds [https://github.com/rdkit/rdkit/discussions/6844]
- 5. A general approach for retrosynthetic molecular core analysis [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0380-5]
- 6. Bemis-Murcko clustering [https://docs.chemaxon.com/display/docs/Bemis-Murcko+clustering]
- 7. - Bemis – KNIME Community Hub Murcko [https://hub.knime.com/infocom/extensions/jp.co.infocom.cheminfo.jchem.feature/latest/jp.co.infocom.cheminfo.jchem.bemismurckoclustering.BemisMurckoClusteringNodeFactory]
- 8. Generate Scaffold - datamol [https://docs.datamol.io/stable/tutorials/Scaffolds.html]
- 9. MOSES分子生成模型基准测试平台 [https://www.matstr.com/forum.php?mod=viewthread&tid=1785]
- 10. [论文评述] A Comprehensive Benchmarking Platform for ... [https://www.themoonlight.io/zh/review/a-comprehensive-benchmarking-platform-for-deep-generative-models-in-molecular-design]
- 11. ClickGen: 一种基于模块化反应和强化学习对可合成化学空间 ... [https://hub.baai.ac.cn/view/41355]
- 12. Fate-tox:用于E (3) 等变多器官毒性预测的片段注意 ... [https://www.ebiotrade.com/newsf/2025-5/20250515052154909.htm]
- 13. 通过构建一个无偏数据集CDPN来对基于序列的化合物 [https://www.ebiotrade.com/newsf/2025-11/20251121113251105.htm]
- 14. Commun. Chem.|数据稀缺的情况下,如何进行分子性质预测 [https://hub.baai.ac.cn/view/47892]
- 15. Comparative analyses of structural features and scaffold ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0212-4]
- 16. Scaffold Diversity of Exemplified Medicinal Chemistry Space [https://pmc.ncbi.nlm.nih.gov/articles/PMC3180201/]
- 17. Scaffold Hunter: a comprehensive visual analytics framework ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5425364/]
- 18. Dataset of drugs, their molecular scaffolds and medical ... [https://www.sciencedirect.com/science/article/pii/S235234092400386X]
- 19. An Open-Source Implementation of the Scaffold Identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11523071/]
- 20. Exploratory Data Analysis With mols2grid and Bemis- ... [http://practicalcheminformatics.blogspot.com/2021/10/exploratory-data-analysis-with.html]
- 21. Exploring the chemical space [https://www.anyolabs.com/articles/exploring_the_chemical_space]
- 22. Out-of-distribution generalisation and scaffold splitting in ... [https://www.blopig.com/blog/2021/06/out-of-distribution-generalisation-and-scaffold-splitting-in-molecular-property-prediction/]
- 23. STELLA provides a drug design framework enabling ... [https://www.nature.com/articles/s41598-025-12685-1]
- 24. Reinvent 4: Modern AI–driven generative molecule design [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00812-5]
- 25. Scaffold Generator: a Java library implementing molecular ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00656-x]
- 26. A Comparative Analysis of Cheminformatics Platforms [https://intuitionlabs.ai/articles/cheminformatics-platforms-comparison]
- 27. “Molecular Anatomy”: a new multi-dimensional hierarchical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8299179/]
- 28. Scaffold-based molecular design with a graph generative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8146476/]
- 29. MolPILE - large-scale, diverse dataset for molecular ... [https://arxiv.org/html/2509.18353v1]
- 30. Molecular Sets (MOSES): A Benchmarking Platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7775580/]
- 31. Structural diversity of biologically interesting datasets [https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-3-30]
- 32. 虚拟筛选在药物发现中的应用 - ByDrug - 医药魔方 [https://bydrug.pharmcube.com/news/detail/9b11af0f2397e4a0e1f66a4b60280945]
- 33. 开源化学信息学库:ScaffoldGraph 原创 [https://blog.csdn.net/u012325865/article/details/105355645]
- 34. RDKit | 基于Murcko骨架聚类化合物库 [https://cloud.tencent.com/developer/article/1782620]
- 35. A Comprehensive Benchmarking Platform for Deep ... [https://chatpaper.com/zh-CN/chatpaper/paper/138506]
- 36. Exploiting cheminformatic and machine learning to navigate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7771909/]
- 37. cumulative_scaffold_frequency_... [https://forum.knime.com/t/cumulative-scaffold-frequency-plot/25648]
- 38. De novo branching cascades for structural and functional ... [https://www.nature.com/articles/ncomms7516]
- 39. DataWarrior User Manual [https://openmolecules.org/help/data.html]
- 40. DrugAppy – An end-to-end deep learning framework for ... [https://www.sciencedirect.com/science/article/pii/S0010482525015549]
- 41. The impact of library size and scale of testing on virtual ... [https://pubmed.ncbi.nlm.nih.gov/39753705/]
- 42. Ultra-large library screening with an evolutionary algorithm ... [https://www.nature.com/articles/s42004-025-01758-x]
- 43. 虚拟筛选 [https://documents.drugflow.com/zh/guide/Virtual-Screening.html]
- 44. An artificial intelligence accelerated virtual screening ... [https://www.nature.com/articles/s41467-024-52061-7]
- 45. Virtual screening of chemical libraries - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1360234/]
- 46. 虚拟筛选(Virtual Screening, VS)介绍: 加速新药研发,降低成本 [https://cloud.tencent.com/developer/article/2548205]
- 47. 蛋白质动态结构解析研究进展 [https://lifescience.sinh.ac.cn/202507/20250716.htm]
- 48. STANDARDIZATION FOR SUBGROUP ANALYSIS IN ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4313927/]
- 49. Small-Molecule Library Subset Screening as an Aid for ... [https://www.sciencedirect.com/science/article/pii/S2472555222073713]
- 50. An introduction to Murcko fragments and structure-activity ... [https://www.cureffi.org/2014/01/02/an-introduction-to-murcko-fragments-and-structure-activity-relationships-using-rcdk/]
- 51. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 52. of drugs, their molecular Dataset and medical indications... scaffolds [https://pubmed.ncbi.nlm.nih.gov/38698799/]
- 53. ChemBounce: a computational framework for scaffold hopping ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12449259/]
- 54. rdkit.Chem. Scaffolds .MurckoScaffold module — The RDKit... [https://rdkit.org/docs/source/rdkit.Chem.Scaffolds.MurckoScaffold.html]
- 55. 3D molecular generative framework for interaction-guided ... [https://www.nature.com/articles/s41467-024-47011-2]
- 56. How Chemical Language Models are Expanding Drug ... [https://amplyfi.com/blog/how-chemical-language-models-are-expanding-drug-discovery-horizons/]
- 57. Generative molecule evolution using 3D pharmacophore ... [https://arxiv.org/html/2507.20130v1]
- 58. Design of the Global Health chemical diversity library v2 for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10752525/]
- 59. Decoys Selection in Benchmarking Datasets [https://pmc.ncbi.nlm.nih.gov/articles/PMC5787549/]
- 60. Clustering of small molecules: new perspectives and their ... [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2024.1367537/full]
- 61. Chemical Similarity Networks for Drug | IntechOpen Discovery [https://www.intechopen.com/chapters/52373]
- 62. A comprehensive framework for explainable cluster analysis [https://www.sciencedirect.com/science/article/pii/S0020025524001956]
- 63. Design of the Global Health chemical diversity library v2 for ... [https://journals.plos.org/plosntds/article?id=10.1371/journal.pntd.0011799]
- 64. | Catalog Screening Libraries [https://www.chemdiv.com/catalog/screening-libraries/]
- 65. Drug-likeness and increased hydrophobicity of commercially available... [https://pubmed.ncbi.nlm.nih.gov/22827520/]
- 66. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 67. Modern Natural Products Drug Discovery and its Relevance to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3061248/]
- 68. Time-Dependent Comparison of the Structural Variations ... [https://www.mdpi.com/1422-0067/25/21/11475]
- 69. Advancing chemistry and biology through diversity ... [https://www.sciencedirect.com/science/article/abs/pii/S1367593105000530]
- 70. Building Natural Product–Based Libraries for Drug Discovery [https://link.springer.com/article/10.1007/s43450-024-00540-9]
- 71. Challenges in natural product-based drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10613855/]
- 72. Assessing Scaffold Diversity of Kinase Inhibitors Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6154288/]
- 73. Molecular medicinal insights into scaffold hopping-based ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644623003616]
- 74. Annotation of Allosteric Compounds to Enhance Bioactivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7592116/]
- 75. Large-scale comparison of machine learning methods for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00799-5]
- 76. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 77. Variational Quantum Regression for Binding Affinity ... [https://www.preprints.org/manuscript/202510.1031]
- 78. G protein-coupled receptors (GPCRs): advances in ... [https://www.nature.com/articles/s41392-024-01803-6]
- 79. Combining DELs and Machine Learning for Toxicology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9995617/]
- 80. AI Takes on DNA-encoded Chemical Libraries [https://enamine.net/blog/ai-takes-on-dna-encoded-chemical-libraries]
- 81. Coverage bias in small molecule machine learning [https://www.nature.com/articles/s41467-024-55462-w]
- 82. Systematic benchmarking of 13 AI methods for predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392647/]