LC-SPE-NMR: Revolutionizing Natural Product and Pharmaceutical Analysis with Drastic Deuterated Solvent Reduction
This article explores the transformative role of Liquid Chromatography-Solid Phase Extraction-Nuclear Magnetic Resonance (LC-SPE-NMR) as a powerful hyphenated technique that significantly reduces the reliance on expensive deuterated solvents in analytical...
LC-SPE-NMR: Revolutionizing Natural Product and Pharmaceutical Analysis with Drastic Deuterated Solvent Reduction
Abstract
This article explores the transformative role of Liquid Chromatography-Solid Phase Extraction-Nuclear Magnetic Resonance (LC-SPE-NMR) as a powerful hyphenated technique that significantly reduces the reliance on expensive deuterated solvents in analytical workflows. Tailored for researchers and drug development professionals, we cover the foundational principles of LC-SPE-NMR, detail its methodology for efficient analyte trapping and solvent exchange, and provide practical troubleshooting guidance. The discussion extends to a comparative analysis with other hyphenated techniques, validating its critical advantages in cost-efficiency, sensitivity, and application in characterizing complex mixtures like natural products and pharmaceutical impurities, ultimately outlining its future impact on biomedical research.
The High Cost of Clarity: Why Deuterated Solvents Challenge Traditional LC-NMR
Troubleshooting Guides
Q1: Why does NMR spectroscopy have inherently low sensitivity compared to techniques like UV-Vis?
The low sensitivity of NMR stems from the very small energy difference between nuclear spin states, resulting in an extremely small population excess in the lower energy state at thermal equilibrium [1].
- Inherently Small Population Difference: The population ratio between the lower (α) and upper (β) nuclear spin states is approximately 1.000064. For a million spins, there are only about 32 more in the α state than the β state. This tiny population excess is what generates the detectable NMR signal. In contrast, UV-Vis spectroscopy has a population ratio of approximately 1x10^42, meaning essentially all molecules are in the ground state, leading to a much larger signal [1].
- Dependence on Gyromagnetic Ratio and Field Strength: The signal-to-noise ratio (S/N) in NMR is proportional to the gyromagnetic ratio (γ) of the nucleus and the external magnetic field strength (B₀). The relationship is often expressed as S/N ∝ γ⁵/²B₀³/² [2]. This is why insensitive nuclei like ¹³C are more challenging to detect than ¹H, and why higher-field magnets are used to improve sensitivity.
Table 1: Quantitative Comparison of NMR and UV-Vis Sensitivity
| Feature | NMR Spectroscopy | UV-Vis Spectroscopy |
|---|---|---|
| Population Ratio (Ground/Excited) | ~1.000064 [1] | ~1x10^42 [1] |
| Typical Population Excess | 32 per million spins [1] | ~1,000,000 per million spins [1] |
| Energy Transition | Nuclear spin states [2] | Electronic states [1] |
Q2: My solvent peaks are overwhelming my analyte signals. What are the primary suppression methods and when should I use them?
Solvent suppression is critical in bio-molecular NMR and metabolomics where aqueous solutions are common. The choice of method depends on your sample and the information you need [3].
- Presaturation (PreSat): This is the simplest method, involving a weak, continuous radiofrequency (rf) field applied at the solvent resonance frequency during the recycle delay. It is very effective but can partially saturate signals close to the solvent peak and is susceptible to saturation transfer to exchangeable protons (e.g., NH), especially at higher pH [3].
- Pulse Field Gradient Methods (e.g., WET, WATERGATE): These methods use magnetic field gradients to dephase solvent magnetization. WET is excellent for suppressing multiple solvent peaks simultaneously and is robust and easy to implement [4]. WATERGATE is highly effective and independent of lineshape, providing superior suppression without affecting exchanging protons [3].
- 1D NOESY-presat: This popular method in metabolomics combines a recovery delay with presaturation of the water resonance. It often provides an improved baseline and solvent suppression compared to simple presaturation, though newer gradient methods can be superior [3].
Table 2: Comparison of Common Solvent Suppression Techniques
| Method | Principle | Best Use Cases | Key Limitations |
|---|---|---|---|
| Presaturation | Continuous weak RF irradiation during delay [3] | Simple, quick experiments; non-exchangeable protons of interest [3] | Saturates nearby signals; affects exchangeable protons (NH, OH) [3] |
| WET | Combination of selective pulses and pulsed field gradients [4] | Suppressing multiple solvent peaks (e.g., DMF) [4] | Requires good shimming; may suppress very close solute peaks [3] |
| WATERGATE | Bipolar gradient pulses to dephase solvent coherence [3] | High-quality suppression; samples with exchangeable protons [3] | Suppresses signals very close to the water resonance [3] |
| 1D NOESY-presat | Presaturation combined with NOE mixing scheme [3] | Metabolomics studies where legacy spectral databases are used [3] | Can be less effective than newer gradient-based methods [3] |
Q3: How can I improve the signal-to-noise ratio and resolution of my spectra?
Optimizing S/N and resolution is a multi-faceted process involving both sample preparation and instrument operation.
- Ensure Proper Sample Preparation:
- Optimize Instrument Setup:
- Shim the magnet meticulously: Good shimming is paramount for high resolution and effective solvent suppression. The process involves adjusting the currents in shim coils to maximize the homogeneity of the magnetic field (B₀) across your sample. This can be done automatically (e.g.,
topshim) or manually by adjusting Z, Z², X, Y, etc., while observing the lock level rise [4] [6]. - Calibrate pulse widths: An incorrectly calibrated 90° pulse will reduce signal intensity. The 90° pulse is typically calibrated by finding the 180° pulse length (which gives a null or inverted signal) and dividing it by two [4].
- Use an appropriate receiver gain (RG): Set the RG as high as possible without causing ADC overflow errors [7].
- Shim the magnet meticulously: Good shimming is paramount for high resolution and effective solvent suppression. The process involves adjusting the currents in shim coils to maximize the homogeneity of the magnetic field (B₀) across your sample. This can be done automatically (e.g.,
Q4: I am using LC-SPE-NMR. How does this technique specifically address solvent interference and sensitivity?
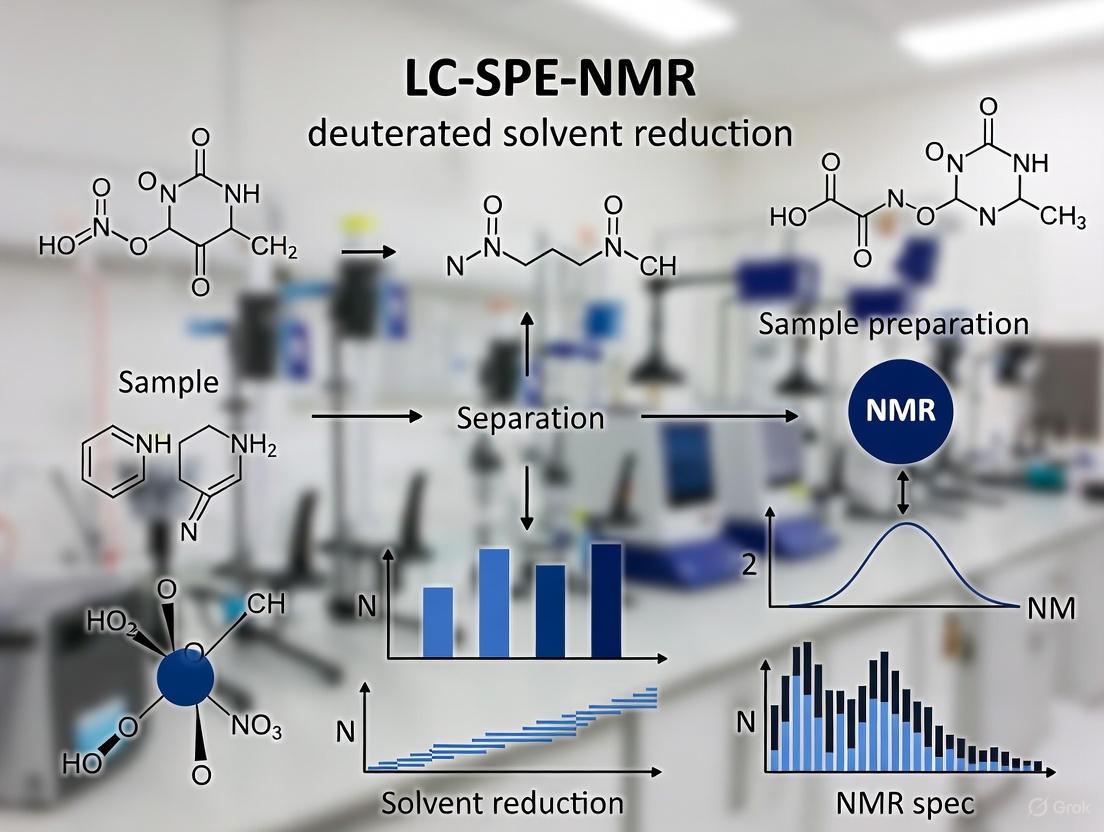
LC-SPE-NMR is a major advancement that directly tackles the core limitations stated in your thesis title by fundamentally changing how the analyte is presented to the NMR spectrometer [8].
- Deuterated Solvent Reduction: The technique uses non-deuterated, standard HPLC-grade solvents for the chromatographic separation. The analyte is trapped on an SPE cartridge and then eluted with a small, pure volume of deuterated solvent (e.g., ~300 µL of acetonitrile-d₃). This drastically reduces the consumption of expensive deuterated solvents and minimizes the residual solvent signal that needs to be suppressed [8].
- Sensitivity Enhancement via Analyte Focusing: The entire analyte from a potentially broad HPLC peak is trapped and focused into a very small volume on the SPE cartridge. When eluted, the entire sample is delivered to the NMR flow cell in a volume close to the detection volume of the probe. This eliminates the dilution that occurs in conventional LC-NMR, leading to a higher effective concentration and improved S/N [8].
- Multiple Trapping: A key feature for sensitivity is the ability to make multiple HPLC injections and trap the same analyte repeatedly on a single SPE cartridge. This pre-concentrates the analyte, substantially increasing the amount of material available for NMR analysis and enabling the acquisition of 2D NMR spectra on minor mixture components [8].
The following diagram illustrates the workflow of the LC-SPE-NMR technique, highlighting how it decouples the separation from the NMR analysis to overcome key limitations [8].
Experimental Protocols
Detailed Methodology: Implementing WET Solvent Suppression
The WET sequence is an effective method for suppressing multiple solvent peaks. The following steps outline how to set it up on a modern spectrometer running TopSpin [4].
Initial Setup and Shimming:
- Prepare your sample and insert it into the spectrometer.
- Lock and shim the sample to achieve optimal magnetic field homogeneity. Good shimming is critical for effective suppression [4].
- Acquire a standard ¹H spectrum to identify the solvent peaks you wish to suppress.
Define Suppression Regions:
- With the ¹H spectrum on the screen, go to the
Acquiremenu, selectOptions, and thenSetup Selective 1D Expts. - Click
Define Regions. Using the integration module, carefully select narrow regions around the center of each solvent peak you want to suppress. Avoid selecting wide regions to prevent suppressing your analyte signals. - Click
Save Region as...and selectSave regions to 'reg'. Then clickSave and Return[4].
- With the ¹H spectrum on the screen, go to the
Create the WET Dataset:
- Back in the
1D Selective Experiment Setupwindow, clickCreate Datasets. - Select
Mult. Solvent Suppr./WET. Enter the number of scans (NS, often 16 is sufficient) and a new experiment number. - A summary window will appear; click
Cancelto proceed with parameter adjustments [4].
- Back in the
Parameter Adjustment and Execution:
- Type
re [expno]to read the parameters of the new WET experiment. - Perform
atmato automatically tune and match the probe for both ¹H and ¹³C (WET suppresses ¹³C satellite peaks). - Increase the relaxation delay (
d1) to 6-10 seconds to ensure full recovery of your analyte magnetization, as WET uses 90° excitation pulses. - Type
rgato set the receiver gain, and thenzgto start the experiment [4].
- Type
Detailed Methodology: The LC-SPE-NMR Experiment
This protocol describes the general workflow for an HPLC-SPE-NMR experiment, which is central to research on deuterated solvent reduction [8].
Chromatographic Separation:
- Perform reversed-phase HPLC separation using standard, non-deuterated solvents (e.g., H₂O and acetonitrile).
- A post-column makeup pump is used to add at least a twofold volume of pure water to the eluate. This reduces the eluting power of the mobile phase, ensuring analytes are retained on the subsequent SPE cartridge [8].
Peak Trapping:
- As chromatographic peaks elute (triggered by UV or MS detection), the flow is directed to individual SPE cartridges (e.g., 2x10 mm or 1x10 mm).
- The analytes are adsorbed onto the SPE material while the diluted mobile phase passes through to waste [8].
Cartridge Processing:
- The SPE cartridges are dried with a stream of pressurized nitrogen gas to remove residual water and solvent.
- The trapped analytes are then eluted from the SPE cartridge using a small, pure volume of a suitable deuterated solvent (e.g., acetonitrile-d₃, typically ~300 µL) directly into the NMR flow cell [8].
NMR Acquisition:
- Once the focused analyte is in the NMR probe, standard 1D or 2D NMR experiments are acquired.
- Due to the analyte focusing and use of pure deuterated solvent, solvent suppression is often minimal or unnecessary, and sensitivity is significantly enhanced. For trace components, multiple trappings of the same peak from repeated HPLC injections can be performed to pre-concentrate the analyte [8].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Advanced NMR Experiments
| Item | Function | Application Notes |
|---|---|---|
| Deuterated Solvents (D₂O, CDCl₃, ACN-d₃) | Provides a lock signal for the spectrometer; defines chemical shift reference [5]. | ACN-d₃ is often preferred in LC-SPE-NMR for its elution strength and low viscosity [8]. |
| SPE Cartridges (e.g., 2x10 mm) | Solid-phase extraction material for trapping HPLC-separated analytes [8]. | Reversed-phase sorption mechanism; choice of phase is critical for trapping efficiency [8]. |
| Internal Standards (TMS, DSS) | Chemical shift reference compound [5]. | TMS for organic solvents; DSS for aqueous solutions [5]. |
| High-Quality NMR Tubes (5 mm) | Holds the sample within the magnetic field [5]. | Use tubes rated for high magnetic fields (≥500 MHz); imperfections degrade resolution [7] [5]. |
| Shigemi Tubes | Matches magnetic susceptibility of specific solvents to limit the active sample volume [2]. | Maximizes signal-to-noise for mass-limited samples by concentrating spins in the detection region [2]. |
A significant economic challenge in modern analytical laboratories, particularly those utilizing LC-NMR and LC-SPE-NMR platforms, is the prohibitive cost of deuterated mobile phases. These solvents are essential for NMR detection as they reduce overwhelming solvent signals that would otherwise obscure analyte signals [9]. However, the routine use of fully deuterated mobile phases is often financially unsustainable, with a single critical run using deuterated acetonitrile costing approximately $100 [9]. This technical support center provides practical solutions and methodologies for researchers seeking to reduce deuterated solvent consumption without compromising analytical capabilities.
Cost Analysis: Deuterated Solvents vs. Traditional Mobile Phases
Table 1: Cost Comparison of HPLC Mobile Phase Components
| Solvent Type | Approximate Cost | Key Application in LC-NMR | Economic Consideration |
|---|---|---|---|
| Deuterated Acetonitrile (CD₃CN) | >$1 per mL [9] | Organic modifier in fully deuterated mobile phases | Cost-prohibitive for routine analysis; used sparingly |
| Deuterated Methanol (CD₃OD) | >$1 per mL (comparable to CD₃CN) | Organic modifier in fully deuterated mobile phases | Similar cost constraints to CD₃CN |
| Deuterated Water (D₂O) | <$0.50 per mL [9] | Aqueous component in mobile phases | Relatively inexpensive; often the only deuterated component in mixed phases |
| Standard HPLC-grade Acetonitrile | Minimal cost relative to deuterated versions | Organic modifier in standard HPLC | Cost-effective but generates strong interfering signals in NMR |
| Standard HPLC-grade Methanol | Minimal cost relative to deuterated versions | Organic modifier in standard HPLC | Cost-effective but generates strong interfering signals in NMR |
Research Reagent Solutions for Cost-Effective Analysis
Table 2: Essential Materials for LC-SPE-NMR Solvent Reduction
| Item / Reagent | Function in Experiment | Role in Cost Reduction |
|---|---|---|
| SPE Cartridges (DVB-type polymers, RP-C18) [10] | Traps and concentrates analytes post-LC separation; enables solvent exchange | Eliminates need for deuterated mobile phases during LC separation |
| Deuterated NMR Solvents (CD₃OD, CD₃CN) [10] | Elutes analytes from SPE cartridges into NMR flow cell | Reduces volume required from ~mL/min flow to <1 mL total per analysis [10] |
| Microcoil NMR Probes [9] | NMR detection with small active volumes (as low as 1.5 μL) | Enables use of highly concentrated samples in minimal deuterated solvent |
| Capillary HPLC System [10] | Chromatographic separation with reduced flow rates | Minimizes total solvent consumption throughout separation process |
| Post-column makeup pump [10] | Adds water to promote analyte retention on SPE cartridges | Facilitates efficient trapping without deuterated solvents in mobile phase |
Experimental Protocols for Deuterated Solvent Reduction
Protocol 1: Standard LC-SPE-NMR Workflow for Solvent Reduction
Purpose: To eliminate deuterated solvents from the LC mobile phase while maintaining NMR compatibility through post-separation solvent exchange.
Materials Required:
- HPLC system with UV or MS detector
- SPE unit with appropriate cartridge (typically DVB-polymer or RP-C18)
- Makeup pump with supply of H₂O
- Deuterated NMR solvent (CD₃OD or CD₃CN)
- NMR spectrometer with flow probe
Procedure:
- HPLC Separation: Perform chromatographic separation using standard (non-deuterated) mobile phases to eliminate deuterated solvent costs during separation [10].
Analyte Trapping: After UV or MS detection, divert analyte peaks to SPE cartridges. Use a makeup flow of H₂O (1-2 mL/min) to promote analyte retention on the SPE stationary phase [10].
Solvent Exchange: Wash trapped analytes with D₂O or H₂O to remove residual non-deuterated HPLC mobile phase [10] [11].
Analyte Elution: Transfer analytes to the NMR flow cell by back-flushing the SPE cartridge with a small volume (<1 mL) of appropriate deuterated solvent (CD₃OD or CD₃CN) [10].
NMR Analysis: Acquire NMR data with the analyte now dissolved in a pure, well-defined deuterated solvent.
Protocol 2: Multiple Trapping for Sensitivity Enhancement
Purpose: To concentrate analytes from multiple HPLC runs for improved NMR sensitivity without increasing deuterated solvent consumption.
Materials Required:
- LC-SPE-NMR system capable of automated multiple injections
- SPE cartridges with high trapping capacity (e.g., GP-phase)
Procedure:
- Initial Trapping: Perform first HPLC separation and trap target analyte on SPE cartridge as described in Protocol 1.
Repeat Injections: Make additional sequential injections, trapping the same analyte peak on the same SPE cartridge [10].
Capacity Monitoring: Monitor trapping efficiency; some stationary phases can retain >100 μg of analyte through multiple trappings [10].
Consolidated Elution: After sufficient analyte accumulation, elute with deuterated solvent directly into NMR flow cell.
Advanced NMR: Use the concentrated sample to acquire heteronuclear NMR experiments (e.g., HSQC, HMBC) overnight [10].
Technical Support Center
Troubleshooting Guides
Table 3: LC-SPE-NMR Troubleshooting for Solvent Reduction Methods
| Problem | Potential Causes | Solutions |
|---|---|---|
| Poor analyte recovery from SPE | Incorrect SPE stationary phase; Inadequate makeup flow; Suboptimal elution solvent | Test different SPE phases (SAX, SCX for polar compounds) [10]; Optimize H₂O makeup flow rate [10]; Ensure deuterated solvent has sufficient elutropic power [10] |
| Inadequate NMR sensitivity | Insufficient analyte concentration; Excessive dilution in NMR flow cell; Probe limitations | Use multiple trapping to concentrate analyte [10]; Employ microcoil probes with small active volumes [9]; Consider cryoprobes for sensitivity enhancement [9] |
| Chromatographic peak broadening in SPE-NMR | Large elution volume; Poor focusing during transfer; Excessive tubing volume | Optimize SPE elution for narrow band formation [10]; Minimize tubing between SPE and NMR cell; Ensure elution volume matches NMR flow cell volume [10] |
| System compatibility issues | Communication errors between modules; Pressure fluctuations; Mobile phase incompatibility | Verify control software integration [10]; Check for leaks or blockages [11]; Ensure all solvents are miscible and compatible [11] |
Frequently Asked Questions (FAQs)
Q1: What is the primary economic benefit of implementing LC-SPE-NMR versus traditional LC-NMR? The primary economic benefit is the dramatic reduction in deuterated solvent consumption. While traditional LC-NMR requires continuous flow of deuterated mobile phases throughout chromatography, LC-SPE-NMR uses deuterated solvents only for the final elution step, reducing consumption to less than 1 mL per analyte [10]. This represents potentially >90% savings in deuterated solvent costs.
Q2: Can I completely eliminate deuterated solvents from my LC-NMR workflow? While complete elimination is challenging due to the fundamental requirements of NMR spectroscopy, LC-SPE-NMR significantly minimizes consumption. The technique allows you to use standard, non-deuterated mobile phases for the chromatographic separation, reserving small, precise volumes of deuterated solvents only for the final NMR analysis step [10].
Q3: What are the key considerations when selecting SPE cartridges for solvent reduction methods? The selection depends on your analyte characteristics. DVB-type polymers and RP-C18 phases work for most applications [10]. For polar or charged analytes like alkaloids or organic acids, consider specialized phases such as SAX (strong anion exchange), SCX (strong cation exchange), or porous carbon materials [10]. Multiple trapping efficiency varies significantly between phases, so empirical testing is recommended.
Q4: How does the multiple trapping technique improve both economics and data quality? Multiple trapping concentrates analyte from several chromatographic runs onto a single SPE cartridge, allowing you to accumulate sufficient material for advanced NMR experiments (such as 2D spectra) without increasing deuterated solvent volume [10]. This improves sensitivity and data quality while maintaining low solvent consumption, making sophisticated structural elucidation more economically feasible.
Q5: What alternative NMR technologies can help reduce operational costs? Microcoil NMR probes with active volumes as low as 1.5 μL enable analysis of highly concentrated samples in minimal deuterated solvent [9]. Additionally, cryogenically cooled probes (cryoprobes) can provide 2-4 fold sensitivity improvements, potentially reducing analysis time or required sample amounts [9].
Workflow Visualization
Troubleshooting Guides
FAQ: Addressing Common Solvent Suppression Issues
Q: Why is my solvent suppression inefficient, leading to poor analyte visibility?
A: Inefficient suppression is often due to magnetic field inhomogeneity or an incorrect suppression frequency. In a highly homogeneous magnetic field, all solvent nuclei have identical resonance frequencies and can be efficiently suppressed when excited at the correct frequency [12]. First, ensure proper shimming has been performed. The final B0 deviation should be below 1 Hz [7]. Second, verify that the suppression sequence is calibrated and targeted at the exact resonance frequency of the solvent peak.
Q: What causes poor magnetic field homogeneity (shimming) in my flowing system?
A: Poor shimming can result from several factors [7]:
- Incorrect Sample Properties: Ensure your sample volume is sufficient and that the solution is homogeneous. The presence of air bubbles or suspended solid particles will distort the magnetic field, causing broad lines and poor shimming.
- System Issues: For high-temperature experiments, ensure the sample has reached full thermal equilibrium before shimming. Fluctuations in air flow can also destabilize the field.
Q: I see an "ADC overflow" error during my experiment. What should I do?
A: An ADC overflow error is typically caused by the receiver gain (RG) being set too high [7]. This can result in poor quality spectra or a complete failure to collect data.
- Immediate Action: Type "ii restart" in your console to reset the hardware after the error occurs.
- Prevention: Manually set the RG to a value in the low hundreds, even if the automatic tuning ("rga") suggests a higher value. Always monitor the first scan of an experiment to ensure no error occurs.
Q: How can I handle samples dissolved in protonated solvents without extensive sample preparation?
A: Using pulsed solvent suppression methods, like the WET sequence, is the key. This technique uses selective frequency excitation to attenuate the large solvent signals before the NMR signal is acquired [12]. This avoids the tedious workup required for solvent exchange, allowing for rapid analysis of samples directly from a synthesis process.
Q: Why is my NMR tube difficult to shim, or does it not fit the spinner correctly?
A: This is likely an issue with NMR tube quality and specifications [7] [13].
- Tube Quality: Low-quality or "high-throughput" NMR tubes have greater variation in outer diameter, which can cause spinner fitment problems and are typically more difficult to shim.
- Solution: Use high-frequency NMR tubes (for spectrometers >=500 MHz) from reputable suppliers. For a tube that is slightly too loose, a temporary fix is to wrap a thin strip of Scotch tape around the tube where the spinner holds it.
Experimental Protocol: WET Solvent Suppression for Aqueous Samples
This protocol details the acquisition of a 1H NMR spectrum for a dilute analyte in a protonated solvent, using water as an example.
1. Principle The WET (Water Suppression Enhanced through T1 effects) sequence employs a series of selective pulses and pulsed magnetic field gradients to selectively saturate the intense solvent signal while minimally affecting the signals of interest from the analyte [12].
2. Materials and Sample Preparation
- Prepare your sample dissolved in a protonated solvent (e.g., neat water).
- While deuterated solvents are standard for locking, this method is designed for protonated solvents on systems with an external hardware lock [12].
- Ensure the sample is homogeneous and free of suspended particles to prevent magnetic field inhomogeneity [13].
3. Instrument Setup
- Calibrate Pulses: Ensure the selective pulses for the solvent suppression sequence are properly calibrated for the solvent's resonance frequency.
- Set Acquisition Parameters:
- Pulsed Field Gradients: Configure the gradient strengths and durations as defined by the WET sequence.
- Receiver Gain (RG): Set manually to a value in the low hundreds to prevent ADC overflow [7].
- Scans (NS): 64 scans or as required for sufficient signal-to-noise.
- Relaxation Delay (D1): 10 seconds to allow for magnetization recovery [12].
4. Data Acquisition and Processing
- Execute the WET solvent suppression sequence.
- After acquisition, process the FID with exponential line broadening (e.g., 0.3 Hz) and apply a Fourier transform.
- The solvent peak should be significantly attenuated (e.g., by a factor of 1000), revealing the analyte signals [12].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key materials and reagents essential for successful solvent suppression experiments, particularly within LC-SPE-NMR workflows focused on deuterated solvent reduction.
| Item | Function & Relevance to Solvent Suppression |
|---|---|
| Deuterated Solvents (e.g., DMSO-d6, CDCl3) | Provides a deuterium signal for the magnetic field lock, which is crucial for maintaining field stability and frequency reproducibility during long or complex experiments [13]. |
| WET Solvent Suppression Sequence | A pulse sequence that uses selective excitation and pulsed field gradients to attenuate large solvent signals. It is the key to analyzing samples in protonated solvents without extensive preparation [12]. |
| High-Quality NMR Tubes | Tubes with tight tolerances (e.g., Precision grade) ensure consistent spinning and are easier to shim, which is a prerequisite for effective solvent suppression [7] [13]. |
| Internal Standard (e.g., TMS, DSS) | Provides a reference peak for chemical shift calibration (δ = 0 ppm). The concentration must be very low to avoid dynamic range issues that can distort the baseline [13]. |
| Benchtop NMR with External Lock | Systems like the Spinsolve ULTRA with an external hardware lock enable the analysis of samples in non-deuterated solvents, as they do not rely on the sample's deuterium signal for field stabilization [12]. |
Workflow Visualization
Solvent Suppression Workflow
LC-SPE-NMR Solvent Reduction
Technical Support Center
This support center provides targeted troubleshooting guides and FAQs to assist researchers in overcoming common challenges in LC-SPE-NMR workflows, with a special focus on methods that reduce deuterated solvent consumption.
Troubleshooting Guides
Table 1: Common LC-SPE-NMR Issues and Solutions
| Problem Area | Specific Issue | Possible Cause | Recommended Solution |
|---|---|---|---|
| NMR Sensitivity | Poor Signal-to-Noise Ratio | Inadequate analyte concentration [9]. | Use LC-SPE to concentrate samples; employ cryoprobes or microcoil probes [9]. |
| Solvent Management | High cost of deuterated solvents [9]. | Using fully deuterated mobile phases. | Use LC-SPE for post-column analyte trapping; elute with minimal volume of deuterated solvent [9] [14]. |
| Chromatography | Retention time shift in LC-NMR | Deuterium isotope effect when using D2O in the mobile phase [9]. | Account for retention time shifts in method development; consider LC-SPE to decouple LC from NMR. |
| System Operation | ADC overflow error on NMR spectrometer |
Receiver gain (RG) set too high [7]. | Set RG to a low hundreds value; type ii restart to reset hardware after error [7]. |
| System Operation | Poor shimming results | Inhomogeneous sample, air bubbles, or poor-quality NMR tube [7]. | Ensure sufficient sample volume and deuterated solvent; use rsh to read a good prior shim file and rerun topshim [7]. |
Frequently Asked Questions (FAQs)
Q1: Why is LC-SPE-NMR considered a major evolution from direct LC-NMR? LC-SPE-NMR addresses the primary limitation of direct on-flow or stop-flow LC-NMR: the inherent low sensitivity of NMR. By trapping chromatographic peaks on solid-phase extraction cartridges, analytes can be concentrated and then eluted with a small, defined volume of deuterated solvent. This process significantly enhances analyte concentration for NMR detection and drastically reduces the consumption of expensive deuterated solvents [9] [14].
Q2: My NMR experiment failed with an automation error on the Bruker spectrometer. What are the first steps I should take?
Stop the automation in IconNMR. In the TopSpin command line, type ii to reinitialize the system. Run this command a few times until no error messages appear. You can then try to manually tune and match the probe (atmm). If successful, restart the automation in IconNMR. If errors persist, restarting TopSpin may be necessary [15].
Q3: Can LC-MS data alone replace the need for LC-NMR or LC-SPE-NMR in structural elucidation? No. LC-MS and LC-NMR provide complementary data. While LC-MS is highly sensitive and provides molecular weight and elemental composition, it often cannot distinguish between isobaric compounds or positional isomers. NMR is essential for providing definitive structural information, including atomic connectivity and stereochemistry. The techniques are synergistic, with LC-MS often used for initial screening and dereplication, and LC-SPE-NMR for complete structure determination [9] [14].
Q4: What is the single biggest factor limiting the sensitivity of online LC-NMR, and how does LC-SPE-NMR mitigate it? The key limiting factor is the low sensitivity of the NMR experiment itself, which requires relatively high concentrations of analyte and long acquisition times. This stems from the small energy difference between nuclear spin states [9]. LC-SPE-NMR mitigates this by trapping and concentrating HPLC peaks, effectively increasing the analyte concentration presented to the NMR probe, which improves the signal-to-noise ratio and reduces the required acquisition time [9] [14].
Experimental Protocols
Detailed Methodology: LC-SPE-NMR for Natural Product Identification
The following protocol, adapted from research on plant secondary metabolites, outlines a standard workflow for analyzing complex plant extracts using LC-SPE-NMR with optimized deuterated solvent use [14].
1. Sample Preparation
- Extraction: Homogenize plant material (e.g., 50-300 mg) and extract with an appropriate solvent. Methanol or methanol-deuterium oxide mixtures are often effective for broad metabolite coverage [16].
- Filtration: Centrifuge the extract and filter the supernatant through a 0.45 µm membrane filter prior to LC injection to prevent column clogging.
2. Liquid Chromatography Separation
- Column: Use a suitable reversed-phase C18 column.
- Mobile Phase: Employ a binary solvent system. To reduce costs, the aqueous phase can be deuterium oxide (D2O), while the organic phase (e.g., acetonitrile or methanol) is used in its protonated form [9].
- Gradient: Optimize a linear gradient for compound separation. The effluent is split post-column, with a minor portion directed to the mass spectrometer and the majority to the SPE unit.
3. Mass Spectrometry Detection
- The split flow is analyzed by MS using electrospray ionization (ESI).
- MS data acquired in real-time is used to make decisions on which chromatographic peaks to trap based on their mass and fragmentation pattern.
4. Solid-Phase Extraction (SPE) Trapping
- Peak Triggering: The system is programmed to trigger a trapping event based on UV or MS signal thresholds.
- Trapping: The HPLC peak is diluted with water to reduce eluting strength and loaded onto a conditioned SPE cartridge (e.g., Hysphere C18 cartridge). Hydrophilic compounds are adsorbed onto the sorbent.
- Drying: An inert gas stream (e.g., nitrogen) passes through the cartridge to remove the protonated solvents completely.
5. NMR Analysis
- Elution: The trapped analyte is eluted from the SPE cartridge directly into the NMR flow cell using a small, precise volume (e.g., 30-150 µL) of deuterated solvent (e.g., CD3OD or [D6]DMSO).
- Data Acquisition: Once the analyte is transferred, standard 1D and 2D NMR experiments (e.g., 1H, COSY, HSQC, HMBC) are performed. The use of cryoprobes or microcoil probes is recommended to maximize sensitivity with the low-volume, concentrated sample [9].
Workflow Visualization
LC-SPE-NMR Workflow
Research Reagent Solutions
Table 2: Essential Materials for LC-SPE-NMR Experiments
| Item | Function / Rationale |
|---|---|
| Deuterated Methanol (CD3OD) | Common elution solvent in LC-SPE-NMR; offers good solubility for a wide range of mid-polarity metabolites. Using it sparingly for final elution is key to cost reduction [9] [16]. |
| Deuterium Oxide (D2O) | Relatively inexpensive deuterated solvent often used as the aqueous component of the LC mobile phase to reduce solvent signal interference in NMR [9]. |
| C18 Reversed-Phase SPE Cartridges | The solid-phase medium used to trap, wash, and concentrate analytes of interest after LC separation, enabling the switch from protonated to deuterated solvents [14]. |
| Methanol (HPLC Grade) | Common protonated solvent for sample extraction and as the organic modifier in the LC mobile phase. 90% methanol with 10% CD3OD can be an effective extraction solvent for NMR [16]. |
| Cryoprobes / Microcoil NMR Probes | Sensitivity-enhanced NMR probes. Cryoprobes reduce electronic noise, while microcoil probes work efficiently with low-volume samples, both crucial for analyzing the low-quantity samples typical in LC-SPE-NMR [9]. |
The LC-SPE-NMR Workflow: A Practical Guide to Efficient Solvent Reduction
Troubleshooting Guide: Resolving Common Issues
Why is my chromatographic separation poor when I use non-deuterated solvents as a precursor to LC-NMR?
Poor separation can stem from various factors unrelated to your solvent choice. The table below outlines common symptoms, their causes, and solutions.
| Symptom | Potential Cause | Solution |
|---|---|---|
| Broad Peaks [17] | System not equilibrated; Injection solvent too strong; Column overload (volume or mass). | Equilibrate column with 10 volumes of mobile phase; Ensure injection solvent is same/weaker strength than mobile phase; Reduce injection volume or sample concentration [17]. |
| Tailing Peaks [17] | Column degradation (old, contaminated, or voided); Injection solvent too strong. | Replace guard cartridge or column; Wash contaminated column; Use a weaker injection solvent [17]. |
| Varying Retention Times [17] | System not equilibrated; Temperature fluctuations; Pump not mixing solvents properly. | Fully equilibrate column; Use a thermostatically controlled column oven; Check pump proportioning valve function [17]. |
| Extra Peaks [17] | Degraded sample; Contaminated solvents or column; "Ghost peaks" from gradient elution. | Inject a fresh sample; Use fresh, high-quality HPLC solvents; Replace guard cartridge; Wash the column [17]. |
How can I manage the strong solvent signal from non-deuterated solvents in NMR analysis?
The intense signal from protonated solvents can overwhelm the weaker signals from your analyte. The primary solution is to use a pulse sequence that suppresses solvent signals [18].
- Technology Used: The WET pulse suppression sequence is an effective method integrated into modern NMR software (e.g., JEOL's No-D NMR) [18].
- How It Works: This technique applies specific radiofrequency pulses to selectively excite and suppress the hydrogen signals from the solvent, allowing the much smaller analyte signals to be detected clearly.
- Consideration: A potential limitation is that analyte signals that overlap with the solvent peak may also be suppressed [18].
What should I do if my NMR system fails to "lock" without deuterated solvent?
Traditional NMR spectrometers use the deuterium signal from the solvent for a process called "field/frequency locking" to maintain magnetic field stability [19].
- Solution: Newer techniques like No-D NMR (No-Deuterium Proton NMR) bypass this requirement. Instead of locking on deuterium, the system performs proton gradient shimming using the strong signal from the protonated solvent itself to stabilize the magnetic field [18]. This makes deuterated solvents unnecessary for the locking process.
Frequently Asked Questions (FAQs)
Can I use any non-deuterated solvent for LC-SPE-NMR?
While you have more flexibility, the choice is not arbitrary. You should select solvents based on:
- Solubility: The solvent must fully dissolve your sample [19].
- Chromatographic Performance: It should provide good separation efficiency (e.g., sharp peaks) [17].
- NMR Signal Interference: The solvent's proton signals should not critically overlap with your analyte's key resonances. Careful solvent selection can minimize this issue [18].
- Compatibility with SPE: The solvent must be compatible with the solid-phase extraction process, which typically uses non-deuterated solvents before the analyte is eluted with a minimal volume of deuterated solvent for the final NMR step [20].
What are the main cost benefits of using non-deuterated solvents?
The primary benefit is a significant reduction in solvent expenses. Deuterated solvents like Acetonitrile-d₃ can cost over $1.00 per mL, whereas their protonated equivalents are orders of magnitude cheaper [9]. For the initial chromatographic separation and SPE trapping steps, which consume the bulk of the solvent, using standard HPLC-grade solvents leads to substantial cost savings without compromising the quality of the separation or the structural information obtained from NMR [20].
How does the LC-SPE-NMR workflow integrate with non-deuterated solvents?
This workflow is specifically designed to minimize the consumption of expensive deuterated solvents [20]. The process is visualized in the diagram below.
Diagram Title: LC-SPE-NMR Solvent Reduction Workflow
- Separation: The mixture is first separated using a standard LC system with non-deuterated solvents [20].
- Trapping: As peaks elute from the column, they are directed and adsorbed onto Solid-Phase Extraction (SPE) cartridges [20].
- Drying: The trapped analytes on the SPE cartridge are dried, typically with a stream of nitrogen gas, to evaporate the non-deuterated solvents [20].
- Elution: The purified analyte is then eluted from the SPE cartridge using a small, controlled volume of deuterated solvent directly into the NMR spectrometer for analysis [20].
Research Reagent Solutions
The table below lists key materials used in the LC-SPE-NMR workflow with non-deuterated solvents.
| Item | Function in the Experiment |
|---|---|
| HPLC-Grade Solvents (e.g., Acetonitrile, Methanol) [20] | The mobile phase for the initial liquid chromatography separation; chosen for their purity and optimal chromatographic performance. |
| Solid-Phase Extraction (SPE) Cartridges [20] | To trap, concentrate, and purify analyte peaks after LC separation, enabling the removal of non-deuterated solvents. |
| Deuterated Solvent (minimal volume) [20] | To elute the purified analyte from the SPE cartridge for the final NMR analysis, providing the deuterium signal for the lock system in standard NMR. |
| Inert Gas (N₂) [20] | To dry the SPE cartridges after trapping the analyte, ensuring complete removal of non-deuterated solvents before NMR elution. |
The following diagram illustrates the complete post-column analyte trapping and focusing process in LC-SPE-NMR.
Troubleshooting Guide
Common Problems and Solutions
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| Low Analyte Recovery | • Eluent strength insufficient• Elution volume too small• Wrong sorbent chemistry | • Increase organic modifier percentage• Use stronger elution solvent• Increase elution volume in increments [21] |
| Poor Reproducibility | • Variable flow rates• Cartridge bed dried out pre-load• Wash solvent too strong | • Control flow to 1-2 mL/min [21]• Keep sorbent wet; re-equilibrate if dry [21]• Weaken wash solvent strength [21] |
| Unsatisfactory Cleanup | • Incorrect purification strategy• Poor solvent selection | • Retain analyte, wash impurities [21]• Re-optimize wash/elution conditions [21] |
| Slow/Variable Flow Rate | • Particulate clogging• High sample viscosity | • Filter/centrifuge sample pre-load [21]• Dilute sample to lower viscosity [21] |
| Failure to Trap Polar Compounds | • Reversed-phase sorption mechanism limitation | • Dilute HPLC eluate with water (2:1) [8]• Explore polar stationary phases [8] |
Frequently Asked Questions (FAQs)
Q1: Why is post-column dilution with water necessary before SPE trapping? Post-column dilution with water decreases the concentration of the organic modifier in the HPLC eluent, reducing its eluting power. This increases the affinity of analytes for the reversed-phase SPE stationary phase, ensuring they are retained on the cartridge instead of passing through to waste [8].
Q2: What is the purpose of drying the SPE cartridge with nitrogen gas before elution? Drying the cartridge with pressurized nitrogen gas removes residual water and the non-deuterated HPLC solvents [8]. This is a critical step to prevent contamination of the final NMR sample with protonated solvents, which would require strong solvent suppression and compromise spectral quality.
Q3: How does this step contribute to the overall sensitivity of LC-SPE-NMR? This step focuses the analyte. The analyte from a potentially broad HPLC peak is trapped and concentrated onto a small SPE cartridge (e.g., with a bed volume of ~8-30 µL) and then eluted with a minimal volume (e.g., <30 µL) of deuterated solvent. This "analyte focusing" results in a much higher sample concentration in the NMR flow cell compared to the original HPLC peak volume [8].
Q4: Can I increase the amount of analyte for NMR analysis? Yes, a key advantage of this method is "multiple trapping." The same analyte from repeated HPLC separations can be trapped onto the same SPE cartridge, significantly increasing the amount of material available for NMR analysis and allowing for the acquisition of more time-consuming 2D NMR experiments [8].
Q5: My very polar analyte is not being trapped. What can I do? The reversed-phase sorption mechanism used in most current HPLC-SPE-NMR setups has a serious limitation in trapping very polar compounds [8]. Potential solutions include optimizing the post-column dilution ratio or, in the future, exploring the use of polar stationary SPE phases, though this is an area requiring further development [8].
Research Reagent Solutions
Essential Materials for Post-Column Trapping
| Item | Function & Key Characteristics |
|---|---|
| SPE Cartridges | Small cartridges (e.g., 1x10 mm or 2x10 mm) with reversed-phase sorbent (e.g., C18) to trap and focus analytes [8]. |
| Deuterated Elution Solvents | High-purity solvents (e.g., Acetonitrile-d₃, Methanol-d₄, Chloroform-d) to quantitatively transfer analyte to NMR tube/probe with minimal signal interference [8] [19]. |
| Post-Column Pump | Delivers a precise flow of pure water to dilute the HPLC eluent, reducing its eluting power and enabling analyte retention on the SPE cartridge [8]. |
| Nitrogen Gas Supply | Provides pressurized gas for drying the SPE cartridges after trapping to remove residual protonated solvents [8]. |
| Automated SPE Interface | System that coordinates trapping triggered by UV/MS, cartridge drying, and elution with deuterated solvent into the NMR flow cell or tube [8]. |
Frequently Asked Questions (FAQs)
Q1: Why is efficient drying of the SPE cartridge critical before solvent exchange? Efficient drying with pressurized nitrogen gas is essential to remove all traces of the non-deuterated HPLC mobile phase (e.g., H₂O, acetonitrile, methanol). Residual protons from these solvents would cause significant interference in the subsequent NMR analysis, leading to large unwanted peaks that require solvent suppression and can obscure analyte signals [10] [8].
Q2: What are the consequences of incomplete solvent exchange on my NMR spectrum? Incomplete exchange results in a mixed solvent system within the NMR flow cell. This leads to multiple, strong solvent peaks, complicated solvent suppression routines, and a increased risk of signal overlap with your analyte of interest. A successful exchange to a pure deuterated solvent provides a well-defined and reproducible NMR environment, simplifying spectral interpretation [10] [8].
Q3: My analyte is not eluting efficiently from the SPE cartridge with the deuterated solvent. What could be wrong? The elution power of the deuterated solvent might be insufficient. Acetonitrile-d₃ is a general-purpose, non-viscous solvent that often provides good results. For more non-polar compounds, chloroform-d can be effective, while methanol-d₄ is better for polar compounds. The strong coordinating properties of DMSO-d₆ can make it difficult to elute analytes as a sharp band, which is why it is less commonly used in online SPE-NMR workflows [8].
Q4: I suspect water contamination in my final sample. How can I confirm and fix this? Water peaks are a common issue. If you observe a large peak for water in your NMR spectrum, it could originate from wet solvent or incomplete drying of the SPE cartridge. To prevent this, ensure your deuterated solvents are anhydrous and that the nitrogen drying step is thorough. NMR solvents can also collect water over time; adding an inert drying agent like molecular sieves to your solvent bottle can help [22] [8].
Q5: How does multiple trapping enhance sensitivity and reduce deuterated solvent use? Multiple trapping involves repeatedly injecting and concentrating the same analyte from successive HPLC runs onto a single SPE cartridge. This strategy accumulates microgram quantities of the analyte, allowing for the acquisition of 2D NMR experiments without overloading a single chromatographic run. Since the entire accumulated sample is eluted with a single, small volume (e.g., < 30 µL) of deuterated solvent, the technique maximizes the analyte concentration in the NMR flow cell while minimizing per-analysis solvent consumption [10] [8].
Troubleshooting Guide
| Problem | Possible Cause | Solution |
|---|---|---|
| Poor NMR signal-to-noise ratio after elution | • Analyte not fully transferred from SPE cartridge• Elution volume too large, diluting the analyte | • Optimize deuterated solvent elution strength [8]• Ensure cartridge size matches NMR cell volume for focused elution [8] |
| Large solvent peaks in NMR spectrum | • Incomplete drying of SPE cartridge• Use of non-deuterated solvent for elution | • Extend nitrogen drying time to remove all protonated solvent [8]• Use pure, high-quality deuterated solvents [19] |
| Analyte recovery is low | • Analyte is too polar for reversed-phase SPE material• Wrong deuterated solvent for elution | • Consider alternative SPE phases (SAX, SCX) for polar analytes [10]• Test different deuterated solvents (CD₃CN, CD₃OD) for optimal elution [8] |
| Broad or distorted peaks in NMR | • Poor magnetic field shimming due to improper sample• Sample is not homogeneous | • Ensure sample is fully dissolved and the cartridge is properly dried [7]• Check that the eluted sample forms a homogeneous solution in the flow cell [22] |
Research Reagent Solutions: Essential Materials
The following table details key reagents and materials essential for the efficient drying and solvent exchange process in LC-SPE-NMR.
| Item | Function in the Protocol |
|---|---|
| SPE Cartridges | The solid-phase medium that traps HPLC-separated analytes. Common types include reversed-phase (C-18, DVB polymer) for most applications, and ion-exchange (SAX, SCX) for charged/polar compounds [10] [8]. |
| Pressurized Nitrogen Gas | An inert gas stream used to dry the SPE cartridge thoroughly after trapping, removing all residual protonated HPLC solvent before elution with deuterated solvent [10] [8]. |
| Deuterated Acetonitrile (CD₃CN) | A preferred elution solvent due to its low viscosity, good eluting power for many compounds, and well-defined residual solvent peak at ~1.94 ppm [8] [19]. |
| Deuterated Methanol (CD₃OD) | A protic deuterated solvent used for eluting more polar compounds. Its residual proton peak is found at ~3.31 ppm [8] [19]. |
| Deuterated Chloroform (CDCl₃) | A standard NMR solvent suitable for eluting non-polar compounds from SPE cartridges. Its residual proton peak is a singlet at 7.26 ppm [8] [19]. |
| Deuterium Oxide (D₂O) | Used as the aqueous component in the HPLC mobile phase and sometimes as a post-column makeup fluid to promote analyte retention on the SPE cartridge [10] [9]. |
Experimental Workflow and Protocol
The following diagram illustrates the key steps for efficient drying and solvent exchange in the LC-SPE-NMR process.
Detailed Protocol for Optimized Solvent Exchange
SPE Cartridge Drying:
- After the target analyte is trapped on the SPE cartridge from the HPLC eluent, activate the nitrogen drying system.
- The drying time must be sufficient to ensure all traces of the protonated mobile phase (e.g., H₂O, CH₃CN, CH₃OH) are evaporated from the cartridge. Inadequate drying is a primary source of large contaminant peaks in the NMR spectrum [8].
Selection of Deuterated Solvent:
- Choose a deuterated solvent based on its elution strength for your analyte and its suitability for NMR. Acetonitrile-d₃ (CD₃CN) is highly recommended as a first choice due to its low viscosity and general effectiveness [8].
- Other common choices are methanol-d₄ (CD₃OD) for polar compounds and chloroform-d (CDCl₃) for non-polar compounds [8] [19].
Elution and Transfer:
- Use the minimal volume of deuterated solvent required to quantitatively elute the analyte from the SPE cartridge as a narrow band. This volume is typically designed to match the active volume of the NMR flow probe (e.g., 30-60 µL for a 30-60 µL flow cell) [10] [8].
- This "peak focusing" is the key to maximizing analyte concentration and sensitivity while conserving expensive deuterated solvent.
Deuterated Solvent Comparison
The table below provides a quantitative comparison of common deuterated solvents used in LC-SPE-NMR elution, highlighting key properties for selection.
| Solvent | Typical Residual ¹H Peak (ppm) | Boiling Point (°C) | Relative Cost | Best Use Case in SPE Elution |
|---|---|---|---|---|
| Acetonitrile-d₃ (CD₃CN) | 1.94 [19] | 82 [19] | Medium | General purpose; excellent for sharp elution bands due to low viscosity [8]. |
| Methanol-d₄ (CD₃OD) | 3.31 [19] | 65 [19] | Medium | Polar compounds that require a protic solvent for elution [8]. |
| Chloroform-d (CDCl₃) | 7.26 [19] | 61 [19] | Low | Non-polar organic compounds [8]. |
| Dimethyl Sulfoxide-d₆ (DMSO-d₆) | 2.50 [19] | 189 [19] | High | Problematic polar compounds; less ideal for online elution due to high viscosity [8] [19]. |
Troubleshooting Guide: Common Issues and Solutions
This guide addresses specific issues that can occur during the elution and data acquisition phase of an LC-SPE-NMR experiment, with a focus on maintaining data quality while minimizing deuterated solvent consumption.
Symptom 1: High Baseline Noise or Drift in the NMR Spectrum
| Symptom | Possible Cause | Solution |
|---|---|---|
| Erratic baseline noise [23] | Air bubble in the LC system or NMR flow cell. | Purge the entire system, including the LC pump and the NMR flow cell, with fresh, degassed mobile phase. |
| Regular, cyclic noise pattern in the baseline [24] [23] | Inconsistent pump operation causing fluctuations in mobile phase composition or flow rate. | Perform routine maintenance on pump check valves and seals. Verify that the deuterated solvent composition is consistent and correctly mixed. |
| Overall bad baseline with high noise [23] | General system contamination. | Perform a thorough system cleaning, flushing both the LC and NMR flow path with appropriate solvents. For UV detectors, a noisy baseline can also indicate a failing lamp or flow cell [23]. |
| Baseline drift during gradient elution [24] | Detector response to a major mobile phase component (e.g., formic acid absorbing strongly at low UV wavelengths). | Ensure the additive is present in both solvent A and B to maintain a consistent concentration, or use a detection wavelength where the additive does not absorb. |
Symptom 2: Poor Peak Shape or Anomalous Peaks in the NMR Spectrum
| Symptom | Possible Cause | Solution |
|---|---|---|
| Peak tailing [23] | - Column overloading- Contamination- Interactions with active sites on the trapping column | - Dilute the sample or reduce the injection volume.- Flush the SPE cartridge and analytical column with strong solvent.- Ensure the trapping chemistry is compatible with your analyte. |
| "Ghost peaks" (peaks not from the analyte) [24] [25] | Elution of highly retained impurities from the mobile phase or system. | Use high-purity, LC-MS grade solvents and additives. Run a blank gradient to identify and flush out impurities. In one case, changing the supplier of isopropanol immediately resolved the issue [24]. |
| Artifact peaks from contamination [25] | Leachables from system components (e.g., septa, tubing) contacting the sample or solvent. | Identify and replace the source of contamination. One study found artifact peaks originated from the sample vial septum [25]. |
| Broadened or split peaks [23] | - Incompatibility between sample solvent and initial mobile phase.- Excessive system volume. | - Dilute the sample in a solvent that matches the initial mobile phase composition.- Use shorter, narrower internal diameter (I.D.) tubing to connect the LC-SPE to the NMR flow cell to reduce post-column volume. |
Symptom 3: Reduced Sensitivity or Signal-to-Noise Ratio
| Symptom | Possible Cause | Solution |
|---|---|---|
| Low signal intensity across all peaks [23] | - Incorrect sample concentration or injection volume.- Analyte adsorption to active sites. | - Verify sample preparation steps and dilution calculations.- Condition the system with a few preliminary injections to passivate active sites. |
| Poor magnetic field homogeneity (line shape) | Inadequate shimming due to improper sample conditions or unstable lock signal. | Ensure the deuterated solvent used for elution is of high isotopic purity (≥99.8%) to provide a strong and stable lock signal for the spectrometer [19]. |
| Overwhelmed NOE effects or "Oversaturation" [26] | Use of overly intense RF saturation pulses during NMR experiments, which can smear out cross-relaxation information. | Optimize the amplitude (γB1/2π) of the saturation pulse. Start with weaker values (e.g., ~10 Hz) and increase only as needed to avoid broadening the signals of interest [26]. |
Frequently Asked Questions (FAQs)
Q1: What are the most critical factors for selecting a deuterated solvent for the final elution in LC-SPE-NMR? The key factors are solubility compatibility (the solvent must fully dissolve your target analyte from the SPE cartridge), chemical compatibility (the solvent should not cause unwanted proton exchange or chemical shifts), and deuterium purity. High isotopic purity (≥99.8%) is crucial for a stable field/frequency lock and for minimizing the large solvent peak that can interfere with your spectrum [19].
Q2: Why might I see unexpected "cross-peaks" in my NOE or SMT NMR spectra after elution? Unexpected cross-peaks can be artifacts from "spill-over" effects. This occurs when a long or intense selective saturation pulse unintentionally affects the signals of nearby protons, making them appear as if they are cross-relaxing [26]. To avoid this, ensure your saturation pulse parameters (power, duration, and frequency) are correctly calibrated for your specific sample and isolated peak.
Q3: How can I reduce deuterated solvent consumption in my LC-SPE-NMR workflow without compromising data quality? Optimizing the elution volume is essential. Use the minimum volume of deuterated solvent needed to quantitatively transfer the analyte from the SPE cartridge to the NMR flow cell. This can be determined experimentally during method development. Furthermore, using a solvent with a high boiling point (like DMSO-d₆) can be beneficial if you need to recover your sample, but it may be harder to remove from the system later [19].
Q4: The baseline in my NMR spectrum is unstable after elution. What LC-related issues should I investigate? This often points to mobile phase inconsistencies. Check that your pump is delivering a consistent flow and that your solvent composition is stable. A failing pump seal or a sticky check valve can cause composition fluctuations that manifest as baseline noise or drift in the NMR detector [24] [23]. Always use high-purity solvents and additives to minimize chemical baseline contributions.
Experimental Protocol: Optimizing the Elution-to-Acquisition Workflow
The following protocol is designed to ensure a robust transfer of the isolated analyte into the NMR flow cell for high-quality data acquisition.
Objective: To reliably elute a target compound from an SPE cartridge using a minimal volume of deuterated solvent and acquire a high-fidelity NMR spectrum.
Materials and Reagents:
- LC-SPE system with appropriate switching valves
- NMR spectrometer with a flow probe (flow cell)
- High-purity deuterated elution solvent (e.g., CD₃CN, CD₃OD, DMSO-d₆)
- Standard sample for system performance verification
Procedure:
- System Priming: Pre-condition the entire fluidic path from the SPE cartridge to the NMR flow cell with the selected deuterated elution solvent. This removes any residual protonated solvent and ensures a homogeneous environment for the analyte.
- Elution Trigger: Activate the method step that switches the valve to direct the flow of deuterated solvent through the SPE cartridge. The flow rate and elution volume should be optimized to ensure quantitative transfer of the analyte. A typical flow rate is 0.5 - 1.0 mL/min.
- Transfer to Flow Cell: The eluted band, now in deuterated solvent, is transferred directly into the NMR flow cell. The transfer tubing should be as short and narrow in I.D. as possible to minimize peak broadening [23].
- NMR Probe Tuning: Once the elution peak is detected as being within the flow cell (often via a UV signal), stop the flow. Manually or automatically tune and match the NMR probe to the sample/solvent combination.
- Magnetic Field Lock and Shimming: Engage the deuterium field-frequency lock using the signal from the deuterated solvent. Perform automated shimming (e.g., gradient shimming) to optimize the magnetic field homogeneity for the best possible spectral line shape.
- Data Acquisition: Begin the NMR experiment (e.g., 1D ( ^1H )) using parameters optimized for your sample. For NOE-based experiments, carefully calibrate saturation power and duration to avoid artifacts like "oversaturation" [26].
- System Cleanup: After acquisition, flush the system thoroughly with a protonated solvent (e.g., acetonitrile/water) to prepare for the next run and to conserve expensive deuterated solvents.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in LC-SPE-NMR | Key Consideration |
|---|---|---|
| Deuterated Elution Solvents (CD₃CN, CD₃OD, DMSO-d₆) [19] | Dissolves and transfers the analyte from the SPE cartridge to the NMR flow cell while providing a deuterium signal for the field-frequency lock. | Select based on analyte solubility and chemical compatibility. Higher deuterium purity (≥99.8%) provides a more stable lock and cleaner baseline [19]. |
| SPE Cartridges | Traps and concentrates the target compound from the LC mobile phase, allowing for solvent exchange to deuterated NMR solvent. | The stationary phase must be orthogonal to the LC column to ensure effective trapping and must be compatible with the deuterated elution solvent. |
| LC-MS Grade Solvents & Additives [24] [23] | Used in the initial LC separation and SPE trapping steps to minimize UV-absorbing or NMR-active impurities that cause ghost peaks or high baseline. | Essential for preventing contamination that can be concentrated on the SPE cartridge and eluted into the NMR, complicating spectral interpretation. |
| NMR Reference Compound (e.g., TMS) [27] | Provides a known internal standard (0 ppm) for precise chemical shift referencing in the final NMR spectrum. | Can be added in small quantities to the deuterated elution solvent if needed, though residual solvent peaks are often used as a secondary reference. |
LC-SPE-NMR Elution & Acquisition Workflow
The diagram below illustrates the core pathway and key decision points for transitioning a sample from the solid-phase extraction (SPE) cartridge to high-quality NMR data acquisition.
Troubleshooting Guides
HPLC Method Development for Impurity Profiling
Problem: Inadequate separation of impurities from the main drug substance. Separation is the foundation of reliable impurity profiling. Poor selectivity can lead to co-elution, making accurate quantification impossible.
- Diagnosis: Closely eluting peaks or peaks with shoulder(s) observed in the chromatogram; spectral purity analysis (PDA) indicates potential co-elution [28].
- Solution:
- Select Dissimilar Chromatographic Columns: Screen the impurity mixture on 4-5 reversed-phase columns with different selectivity (e.g., C18, phenyl, cyano). Use chemometric approaches or column characterization parameters to select the most orthogonal columns [29].
- Optimize Mobile Phase pH: Screen a pH range of 2–9 (within column stability limits) in combination with the selected columns. Model the retention time (tR) of each impurity as a function of pH to predict the pH that provides the maximum minimal resolution (Rsmin) between consecutive peaks [29].
- Reconsider Gradient Slope: If the overall Rsmin remains low after pH optimization, adapt the gradient's start and end conditions or the gradient time to spread the peaks over a wider retention window [29].
- Prevention: Follow a structured method development approach, sequentially optimizing the stationary phase, pH, organic modifier, and temperature based on their influence on selectivity [29].
Problem: Drifting retention times and peak shape issues. Retention time stability is critical for reproducible results and accurate peak identification.
- Diagnosis: Consistent drift in retention time across injections; poor peak symmetry (tailing or fronting) [30].
- Solution:
- Verify Buffer Capacity: Ensure the buffer concentration is sufficient (typically 10-100 mM, with 25 mM being common) to control pH effectively throughout the analysis. A salt solution without adequate buffering capacity at the desired pH will lead to instability [30].
- Check HPLC Instrumentation: Monitor system pressure for consistency. Check for pump malfunctions, faulty sealings, or issues with the solvent mixing device [30].
- Confirm Sample Stability: Some analytes, like omeprazole, are unstable under acidic conditions or when exposed to light or prolonged sonication. Ensure samples are stored in stable, buffered conditions and protected from light [30].
- Prevention: Regularly calibrate and maintain the HPLC system. Use a column oven for stable temperature control and always employ a buffer with adequate capacity at the target pH [30].
LC-SPE-NMR and Structural Elucidation
Problem: Inaccurate quantitative evaluation in ¹H NMR. Accurate integration is essential for determining compound purity, ratios in mixtures, and quantitative structure assessments.
- Diagnosis: Inconsistent integration values between proton environments; large errors (>3%) when comparing different proton types (e.g., aromatic CH to a methyl group) [31].
- Solution:
- Optimize Relaxation Delay: Set an adequate delay (5–10 seconds) between scans to avoid saturation effects, especially for small molecules with long T1 relaxation times [31].
- Ensure Proper Digital Resolution: Acquire the spectrum with enough data points to accurately define each peak. A resonance with a width of 0.5 Hz should be sampled every 0.25 Hz for <1% error [31].
- Account for Isotopic Satellites: ¹³C satellites can contribute 1.1% to a peak's total area. Use ¹³C decoupling or ensure satellites from intense peaks do not overlap with the signal being integrated [31].
- Correct the Baseline: Use NMR software routines to adjust for baseline slant or curvature before integration [31].
- Prevention: For high-accuracy quantitation (<1% error), routinely measure T1 relaxation times to calculate the optimal relaxation delay and ensure excellent signal-to-noise [31].
Problem: Difficulty identifying isomeric impurities or confirming stereochemistry with LC-MS. Mass spectrometry may not distinguish compounds with identical molecular weights but different structural arrangements.
- Diagnosis: LC-MS data shows a single mass for a peak, but other data suggests a mixture or unknown stereochemistry.
- Solution:
- Employ 2D NMR Techniques: After isolation via LC-SPE, use a suite of NMR experiments [32]:
- COSY: Identifies proton-proton coupling networks.
- HSQC/HMQC: Maps direct correlations between protons and their attached carbon atoms.
- HMBC: Detects long-range (2-3 bond) proton-carbon couplings, establishing connectivity.
- NOESY/ROESY: Provides spatial proximity information critical for determining 3D configuration and stereochemistry [32].
- Use Chiral NMR Techniques: Apply chiral solvating agents or shift reagents to resolve and assign chiral centers [32].
- Employ 2D NMR Techniques: After isolation via LC-SPE, use a suite of NMR experiments [32]:
- Prevention: Integrate NMR as an orthogonal technique to LC-MS in impurity profiling workflows, as it is uniquely sensitive to isomeric differences and spatial structure [32].
Frequently Asked Questions (FAQs)
1. How do I assess peak purity in HPLC, and why can't I rely on retention time alone? A photodiode array (PDA) detector is the most common tool for assessing peak purity. It measures UV absorbance across a peak and identifies spectral variations that may indicate co-elution. Software calculates metrics like purity angle and threshold, but these should never be used alone. Always manually review spectral overlays, especially at the peak edges, as co-elution can compromise quantification even when a peak appears well-resolved. For definitive assessment, LC-MS detects co-elution based on mass differences [28].
2. What are the key factors to consider when developing an impurity profiling method? The development follows a sequential optimization of factors based on their influence on selectivity [29]:
- Stationary Phase: Has the largest influence. Start by screening a set of dissimilar (orthogonal) columns.
- Mobile Phase pH: Critically affects the selectivity for ionizable compounds. Optimize next.
- Organic Modifier: The type (e.g., acetonitrile vs. methanol) and composition can be fine-tuned.
- Gradient and Temperature: These factors are optimized last for final method tuning.
3. My sample's impurity profile changes over time. What could be causing this? This is a common stability issue. Factors include [30]:
- Light Exposure: UV light can catalyze photodegradation reactions.
- Solution pH: The sample may be unstable at the storage pH. For example, omeprazole degrades in acid.
- Sonication: Extended sonication can provide enough energy to degrade compounds or promote reactions. To prevent this, store samples in amber vials, ensure the solution pH is stable, and minimize sonication time.
4. When is NMR superior to LC-MS for impurity identification? NMR is particularly powerful in these scenarios [32]:
- Isomeric Impurities: It can distinguish positional isomers, tautomers, and stereoisomers that have identical masses.
- Non-Ionizable Compounds: It detects compounds that do not ionize well and may be missed by MS.
- Full Structural Elucidation: It provides the complete molecular framework, including functional groups and relative stereochemistry, without the need for reference standards.
- Chiral Analysis: It can resolve and assign chiral centers.
5. How does buffer concentration affect my HPLC analysis? Buffer concentration plays a key role in method robustness [30]:
- Too Low (<10 mM): Can lead to peak shape problems (tailing) and retention time variability because the mobile phase pH is poorly controlled.
- Adequate (10-100 mM): Provides stable pH control, ensuring consistent ionization states, retention times, and peak shapes. 25 mM is a common starting point.
- Too High: May exceed the salt's solubility in the mobile phase (e.g., causing phosphate salts to precipitate with acetonitrile) and potentially reduce column lifespan.
Workflow and Relationship Diagrams
Diagram 1: Impurity Profiling Method Development Workflow
Diagram 2: LC-SPE-NMR for Impurity Identification
Research Reagent Solutions
This table lists key materials and their functions in developing robust impurity profiling methods.
| Reagent/Material | Function in Impurity Profiling |
|---|---|
| Dissimilar HPLC Columns (e.g., C18, Phenyl, Cyano) | Screening columns with different selectivities to maximize the chance of resolving all impurities from the main compound and from each other [29]. |
| Buffers (e.g., Phosphate, Formate, Acetate) | Control the pH of the mobile phase to manipulate the ionization and thus the retention of ionizable analytes, which is a primary factor affecting selectivity [29] [30]. |
| Deuterated Solvents (e.g., CD₃OD, D₂O, CDCl₃) | The NMR-active solvent for structure elucidation after LC-SPE isolation. Allows for locking and shimming the NMR magnet and does not produce interfering signals in the ¹H spectrum [32]. |
| Chiral Derivatizing Agents | Chemicals that react with chiral analytes to form diastereomers, which can then be separated by standard HPLC or analyzed by NMR to determine enantiomeric purity and absolute configuration [32]. |
| Solid Phase Extraction (SPE) Cartridges | Used in the LC-SPE interface to trap HPLC eluates, remove non-deuterated solvents, and concentrate the analyte for subsequent NMR analysis, significantly enhancing sensitivity [32]. |
Frequently Asked Questions
What is multi-trapping 2D-LC and what is its primary advantage? Multi-trapping two-dimensional liquid chromatography (2D-LC) is an advanced analytical technique where a sample is first separated in a first dimension (1D) column, after which specific analyte fractions are temporarily stored, or "trapped," on a secondary column. The primary advantage is the ability to perform multiple trapping cycles from successive HPLC runs, concentrating low-level analytes to significantly improve the signal-to-noise ratio for detection and quantification [33].
Which low-level impurities can be monitored with this technique? This technique is particularly suited for enriching and quantifying challenging low-level impurities in pharmaceutical development, including:
- Unknown impurities at sub-parts-per-million (ppm) levels [34].
- Co-eluting impurities that can lead to undesired summation above specification limits [34].
- Potential mutagenic impurities at ppm levels, even in poorly soluble substrates [33] [34].
What are the quantitative performance characteristics of multi-trapping 2D-LC? When properly configured, the technique demonstrates excellent quantitative performance. Studies show it can achieve a recovery rate of over 97.0% with a relative standard deviation (RSD) of lower than 3.0%, confirming its accuracy and precision for impurity analysis. The system also demonstrates linear enrichment for up to 20 trapping cycles [33] [34].
Troubleshooting Guides
Poor Recovery of Target Analytes
Problem: Inconsistent or low recovery of analytes from the trap column.
Solution:
- Verify Trap Column Sorbent: Ensure the trap column sorbent is compatible with your target analytes and the mobile phases used in both dimensions. The sorbent must efficiently trap and subsequently release the analytes [34].
- Check Online Dilution: For methods that use online dilution to ensure the 1D eluent is compatible with the trapping process, confirm the dilution ratio and mixing efficiency. Inadequate dilution can lead to poor trapping efficiency [34].
- Review Valve Switching Timing: Precise timing of the switching valves is critical to ensure the entire fraction of interest is transferred to the trap column and later to the 2D column for analysis. Even minor timing errors can cause significant analyte loss [34].
Inconsistent Enrichment Linearity Over Multiple Cycles
Problem: The increase in analyte signal is not linear with the number of trapping cycles.
Solution:
- Confirm System Stability: Before starting a multi-trapping sequence, ensure the 1D separation is highly reproducible. Any retention time drift in the 1D separation will result in the heart-cut window missing the target analyte in subsequent cycles [34].
- Inspect Trap Column Capacity: Verify that the mass of the analyte being loaded over multiple cycles does not exceed the binding capacity of the trap column. Overloading will lead to analyte breakthrough and loss [33].
- Validate Fluidics: Check for any leaks or inconsistencies in the pumping systems that could affect the transfer volume and consistency between cycles [34].
Experimental Performance Data
The following table summarizes the quantitative performance of multi-trapping 2D-LC as demonstrated in real-world pharmaceutical case studies [33] [34].
| Application / Analyte | Impurity Level | Number of Trapping Cycles | Recovery (%) | Precision (RSD) |
|---|---|---|---|---|
| Unknown impurities (material discoloration) | Sub-ppm | 10 | >97.0 | <3.0% |
| Co-eluting impurity | 0.05% (w/w) | Information not specified | >97.0 | <3.0% |
| Potential mutagenic impurity | 10 ppm | Information not specified | >97.0 | <3.0% |
| Standard test mixture (varied compounds) | Up to 0.15% (w/w) | 20 | >97.0 | <3.0% |
Detailed Experimental Protocol: Multi-Trapping 2D-LC for Impurity Enrichment
This protocol outlines the methodology for enriching low-level impurities using a heart-cutting trapping mode 2D-LC system, built from standard, GMP-ready instrument modules [34].
1. Instrument Configuration
- System: A 2D-LC system configured with an additional binary pump for the second dimension.
- Valves: Two 6-port/2-position switching valves are used to control the heart-cutting and trapping processes.
- Columns:
- 1D Column: A suitable fit-for-purpose column for the initial separation.
- Trap Column: A short column selected for its ability to focus and retain the target analytes.
- 2D Column: A suitable analytical column for the final separation of the enriched analytes.
- Detection: UV and/or Mass Spectrometry detectors after the 2D column [34].
2. Procedure
- Step 1: First Dimension Separation. The sample is injected and separated on the 1D column. The mobile phase is optimized for high-resolution separation of the complex mixture.
- Step 2: Heart-Cutting and Trapping. As the peak(s) of interest elute from the 1D column, the switching valve is activated to divert the fraction containing the analyte onto the trap column. The trap column captures and focuses the analyte while incompatible solvents are washed to waste. This step is repeated for multiple injections to accumulate the analyte.
- Step 3: Second Dimension Elution. After the desired number of trapping cycles, the switching valve reconfigures the flow path. The 2D pump delivers a strong mobile phase to elute the concentrated analyte from the trap column onto the 2D analytical column.
- Step 4: Second Dimension Separation and Detection. The analyte undergoes a final separation on the 2D column under optimized conditions and is then directed to the detector (UV or MS) for sensitive quantification [33] [34].
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key materials and their functions for implementing a multi-trapping 2D-LC workflow based on the cited research.
| Item | Function / Application |
|---|---|
| Standard Test Mixture (STM) | A diverse set of compounds used to evaluate system performance, trapping efficiency, and quantitative capabilities for analytes with varied properties [34]. |
| Trap Column Sorbent | A short column with selective sorbent that temporarily retains and focuses the heart-cut fraction from the 1D separation, enabling cleaning and enrichment [34]. |
| Off-the-shelf GMP-ready 2D-LC System | Instrumentation comprising standard modules (pumps, valves, detectors) controlled by native software, ensuring the method is suitable for quality-control environments [34]. |
| UPLC/HPLC Grade Solvents | High-purity solvents (e.g., acetonitrile, methanol, water) with additives like trifluoroacetic acid (TFA) or formic acid for mobile phase preparation to ensure optimal separation and MS compatibility [34]. |
Maximizing Efficiency: Overcoming Common LC-SPE-NMR Implementation Challenges
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: My target analytes are not being retained on my reversed-phase SPE cartridge. What could be the problem? This lack of retention is often due to an overly polar sample matrix or mobile phase. Ensure your sample is loaded in a weak, aqueous solvent. For very hydrophilic compounds, consider switching to a mixed-mode phase or a phase with higher carbon loading, which provides stronger hydrophobic retention [35].
Q2: I am experiencing low recovery of my target compounds from a Strong Anion Exchange (SAX) cartridge. How can I improve this? Low recovery from SAX phases can result from an incorrect elution solvent. SAX phases require a strong elution buffer with a high ionic strength (e.g., a concentrated salt solution) or a change in pH to disrupt the ionic interaction. Ensure the target analytes are ionized and carry a negative charge during the loading and washing steps [35].
Q3: After using my Strong Cation Exchange (SCX) cartridge, I see co-elution of interfering compounds. How can I enhance the cleanup? Co-elution in SCX methods often means the washing step was not stringent enough. Optimize the wash solvent with a slightly higher ionic strength buffer to remove weakly bound, interfering cationic compounds without eluting your targets. Using a mixed-mode SCX cartridge, which combines ionic and reversed-phase mechanisms, can also significantly improve selectivity [35].
Q3: My final NMR analysis shows poor signal-to-noise after LC-SPE concentration. What steps should I check? First, verify the efficiency of the SPE elution step. A poor NMR signal can indicate that the target compounds were not fully transferred from the SPE cartridge to the NMR tube. Ensure the elution solvent is strong enough and fully deuterated to maintain the NMR lock. Method development should use LC-MS to confirm high SPE recovery rates before proceeding to NMR analysis [16].
Troubleshooting Common SPE Issues
The table below summarizes common problems and their solutions across different SPE phases.
| SPE Phase | Common Problem | Possible Cause | Suggested Solution |
|---|---|---|---|
| Reversed-Phase (e.g., C18) | Poor analyte retention | Sample solvent is too organic/strong | Dilute sample with water; ensure environment is aqueous [35] |
| Reversed-Phase (e.g., C18) | Low recovery | Elution solvent is too weak | Use a stronger organic solvent (e.g., acetonitrile, methanol) [35] |
| Strong Anion Exchange (SAX) | Low recovery | Insufficient elution strength | Use a high-ionic-strength buffer or pH-adjusted solvent to neutralize charges [35] |
| Strong Cation Exchange (SCX) | Co-elution of impurities | Incomplete washing | Optimize wash buffer ionic strength to remove weakly bound contaminants [35] |
| All Phases | Inconsistent results | Column overloading or clogging | Reduce sample load or pre-filter/centrifuge the sample [35] |
Experimental Protocols for Method Development
Protocol 1: Screening SPE Phases for an Unknown Mixture
This protocol is designed for the initial selection of the most suitable SPE phase when analyzing complex, unknown samples, such as botanical extracts, within an LC-SPE-NMR workflow [16].
- Sample Preparation: Prepare a standardized extract of your sample. As an example, for botanical ingredients like Camellia sinensis or Cannabis sativa, homogenize the material and extract 50 mg (±1 mg) with 1 mL of a 90% methanol/10% deuterated methanol solution. This solvent provides broad metabolite coverage for both LC-MS and subsequent NMR analysis [16].
- LC Condition Optimization: Use a reversed-phase UPLC method for initial separation. A C18 column with a particle size of <2 µm is recommended for fast, high-resolution separation. Employ a water-acetonitrile gradient with 0.1% formic acid [35] [16].
- Parallel SPE Screening: Set up identical SPE stations for Reversed-Phase (C18), SAX, and SCX phases. Using the same LC effluent, direct the flow to the different SPE cartridges in parallel runs.
- Elution and Analysis: Elute the trapped compounds from each SPE phase into a vial using a small volume (e.g., 50-100 µL) of a fully deuterated elution solvent (e.g., CD3OD). The choice of deuterated solvent is critical for minimizing solvent usage in NMR [16].
- LC-MS Analysis: Analyze the eluates from each SPE phase using LC-MS. Compare the number of detected metabolites, signal intensity, and the presence of key target compounds.
- Phase Selection: The SPE phase that yields the highest number of metabolites and the strongest signals for your compounds of interest, as determined by LC-MS, should be selected for the final LC-SPE-NMR method.
Protocol 2: Optimizing SPE for Deuterated Solvent Reduction in NMR
This protocol focuses on integrating the selected SPE phase into a system designed to reduce the consumption of expensive deuterated solvents in NMR.
- LC Separation: The LC method from Protocol 1 is used. The key is to use standard, non-deuterated solvents (e.g., H2O, CH3CN) for the mobile phase.
- Post-Column Splitting and Dilution: After the LC detector, the flow is split. A small fraction can be directed to the mass spectrometer for real-time monitoring, while the majority is diluted with a make-up solvent (e.g., water). This dilution weakens the eluent strength, ensuring optimal retention of the target peak on the subsequent SPE cartridge [16].
- SPE Trapping: The diluted LC effluent is directed to the selected and pre-conditioned SPE cartridge. The target analyte is trapped and concentrated on the SPE phase while non-deuterated solvents are sent to waste.
- Analyte Elution to NMR: Once the trapping is complete, the target compound is eluted from the SPE cartridge directly into the NMR tube using a minimal volume of a fully deuterated solvent. This step efficiently transfers the analyte into an NMR-compatible environment using the smallest possible amount of deuterated solvent.
- NMR Acquisition: The NMR tube is placed in the spectrometer for data acquisition. The use of a deuterated solvent for elution provides the lock signal required for stable and high-quality NMR data [16].
SPE Phase Selection Workflow
The following diagram illustrates the logical decision process for selecting the appropriate Solid-Phase Extraction (SPE) phase.
Research Reagent Solutions
The table below lists key materials and reagents essential for developing and executing LC-SPE-NMR methods.
| Item | Function/Description | Application Note |
|---|---|---|
| Methanol (with 10% CD3OD) | Effective extraction solvent for broad metabolite coverage in botanical samples, compatible with both LC-MS and NMR [16]. | Provides the broadest metabolite coverage for fingerprinting; the small percentage of deuterated solvent aids the NMR lock without high cost [16]. |
| Deuterated Methanol (CD3OD) | Deuterated elution solvent for transferring analytes from SPE to the NMR tube. | A strong elution solvent for reversed-phase SPE; its deuterium provides the necessary lock signal for NMR analysis [16]. |
| C18 SPE Phase | Reversed-phase material with strong hydrophobicity for retaining non-polar to moderately polar compounds [35]. | The most widely used phase; ideal for concentrating analytes from aqueous LC streams before NMR. |
| Strong Anion Exchange (SAX) Phase | SPE material with quaternary ammonium groups to retain and separate acidic, anionic compounds [35]. | Used to selectively isolate acids and other anions from complex matrices, simplifying the NMR spectrum. |
| Strong Cation Exchange (SCX) Phase | SPE material with sulfonic acid groups to retain and separate basic, cationic compounds [35]. | Used to selectively isolate bases and other cations from complex matrices, simplifying the NMR spectrum. |
| Phosphate Buffer in D2O | Buffer solution used to control pH in NMR samples, improving spectral consistency and chemical shift reproducibility [16]. | Mitigates chemical shift variations caused by pH differences, which is critical for database matching and authentication. |
Frequently Asked Questions (FAQs)
Q1: What is the primary function of the make-up flow in an LC-SPE-NMR system? The make-up flow is introduced post-LC column to promote quantitative analyte retention on the Solid Phase Extraction (SPE) cartridge. It works by diluting the HPLC mobile phase and reducing its elutropic strength, creating conditions that favor the binding of analytes to the SPE stationary phase. This process is critical for post-HPLC focusing of analyte peaks to match the volume of the NMR probe flow cell, which significantly enhances sensitivity by increasing analyte concentration [10].
Q2: Why might my analytes not be retaining on the SPE cartridge, and how can I troubleshoot this? Poor analyte retention typically stems from an incorrectly configured make-up flow. Key parameters to investigate are:
- Composition and Flow Rate: The most commonly used make-up solvent is pure H₂O, delivered at a flow rate of 1-2 mL/min. This effectively reduces the organic solvent concentration from the LC eluent, facilitating analyte binding to hydrophobic SPE phases [10].
- SPE Stationary Phase Mismatch: The standard stationary phases (e.g., DVB-type polymers or RP-C18 silica) may not be optimal for all compounds. For charged or highly polar analytes like alkaloids or organic acids, you should consider switching to modified SPE phases such as Strong Anion Exchange (SAX), Strong Cation Exchange (SCX), or porous carbon materials to achieve quantitative retention [10].
Q3: How does the make-up flow contribute to reducing deuterated solvent consumption? The make-up flow enables a comprehensive solvent exchange. After the analyte is trapped on the SPE cartridge, the HPLC mobile phase (which may contain non-deuterated organic solvents) is removed by prolonged washing with a solvent of low elutropic strength, such as D₂O. The analyte is then eluted into the NMR spectrometer with a minimal, defined volume of deuterated solvent (e.g., CD₃OD or CD₃CN). This process avoids the need for using prohibitively expensive deuterated solvents throughout the entire HPLC separation, leading to significant cost savings [10] [9].
Q4: Can I use the same SPE conditions for all my analytes when performing multiple trappings? No, SPE trapping and elution conditions often need to be optimized for each analyte class. Pronounced differences in trapping efficacy between different stationary phases have been observed. For instance, one study demonstrated that approximately 100 µg of scopoletin could be accumulated on a GP phase cartridge after seven trappings, whereas only about 20 µg was retained on a C-18 phase under identical conditions. Therefore, the make-up flow composition and SPE phase must be selected based on the physicochemical properties of your target analytes [10].
Experimental Protocol: Establishing Make-Up Flow Parameters
This protocol provides a detailed methodology for optimizing the make-up flow to ensure quantitative analyte retention on SPE cartridges in an LC-SPE-NMR setup.
2.1 Materials and Equipment
- LC system with a post-column make-up flow capability
- SPE unit (e.g., automated system with a 96-well plate format for SPE cartridges)
- Standard SPE cartridges (e.g., 2 x 10 mm, packed with DVB-polymer or RP-C18 silica)
- Specialized SPE cartridges (SAX, SCX, porous carbon)
- Make-up solvent: High-purity water (H₂O)
- Test analytes and parent compound
- Syringe pump (if required for system configuration)
2.2 Procedure
- Initial System Configuration: Connect the make-up flow line immediately after the LC column outlet and before the SPE cartridge inlet. Use a low-dead-volume T-union to ensure efficient mixing.
- Set Baseline Make-Up Flow Conditions: Initiate with a make-up flow of pure H₂O at a flow rate of 1.5 mL/min. This is a standard starting point found to be effective for a wide range of analytes [10].
- Perform Analytical LC Run: Inject the test analyte and run the optimized LC method. The make-up flow will mix with the column eluent and direct the diluted analyte to the SPE cartridge.
- Evaluate Trapping Efficiency: Monitor the process via UV or MS triggers. A failure to retain the analyte on the SPE cartridge suggests the make-up flow conditions or SPE phase are not optimal.
- Troubleshoot and Optimize:
- If retention is poor, systematically adjust the make-up flow rate (e.g., between 1-2 mL/min) to find the optimal dilution factor.
- If changing the flow rate is insufficient, consider the chemistry of your analyte. For ionic compounds, switch to an ion-exchange SPE phase (SAX for acids, SCX for bases). For highly polar neutral compounds, a porous carbon phase may be more effective [10].
- Elute to NMR: Once quantitative retention is confirmed, wash the SPE cartridge with D₂O to remove residual salts and protonated solvents. Subsequently, elute the trapped analyte with a minimal volume (typically < 1 mL) of an appropriate deuterated solvent (e.g., CD₃OD or CD₃CN) directly into the NMR flow probe [10].
Workflow Diagram: The LC-SPE-NMR Process with Make-Up Flow Integration
The following diagram illustrates the complete workflow, highlighting the critical role of the make-up flow in bridging the LC separation stage with the NMR detection stage.
Research Reagent Solutions
The table below lists key materials and their functions for optimizing the make-up flow and SPE process.
| Item | Function in the Experiment |
|---|---|
| DVB-type Polymer SPE Cartridge | A standard stationary phase for trapping a wide range of analytes; useful for method development [10]. |
| RP-C18 Silica SPE Cartridge | Another common reversed-phase material for retaining hydrophobic compounds [10]. |
| Ion-Exchange SPE Cartridges (SAX/SCX) | Essential for the quantitative retention of charged or highly polar analytes (e.g., alkaloids, organic acids) that poorly retain on standard phases [10]. |
| High-Purity H₂O | The standard make-up solvent used to dilute the LC eluent, reducing its elutropic strength and promoting analyte binding to the SPE cartridge [10]. |
| Deuterated NMR Solvents (CD₃OD, CD₃CN) | Used in minimal volumes to elute the analyte from the SPE cartridge into the NMR probe, enabling high-quality spectra while conserving costly deuterated solvents [10] [9]. |
| Hexylamine / Acetic Acid | An ion-pair reagent combination that can be added to the mobile phase or make-up flow to improve the retention of very hydrophilic metabolites on reversed-phase systems, aiding their subsequent trapping [36]. |
In LC-SPE-NMR, the choice of elution solvent is a critical bridge between effective chromatographic separation and high-quality nuclear magnetic resonance (NMR) analysis. This technical guide addresses the core challenge of selecting a solvent with sufficient power to release analytes from solid-phase extraction (SPE) cartridges while also being compatible with NMR spectroscopy, primarily through the use of deuterated solvents. The following FAQs and troubleshooting guides are designed to help researchers navigate this balance, enabling efficient analyte recovery and optimal spectral clarity.
Frequently Asked Questions (FAQs)
1. Why can't I use protonated solvents for the NMR step in LC-SPE-NMR? Using protonated solvents for NMR results in intense solvent signals that overwhelm the much weaker signals from your analytes. This causes issues like receiver saturation, poor digitization of solute resonances, and baseline distortions, making it difficult or impossible to interpret the spectrum [37]. The LC-SPE-NMR setup specifically uses a solvent exchange step to replace the protonated HPLC mobile phase with a deuterated NMR solvent, thereby circumventing the need for complex solvent suppression techniques and providing well-defined NMR conditions for reliable spectral comparison [10] [38].
2. Which deuterated solvents are most suitable for eluting analytes from SPE cartridges? Methanol-d4 (CD₃OD) and acetonitrile-d3 (CD₃CN) are the most frequently used and recommended deuterated solvents for eluting analytes from SPE cartridges in LC-SPE-NMR systems [10]. Their elutropic power and hydrogen-bonding capacity are effective for releasing a wide range of trapped compounds. In contrast, highly viscous solvents like DMSO-d6 and deuterated pyridine (C5D5N), common in tube-based NMR, are rarely used in this hyphenated technique, and deuterated chloroform (CDCl₃) sees only occasional application [10].
3. How does solvent selection impact my ability to recover my sample post-NMR? The solvent's boiling point directly affects the ease of sample recovery. Low-boiling-point solvents like CDCl₃ (61.2°C) are easily removed via evaporation [39]. High-boiling-point solvents like DMSO-d6 (189°C) were historically difficult to remove, often forcing sample discard. However, modern tools like evaporators using novel "Spiral Plug" technology now enable the recovery of samples even from high-boiling-point solvents directly from the vial [39].
Troubleshooting Guides
Problem: Poor Analyte Elution from SPE Cartridge
Symptoms
- Weak or absent NMR signals despite successful HPLC separation and trapping.
- Incomplete transfer of the analyte band to the NMR flow cell.
Possible Causes and Solutions
- Cause 1: Insufficient Elutropic Power. The chosen deuterated solvent lacks the strength to displace the analyte from the specific SPE stationary phase.
- Solution: Switch from CD₃CN to the stronger eluting solvent CD₃OD, or vice-versa, as their elution performances can vary significantly for different analyte classes [10].
- Cause 2: Suboptimal SPE Stationary Phase. The standard C18 or DVB-type polymer phases may not be optimal for charged or highly polar analytes like alkaloids or organic acids.
- Solution: Investigate alternative SPE phases, such as strong anion exchange (SAX), strong cation exchange (SCX), or porous carbon materials, which may provide better trapping and release characteristics for challenging compounds [10].
Problem: Solvent Peak Interference in NMR Spectrum
Symptoms
- Large residual solvent peaks obscure analyte signals of interest.
- Difficulty performing automatic shimming or baseline correction.
Possible Causes and Solutions
- Cause 1: Overlap with Analyte Resonances. The residual proton peak of the solvent (e.g., CDCl₃ at 7.26 ppm) overlaps with key analyte signals [19].
- Solution: Consult solvent data sheets and select an alternative deuterated solvent whose residual peaks do not interfere with your analyte's expected chemical shift regions. For example, CD₃CN has a residual peak at 1.94 ppm, which may offer a clearer spectral window [19] [40].
- Cause 2: High Water Content. Water contamination can introduce a large, variable peak that degrades spectral quality.
- Solution: Minimize water peaks by using single-use ampoules, handling solvents in a dry atmosphere, and thoroughly drying NMR tubes and pipettes before use [40].
Research Reagent Solutions
The following table details key materials essential for the LC-SPE-NMR workflow, with a focus on the elution and NMR analysis phase.
| Item | Function in LC-SPE-NMR |
|---|---|
| Deuterated Methanol (CD₃OD) | A prime deuterated solvent for eluting analytes from SPE cartridges; offers a good balance of elutropic power and hydrogen-bonding capacity [10]. |
| Deuterated Acetonitrile (CD₃CN) | Another primary elution solvent; favored for its thermal stability and predictable chemical shifts, often serving as an alternative to CD₃OD [10]. |
| SPE Cartridges (C18/DVB) | The solid-phase medium for trapping HPLC-separated analytes; allows for solvent exchange from protonated mobile phase to deuterated NMR solvent [10]. |
| SPE Cartridges (SAX/SCX) | Specialized stationary phases used as alternatives to C18 for the trapping and release of charged or highly polar analytes [10]. |
| NMR Flow Probe | The detection unit where the eluted and focused analyte band is transferred for NMR measurement; cell volumes typically range from 60–250 μL [10]. |
Experimental Protocols and Data Presentation
Protocol: Method Development for SPE Elution
- SPE Conditioning: Pre-equilibrate the selected SPE cartridge (e.g., C18, DVB, SAX) with a solvent of low elutropic strength, such as D₂O or H₂O [10].
- Analyte Trapping: Load the analyte onto the cartridge from the HPLC system, often using a post-column makeup flow of water (1-2 mL/min) to promote binding [10].
- Drying: Wash the cartridge extensively with the same weak solvent to remove residual HPLC mobile phase completely.
- Elution Optimization: Back-flush the cartridge with a narrow band (typically <1 mL) of your test deuterated NMR solvent (e.g., CD₃OD or CD₃CN) to elute the analyte directly into the NMR flow probe [10].
- Evaluation: Assess success based on the narrowness of the elution band and the signal-to-noise ratio in the resulting NMR spectrum.
Quantitative Solvent Properties Table
The table below summarizes key properties of common deuterated solvents to guide selection based on NMR and practical considerations.
| Solvent | Typical Residual ¹H Peak (ppm) | Boiling Point (°C) | Key NMR Advantages | Key NMR Limitations |
|---|---|---|---|---|
| CDCl₃ | 7.26 | 61.2 | Affordable, versatile, easy to remove for sample recovery [19] [39] | Peak may overlap aromatic signals [19] |
| DMSO-d₆ | 2.50 | 189 | Excellent for polar compounds and polymers [19] | High boiling point, difficult to remove, can coordinate with samples [19] [39] |
| CD₃OD | 3.31 | 64.7 | Good for protic environments, enables H-exchange studies [19] | Residual peak sensitive to impurities [19] |
| CD₃CN | 1.94 | 81.6 | Thermally stable, predictable shifts, ideal for temperature studies [19] | Limited solubility for highly polar substances [19] |
| D₂O | Variable (HOD) | 101.4 | Ideal for polar/ionic samples, identifies exchangeable protons [19] | Poor for organic compounds, sensitive reference signal [19] |
Workflow and Relationship Visualizations
Diagram 1: LC-SPE-NMR Solvent Selection Workflow. This diagram illustrates the critical decision point at the elution stage and the consequences of solvent choice on NMR compatibility and sample recovery.
Diagram 2: Key Factors for Solvent Selection. This diagram logically groups the primary factors that must be balanced when choosing an elution solvent for LC-SPE-NMR, highlighting the interconnectedness of chromatographic, spectroscopic, and practical requirements.
Technical Support Center
Troubleshooting Guides
Guide 1: Poor Retention of Polar Compounds in Reversed-Phase LC
- Problem: Highly polar analytes elute at or near the void volume on standard C18 columns, leading to inadequate separation.
- Cause: Traditional reversed-phase stationary phases (e.g., C18) are designed for non-polar compounds and offer minimal interaction with polar molecules.
- Solutions:
- Switch to a HILIC Column: Use hydrophilic interaction liquid chromatography. This technique employs a polar stationary phase (e.g., silica or zwitterionic) and an acetonitrile-rich mobile phase (>80%). Analytes are retained based on their hydrophilicity and elute in order of increasing polarity [41].
- Use a Specialized Reversed-Phase Column: Select columns specifically engineered for polar compounds, such as those with lower ligand density and larger pore sizes (e.g., T3 columns), which reduce "dewetting" and enhance aqueous retention [41].
- Employ Mixed-Mode Chromatography: Utilize a column that combines reversed-phase and ion-exchange mechanisms (e.g., reversed-phase ion exchange). This allows you to modulate retention by adjusting mobile-phase pH, ionic strength, and organic solvent content to suit charged polar analytes [41].
Guide 2: Signal Suppression or Poor Sensitivity in LC-MS for Polar Compounds
- Problem: Low signal-to-noise ratio for polar analytes during LC-MS analysis, often due to ion suppression.
- Cause: Co-eluting matrix components from complex samples (e.g., proteins, lipids) can compete for charge during the electrospray ionization process, suppressing the analyte signal [42].
- Solutions:
- Improve Sample Cleanup: Implement more rigorous sample preparation to remove matrix interferences. Techniques like solid-phase extraction (SPE) are recommended, though note that highly polar metabolites may not retain on conventional SPE cartridges [42] [43].
- Optimize MS Source Parameters: Fine-tune parameters like desolvation temperature and nebulizer gas flow based on your specific LC method and analytes. A 20% increase in response has been demonstrated through such optimization [42].
- Consider APCI: If analytes are thermally stable, switch from electrospray ionization (ESI) to atmospheric pressure chemical ionization (APCI). Matrix effects are generally less extensive in APCI because ionization occurs via gas-phase reactions [42].
Guide 3: Managing Solvent Costs and Compatibility in LC-NMR
- Problem: The high cost of fully deuterated solvents for continuous-flow LC-NMR and potential solvent signal interference.
- Cause: Using deuterated solvents as the mobile phase is prohibitively expensive, while protonated solvents create large interfering signals in the NMR spectrum [10].
- Solution - Implement LC-SPE-NMR: This is a key strategy for deuterated solvent reduction. The HPLC effluent, using a protonated mobile phase, is passed through a solid-phase extraction (SPE) cartridge after a makeup flow is added to promote analyte retention. The trapped analytes are dried, then eluted with a small volume (typically < 1 mL) of a deuterated solvent into the NMR flow probe. This drastically reduces deuterated solvent consumption and provides a clean, well-defined NMR spectrum [10].
Frequently Asked Questions (FAQs)
FAQ 1: What is the most effective way to separate a mixture of very polar metabolites?
For complex mixtures of highly polar metabolites, no single technique is perfect. A multi-technique approach is recommended:
- HILIC-MS: Ideal for retaining and separating a wide range of polar compounds and is highly compatible with MS detection [41] [43].
- Capillary Electrophoresis-MS (CE-MS): A powerful complementary technique, especially for charged polar ions. It uses a different separation mechanism (electrophoretic mobility) and can resolve compounds that are challenging for LC [43].
- GC-MS: Can be used for volatile polar compounds or those that can be made volatile through derivatization [43].
FAQ 2: When should I use NMR over MS for the identification of an unknown polar compound?
MS and NMR provide complementary information. NMR is indispensable when:
- You need to identify isomeric compounds that have identical mass-to-charge ratios (m/z) but different atom connectivity [44].
- Unequivocal structural elucidation is required, as NMR can distinguish between different functional groups and establish atomic connectivity through 2D experiments [44] [45].
- You are working with a novel compound not found in mass spectral databases [44].
FAQ 3: What is the best solvent for extracting a broad range of polar metabolites from botanical samples for NMR and LC-MS analysis?
Research across multiple botanical species indicates that methanol is the most versatile and effective solvent. Using 100% methanol or a mixture of methanol and deuterium oxide (for NMR locking purposes) provides the broadest metabolite coverage, successfully extracting amino acids, sugars, and phenolic compounds [46].
FAQ 4: Why is my NMR spectrum for a polar compound noisy or of poor quality?
This can result from several factors related to sample preparation:
- Insufficient Sample Concentration: For 1H NMR of small molecules, 5-25 mg in 0.6 mL of solvent is typical. Too little sample results in a weak signal [47].
- Particulate Matter: Solid particles in the NMR tube can distort the magnetic field. Always filter your sample solution before transferring it to the tube [48] [47].
- Paramagnetic Impurities: Trace oxygen or metal ions can broaden NMR signals. For sensitive experiments, degas the sample or use an inert atmosphere [47].
Experimental Protocols & Data
Protocol: HPLC-SPE-NMR for Analyzing Polar Natural Products
This protocol is central to research on deuterated solvent reduction [10].
- HPLC Separation: Inject the crude extract onto an HPLC system using standard protonated solvents (e.g., H2O and ACN with 0.1% formic acid). Use a UV or MS detector to trigger the next step.
- SPE Trapping: Post-column, add a makeup flow of water (e.g., 1-2 mL/min) to dilute the organic solvent and promote analyte retention on an SPE cartridge (typically DVB-polymer or RP-C18).
- Cartridge Drying: Use a stream of nitrogen or air to dry the SPE cartridge thoroughly, removing all protonated HPLC solvent.
- Analyte Elution to NMR: Back-flush the trapped analyte from the SPE cartridge using a minimal volume (e.g., 50-100 µL) of deuterated solvent like CD3OD or CD3CN. This elutes the analyte as a concentrated band directly into the NMR flow cell.
- NMR Data Acquisition: Acquire your NMR spectra. The analyte is now in a pure deuterated solvent, providing excellent signal resolution without the need for solvent suppression.
Table 1. Optimization of SPE Phase for Different Analytic Classes in LC-SPE-NMR
| Analyte Class | Recommended SPE Phase | Key Consideration |
|---|---|---|
| General Natural Products | DVB-Polymer | Robust retention for a wide range of mid-polarity compounds [10]. |
| General Natural Products | RP-C18 Silica | Standard phase; good for many applications [10]. |
| Polar Acids/Bases | SAX or SCX | Provides ion-exchange mechanism for charged analytes that poorly retain on RP phases [10]. |
Table 2. Comparison of Chromatographic Techniques for Polar Compound Analysis
| Technique | Mechanism | Best For | Key Advantage | Consideration |
|---|---|---|---|---|
| Reversed-Phase (T3) | Hydrophobic interaction | Polar compounds with some hydrophobicity. | Compatible with 100% aqueous mobile phases; reduces dewetting [41]. | May still fail for highly polar/ionic species. |
| HILIC | Partitioning to water layer on polar stationary phase | Very polar, hydrophilic analytes (sugars, amino acids) [41]. | Excellent retention of polar compounds; MS-compatible [41] [43]. | Requires high organic mobile phase; longer equilibration. |
| Mixed-Mode | RP + Ion Exchange | Charged polar analytes (acids, bases, peptides). | Tunable selectivity via pH and ionic strength [41]. | Method development can be more complex. |
| Capillary Electrophoresis | Electrophoretic mobility | Charged polar/ionic metabolites in complex matrices [43]. | High separation efficiency; very small sample volumes [43]. | Lower loading capacity; buffer compatibility with MS can be challenging [43]. |
Workflow Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 3. Essential Materials for Polar Compound Analysis
| Item | Function/Application |
|---|---|
| HILIC Columns (e.g., BEH Z-HILIC) | Retains highly polar analytes using a polar stationary phase and acetonitrile-rich mobile phase [41]. |
| Specialized RP Columns (e.g., T3, Atlantis Premier BEH C18 AX) | Enhanced retention for polar compounds in reversed-phase mode; reduces nonspecific adsorption and dewetting [41]. |
| Deuterated Methanol (CD₃OD) | Common deuterated solvent for NMR; also an effective extraction solvent for broad metabolite profiling in botancials [46] [10]. |
| Deuterated Acetonitrile (CD₃CN) | Deuterated solvent for NMR; often used to elute analytes from SPE cartridges in LC-SPE-NMR due to its elution power and low viscosity [10]. |
| SPE Cartridges (DVB, C18, SAX/SCX) | Used for post-column analyte trapping and concentration in LC-SPE-NMR, enabling deuterated solvent exchange [10]. |
| Methanol (CH₃OH) | Versatile and effective solvent for extracting a wide range of polar metabolites from biological and botanical samples for both LC-MS and NMR analysis [46]. |
Technical Support Center: Troubleshooting LC-SPE-NMR Workflows
This technical support center provides targeted troubleshooting guides and FAQs to help researchers address specific challenges in high-throughput LC-SPE-NMR workflows, particularly within the context of deuterated solvent reduction research.
Liquid Chromatography-Solid Phase Extraction-Nuclear Magnetic Resonance (LC-SPE-NMR) is a powerful hyphenated technique that combines the separation power of LC, the purification and concentration capabilities of SPE, and the structural elucidation strengths of NMR. A primary research focus in this field is reducing the consumption of expensive deuterated solvents, which are essential for NMR locking and shimming but represent a significant operational cost [16] [49].
The following diagram illustrates the ideal automated workflow for a high-throughput LC-SPE-NMR system with integrated deuterated solvent reduction strategies.
Automated LC-SPE-NMR with Troubleshooting
Troubleshooting Guide: Common Issues and Solutions
This section addresses frequent problems encountered in LC-SPE-NMR workflows, with a focus on issues related to deuterated solvent reduction.
Table 1: LC-SPE-NMR Troubleshooting Guide
| Problem Area | Observed Symptom | Potential Root Cause | Recommended Solution |
|---|---|---|---|
| Liquid Chromatography (LC) | Peak Tailing or Fronting [50] | - Secondary interactions with stationary phase- Column overload- Injection solvent mismatch [50] | - Reduce sample load or dilution- Ensure sample solvent compatibility with mobile phase- Use a column with less active residual sites [50] |
| Ghost Peaks [50] | - Carryover from prior injections- Contaminants in mobile phase or system- Column bleed [50] | - Run blank injections to identify contaminants- Clean autosampler and injection path- Use fresh, high-purity mobile phase; replace column if needed [50] | |
| Retention Time Shift [50] | - Change in mobile phase composition/pH- Pump flow rate variance- Column temperature fluctuation [50] | - Verify mobile phase preparation and flow rate- Ensure column thermostat is stable- Compare with historical controls; check for column aging [50] | |
| Solid Phase Extraction (SPE) | Poor Recovery [51] | - Analyte breakthrough during loading/wash- Incomplete elution- Analyte instability or protein binding [51] | - Check solvent compatibility in all SPE steps; alter sample solvent or wash steps to enhance retention- Increase elution solvent strength; confirm it addresses secondary interactions [51] |
| Poor Reproducibility [51] | - Inconsistent sample preparation- SPE sorbent lot-to-lot variability- Signal suppression/enhancement from matrix (in LC-MS) [51] | - Verify analytical system function with standards- Compare sorbent lot numbers- Improve sample cleanup via wash protocol modification or sorbent change [51] | |
| Insufficiently Clean Extracts [51] | - Inadequate wash steps failing to remove interferences | - Use a wash solvent with the strongest elution strength that does not elute the analyte- Consider using water-immiscible solvents for nonpolar mechanisms- Switch to a less retentive sorbent or a mixed-mode mechanism [51] | |
| NMR with Reduced Deuterated Solvents | Poor Deuterium Lock | - Insufficient level of deuterium in the solvent- Contaminants in recovered/reduced solvent | - Optimize solvent mixture (e.g., 1:1 methanol-deuterium oxide) for balance of cost and performance [16]- Ensure solvent purity and filter if necessary |
| Low Sensitivity/Noise | - Inadequate analyte concentration from SPE- Solvent artifacts or impurities | - Confirm SPE elution efficiency and concentration factor- Use high-purity solvents; check for chemical or particulate contamination [49] |
Frequently Asked Questions (FAQs)
Q1: What are the most significant operational challenges when reducing deuterated solvent use in high-throughput NMR? The primary challenges are maintaining a stable deuterium lock for the NMR spectrometer and ensuring sufficient solubility for the analytes, both of which can be compromised when minimizing expensive deuterated solvents. Using optimized solvent mixtures, such as methanol-deuterium oxide (1:1), can provide a practical balance between cost and analytical performance [16]. Contamination of recovered or reduced solvents is another major risk that can lead to spectral artifacts and poor locking.
Q2: How can we improve the recovery of target analytes from SPE cartridges before NMR analysis? To improve SPE recovery, systematically analyze where analyte loss occurs. Process standards through the entire protocol and collect fractions from each step (load, wash, elute) for analysis [51]. If breakthrough occurs during loading, strengthen the sample solvent or sorbent retention conditions. If analytes are retained but not eluted, increase the elution solvent strength or volume. Also, verify that analytes are not precipitating with proteins or becoming unstable in the sample matrix.
Q3: Our LC peaks are tailing, which complicates fraction collection for SPE-NMR. What are the quickest fixes? The most common fixes for peak tailing are to reduce the sample mass or injection volume (to address column overload) and to ensure the sample is dissolved in a solvent that is not stronger than the initial mobile phase [50] [52]. If the problem persists across all peaks, it may indicate a physical issue with the column, such as a void, which requires column inspection or replacement.
Q4: Can NMR effectively detect impurities that might be missed by LC-MS in our purified samples? Yes, NMR is an excellent orthogonal technique to LC-MS and is particularly adept at detecting isomeric impurities (e.g., positional isomers, tautomers), non-ionizable compounds, residual solvents, and degradation products that have distinct structural fingerprints but similar masses [32]. This makes it invaluable for comprehensive impurity profiling in pharmaceutical development.
Q5: What is the recommended solvent for cross-species metabolite fingerprinting that is also compatible with deuterated solvent reduction goals? Research indicates that methanol, including variants with 10% deuterated methanol (CD₃OD), is a highly effective and versatile extraction solvent for metabolite fingerprinting across multiple botanical species using both NMR and LC-MS [16]. It provides broad metabolite coverage and is a practical choice for developing standardized protocols.
Experimental Protocol: Optimized LC-SPE-NMR with Solvent Reduction
Title: Protocol for Automated LC-SPE-NMR Analysis with Minimized Deuterated Solvent Consumption
1. Sample Preparation (LC-MS Compatible)
- Homogenization: Homogenize plant or biological material to ensure uniformity [16].
- Extraction: Extract 50-300 mg of material with 1-2 mL of 90% methanol + 10% CD₃OD or a 1:1 mixture of methanol-deuterium oxide. The mass-to-solvent ratio should be optimized for the specific matrix [16].
- Clarification: Centrifuge the extract and filter the supernatant using a 0.45 µm or 0.2 µm syringe filter to remove particulates that could clog the LC or SPE system [52].
2. Liquid Chromatography Separation
- Column Selection: Use a suitable reversed-phase column (e.g., C18).
- Mobile Phase: Employ MS-compatible solvents (e.g., water and acetonitrile, both with 0.1% formic acid) in a gradient elution optimized for the compound class of interest.
- Injection: The injection volume and sample concentration should be calibrated to avoid column overload, which causes peak tailing [50].
- Post-Column Dilution: Implement a post-column tee to add a make-up solvent (e.g., water). This reduces the eluting strength of the LC solvent before it reaches the SPE cartridge, ensuring optimal retention of the target analytes on the SPE sorbent.
3. Solid Phase Extraction Trapping and Desalting
- SPE Sorbent: Select an appropriate sorbent chemistry (e.g., C18, C8) based on the analyte's hydrophobicity.
- Peak Triggering: Use the LC's UV or MS signal to automatically trigger the trapping of specific peaks of interest onto individual SPE cartridges.
- Desalting/Wash: Wash the trapped cartridge with a weak, volatile solvent (e.g., water or a mild aqueous buffer) to remove salts and highly polar contaminants from the LC mobile phase. The wash solvent must be strong enough to remove interferences but weak enough to retain the analyte [51].
4. Solvent Swap and Analyte Elution
- Drying: Pass a stream of inert gas (e.g., nitrogen) through the SPE cartridge to evaporate residual protic solvents.
- Elution for NMR: Elute the purified and concentrated analyte directly into the NMR flow cell using a minimized volume (e.g., 50-150 µL) of a deuterated solvent. The choice of solvent can be optimized; for example, a 1:1 CD₃OD:D₂O mixture may be sufficient for many applications, significantly reducing pure deuterated solvent use [16].
5. NMR Data Acquisition and Processing
- Automated Locking and Shimming: The NMR spectrometer automatically locks on the deuterium signal of the eluent and shims the magnet.
- Acquisition: Run predefined, automated NMR experiments (e.g., 1D ¹H, 1D ¹³C, or 2D experiments like COSY and HSQC).
- Data Processing: Use automated software to process the data (Fourier transformation, phasing, baseline correction) and, if available, perform database matching for initial structural identification [32].
Research Reagent Solutions for LC-SPE-NMR
Table 2: Essential Materials for LC-SPE-NMR Workflows
| Item/Category | Function/Purpose | Examples & Selection Notes |
|---|---|---|
| Deuterated NMR Solvents | Provides the deuterium lock signal for the NMR spectrometer; dissolves the sample for analysis. | CD₃OD (Deuterated Methanol): A common, versatile choice. D₂O (Deuterium Oxide): Often used in mixtures to reduce cost. Optimized Mixtures (e.g., 1:1 CH₃OH:D₂O or 90:10 CH₃OH:CD₃OD) can provide a balance of cost, lock stability, and extraction efficiency [16]. |
| SPE Sorbents & Cartridges | Isolate, purify, and concentrate target analytes from the LC eluent; enable solvent exchange. | Reversed-Phase (C18, C8, C4): For non-polar to medium-polarity compounds. Mixed-Mode Sorbents: Combine reversed-phase and ion-exchange mechanisms for superior cleanup of complex matrices, especially for ionizable analytes [51]. |
| LC Columns & Mobile Phases | Separate complex mixtures of compounds before they enter the SPE/NMR system. | Reversed-Phase Columns (e.g., C18). MS-Grade Solvents & Buffers (e.g., water, acetonitrile, methanol with 0.1% formic acid or ammonium buffers) ensure compatibility with in-line MS detection and prevent system blockages [50]. |
| Sample Filtration Supplies | Removes particulate matter from samples to protect LC and SPE components from clogging. | Syringe Filters (0.2 µm or 0.45 µm pore size), preferably made of materials compatible with the sample solvent (e.g., PTFE, nylon, cellulose) [52]. |
| Automation & Data Software | Orchestrates the entire hyphenated system, triggers actions based on data, and processes results. | LIMS (Laboratory Information Management System): Tracks samples and data. Automated Workflow Platforms: Control instruments and enable peak-based triggering of SPE trapping. NMR Processing Software: For structural elucidation and verification [32]. |
Workflow Logic for Solvent Reduction
The following diagram details the logical decision process for implementing deuterated solvent reduction in an LC-SPE-NMR protocol, helping to balance cost-saving goals with analytical data quality.
Solvent Reduction Decision Logic
LC-SPE-NMR in the Analytical Arsenal: A Comparative Advantage
Technical Support Center
Troubleshooting Guides & FAQs
FAQ: What are the most common issues when moving from Direct LC-NMR to LC-SPE-NMR?
| Issue | Likely Cause | Solution |
|---|---|---|
| Poor NMR sensitivity in LC-SPE-NMR | Analyte not fully eluting from SPE cartridge | Optimize the deuterated solvent used for elution; CD3CN is often effective [8]. |
| Failed trapping of analytes on SPE cartridge | Analyte is too polar for reversed-phase SPE sorption [8] | Dilute HPLC eluent with more water post-column to reduce elution strength; for highly polar compounds, method may not be suitable [53] [8]. |
| Broad or distorted chromatographic peaks in Direct LC-NMR | Overloaded HPLC column [8] | In LC-SPE-NMR, the separation can be run without overloading; the analyte will still be focused into a small elution volume [8]. |
FAQ: I am considering LC-SPE-NMR to reduce costs. What are the primary factors affecting deuterated solvent consumption?
The primary factor is the LC mobile phase. In Direct LC-NMR, the entire separation requires deuterated solvents, often including expensive deuterated organic modifiers like acetonitrile [9]. LC-SPE-NMR uses standard, non-deuterated HPLC-grade solvents for the separation. Deuterated solvent (e.g., CD3OD, CD3CN) is only used to elute the trapped analyte from the SPE cartridge into the NMR probe, typically requiring a volume of about 300 µL or less [8]. This can reduce deuterated solvent consumption by over 95% for a typical analysis.
FAQ: I'm getting an "ADC Overflow" error during my NMR experiment. What should I do?
This error means the NMR signal is too strong for the receiver. Solutions include [7] [54]:
- Reduce the receiver gain (
RG). Manually set it to a lower value (e.g.,gain=24). - Reduce the pulse width (
pw). Halving the value (e.g.,pw=pw/2) decreases the signal. Do not reducepwbelow ~1 microsecond. - Reduce the transmitter power (
tpwr). Reducing it by 6 dB has a similar effect to halving the pulse width.
FAQ: My sample is stuck in the magnet and won't eject. What should I do?
Never attempt to extract the sample by reaching into the magnet with any object [54]. First, check if the issue is software or hardware. If you hear a click or change in airflow when you try to eject, it's a hardware issue. Check that the VT gas line is properly connected and airflow is set correctly [54]. If there is no audible change, it is likely a software problem. Try restarting the acquisition process or consult facility staff [54] [55].
Experimental Protocols
Protocol: Standard Workflow for LC-SPE-NMR Analysis
This protocol outlines the steps for analyzing a mixture using the LC-SPE-NMR platform to achieve significant reduction in deuterated solvent usage [8].
- Sample Preparation: Prepare the sample extract in a suitable HPLC-compatible solvent (e.g., methanol). For botanicals, a common effective solvent is methanol, optionally with 10% deuterated methanol (
CD3OD) to aid the NMR lock at this stage [16]. - HPLC Separation:
- Column: Use an analytical or semi-preparative reversed-phase column.
- Mobile Phase: Use standard, non-deuterated solvents (e.g.,
H2O,CH3CN,CH3OH). The addition of 0.1% formic acid can improve chromatography. - Detection: Use in-line UV or MS detection to trigger peak trapping.
- Post-Column Dilution & SPE Trapping:
- Connect a post-column pump to dilute the HPLC eluent with at least two parts water. This reduces the elution strength, allowing analytes to be retained on the SPE cartridge [8].
- When a peak of interest is detected, the flow is directed to an SPE cartridge. Multiple trappings of the same analyte from repeated runs can be performed to increase concentration [8].
- Cartridge Drying: After trapping, purge the SPE cartridge with pressurized nitrogen gas to remove residual, non-deuterated solvents [8].
- NMR Analysis:
- Elute the purified analyte from the SPE cartridge directly into the NMR flow cell using a small volume (e.g., ~30 µL) of pure deuterated solvent (e.g.,
CD3CNorCD3OD). - Acquire NMR data without the need for strong solvent suppression techniques [8].
- Elute the purified analyte from the SPE cartridge directly into the NMR flow cell using a small volume (e.g., ~30 µL) of pure deuterated solvent (e.g.,
LC-SPE-NMR Workflow
The Scientist's Toolkit
Research Reagent Solutions for LC-SPE-NMR
| Item | Function in LC-SPE-NMR |
|---|---|
Deuterated Elution Solvents (e.g., CD3CN, CD3OD) |
Small volumes are used to transfer purified analytes from SPE cartridges to the NMR probe. CD3CN is often preferred for its low viscosity and sharp NMR signals [8] [19]. |
Non-deuterated HPLC Solvents (e.g., CH3CN, CH3OH, H2O) |
Standard, inexpensive solvents used for the entire liquid chromatography separation, drastically reducing cost compared to Direct LC-NMR [8]. |
| Reversed-Phase SPE Cartridges | Small cartridges (e.g., 2x10 mm) that trap HPLC-separated analytes. This key component disjoints the non-deuterated LC system from the NMR spectrometer [8]. |
| Post-column Pump | A pump used to add water to the HPLC eluent before it reaches the SPE cartridge, reducing the eluent's strength and ensuring analyte retention [8]. |
| Cryogenically Cooled NMR Probe | A flow probe where the electronics are cryogenically cooled. This reduces electronic noise and can provide a 3-4 fold increase in sensitivity, which is crucial for analyzing low-concentration analytes [9] [56]. |
Technical Support Center
Troubleshooting Guides
Guide 1: Addressing Low NMR Sensitivity in LC-SPE-NMR
- Problem: Poor signal-to-noise ratio in NMR spectra, preventing confident structural elucidation.
- Explanation: NMR is inherently less sensitive than MS. This issue is pronounced when analyzing low-concentration analytes from complex mixtures [9].
- Solution:
- Implement Multiple Trapping: Concentrate the analyte by repeatedly loading the same chromatographic peak onto the SPE cartridge. This can increase the amount of analyte transferred to the NMR flow cell [10].
- Optimize SPE Cartridge Phase: If analyte recovery is low, switch the SPE stationary phase. Divenylbenzene (DVB)-type polymers often provide better trapping efficiency for many natural products than standard C-18 phases [10].
- Verify Make-up Solvent: Ensure a post-column make-up flow of water (1-2 mL/min) is used to promote optimal analyte binding to the SPE cartridge [10].
Guide 2: Managing Solvent Interference and Costs
- Problem: Strong solvent signals overwhelm analyte signals in NMR spectra, and the cost of fully deuterated mobile phases is prohibitive.
- Explanation: Protons in standard HPLC solvents (e.g., acetonitrile, methanol) generate intense NMR signals that can mask analyte signals [9].
- Solution:
- Employ SPE Solvent Exchange: This is the core advantage of LC-SPE-NMR. After trapping, the analyte is washed with water to remove the original mobile phase and is then eluted with a pure, deuterated solvent (e.g., CD₃OD or CD₃CN). This provides a well-defined NMR solvent at a fraction of the cost of using deuterated solvents for the entire LC run [10].
- Select Appropriate Deuterated Solvent: Deuterated methanol and acetonitrile are prime candidates due to their good eluting power and hydrogen bonding capacity. Note that highly viscous solvents like DMSO-d6 are not typically used in online SPE-NMR elution [10].
Guide 3: Overcoming Challenges in Structural Isomer Differentiation
- Problem: LC-MS/MS data suggests a molecular formula, but several structural isomers are possible and cannot be distinguished by fragmentation patterns alone.
- Explanation: MS can struggle to differentiate isobaric compounds and positional isomers, as they often yield identical molecular masses and very similar fragmentation patterns [9] [57].
- Solution:
- Acquire 2D NMR Spectra: Use the analyte concentrated via SPE to perform heteronuclear NMR experiments like HSQC and HMBC. These experiments establish direct atom-atom connectivities and can unequivocally distinguish between isomers [9] [10].
- Analyze Coupling Constants: In the 1H NMR spectrum, use the J-coupling constants and signal splitting patterns to determine the number of neighboring hydrogens and the dihedral angles between them, which are characteristic of the molecular geometry [58].
Frequently Asked Questions (FAQs)
FAQ 1: Why can't I just use LC-MS for complete structure elucidation? LC-MS is excellent for determining molecular weight and formula, and provides fragmentation clues. However, it often cannot distinguish between structural isomers, and definitive structural identification typically requires comparison with an authentic standard [9] [57]. NMR is required for unambiguous determination of atom connectivity and spatial arrangement within the molecule [9].
FAQ 2: What are the key advantages of the SPE step in LC-SPE-NMR? The SPE (Solid Phase Extraction) step provides two critical benefits:
- Analyte Concentration: It focuses the chromatographic peak into a very small volume of deuterated solvent, matching the volume of the NMR flow cell and significantly enhancing sensitivity [10].
- Solvent Exchange: It enables complete replacement of the protonated LC mobile phase with a deuterated NMR solvent. This eliminates strong solvent signals in the NMR spectrum and allows for well-defined, reproducible NMR conditions ideal for database comparisons [10].
FAQ 3: My analyte isn't trapping efficiently on the SPE cartridge. What could be wrong? Trapping efficiency is highly dependent on the chemistry of your analyte and the SPE stationary phase. For charged or highly polar analytes like organic acids or alkaloids, standard reversed-phase (C-18) cartridges may be ineffective. Consider switching to a stationary phase with different selectivity, such as ion-exchange (SAX, SCX) or porous carbon materials [10].
FAQ 4: What is the typical sample requirement for obtaining a full set of 2D NMR data via LC-SPE-NMR? With multiple trapping, several dozen micrograms of an analyte can be accumulated on a single SPE cartridge. This quantity is typically sufficient to record a full set of structure-elucidating 2D NMR spectra (such as COSY, HSQC, and HMBC) within a few hours on a standard NMR spectrometer [10].
Experimental Data & Protocols
Table 1: Orthogonal Information from LC-MS and NMR for Structural Elucidation
| Structural Feature | LC-MS/MS Capability | NMR Capability |
|---|---|---|
| Molecular Formula | Excellent (via high-resolution MS) | Not directly determined |
| Functional Groups | Can identify some (e.g., sulfates via mass loss) | Excellent (via chemical shift) |
| Isomer Differentiation | Poor for many positional isomers | Excellent (via chemical shift and J-coupling) |
| Atom Connectivity | Inferred from fragmentation | Directly determined (via 2D experiments like COSY, HMBC) |
| Quantitation | Can be semi-quantitative; suffers from matrix effects | Inherently quantitative |
| Sensitivity | Excellent (femtomole level) | Moderate (microgram level) |
Table 2: Essential Research Reagent Solutions for LC-SPE-NMR
| Reagent / Material | Function in the Experiment |
|---|---|
| Divenylbenzene (DVB) SPE Cartridges | Solid-phase extraction material for trapping a wide range of organic analytes after LC separation. Often provides higher trapping efficiency for natural products than C-18 [10]. |
| Deuterated Methanol (CD₃OD) | Used to elute trapped analytes from the SPE cartridge to the NMR probe. A pure, defined NMR solvent that avoids signal interference [10]. |
| Deuterated Water (D₂O) | Used as a post-column make-up solvent to promote analyte binding to the SPE cartridge and to wash away residual protonated solvent [10]. |
| Deuterated Acetonitrile (CD₃CN) | An alternative elution solvent for the SPE cartridge, useful due to its different elutropic power and hydrogen bonding capacity [10]. |
Protocol: Standard Workflow for De Novo Structure Elucidation using LC-SPE-NMR
- LC Separation: Inject the complex sample (e.g., natural product extract) onto the HPLC system. Use UV or MS detection to monitor the separation and trigger subsequent steps [10].
- Peak Trapping & Solvent Exchange: As the peak of interest elutes, mix it with a make-up flow of water and direct it to an SPE cartridge. The analyte binds to the stationary phase while the protonated mobile phase is washed to waste [10].
- Analyte Elution to NMR: After washing, back-flush the SPE cartridge with a small volume (e.g., < 1 mL) of pure deuterated solvent (e.g., CD₃OD) to transfer the concentrated analyte to the NMR flow cell [10].
- NMR Data Acquisition: Acquire NMR data. Start with a 1H NMR spectrum. For unknown structures, proceed to collect 2D experiments such as COSY (H-H correlations), HSQC (H-C direct bonds), and HMBC (H-C long-range couplings) to establish the structural framework [9] [10].
- Data Integration & Analysis: Combine the molecular formula and fragmentation information from LC-MS with the detailed connectivity and stereochemistry information from NMR to unambiguously elucidate the complete molecular structure.
Workflow and Relationship Diagrams
FAQs: Enhancing Sensitivity in LC-SPE-NMR
1. What are the primary technological solutions for improving sensitivity and Lowering the Limit of Detection (LOD) in NMR-based methods?
The primary technological advancements for boosting sensitivity are cryogenically cooled probes (cryoprobes) and microcoil probe technology. Cryoprobes work by cooling the receiver coils and preamplifiers to very low temperatures (15-30 K), which reduces electronic noise and can increase the signal-to-noise ratio (S/N) by a factor of up to 4 [59]. Microcoils, on the other hand, focus on increasing mass sensitivity by using a solenoid coil design and a much smaller detection volume (e.g., 2.5-5 µL versus 40-120 µL for traditional probes) [59]. When these two technologies are combined, they can work synergistically to increase sensitivity and lower the detection limit by over 20-fold compared to conventional NMR probes [59].
2. How does the LC-SPE-NMR technique itself contribute to sensitivity gains?
The LC-SPE-NMR (Liquid Chromatography-Solid Phase Extraction-Nuclear Magnetic Resonance) workflow is a major advancement. It concentrates the analyte after chromatographic separation, leading to significant sensitivity gains [60] [61]. Key steps include:
- Analyte Trapping: Peaks of interest eluting from the HPLC column are trapped onto individual SPE cartridges, removing the original, often protonated, mobile phase [60] [61].
- Analyte Concentration: The trapped analytes are eluted from the SPE cartridge with a small volume (e.g., < 30 µL) of deuterated solvent directly into the NMR flow cell. This focusing effect can lead to a 2 to 4-fold increase in sensitivity, and for broader chromatographic peaks, the gain is even more substantial [61].
- Multiple Trapping: The same analyte from repeated LC injections can be trapped onto the same SPE cartridge, concentrating the sample and potentially improving the S/N by a factor of 10 or more [60] [61].
3. What practical issues can lead to lower-than-expected sensitivity, and how can they be troubleshooted?
Unexpected sensitivity loss can often be traced to sample preparation and hardware maintenance issues.
- Sample-Related Issues: Ensure your sample is perfectly clean. Particulates can clog the SPE cartridges or the NMR flow probe. Incompletely dissolved samples will lead to inaccurate concentration estimates and poor performance. For LC-SPE-NMR, optimize the post-column dilution with water to ensure quantitative retention of your analyte on the SPE cartridge [60].
- Hardware and Setup Issues: Check for physical problems like a clogged injection needle or blocked inline filters/frits, which can reduce the amount of sample reaching the system [50]. Carryover from prior injections can also contaminate your sample and distort data; perform blank injections and clean the autosampler and injection needle regularly [50]. Always use high-quality, clean, and fresh deuterated solvents for the final elution in SPE to avoid introducing contaminants [50].
Sensitivity Comparison of NMR Technologies
Table 1: Impact of different technologies on NMR sensitivity and application in LOD reduction.
| Technology | Key Mechanism | Reported Sensitivity Gain | Impact on LOD | Compatibility with LC-SPE-NMR |
|---|---|---|---|---|
| Cryoprobes | Cools receiver coils & preamplifiers to ~15-30 K to reduce electronic noise [59]. | Up to 4-fold increase in S/N [59]. | Significantly lowers LOD, enabling detection of smaller sample amounts. | Excellent; a core technology for modern high-sensitivity LC-(SPE)-NMR systems [59]. |
| Microcoils | Uses smaller detection volumes (e.g., 2.5-5 µL) to increase mass sensitivity [59] [60]. | Higher mass sensitivity per unit sample. | Allows for the analysis of very limited samples, as in CapNMR [59]. | Excellent for on-line (microcoil HPLC-NMR) and off-line (CapNMR) analyses [59]. |
| Combined Cryo & Microcoil | Integrates the noise reduction of cryogenics with the mass sensitivity of microcoils [59]. | Over 20-fold increase in S/N and LOD improvement [59]. | Dramatically lowers LOD, enabling heteronuclear experiments on LC timescales [59]. | The pinnacle of sensitivity for direct and indirect hyphenation. |
| LC-SPE Workflow | Post-column trapping and concentration of analytes using SPE before NMR analysis [60] [61]. | 2-4x S/N gain per run; >10x with multiple trapping [61]. | Major reduction in practical LOD by loading more analyte into the probe. | The foundational methodology for this technique. |
Table 2: Exemplary LOD and LOQ values achieved with sensitivity-enhanced NMR in related fields.
| Analytical Technique | Application Context | Analyte | Reported LOD | Reported LOQ | Key Enabling Technology |
|---|---|---|---|---|---|
| qNMR Spectroscopy [62] | Quantification of aged microplastics | Polystyrene (PS), Polyvinyl chloride (PVC), Polyethylene terephthalate (PET) | 0.87 - 2.79 µg/mL | 2.89 - 9.29 µg/mL | 600 MHz spectrometer with a QCI-P CryoProbe [62] |
| Pyrolysis-GC-MS [63] | Detection of tire wear particles in soil/sediment | Styrene butadiene rubber (SBR), Natural rubber (NR) | 3.8 - 7.7 µg/g | Not specified | Method optimization and Single Ion Monitoring (SIM) [63] |
Troubleshooting Guide: Sensitivity and LOD Issues
Problem: Inconsistent or Poor Signal-to-Noise Ratio Despite Using a Cryoprobe
- Potential Cause 1: Insufficient Sample Concentration or Poor Trapping on SPE Cartridge.
- Solutions: Verify the concentration of your sample solution. For LC-SPE-NMR, ensure the post-column dilution with water is optimized for your compound's hydrophobicity to ensure quantitative retention on the SPE cartridge [60]. Consider using multiple trapping (repeated injections) to increase the amount of analyte [60].
- Potential Cause 2: Hardware Issues or Contamination.
- Solutions: Check the NMR flow cell or probe for air bubbles. Run system suitability tests with a standard compound. Inspect and clean or replace SPE cartridges and in-line filters if they are clogged with particulates [50]. Ensure the cryoprobe is maintaining its correct temperature.
Problem: Unexpected Ghost Peaks or High Baseline in Chromatogram-NMR Data
- Potential Cause 1: Carryover from Previous Injections or Contaminated Solvents.
- Solutions: Perform a series of blank injections (with your strong solvent) to identify and flush out carryover. Clean the autosampler, injection needle, and loop thoroughly [50]. Prepare fresh mobile phases and use high-purity solvents.
- Potential Cause 2: Deuterated Solvent Impurities or Column Bleed.
- Solutions: Use fresh, high-quality deuterated solvents for the SPE elution step. If the ghost peaks increase with column usage, it may indicate degradation of the analytical or SPE column; replace the guard column or the column itself [50].
Experimental Protocol: Sensitivity Assessment via qNMR
This protocol is adapted from methods used to quantify aged microplastics, demonstrating how to establish a calibration curve and determine the LOD/LOQ for an analyte using a sensitivity-enhanced NMR system [62].
1. Materials and Instrumentation
- NMR Spectrometer: Equipped with a cryogenically cooled probe (e.g., Bruker Ascend 600 MHz with a QCI-P CryoProbe) [62].
- Deuterated Solvent: Choose based on analyte solubility (e.g., THF-d~8~, CDCl~3~) [62].
- Internal Standard: A high-purity compound with a sharp, non-overlapping signal and known concentration (e.g., Dimethyl sulfone, DMSO~2~) [62].
- Analyte: Your compound of interest (e.g., a deuterated molecule from LC-SPE-NMR research).
2. Sample Preparation
- Prepare a stock solution of your analyte in the chosen deuterated solvent.
- Prepare a series of standard solutions with known concentrations of the analyte, each containing the same, precise concentration of the internal standard [62]. The concentration range should bracket your expected LOD/LOQ.
3. NMR Acquisition Parameters
- Temperature: 298 K (room temperature) [62].
- Use a sufficiently long relaxation delay (d1) > 5-7 times the T1 of the slowest relaxing signal of interest to ensure quantitative conditions.
- Number of scans: Adjusted based on required S/N, but kept consistent for all standard samples.
4. Data Processing and Calculation
- Process all spectra with identical parameters (e.g., line broadening, phasing, baseline correction).
- For each standard spectrum, integrate the selected resonance peak for the analyte (I~a~) and the internal standard (I~is~).
- Generate a Calibration Curve: Plot the known concentration ratio (Analyte/IS) against the measured signal intensity ratio (I~a~/I~is~). A strong linearity (R² > 0.99) indicates a robust method [62].
- Calculate LOD and LOQ: LOD = 3.3 * σ / S and LOQ = 10 * σ / S, where σ is the standard deviation of the response (y-intercept of the calibration curve can be used) and S is the slope of the calibration curve.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential materials and reagents for high-sensitivity LC-SPE-NMR experiments.
| Item | Function / Role | Key Consideration |
|---|---|---|
| SPE Cartridges (e.g., 2x10 mm) | Post-column trapping and concentration of HPLC-eluted analytes; key to the SPE workflow [60] [61]. | Choice of sorbent (e.g., polystyrene-divinylbenzene, C18-bonded silica) must be compatible with the analyte's chemistry for high retention and elution efficiency [60]. |
| Deuterated Elution Solvents (e.g., CD~3~CN, CD~3~OD) | To elute trapped analytes from SPE cartridges in a small, concentrated volume for high-sensitivity NMR detection [60] [61]. | Purity is critical to avoid background signals. Using a minimal volume (< 30 µL) is key to the concentration effect [61]. |
| Internal Standard for qNMR (e.g., DMSO~2~) | A compound with known concentration used for quantitative concentration determination of the analyte via signal intensity ratios [62]. | Must be highly pure, chemically stable, and have a sharp, non-overlapping NMR signal in a clear region of the spectrum [62]. |
| Post-column Dilution Pump | Adds water to the HPLC eluent to reduce its eluting strength, ensuring analytes are retained on the SPE cartridges [60]. | The dilution ratio is a critical optimization parameter for quantitative trapping of analytes with varying polarities [60]. |
This technical support center provides targeted guidance for researchers utilizing the LC-SPE-NMR (Liquid Chromatography-Solid Phase Extraction-Nuclear Magnetic Resonance) technique, a powerful hyphenated technology for the structure elucidation of complex molecules. A primary advantage of this method is its significant reduction in the consumption of expensive deuterated solvents, a major operational cost in pharmaceutical R&D. The following FAQs, troubleshooting guides, and case studies are designed to help you optimize your experiments, overcome common challenges, and understand the proven impact of this technology on accelerating drug development and regulatory submissions.
FAQs: LC-SPE-NMR and Deuterated Solvent Reduction
1. How does LC-SPE-NMR specifically reduce deuterated solvent consumption compared to traditional NMR?
In traditional NMR, samples are dissolved in several hundred microliters of deuterated solvent. LC-SPE-NMR revolutionizes this process by trapping chromatographic peaks on an SPE cartridge after separation. The key steps are [10]:
- HPLC Separation: The sample mixture is separated using a standard (non-deuterated) HPLC mobile phase.
- Analyte Trapping: Single analyte peaks are concentrated and trapped on a solid-phase extraction (SPE) cartridge.
- Solvent Exchange: The non-deuterated HPLC mobile phase is removed by washing with a solvent of low elutropic strength (e.g., H₂O).
- Efficient Elution: The purified analyte is then backflushed from the SPE cartridge into the NMR flow probe using a small volume (typically less than 1 mL) of a deuterated solvent like CD₃OD or CD₃CN [10].
This process avoids the need for deuterated solvents in the mobile phase and minimizes the volume required for the final analysis, leading to substantial cost savings.
2. What are the sensitivity gains from using LC-SPE-NMR, and how do they impact data quality?
The primary sensitivity gain comes from post-HPLC focusing of the analyte. By trapping a chromatographic peak on an SPE cartridge and eluting it in a minimal volume of deuterated solvent, the analyte concentration in the NMR flow cell is dramatically increased. The observed signal-to-noise ratio (S/N) gains are inversely proportional to the chromatographic peak volume [10]. Furthermore, the technique allows for multiple trapping—repeatedly injecting and trapping the same analyte to accumulate several dozen micrograms on a single SPE cartridge. This enables the acquisition of essential 2D NMR spectra (such as COSY, HSQC, and HMBC) necessary for de novo structure elucidation within a few hours on a standard spectrometer [10].
3. Can LC-SPE-NMR be used for regulatory submissions, and what are the data integrity considerations?
Yes, structure elucidation data generated via NMR is critical for regulatory submissions to agencies like the FDA and EMA, particularly for the identification and confirmation of Active Pharmaceutical Ingredients (APIs) and impurities [32]. To ensure data integrity for submissions:
- Use GLP-compliant workflows: Ensure your laboratory follows Good Laboratory Practice standards.
- Maintain detailed documentation: Document all procedures, including SPE cartridge types, elution solvents, and NMR parameters.
- Ensure data traceability: From the raw chromatogram to the final NMR spectrum, the data flow must be clear and auditable.
- Validate methods: Where applicable, methods should be validated to demonstrate their reliability and robustness [32].
4. What is the "No-D NMR" method and how does it compare to LC-SPE-NMR?
"No-D NMR" is a complementary approach that allows NMR data acquisition without using any deuterated solvent. It uses the protonated solvent's signal for gradient shimming and employs pulse sequences like WET (Water Suppression Enhanced through T1 effects) to suppress the large solvent peaks in the proton spectrum [64]. The table below compares the two techniques:
Table: Comparison of Deuterated Solvent Reduction Techniques
| Feature | LC-SPE-NMR | No-D NMR |
|---|---|---|
| Deuterated Solvent Use | Minimal volume (~1 mL) for elution [10] | None required [64] |
| Primary Goal | Analyte concentration and purification; solvent exchange | Direct analysis in protonated reaction solvents |
| Best For | Full structure elucidation of components in a mixture, especially mass-limited samples | Reaction monitoring, quick checks, and quantitative analysis when solvent peaks don't overlap [64] |
| Sensitivity | Very high, can be boosted by multiple trapping [10] | Can be lower due to the dynamic range challenge of solvent suppression |
| Key Limitation | Requires optimization of SPE trapping/elution conditions [10] | Solvent suppression may weaken nearby analyte peaks [64] |
Troubleshooting Guide: Common LC-SPE-NMR Issues
Table: LC-SPE-NMR Troubleshooting Guide
| Problem | Potential Root Cause | Recommended Solution |
|---|---|---|
| Poor Trapping Efficiency | Incorrect SPE stationary phase for analyte class; insufficient make-up flow [10]. | - For most analytes, use DVB-type polymers or RP-C18 phases.- Optimize post-column make-up flow (often H₂O at 1-2 mL/min).- For polar/charged analytes (e.g., alkaloids), test SAX or SCX phases [10]. |
| Low NMR Signal/Noise | Incomplete analyte elution from SPE; elution volume too large [10]. | - Ensure deuterated solvent (e.g., CD₃OD, CD₃CN) has sufficient elutropic power.- The goal is to elute the analyte in a volume matching the NMR flow cell volume for maximum concentration. |
| Broad or Distorted Peaks | Poor chromatography; peak diffusion during transfer to NMR; magnetic field inhomogeneity. | - Optimize HPLC separation first.- Ensure a narrow elution band from the SPE cartridge.- Use the solvent peak for effective gradient shimming [64]. |
| High Backpressure | Blocked in-line filter or SPE cartridge frit [65]. | - Replace the in-line filter or the specific SPE cartridge.- Always filter solvents and samples before use to prevent blockages. |
Experimental Protocols & Workflows
Standard LC-SPE-NMR Workflow for Natural Product Extract Analysis
This protocol is adapted from applications for characterizing secondary metabolites in plant extracts [10].
1. Sample Preparation:
- Dissolve the crude extract in a solvent compatible with the HPLC mobile phase.
- Filter through a 0.45 µm membrane filter to remove particulates.
2. HPLC Separation:
- Use an optimized HPLC-UV or HPLC-MS method to separate the components.
- The mobile phase is a standard, non-deuterated mixture (e.g., Acetonitrile/H₂O with modifiers).
3. SPE Trapping:
- Post-column, add a makeup solvent (often H₂O) to promote analyte retention on the SPE cartridge.
- The system is triggered by UV or MS signal to divert a single chromatographic peak onto a pre-equilibrated SPE cartridge (e.g., 2 x 10 mm GP resin or C18).
- For trace components, employ multiple trapping by repeated injections to accumulate enough material on one cartridge [10].
4. Solvent Exchange:
- After trapping, wash the SPE cartridge with a prolonged flow of H₂O or D₂O to remove the original HPLC mobile phase salts and buffers.
5. NMR Analysis:
- Elute the purified and concentrated analyte from the SPE cartridge into the NMR flow probe using a small volume (e.g., ~30-50 µL) of deuterated solvent like CD₃OD or CD₃CN.
- Acquire NMR data. Start with 1D ¹H, then proceed to necessary 2D experiments (COSY, HSQC, HMBC) for structure elucidation.
The workflow is summarized in the diagram below:
Protocol for Method Development: SPE Condition Optimization
A critical step for success is ensuring your analyte of interest is efficiently trapped and released from the SPE cartridge.
Materials:
- Standard HPLC system with UV/VIS or MS detector
- LC-SPE interface unit
- Selection of SPE cartridges (e.g., DVB polymer, C18, C8, SAX, SCX)
- Makeup solvent (typically H₂O)
- Deuterated elution solvents (CD₃OD, CD₃CN)
Method:
- Trapping Test: Inject a standard of your analyte and monitor the UV effluent after the SPE cartridge. A successful trap will show no analyte signal passing through the cartridge.
- Elution Test: After trapping, elute the analyte into a vial (not the NMR) and analyze the eluent via HPLC-UV to quantify the recovery percentage. A good method should have >90% recovery.
- Solvent Selection: Test different deuterated solvents for elution efficiency. CD₃CN and CD₃OD are most common due to their good elution power and low viscosity [10].
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Materials for LC-SPE-NMR Experiments
| Item | Function / Explanation | Application Notes |
|---|---|---|
| SPE Cartridges (DVB Polymer) | The solid phase for trapping analytes; DVB (divinylbenzene) has shown high trapping efficiency for a wide range of compounds [10]. | Preferred for multiple trapping due to high capacity. |
| SPE Cartridges (RP-C18 Silica) | Alternative stationary phase for reversed-phase trapping. | A versatile choice; may have lower capacity for some compounds compared to DVB [10]. |
| Deuterated Methanol (CD₃OD) | NMR elution solvent. Effective elution power and commonly used for natural products. | A prime candidate for eluting a wide range of medium-polarity molecules from SPE cartridges [10]. |
| Deuterated Acetonitrile (CD₃CN) | NMR elution solvent. Different elutropic and hydrogen-bonding capacity compared to methanol. | Another prime candidate; useful if CD₃OD provides poor elution or for method differentiation [10]. |
| Make-up Solvent (H₂O) | Added post-column to dilute the organic mobile phase and promote strong retention on the SPE cartridge. | Critical for ensuring the analyte "sticks" to the SPE material and is not lost during loading [10]. |
| 0.5 µm In-line Filter | Placed post-column/pre-SPE to protect the cartridge and system from particulates. | Prevents blockages and high backpressure, a common source of system failure [65]. |
For researchers using LC-SPE-NMR, deuterated solvents represent a significant and recurring operational cost. This technical support center provides targeted guidance to help scientists and drug development professionals dramatically reduce deuterated solvent consumption without compromising data quality, directly supporting the goals of sustainable and cost-effective laboratory operations.
Quantitative Savings: Data at a Glance
The financial and consumption scale of deuterated solvents makes reduction strategies critically important for modern labs.
Table 1: Global Deuterated Solvents Market Overview [66] [67]
| Metric | Value (2023-2024) |
|---|---|
| Global Market Size | USD 2514.2 million (2024) |
| Projected Market Size (2031) | USD 7105.7 million |
| Compound Annual Growth Rate (CAGR) | 16.00% |
| Pharmaceutical Industry Demand Share | >60% |
| Annual Deuterated Solvent Consumption | >15,000 tons |
Table 2: Cost Comparison of Common Deuterated Solvents [67]
| Solvent | Typical Cost Multiplier (vs. Protonated) | Key Application in NMR |
|---|---|---|
| Deuterated Chloroform (CDCl₃) | 4x to 6x higher | Routine organic compound analysis |
| Deuterated DMSO (DMSO-d₆) | Similar premium | Polar molecules, challenging samples |
| Deuterated Benzene (C₆D₆) | Similar premium | Advanced NMR applications |
Experimental Protocols for Solvent Reduction
Protocol 1: No-D NMR with WET Solvent Suppression
This method enables the acquisition of usable NMR spectra directly from reaction mixtures using protonated solvents, eliminating the need for deuterated solvents during initial screening and reaction monitoring [64].
Detailed Methodology:
- Sample Preparation: Dissolve the reaction mixture aliquot directly in the protonated reaction solvent (e.g., methanol). No deuterated solvent is added.
- Magnetic Field Shimming: Utilize the strong proton signal of the solvent itself for gradient shimming. A selective excitation pulse is applied to excite only the solvent peak during the shimming experiment, ensuring magnetic field homogeneity without a deuterium lock [64].
- Data Acquisition with Suppression:
- Acquire a conventional 1-scan ¹H NMR spectrum to identify the chemical shifts of the large solvent peaks.
- Employ the WET (Water Suppression Enhanced through T1 effects) pulse sequence to suppress these identified solvent peaks. The system can be configured to automatically detect and suppress the tallest peaks and perform ¹³C decoupling if needed [64].
- The spectrum is automatically referenced using the known chemical shift of the solvent proton peak.
Limitations and Considerations: Solvent suppression may slightly attenuate analyte signals very close to the solvent peaks (e.g., within ~1 ppm). Quantitative analysis (qNMR) is possible only for peaks unaffected by the suppression pulses [64].
Protocol 2: Purity Assessment with DSC and qNMR
For quantifying volatile deuterated compounds like benzene-d₆ in a reference material context, combining DSC with qNMR minimizes the need for extensive use of other deuterated solvents [68].
Detailed Methodology:
- DSC Purity Analysis:
- Use a Differential Scanning Calorimeter (DSC) calibrated with Certified Reference Materials (CRMs) of Indium and Zinc.
- Seal the volatile sample (e.g., benzene-d₆) in a hermetic crucible.
- Cool the sample to -20°C to ensure complete solidification.
- Perform melting curve analysis at a low heating rate (e.g., 0.3 K/min) to ensure thermodynamic equilibrium and reproducible results. The purity is calculated from the melting point depression using the Van't Hoff equation [68].
- qNMR for Isotopic Impurity:
- Since DSC cannot distinguish isotopic compounds, use quantitative ¹H NMR (qNMR) to specifically quantify the protiated benzene impurity in the benzene-d₆ sample.
- The final purity of the deuterated compound is calculated by subtracting the qNMR-determined impurity content from the DSC-derived purity value [68].
This hybrid approach was successfully verified against the traditional mass balance method, confirming its effectiveness for volatile deuterated compounds [68].
Troubleshooting Guide and FAQs
Frequently Asked Questions
Q1: How can I stabilize the magnetic field without a deuterated solvent for the lock signal? Modern superconducting magnets have very low drift rates (4-15 Hz/hour). For short experiments (e.g., a standard ¹H NMR taking a few minutes), this drift is negligible and a deuterium lock is not strictly necessary. For longer experiments, shimming on the proton signal of the solvent itself provides sufficient magnetic field homogeneity for many applications [64].
Q2: Can I perform 2D NMR experiments (like COSY or HSQC) without deuterated solvents? Yes, it is possible. The primary challenge for 2D experiments, which have longer acquisition times, is magnetic field drift. If the drift rate is low enough over the experiment duration, meaningful 2D data can be acquired using the No-D NMR methodology with proper proton-based shimming [64].
Q3: My NMR sample has a poor lineshape after using alternative methods. What should I check? Poor resolution can often be traced to sample preparation. Ensure your sample is homogeneous and free of air bubbles or insoluble particles. Verify that the sample volume is sufficient for the NMR tube being used. For high-temperature experiments, allow the sample to reach full thermal equilibrium and re-shim before data acquisition [7].
Q4: Are there impurities that LC-MS might miss that these alternative methods can detect? Absolutely. NMR is orthogonal to LC-MS and is particularly adept at detecting isomeric impurities (positional isomers, tautomers), non-ionizable compounds, and residual solvents that may not be easily visible by mass spectrometry [32].
Research Reagent Solutions
Table 3: Essential Materials for Solvent-Reduced NMR [68] [64]
| Reagent / Material | Function in Experiment |
|---|---|
| Protonated Solvents (e.g., Methanol) | Serves as the low-cost solvent for No-D NMR; the target for signal suppression. |
| WET NMR Pulse Sequence | Key software/hardware capability for suppressing large solvent signals in protonated solvents. |
| Hermetic Sealed Crucibles | Essential for DSC analysis of volatile deuterated compounds to prevent sample loss. |
| Certified Reference Materials (CRMs) | Used for calibrating DSC (e.g., Indium, Zinc) and qNMR (e.g., pure benzene) for accurate quantification. |
| qNMR Internal Standard | A substance of known purity used for quantitative calibration in NMR experiments. |
Workflow Visualization
The following diagram illustrates the logical decision pathway for choosing the appropriate solvent-reduction strategy based on your experimental goal.
Decision Workflow for Solvent Reduction
Conclusion
LC-SPE-NMR has unequivocally evolved from an academic curiosity into a robust, indispensable analytical tool that directly addresses one of the most significant practical constraints in NMR-based analysis: the high cost of deuterated solvents. By decoupling the chromatographic separation from NMR detection through an intelligent SPE interface, this technique delivers substantial cost savings, enhanced sensitivity via analyte focusing, and superior spectral quality in pure deuterated solvents. Its proven utility in de novo structure elucidation of natural products, impurity profiling, and metabolite identification is accelerating discovery and development cycles in pharmaceuticals. Future advancements will likely focus on expanding the range of analyzable compounds—particularly very polar molecules—through new SPE materials, further miniaturization and automation, and deeper integration with mass spectrometry. As these innovations mature, LC-SPE-NMR is poised to become an even more central pillar in the analytical workflows of biomedical and clinical research, making high-quality structural data more accessible and affordable than ever before.
References
- 1. Why does NMR have an inherently low sensitivity? [https://www.nanalysis.com/nmready-blog/2022/12/14/why-nmr-has-an-inherent-low-sensitivity-in-comparison-with-other-spectroscopic-methods]
- 2. Sensitivity Enhancement in Solution NMR - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3967054/]
- 3. Solvent Suppression - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/solvent-suppression]
- 4. UMass Nuclear Magnetic Resonance (NMR) Labs [https://websites.umass.edu/weiguoh/?cat=160]
- 5. How To Prepare And Run An NMR Sample - Blogs - News [https://www.alwsci.com/news/how-to-prepare-and-run-an-nmr-sample-85160307.html]
- 6. Expert Advice: Performing and analyzing nuclear magnetic ... [https://www.drugdiscoverynews.com/expert-advice-solving-the-protein-nmr-puzzle-15852]
- 7. Troubleshooting | Department of Chemistry and Biochemistry [https://chem.umd.edu/research/core-facilities/biomolecular-nmr/user-guide/troubleshooting]
- 8. Solid Phase Extraction NMR - an overview [https://www.sciencedirect.com/topics/chemistry/solid-phase-extraction-nmr]
- 9. The integration of LC-MS and NMR for the analysis of low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6339611/]
- 10. HPLC–SPE–NMR — A Novel Hyphenation Technique [https://www.chromatographyonline.com/view/hplc-spe-nmr-novel-hyphenation-technique]
- 11. HPLC Troubleshooting Guide [https://scioninstruments.com/us/blog/hplc-troubleshooting-guide-2/]
- 12. Solvent suppression: the key to measure your samples ... [https://magritek.com/2022/03/24/solvent-suppression-the-key-to-measure-your-samples-immediately-while-they-are-synthesized/]
- 13. NMR Sample Preparation [https://cse.umn.edu/chem/nmr-sample-preparation]
- 14. Application of LC–MS and LC–NMR Techniques for ... [https://www.sciencedirect.com/science/article/pii/B9780123979223000022#!]
- 15. Common Questions & Problems | Baylor Sciences Building [https://bsb.research.baylor.edu/core-research-centers/center-nmr-spectroscopy/common-questions-problems]
- 16. Optimization of Extraction Methods for NMR and LC-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388471/]
- 17. Chromatography Troubleshooting Guides-Liquid ... [https://www.thermofisher.com/us/en/home/industrial/chromatography/chromatography-learning-center/chromatography-consumables-resources/chromatography-troubleshooting-guides/chromatography-troubleshooting-guides-liquid-chromatography.html]
- 18. Running NMR Experiments Using Non-Deuterated Solvents [https://www.jeolusa.com/RESOURCES/Analytical-Instruments/No-D-NMR]
- 19. Deuterated Solvents for NMR: Guide [https://allanchem.com/deuterated-solvents-nmr-guide/]
- 20. LC–NMR for Natural Product Analysis: A Journey from an ... [https://www.mdpi.com/2413-4155/3/1/6]
- 21. Common Problems in Solid-Phase Extraction: A Practical ... [https://www.welch-us.com/blogs/knowleage-base/common-problems-in-solid-phase-extraction-a-practical-troubleshooting-guide]
- 22. Troubleshooting 1 H NMR Spectroscopy [http://www.chem.rochester.edu/notvoodoo/pages/troubleshooting/proton_nmr.php]
- 23. Effective LC Troubleshooting: Symptom-Based Strategies ... [https://www.restek.com/articles/effective-lc-troubleshooting-symptom-based-strategies-and-solutions?srsltid=AfmBOoq2URoF3pFCouBrYUlDpOr7TRBwRhbr75wIv4GHy1oCDjYEtcNQ]
- 24. Essentials of LC Troubleshooting, Part IV: What Is Going ... [https://www.chromatographyonline.com/view/essentials-of-lc-troubleshooting-part-iv-what-is-going-on-with-the-baseline-]
- 25. Investigations of artifact peaks in sensitive high ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967399011358]
- 26. Identifying and Overcoming Artifacts in 1 H-Based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10037324/]
- 27. Optimizing NMR Processing: Techniques and Best Practices [https://www.jeolusa.com/NEWS-EVENTS/Blog/optimizing-nmr-processing-techniques-and-best-practices]
- 28. Peak Purity in HPLC: Assessment, Troubleshooting, and ... [https://www.sepscience.com/peak-purity-in-hplc-assessment-troubleshooting-and-best-practices-11506]
- 29. Method Development for Drug Impurity Profiling: Part 1 [https://www.chromatographyonline.com/view/method-development-drug-impurity-profiling-part-1]
- 30. Problem with Impurity.Please help me! [http://www.chromforum.org/viewtopic.php?t=17801]
- 31. How to Reduce 1H NMR – Quantitative Evaluation Errors [https://www.aiinmr.com/nmr-spectroscopy-articles-case-studies/how-to-reduce-1h-nmr-quantitative-evaluation-errors/]
- 32. 2025 Pharma Trends: Structure elucidation services by NMR [https://resolvemass.ca/structure-elucidation-services-by-nmr/]
- 33. Trapping Mode Two-Dimensional Liquid Chromatography [https://www.chromatographyonline.com/view/trapping-mode-two-dimensional-liquid-chromatography-low-level-impurity-enrichment-pharmaceutical-development]
- 34. Trapping mode two-dimensional liquid chromatography for ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967323002698]
- 35. HPLC Column Selection Guide [https://www.auroraprosci.com/HPLC-column-selection-guide?srsltid=AfmBOoorQRQJBSZfS8EsdyI9prM3kdK2v9Rf1b0SzkAN1UlAU_Zdf_er]
- 36. Quantitative structure-retention relationship for reliable ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914021009814]
- 37. Solvent signal suppression in NMR [https://www.sciencedirect.com/science/article/abs/pii/S007965651000021X]
- 38. LC-UV-solid-phase extraction-NMR-MS combined with a ... [https://pubmed.ncbi.nlm.nih.gov/14616013/]
- 39. NMR solvent selection - that also allows sample recovery [https://biochromato.com/nmr-solvent-selection/]
- 40. Selection Guide on Deuterated Solvents for NMR [https://labinsights.nl/en/article/selection-guide-on-deuterated-solvents-for-nmr]
- 41. Improving, Retaining, and Separating Polar Compounds ... [https://www.waters.com/blog/improving-retaining-and-separating-polar-compounds-using-chromatographic-techniques/?srsltid=AfmBOooh6qgJU9A6sHC9AdS8vfceeJ0fPW0y_2OGRMJY_gU8DLVjKH9q]
- 42. LC–MS Sensitivity: Practical Strategies to Boost Your ... [https://www.chromatographyonline.com/view/lc-ms-sensitivity-practical-strategies-boost-your-signal-and-lower-your-noise]
- 43. Separation Technique for the Determination of Highly Polar ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901216/]
- 44. NMR-Based Chromatography Readouts: Indispensable Tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9658744/]
- 45. Liquid Chromatography Nuclear Magnetic Resonance [https://www.sciencedirect.com/topics/chemistry/liquid-chromatography-nuclear-magnetic-resonance]
- 46. Optimization of Extraction Methods for NMR and LC-MS ... [https://www.mdpi.com/1420-3049/30/16/3379]
- 47. NMR Sample Preparation - Chemical Instrumentation Facility [https://www.cif.iastate.edu/nmr/nmr-tutorials/sample-preparation]
- 48. How to make an NMR sample [https://chem.ch.huji.ac.il/nmr/preparation/preparation.html]
- 49. How Deuterated NMR Solvents Works — In One Simple ... [https://www.linkedin.com/pulse/how-deuterated-nmr-solvents-works-one-simple-flow-rks2f/]
- 50. LC Troubleshooting Essentials: A Guide to Common ... [https://www.chromatographyonline.com/view/lc-troubleshooting-essentials-a-guide-to-common-problems-and-solutions-for-peak-tailing-ghost-peaks-and-pressure-spikes]
- 51. Three Common SPE Problems | LCGC International [https://www.chromatographyonline.com/view/three-common-spe-problems-0]
- 52. Some Common Issues With Liquid Chromatography And ... [https://www.alwsci.com/news/some-common-issues-with-liquid-chromatography-84962676.html]
- 53. Comparison of analytical and semi-preparative columns for ... [https://www.sciencedirect.com/science/article/abs/pii/S002196730502217X]
- 54. Trouble Shooting Page [http://nmr.ucsd.edu/troubleshooting.html]
- 55. Troubleshooting 101 [https://www.emory.edu/NMR/troubleshooting.html]
- 56. Fully Integrated Analysis of Metabolites, Impurities, and ... [https://www.spectroscopyonline.com/view/fully-integrated-analysis-metabolites-impurities-and-degradants-using-lc-nmr-ms]
- 57. Mass Spectrometry for Natural Products Research [https://www.chromatographyonline.com/view/mass-spectrometry-natural-products-research-challenges-pitfalls-and-opportunities]
- 58. How to read NMR spectra from the basics (chemical shift, ... [https://www.jeol.com/column/detail005.php]
- 59. Research Assistant - Bohrium | AI for Science with Global Scientists [https://www.bohrium.com/paper-details/recent-advancements-in-hplc-nmr-and-applications-for-natural-product-profiling-and-identification/811634972153610241-3640]
- 60. Thieme E-Journals - Planta Medica / Full Text [https://www.thieme-connect.de/products/ejournals/html/10.1055/s-2005-873114]
- 61. - LC - SPE reviews NMR [https://www.selectscience.net/product/lc-spe-nmr]
- 62. Overcoming the challenge of quantifying aged microplastic by ... [https://pubs.rsc.org/en/content/articlehtml/2025/em/d5em00393h]
- 63. Improvement and suitability of analytical method for the ... [https://www.sciencedirect.com/science/article/pii/S3051060025000137]
- 64. Performing NMR Spectroscopy Without Deuterated Solvents [https://www.azom.com/article.aspx?ArticleID=19599]
- 65. LC Troubleshooting: The Basics | LCGC International [https://www.chromatographyonline.com/view/lc-troubleshooting-basics-0]
- 66. The global deuterated NMR solvents market size will be ... [https://www.cognitivemarketresearch.com/deuterated-nmr-solvents-market-report]
- 67. Deuterated Compounds Market Size & Forecast [2033] [https://www.marketgrowthreports.com/market-reports/deuterated-compounds-market-102861]
- 68. Quantification of a volatile deuterated compound by the ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914022003344]