Decoding Molecular Complexity: The Scaffold Tree Methodology for Hierarchical Ring Analysis in Modern Drug Discovery
This article provides a comprehensive overview of the scaffold tree methodology for hierarchical ring analysis, tailored for researchers, scientists, and drug development professionals.
Decoding Molecular Complexity: The Scaffold Tree Methodology for Hierarchical Ring Analysis in Modern Drug Discovery
Abstract
This article provides a comprehensive overview of the scaffold tree methodology for hierarchical ring analysis, tailored for researchers, scientists, and drug development professionals. We explore the foundational concepts, historical evolution, and core principles of scaffold trees, detailing the step-by-step algorithmic implementation and its applications in drug discovery, such as scaffold hopping and chemical space visualization. The article addresses common troubleshooting issues and optimization strategies, including AI integration, and validates the methodology through comparative analysis with alternative approaches. Finally, we discuss future directions for biomedical and clinical research.
Foundations of Scaffold Trees: Origins, Principles, and Chemical Intuition
Core Concepts and Definitions in Scaffold Analysis
The systematic analysis of molecular scaffolds is foundational to modern cheminformatics and drug discovery. This methodology enables researchers to classify compound libraries, visualize chemical space, and derive meaningful structure-activity relationships (SAR) by focusing on core molecular architectures [1].
1.1 Foundational Scaffold Definitions The field is built upon several key, hierarchically related definitions:
- Bemis-Murcko Framework: The cornerstone definition, which identifies the scaffold as the union of all ring systems and the linker atoms that connect them, with all side chains removed [2] [3]. This provides a concrete, chemically detailed core structure.
- Graph Framework (Murcko Scaffold): An abstraction of the Bemis-Murcko framework where atom types and bond orders are disregarded, leaving only the topological skeleton [2] [3]. This clusters molecules with similar shape but different atomic compositions.
- Scaffold (Oprea) Topology: A further abstraction obtained by iteratively replacing vertices of degree two with a single edge, resulting in a minimal graph that describes the ring structure's connectivity [2].
1.2 The Evolution to Hierarchical Systems While powerful, single-scaffold definitions have limitations, such as clustering molecules with minor structural differences into separate groups [3]. This led to the development of hierarchical systems that relate scaffolds through deconstruction rules:
- Hierarchical Scaffold Clustering (HierS): Generates a hierarchy by dissecting a framework into all possible parent scaffolds through the stepwise removal of entire ring systems. A child scaffold can have multiple parents in this network [2] [3].
- The Scaffold Tree: A deterministic, rule-based algorithm that iteratively removes one ring at a time from a scaffold according to a set of chemical priorities (e.g., ring complexity, heteroatom content). This creates a unique, linear path of scaffolds from the original molecule to a single-ring root, forming a strict tree hierarchy [4] [3].
- Scaffold Networks: An exhaustive approach that, like HierS, generates all possible parent scaffolds via ring removal but without applying prioritization rules. This creates a complex network with multi-parent relationships, offering a more comprehensive exploration of chemical space at the cost of complexity [3].
- Multi-Dimensional Frameworks (Molecular Anatomy): A recent, flexible approach that defines nine different molecular representations at varying abstraction levels. It combines multiple fragmentation rules to create a multi-dimensional network of interconnected frameworks, aiming to capture SAR information more effectively than single-rule methods [1].
Table 1: Comparative Analysis of Hierarchical Scaffold Methodologies
| Methodology | Core Principle | Hierarchy Type | Key Advantage | Primary Limitation |
|---|---|---|---|---|
| Bemis-Murcko Framework [2] [3] | Isolation of rings and linkers. | Single level, no hierarchy. | Simple, intuitive, chemically detailed. | Can separate highly similar molecules. |
| HierS (Hierarchical Scaffolding) [2] [3] | Removal of entire ring systems. | Network (multi-parent). | Captures all sub-structures. | Can be complex; not a unique tree. |
| Scaffold Tree [4] [3] | Rule-based, iterative single-ring removal. | Unique, deterministic tree. | Clear, interpretable hierarchy; efficient navigation. | Rule-dependent; may not generate all relevant sub-cores. |
| Scaffold Network [3] | Exhaustive single-ring removal. | Complex network. | Explores all possible sub-structures; good for activity cliff analysis. | Can become very large and difficult to visualize. |
| Molecular Anatomy [1] | Multiple scaffold definitions & fragmentation rules. | Multi-dimensional network. | Flexible, captures SAR from diverse chemotypes. | Higher computational and conceptual complexity. |
Computational Protocols for Scaffold Generation and Analysis
2.1 Protocol: Generating a Bemis-Murcko Framework This is the fundamental first step for most scaffold analyses [5].
- Input Preparation: Provide molecular structures in a standard format (e.g., SMILES, SDF).
- Preprocessing: Standardize structures (e.g., neutralize charges, remove solvents) using a toolkit like RDKit or OpenBabel.
- Side Chain Removal: Algorithmically identify and detach all acyclic atoms that are not part of a linker connecting two ring systems.
- Framework Output: Return the remaining connected structure consisting of rings and inter-ring linkers. Optionally, generate the graph framework by converting all atoms to carbon and all bonds to single bonds.
2.2 Protocol: Constructing a Scaffold Tree The following steps outline the rule-based algorithm to build a deterministic scaffold hierarchy [3]:
- Input: Start with a Bemis-Murcko framework (including atoms connected via double bonds to preserve hybridization).
- Ring Perception: Identify all individual rings using a Smallest Set of Smallest Rings (SSSR) algorithm.
- Iterative Ring Removal: While the scaffold has more than one ring: a. Identify removable "terminal" rings (whose removal does not disconnect the scaffold). b. Apply a series of prioritization rules to select exactly one ring for removal. Key rules typically favor retaining, in order: i) Rings with heteroatoms, ii) Larger rings, iii) Aromatic rings, iv) Rings with more complex substitution patterns. c. Remove the selected ring and any attached linker atoms that become non-connecting. d. Record the new parent scaffold.
- Tree Assembly: Link each child scaffold to its single, rule-determined parent scaffold from the step above. The final single-ring scaffold is the leaf node.
2.3 Protocol: Conducting a Scaffold-Based SAR Analysis (HDAC7 Case Study) This protocol, based on a published HTS analysis [1], details how to identify active chemotypes.
- Dataset Curation: Use a dataset with associated bioactivity data. Example: 26,092 commercial compounds tested for HDAC7 inhibition, stratified by percent inhibition (e.g., Inactive: <19%, Very Strong: >80%) [1].
- Scaffold Generation: Apply the Bemis-Murcko algorithm to all compounds to extract molecular frameworks.
- Activity Annotation: Assign each scaffold an activity score based on the compounds it represents. Common metrics include:
- Active Hit Rate: (Number of active compounds containing scaffold / Total number of compounds containing scaffold).
- Average Potency of active compounds per scaffold.
- Hierarchical Enrichment Analysis: Build a Scaffold Tree or Network from the frameworks. Propagate activity annotations upward from child to parent scaffolds to identify enriched branches or sub-structures common to active compounds.
- Visualization & Interpretation: Use a tool like Scaffold Hunter or a custom treemap [2] to visualize the hierarchy, coloring nodes by activity metrics to quickly identify promising active cores for lead optimization.
Applications in Drug Discovery and Chemical Biology
3.1 Mapping Chemical Space and Library Design Scaffold analysis is critical for understanding the coverage and diversity of compound libraries. By organizing libraries into a scaffold hierarchy, researchers can ensure broad coverage of chemical space or, conversely, focus on a specific region enriched for a target class [1] [2]. The analysis of the PubChem database to create a background scaffold hierarchy for visualization is a prime example of mapping empirical chemical space [2].
3.2 Identifying Privileged Substructures and Scaffold Hopping A core application is the data-mining of known drugs or bioactive molecules to identify "privileged scaffolds"—core structures that appear frequently in compounds active against a particular target family [3]. Furthermore, scaffold hierarchies enable scaffold hopping, the intentional design of novel active compounds with a different core but similar spatial orientation of functional groups [3] [6]. A recent study successfully designed a novel glycosyl-based α-glucosidase inhibitor scaffold using scaffold hopping informed by pharmacophore and 3D-QSAR models [6].
3.3 Analysis of High-Throughput Screening (HTS) Data In HTS triage, scaffold-based clustering groups actives sharing a common core, helping to distinguish true SAR from noisy assay data. The "Molecular Anatomy" approach demonstrated superior performance in clustering active molecules from different structural classes and capturing SAR in a COX-2 inhibitor dataset and a large HDAC7 HTS campaign [1].
3.4 Enabling Explainable Machine Learning Incorporating scaffold knowledge addresses the "black box" limitation of many deep learning models in drug discovery. By using a scaffold-based split (ensuring training and test sets share no common scaffolds), researchers can better evaluate a model's ability to generalize to novel chemotypes [7]. Furthermore, knowledge graphs that integrate elemental and functional group information with molecular graphs can provide chemically sound explanations for model predictions [7].
Table 2: The Scientist's Toolkit for Scaffold Research
| Tool/Reagent | Category | Primary Function in Scaffold Analysis | Key Features / Examples |
|---|---|---|---|
| RDKit [8] | Open-Source Cheminformatics Library | Core library for reading molecules, performing substructure searches, and generating Bemis-Murcko frameworks. | Python/C++ library; widely used for prototyping. |
| Scaffold Generator [3] | Open-Source Java Library | Dedicated library for generating scaffold trees, networks, and hierarchies from molecular datasets. | Built on CDK; highly customizable with multiple framework definitions. |
| Scaffold Hunter [2] [4] | Visualization Software | Interactive visualization and exploration of chemical datasets using scaffold trees and other hierarchies. | Enables intuitive navigation of chemical space linked to properties. |
| Scaffvis [2] | Web-Based Visualization Tool | Hierarchical, treemap-based visualization of compound sets on a background of known chemical space (e.g., PubChem). | Provides context by showing scaffold frequency in a reference database. |
| Molecular Anatomy Web Interface [1] | Web Application | Implements the multi-dimensional scaffold network generation and analysis for HTS data. | Applies nine scaffold representations; useful for complex SAR analysis. |
| ChEMBL Database [1] [8] | Bioactivity Database | Source of curated molecules and bioactivity data for validating scaffold analysis methods and identifying privileged structures. | Contains scaffolds and indications of known drugs [8]. |
Data, Metrics, and Advanced Integrations
4.1 Quantitative Analysis of Scaffold Diversity Key metrics are used to quantify the scaffold composition of a compound collection [1]:
- Scaffold Frequency Distribution: The number of compounds represented by each unique scaffold. Typically follows a power-law distribution, with few scaffolds representing many compounds and many scaffolds ("singletons") representing a single compound.
- Scaffold Hit Rate: A crucial metric in HTS analysis, calculated as the percentage of compounds containing a given scaffold that show bioactivity above a threshold.
- Hierarchical Enrichment Factor: Measures whether a particular branch of a scaffold tree is statistically enriched with active compounds compared to the random expectation for the dataset.
Table 3: Scaffold Analysis of Sample Datasets
| Dataset | Source | Number of Compounds | Key Scaffold Analysis Finding | Reference |
|---|---|---|---|---|
| Clinical COX-2 Inhibitors | Integrity Database | 816 | Multi-representation "Molecular Anatomy" approach effectively clustered actives from different structural classes. | [1] |
| HDAC7 HTS Library | Commercial & Internal | 26,092 | Scaffold-based analysis identified chemotypes enriched in strong and very strong inhibitors. | [1] |
| PubChem Compound Database | PubChem | ~100 million (background) | Large-scale analysis defined an empirical scaffold hierarchy used as a universal background for visualization. | [2] |
| Collection of Open Natural Products (COCONUT) | COCONUT DB | >450,000 | Scaffold network generation completed within one day, demonstrating scalability of modern tools. | [3] |
4.2 Integration with Knowledge Graphs and AI The frontier of scaffold analysis involves its integration with advanced artificial intelligence. Knowledge graphs that encode chemical prior knowledge—such as element properties, functional groups, and known scaffold-bioactivity relationships—can be used to enhance deep learning models [7]. This integration guides models to learn chemically meaningful representations, improves generalization across scaffold hops, and increases the interpretability of predictions by tracing model attention back to specific substructures or scaffold rules.
The Scaffold Tree algorithm, introduced by Schuffenhauer et al. in 2007, established a foundational methodology for the systematic and hierarchical organization of chemical space [9]. Within the broader thesis of scaffold tree methodology for hierarchical ring analysis, this algorithm represents a critical evolution from simple scaffold identification to a deterministic classification system. It transforms molecular frameworks into a unique tree hierarchy through iterative ring removal, enabling researchers to navigate complex datasets intuitively [4]. This approach addressed a key need in medicinal chemistry and drug development: moving beyond flat, list-based comparisons of compounds to understanding inheritance relationships and structural ancestry within large-scale screening data [10]. The algorithm's design, which is data-set-independent and scales linearly with the number of compounds, provided a robust tool for visualizing the scaffold universe, clustering compounds, and identifying novel bioactive molecules [11].
Algorithmic Foundation and Core Principles
The core operation of the Scaffold Tree algorithm is the stepwise simplification of a molecular framework (the Murcko scaffold) into a series of parent scaffolds, culminating in a single root ring [9]. This process is governed by a series of chemically meaningful prioritization rules applied during each ring-removal step, ensuring that the most characteristic rings of the molecule are retained for as long as possible [10].
Hierarchy Generation Workflow: The tree is built from the leaf nodes (the full molecular frameworks) upward toward a root. For each molecule:
- The molecular framework is generated by removing all terminal side chains (acyclic appendages).
- Rings are iteratively removed one at a time from this framework according to a fixed set of rules.
- Each removal creates a new, simpler scaffold that becomes the parent node of the previous, more complex scaffold.
- This process continues until only a single ring remains, which serves as the root scaffold for that particular branch [4] [11].
Prioritization Rules for Ring Removal: The order of ring removal is deterministic and based on the following hierarchy (applied sequentially until a decision is made):
- Bridged ring systems are retained; rings are removed from systems without bridged rings first.
- Spiro rings are retained; rings are removed from systems without spiro rings first.
- Heteroatom content: Rings with fewer heteroatoms are removed before rings with more heteroatoms.
- Ring size: Smaller rings are removed before larger rings.
- Aromaticity: Aliphatic rings are removed before aromatic rings.
- If ties remain, a predefined canonical order is applied [10].
This rule set ensures that peripheral, simpler, and less characteristic rings are pruned first, preserving the core pharmacophoric features of the molecule at higher levels of the tree [9].
Diagram 1: Scaffold Tree Generation Workflow (94 chars)
Diagram 2: Ring Removal Prioritization Rule Hierarchy (95 chars)
Quantitative Applications and Analysis
The Scaffold Tree algorithm's utility is demonstrated through its application to large, real-world chemical databases. Its deterministic nature allows for consistent analysis and comparison across different studies.
Table 1: Key Algorithmic Properties from Original Publication [9] [10]
| Property | Description | Implication |
|---|---|---|
| Determinism | Unique, reproducible tree for any given input molecule. | Enables consistent analysis and sharing of results. |
| Data-Set Independence | Tree generation depends only on the molecule's structure, not on the surrounding dataset. | Trees remain stable when compounds are added to or removed from an analysis. |
| Scalability | Computational complexity scales linearly (O(n)) with the number of compounds. | Capable of processing large-scale databases (e.g., >1 million compounds). |
| Chemical Intuitiveness | Prioritization rules preserve chemically characteristic rings (bridged, spiro, heteroatom-rich). | Resulting hierarchy aligns with medicinal chemists' intuition about molecular cores. |
Table 2: Analysis of PubChem Database Using Scaffold Hierarchy (Post-2007 Application) [2]
| Analysis Dimension | Finding | Significance for Hierarchical Ring Analysis |
|---|---|---|
| Hierarchy Structure | A 9-level rooted tree (8 scaffold levels + molecule leaves) was sufficient to map the PubChem chemical space. | Defines a practical depth for comprehensive hierarchical visualization of vast empirical chemical space. |
| Branching Factor | Native Scaffold Trees often have highly variable branching, complicating visualization. | Motivated the development of modified hierarchies (e.g., in Scaffvis) for more homogeneous visual layouts. |
| Background Mapping | User datasets can be visualized against the background of the pre-computed PubChem scaffold hierarchy. | Enables contextual analysis by showing how a target compound set relates to the broader, known chemical universe. |
| Visualization | Implemented in the web tool Scaffvis as an interactive, zoomable treemap. | Translates hierarchical ring analysis into an intuitive visual exploration tool for drug discovery professionals. |
Experimental Protocols and Methodologies
Protocol 1: Generating a Scaffold Tree for a Novel Compound Set Objective: To classify a library of novel bioactive compounds or a HTS (High-Throughput Screening) hit list using the Scaffold Tree algorithm to identify core structural classes and their relationships. Materials: Compound structures (e.g., in SMILES or SDF format), computing infrastructure, and Scaffold Tree implementation software (e.g., original scripts, RDKit toolkit, or Scaffold Hunter). Procedure:
- Data Preparation: Standardize input molecular structures (neutralize charges, remove salts, generate canonical tautomers).
- Framework Generation: For each molecule, generate its Murcko scaffold by pruning all terminal acyclic side chains [9].
- Tree Construction: For each unique Murcko scaffold, apply the iterative ring-removal algorithm: a. Identify all rings in the current scaffold. b. Apply the prioritization rules (bridged > spiro > heteroatom count > size > aromaticity > canonical) to select the single ring for removal. c. Remove the selected ring and its attached linkers, ensuring the remaining structure is a valid, connected scaffold. d. Register this new scaffold as the parent node. e. Repeat steps a-d using the new scaffold as the input until a single ring remains.
- Tree Merging: Merge the individual chains from each molecule into a single global tree structure by unifying identical scaffold nodes at each level.
- Annotation & Analysis: Annotate tree nodes with properties from the child molecules (e.g., bioactivity mean, count). Visually analyze the tree to identify scaffolds enriched with activity [10].
Protocol 2: Hierarchical Visualization with Background Chemical Space (Using Scaffvis) [2] Objective: To visualize a proprietary compound library in the context of the known public chemical space to assess its novelty and distribution. Materials: The Scaffvis web application, public pre-computed scaffold hierarchy (e.g., from PubChem Compound), and the proprietary compound set. Procedure:
- Background Loading: The pre-computed scaffold hierarchy tree, derived from millions of PubChem compounds, serves as a fixed background map.
- Input Processing: Upload the proprietary compound set. The tool calculates the corresponding scaffold hierarchy for each compound.
- Hierarchy Mapping: Each compound is mapped onto the background tree. Its path from the root to its leaf scaffold is highlighted within the static global hierarchy.
- Treemap Visualization: The tool generates a zoomable treemap visualization. a. Each rectangle represents a scaffold node from the background hierarchy. b. The size of the rectangle encodes the relative frequency of that scaffold in the background database (e.g., PubChem). c. The color of the rectangle encodes the frequency or a computed property (e.g., average potency) of the scaffold within the uploaded proprietary set.
- Interactive Exploration: Researchers can zoom into dense regions of the map, click on scaffolds to list associated compounds, and identify areas where their library is over- or under-represented compared to public chemical space.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Software Tools and Resources for Scaffold Tree Analysis
| Tool/Resource Name | Type | Primary Function in Scaffold Tree Research | Access / Reference |
|---|---|---|---|
| RDKit | Open-Source Cheminformatics Library | Provides functions for generating Murcko scaffolds, ring perception, and implementing custom tree-building algorithms. | https://www.rdkit.org |
| Scaffold Hunter | Standalone Software Application | Enables interactive creation, visualization, and analysis of Scaffold Trees from molecular datasets. Integrates bioactivity data [2]. | https://scaffoldhunter.sourceforge.io |
| Scaffvis | Web-Based Client-Server Application | Specializes in visualizing user compound sets hierarchically on a background (e.g., PubChem) using a zoomable treemap [2]. | https://github.com/chemdb/Scaffvis |
| PubChem Compound Database | Public Chemical Structure Database | Source of millions of structures for building reference background hierarchies and for benchmarking analyses [2] [9]. | https://pubchem.ncbi.nlm.nih.gov |
| SMILES/SDF Formats | Data Standards | Universal text-based formats (SMILES) or structural data files (SDF) for representing input molecules and exchanging scaffold data. | IUPAC Standard [11] |
| Original Algorithm Scripts | Reference Code | The canonical implementation of the 2007 algorithm rules; serves as a gold standard for validation. | Described in J Chem Inf Model, 2007, 47, 47-58 [9]. |
This protocol details the application of the Scaffold Tree algorithm, a deterministic and chemically intuitive method for hierarchically organizing molecular datasets based on their core ring systems [4]. The methodology is founded on two interdependent core principles: the iterative removal of rings from complex molecular frameworks and the application of a chemically meaningful set of prioritization rules to guide this deconstruction in a consistent, data-set-independent manner [9]. By systematically pruning peripheral rings to reveal central, characteristic scaffolds, the algorithm generates a unique tree hierarchy where leaf nodes represent molecular frameworks and the root is a single ring [12]. These Application Notes provide a detailed experimental workflow for implementing scaffold tree analysis, from molecular standardization to tree visualization and interpretation, framed within broader research on hierarchical ring analysis for drug discovery and chemical space navigation [13].
The scaffold tree algorithm was developed to address the need for a systematic, chemically intuitive classification of molecular scaffolds—the core ring systems and linkers that define a compound's shape [13]. In contrast to similarity-based clustering or other hierarchy methods that can be dataset-dependent, the scaffold tree provides a deterministic and unique hierarchy [9]. Its primary function is to organize large chemical libraries, enabling researchers to visualize chemical space, cluster compounds, and identify novel bioactive scaffolds by revealing relationships between complex structures and their simpler constituent rings [4].
The algorithm is defined by its two-stage process on a per-molecule basis. First, the molecular framework (or Murcko scaffold) is generated by removing all terminal side chains [9]. Second, this framework is deconstructed through iterative ring removal, guided by strict prioritization rules, until a single-ring root scaffold remains [12]. When applied to a collection of molecules, the union of all individual decomposition paths forms a connected scaffold tree, providing a global map of scaffold relationships within the set [2].
Core Principles and Quantitative Foundations
The Iterative Ring Removal Engine
The deconstruction process is an iterative cycle of ring perception, candidate identification, rule-based selection, and excision. It employs a Smallest Set of Smallest Rings (SSSR) perception to identify all rings within the current scaffold [13]. A "removable" or "terminal" ring is defined as one whose removal does not disconnect the remaining scaffold graph. From the set of terminal rings, one is selected for removal based on the prioritization rules detailed in Section 2.2. The selected ring and any linker atoms that become acyclic side chains after its removal are pruned. This cycle repeats on the newly generated, simpler parent scaffold.
Table 1: Performance and Scalability of Scaffold Tree Generation
| Dataset | Source | Number of Compounds | Reported Processing Time | Key Metric |
|---|---|---|---|---|
| Natural Products (NP) | COCONUT Database [13] | >450,000 | < 24 hours | Scaffold network generation |
| Drug Molecules | DrugBank [13] | Not Specified | Performance snapshot reported | Library validation |
| Clinical Trial Compounds | Analysis by Pitt et al. [14] | ~450,000 unique ring systems from 2.24B molecules | Not Specified | Size of explored space |
| Scaffold Hopping Validation | ChemBounce Tool [15] | Diverse set (e.g., peptides, macrocycles, small molecules) | 4 seconds to 21 minutes per structure | Varies by molecular complexity |
Chemically Meaningful Prioritization Rules
The chemical intelligence of the algorithm is encoded in its prioritization rules, which ensure the most characteristic, central ring is preserved longest. The rules are applied in sequence; if a decision cannot be made with the first rule, the algorithm proceeds to the next [9] [13].
Table 2: Hierarchy of Chemically Meaningful Prioritization Rules for Ring Removal [9] [13]
| Priority Order | Rule Name | Chemical Rationale & Objective |
|---|---|---|
| 1 | Heteroatom Content | Remove rings with the fewest heteroatoms first. Preserves heterocycles, which are often pharmacophorically important. |
| 2 | Ring Size | Remove the largest ring first. Prefers to retain smaller, often more strained and characteristic ring systems. |
| 3 | Aromaticity | Remove aliphatic rings before aromatic rings. Aromatic systems are considered more central to scaffold identity. |
| 4 | Saturation | Remove rings with the highest degree of saturation. Prefers unsaturated systems. |
| 5-13 | Further Discriminators | Includes rules based on bond count, adjacency to heteroatoms, and other topological features to break remaining ties deterministically. |
The result is a linear, unique path of scaffolds from the original molecule to a single-ring root, enabling a unambiguous hierarchical classification [13].
Diagram Title: Scaffold Tree Generation Workflow (86 characters)
Detailed Experimental Protocols
Protocol 1: Standardized Molecular Input Preparation
Objective: Generate consistent, QSAR-ready molecular structures from raw input data (e.g., SMILES, SDF) for reliable scaffold analysis.
- Data Acquisition: Obtain molecular structures in SMILES format from public databases (e.g., PubChem [2], ChEMBL [15]) or proprietary sources.
- Standardization: Process all SMILES using a cheminformatics toolkit (e.g., RDKit, CDK). Key steps include:
- Remove Salts/Disconnected Fragments: Eliminate counterions and solvents separated by a "." in the SMILES string [15].
- Aromatization: Apply consistent aromaticity models (e.g., RDKit's default).
- Explicit Hydrogen Handling: Standardize hydrogen counts.
- Stereochemistry: Check and clean stereochemical descriptors.
- Validation: Filter out molecules that fail parsing or that are purely acyclic (if using the standard Murcko framework definition which requires at least one ring) [13].
- Output: A clean list of canonical SMILES strings or a standardized SDF file.
Protocol 2: Core Scaffold Tree Construction
Objective: Execute the iterative ring removal algorithm to build a scaffold tree from a prepared molecular dataset.
- Framework Extraction: For each standardized molecule, generate its Murcko framework (all rings and the linkers connecting them, with all terminal side chains removed) [9] [14].
- Scaffold Deconstruction: For each unique Murcko framework: a. Initialize the current scaffold as the framework. b. While the current scaffold contains more than one ring: i. Perform SSSR analysis to list all rings. ii. Identify all terminal rings from the SSSR set. iii. Apply the hierarchy of prioritization rules (Table 2) to select the single ring for removal. iv. Remove the selected ring and any linker atoms that become non-cyclic after removal. v. Record the new, simpler scaffold as the parent of the previous one. c. The final single-ring scaffold is the root for this branch.
- Hierarchy Assembly: Merge all recorded parent-child relationships from all molecules into a single directed graph. This forms the complete scaffold tree, where nodes are scaffolds and edges represent the "is-a-parent-of" relationship [4].
- Tool Implementation: This can be performed using the
ScaffoldGeneratorlibrary in the Chemistry Development Kit (CDK) [13] or other specialized software likeScaffoldGraph[15].
Protocol 3: Analysis & Visualization for SAR Exploration
Objective: Annotate and visualize the scaffold tree to identify clusters of bioactivity and promising scaffold hops.
- Data Annotation: Map experimental data (e.g., bioactivity pIC50, toxicity flags, physicochemical properties) from the original molecules onto their corresponding leaf-node scaffolds and all parent scaffolds in their decomposition path.
- Tree Pruning & Focus: To manage complexity, prune branches that contain only inactive compounds or are not of interest. Alternatively, focus analysis on a specific subtree rooted at a scaffold of high interest [2].
- Visualization: Use visualization tools (e.g., Scaffold Hunter [2], Scaffvis [2], or DataGrok [12]) to create interactive tree maps.
- Node Size: Encode the frequency of a scaffold in the dataset.
- Node Color: Encode the average bioactivity or other property of compounds sharing that scaffold.
- Tooltip: Display detailed scaffold structure and aggregated data.
- Identification of Privileged Scaffolds: Visually identify nodes (scaffolds) that are:
Diagram Title: Computational Scaffold Hopping Protocol (63 characters)
Applications in Hierarchical Ring Analysis Research
The scaffold tree methodology serves as a foundational tool for several advanced research applications in drug discovery.
Visualizing Chemical Space & Diversity: The tree provides a navigable map of ring system relationships in large databases like PubChem or corporate collections, revealing overrepresented scaffolds and voids in coverage [4] [2]. For example, analysis shows molecules in clinical trials utilize only about 0.1% of the estimated 450,000 unique ring systems available in synthesized chemical space, highlighting vast areas for exploration [14].
Scaffold Hopping & Lead Optimization: The hierarchical classification directly enables scaffold hopping by identifying structurally distinct yet closely related parent or sibling scaffolds in the tree that may retain bioactivity [4]. Modern computational frameworks like ChemBounce operationalize this by replacing a query scaffold with similar ones from a large library, followed by filtering for synthetic accessibility (SAscore) and drug-likeness (QED) [15]. This approach can generate novel, patentable candidates while preserving pharmacophores.
Trend Analysis in Drug Discovery: Tracking the appearance and success of scaffolds through the tree hierarchy over time can inform on trends. Research indicates that approximately 67% of small molecules in clinical trials are composed solely of ring systems already found in marketed drugs, underscoring the reuse and recombination of known, "privileged" systems [14].
Integration with Machine Learning: The deterministic, structure-based hierarchy of the scaffold tree is ideal for creating meaningful splits in datasets for machine learning model training and validation, ensuring scaffolds in the test set are structurally distinct from those in the training set [13].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software Tools and Libraries for Scaffold Tree Analysis
| Tool/Resource Name | Type | Primary Function in Scaffold Analysis | Key Feature/Reference |
|---|---|---|---|
| Scaffold Generator | Java Library | Core implementation of scaffold tree, network, and other hierarchy generation within the CDK. | Highly customizable, supports multiple framework definitions [13]. |
| ChemBounce | Python Tool/Cloud Notebook | Computational framework for scaffold hopping using a large, curated scaffold library. | Integrates synthetic accessibility (SAscore) and shape similarity filtering [15]. |
| ScaffoldGraph | Python Library | Graph-based handling of scaffold hierarchies and molecular fragmentation. | Implements the HierS algorithm for fragmentation [15]. |
| RDKit | Cheminformatics Toolkit | Molecular standardization, SMILES parsing, fingerprint generation, and general cheminformatics operations. | Open-source, widely used for preprocessing and descriptor calculation. |
| Scaffvis | Web Visualization Tool | Interactive, zoomable treemap visualization of scaffold hierarchies on a PubChem background. | Enables visualization against empirical chemical space [2]. |
| ChEMBL Database | Chemical Database | Source of synthesis-validated bioactive compounds for building curated scaffold libraries. | Provides over 3 million unique scaffolds for hopping exercises [15]. |
| PubChem Compound | Chemical Database | Large-scale public repository for background chemical space analysis and diversity assessment. | Used for large-scale scaffold frequency analysis [2]. |
Diagram Title: Hierarchy of Scaffold Abstraction Levels (62 characters)
The Role in Chemical Space Navigation and Drug Discovery
The systematic navigation of drug-like chemical space is a foundational challenge in modern drug discovery. With an estimated 10⁶⁰ synthesizable organic molecules constituting this vast space, efficient strategies are required to identify novel, potent, and synthetically accessible leads [16]. Central to this endeavor is the scaffold tree methodology, which provides a hierarchical framework for deconstructing molecules into their core ring systems and analyzing structural relationships [15]. This approach transforms the overwhelming complexity of chemical space into a navigable map of privileged scaffolds and their derivatives, enabling targeted exploration for new bioactive compounds.
The integration of generative artificial intelligence (AI) with scaffold-based analysis marks a paradigm shift. Contemporary generative models, including variational autoencoders (VAEs), generative adversarial networks (GANs), and Transformers, can now propose novel molecular structures that transcend traditional similarity-based searches [16]. These models navigate chemical space by learning latent representations of molecular properties and bioactivity, allowing for the de novo design of compounds optimized for specific targets. However, the practical success of these AI-generated molecules hinges on their synthetic feasibility and alignment with medicinal chemistry principles, areas where scaffold-based reasoning provides essential constraints and validation [15] [17].
This document presents application notes and detailed protocols for implementing scaffold tree methodology and complementary computational techniques within a cohesive drug discovery workflow. Framed within a broader thesis on hierarchical ring analysis, the content is designed for researchers and scientists aiming to bridge cutting-edge computational navigation with experimentally grounded scaffold hopping and optimization.
Key Concepts and Quantitative Landscape
The Scaffold and Ring System Universe
Scaffolds, defined as the core cyclic structures of molecules after removal of side chains and linkers, form the architectural backbone of drug-like chemical space. Analyses reveal a highly focused utilization of ring systems in successful drugs.
Table 1: Analysis of Ring Systems in Medicinal Chemistry
| Analysis Parameter | Findings | Implication for Drug Discovery |
|---|---|---|
| Total Unique Medicinal Chemistry-Relevant Ring Systems [18] | A database of ~4 million ring systems has been compiled. | Provides a near-comprehensive library for bioisosteric replacement and scaffold hopping in generative chemistry. |
| Ring Popularity in Drugs & Clinical Trials [19] | 67% of small molecules in clinical trials contain only ring systems already present in marketed drugs. | Highlights conservative exploration but also an opportunity for innovation with novel, validated ring systems. |
| Critical Scaffolds for c-MET Inhibitors [20] | Analysis of 2,278 molecules identified common scaffolds (e.g., M5, M7, M8) and key fragments (pyridazinones, triazoles, pyrazines). | Reveals "safe bet" structural motifs for a specific target class, guiding focused library design. |
| Structural Determinants of c-MET Activity [20] | Active inhibitors are characterized by: ≥3 aromatic heterocycles, ≥5 aromatic nitrogen atoms, ≥8 N−O bonds. | Provides quantifiable, interpretable design rules for machine learning models and medicinal chemists. |
Performance of Scaffold Hopping Tools
Scaffold hopping is a critical strategy for generating novel intellectual property while maintaining biological activity. The performance of computational tools is benchmarked across multiple parameters.
Table 2: Comparative Analysis of Scaffold Hopping Tool Performance
| Tool / Framework | Core Methodology | Key Performance Metrics | Reference / Availability |
|---|---|---|---|
| ChemBounce [15] | Fragment replacement from a curated library of 3.2M ChEMBL scaffolds with ElectroShape similarity filtering. | Generates compounds with higher synthetic accessibility (lower SAscore) and better drug-likeness (higher QED) vs. commercial tools. Processing time: 4 sec to 21 min per molecule. | Open-source (GitHub, Google Colab). |
| Generative AI Models (RNNs, VAEs, GANs, etc.) [16] | Learn latent chemical space representations to generate novel structures beyond direct similarity. | Excels in novelty and exploration of uncharted chemical space. Challenges remain in ensuring synthetic accessibility and precise property control. | Various open-source and proprietary platforms. |
| Commercial Tools (e.g., Schrödinger, BioSolveIT) [15] | Proprietary algorithms for core hopping, isosteric matching, and shape-based searching. | Established, user-friendly platforms. May generate structures with lower synthetic accessibility compared to newer data-driven tools like ChemBounce. | Commercial software suites. |
Experimental Protocols
Protocol 1: Implementing Scaffold Hopping with ChemBounce
This protocol details the steps for using the ChemBounce framework to perform scaffold hopping for hit expansion and lead optimization [15].
1. Input Preparation and Validation
- Objective: Prepare a valid SMILES string of the query active compound.
- Procedure:
- Obtain the canonical SMILES for your query molecule from reliable sources (e.g., PubChem).
- Pre-process the SMILES: Remove salts, solvents, or disconnected components (often indicated by a "
." in the SMILES). Retain only the primary active structure. - Validate the SMILES string using a cheminformatics toolkit (e.g., RDKit) to ensure correct syntax, atomic valence, and stereochemistry.
- Critical Notes: ChemBounce will fail on invalid SMILES. Common errors include unbalanced brackets, incorrect ring closure numbers, or invalid atomic symbols.
2. Command-Line Execution and Parameterization
- Objective: Execute ChemBounce to generate novel analogs.
- Procedure:
- Clone the repository:
git clone https://github.com/jyryu3161/chembounce.git - Navigate to the directory and run the core command:
- Clone the repository:
- Parameter Explanation:
-o OUTPUT_DIR: Path to save results.-i INPUT_SMILES: Query molecule SMILES string.-n NUMBER_OF_STRUCTURES: Target number of output molecules per fragment (default 100).-t SIMILARITY_THRESHOLD: Minimum Tanimoto fingerprint similarity between input and output (default 0.5). Increase (e.g.,-t 0.7) for more conservative hopping.--core_smiles SMILES: (Optional) Specify a substructure (e.g., a critical pharmacophore) that must be retained in all output molecules.--replace_scaffold_files FILES: (Optional) Use a custom scaffold library instead of the default ChEMBL-derived one.
3. Post-Processing and Triage of Results
- Objective: Filter and prioritize generated compounds for further study.
- Procedure:
- Calculate Properties: For all output molecules, compute key physicochemical properties (Molecular Weight, LogP, H-bond donors/acceptors, rotatable bonds).
- Apply Filters: Apply relevant filters (e.g., Lipinski's Rule of Five, PAINS alerts) using toolkits like RDKit or open-source filters.
- Assess Synthetic Accessibility: Calculate Synthetic Accessibility (SA) scores. Prioritize compounds with SA scores < 6 [17].
- Diversity Analysis: Cluster the filtered molecules based on molecular fingerprints to select a structurally diverse subset for virtual screening or synthesis.
Protocol 2: Scaffold and SAR Analysis for a Target Family
This protocol outlines a machine learning-guided analysis to identify privileged scaffolds and key structural features for a specific target class, using c-MET kinase inhibitors as a model [20].
1. Dataset Curation and Preparation
- Objective: Assemble a high-quality, annotated dataset of active and inactive molecules for the target.
- Procedure:
- Data Collection: Extract bioactivity data (IC₅₀, Ki, Kd) from ChEMBL [21]. Use standardized
pChEMBLvalues (negative log of the molar concentration). - Define Activity Threshold: Establish a meaningful threshold for "active" vs. "inactive" (e.g., IC₅₀ < 100 nM for active).
- Standardize and Deduplicate: Standardize structures (neutralize, remove duplicates) and curate to ensure a balanced distribution of actives and inactives.
- Data Collection: Extract bioactivity data (IC₅₀, Ki, Kd) from ChEMBL [21]. Use standardized
2. Hierarchical Scaffold Decomposition and Network Construction
- Objective: Map the scaffold-based chemical space of the dataset.
- Procedure:
- Generate Scaffold Trees: For each molecule, apply the HierS algorithm [15] using the ScaffoldGraph library to iteratively remove rings and generate a hierarchy of scaffolds (from the full molecule to the simplest ring system).
- Construct a Chemical Space Network (CSN): Create a network where nodes represent unique scaffolds and edges connect scaffolds if one is a direct subgraph of the other. Annotate nodes with metadata (e.g., average activity of all molecules containing that scaffold).
- Identify Key Scaffolds: Visually and statistically analyze the CSN to locate densely connected clusters of highly active scaffolds (e.g., M5, M7, M8 for c-MET) [20].
3. Machine Learning-Based Feature Extraction and Rule Generation
- Objective: Translate structural patterns into interpretable design rules.
- Procedure:
- Feature Calculation: Compute a comprehensive set of molecular descriptors and fingerprints for all compounds.
- Train a Predictive Model: Train a machine learning classifier (e.g., Random Forest, XGBoost) to distinguish active from inactive compounds.
- Perform Activity Cliff Analysis: Identify pairs of structurally similar molecules with large differences in potency. Analyze the specific substituents or minor structural changes causing the "cliff".
- Extract Interpretable Rules: Use a decision tree model on the most important molecular features to generate human-readable rules. For c-MET, this yielded: "aromatic heterocycles ≥ 3" AND "aromatic nitrogens ≥ 5" AND "N−O bonds ≥ 8" [20]. These rules can directly guide the design or filtering of new compounds.
Visualization of Workflows and Relationships
Scaffold-Based Chemical Space Navigation Workflow
Navigating Chemical Space: A Comparison of Computational Approaches
Hierarchical Ring Analysis Process for SAR Insight
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Resources for Chemical Space Navigation and Scaffold Analysis
| Tool / Resource | Type | Primary Function in Research | Access / Reference |
|---|---|---|---|
| ChemBounce | Computational Framework | Open-source tool for scaffold hopping using a synthesis-validated fragment library and shape-based similarity filtering [15]. | GitHub: jyryu3161/chembounce; Google Colab. |
| ScaffoldGraph | Software Library | Python library for generating scaffold trees and hierarchical networks from molecular datasets, implementing algorithms like HierS [15]. | Open-source (GitHub). |
| ChEMBL Database | Bioactivity Database | Public repository of >24 million bioactivity data points for training predictive models and building target-focused libraries [15] [21]. | https://www.ebi.ac.uk/chembl/ |
| Medicinal Chemistry Ring System Database | Structural Database | A curated set of ~4 million ring systems derived from bioactive molecules, essential for bioisosteric replacement and scaffold inspiration [18]. | Described in Ertl, 2024. |
| RDKit | Cheminformatics Toolkit | Open-source fundamental toolkit for SMILES parsing, molecular fragmentation, fingerprint calculation, and property prediction [15]. | http://www.rdkit.org |
| ODDT / ElectroShape | Shape Similarity Tool | Python library (ODDT) containing the ElectroShape method for calculating 3D molecular shape and charge distribution similarity, critical for pharmacophore retention [15]. |
Open-source (GitHub). |
| PDBbind & CASF Benchmark | Structure-Activity Database | Curated sets of protein-ligand complexes with binding affinity data for benchmarking physics-based and knowledge-based scoring functions [21]. | http://www.pdbbind.org.cn/ |
| Generative Model Libraries (e.g., PyTorch, TensorFlow with Chem-specific packages) | AI/ML Development Framework | Platforms for building and deploying generative AI models (VAEs, GANs, Transformers) for de novo molecular design [16]. | Open-source. |
The scaffold tree methodology provides a deterministic, hierarchical framework for organizing molecular complexity, transforming vast chemical spaces into navigable structures for rational drug design. This application note details the core concepts of virtual scaffolds and ring systems within this classification scheme, presents quantitative analyses of ring system utilization in drug discovery, and provides explicit protocols for implementing scaffold-based virtual screening and hierarchical analysis. The integration of these elements supports the efficient identification of novel bioactive cores and the strategic expansion of medicinal chemistry space.
Background and Thesis Context
A central challenge in modern drug discovery is the efficient navigation of an enormous chemical space to identify novel, bioactive molecular cores or scaffolds. High-throughput screening (HTS) campaigns, particularly against antibacterial targets, have historically suffered from high costs and low hit rates, often failing to deliver structurally diverse lead matter [22]. This highlights a critical bottleneck: the need for intelligent methods to prioritize and analyze chemical libraries.
The broader thesis of scaffold tree methodology addresses this by imposing a chemically intuitive, hierarchical order on molecular datasets. It posits that a deterministic classification of scaffolds—core structures derived by removing terminal side chains—enables researchers to visualize chemical space, identify structure-activity relationships (SAR), and pinpoint rare or virtual scaffolds that represent promising, unexplored chemotypes [23] [10]. This approach moves beyond mere property-based filtering to a structure-centric analysis, which is essential for scaffold hopping and innovation in ring system design, the foundational building blocks of most drugs [14] [24].
Core Terminology and Definitions
- Virtual Scaffolds: These are molecular scaffolds generated during the hierarchical decomposition process (e.g., ring removal) that are not present as original frameworks in the analyzed compound set. They represent plausible, simpler core structures that can inspire the design or acquisition of novel compounds to fill gaps in chemical space or explore new structure-activity landscapes [23] [3].
- Ring Systems: A ring system is defined as a single ring or multiple rings connected by fusion (sharing atoms/bonds) or spiro linkages. It excludes linker atoms and side chains. Ring systems are the primary determinants of a molecule's shape, physicochemical properties, and are the central unit of analysis in scaffold classification [24].
- Deterministic Classification (Scaffold Tree): A rule-based algorithm that reduces a molecular scaffold to a single ring through the iterative, prioritized removal of rings. The priority rules (e.g., remove smaller rings before larger ones, remove aliphatic before aromatic, remove rings with fewer heteroatoms first) ensure a unique, reproducible, and dataset-independent hierarchical tree for every molecule. This creates a predictable parent-child relationship between scaffolds [3] [10].
Application Notes and Protocols
Quantitative Landscape of Ring Systems in Drug Discovery
Analysis of clinical trial compounds and approved drugs reveals a conservative yet evolving use of ring systems, as summarized in Table 1.
Table 1: Prevalence and Novelty of Ring Systems in Drug Discovery
| Metric | Clinical Trial Compounds | Approved Drugs | Source/Implication |
|---|---|---|---|
| Using known drug ring systems | 67% | ~70% (annual new drugs) | High reliance on pre-validated systems [14]. |
| Unique systems available | ~450,000 (estimated in synthetic space) | Not Applicable | Vast pool of untapped potential [14]. |
| Unique systems utilized | ~0.1% of available pool | Fewer than in trials | Extreme concentration on a tiny fraction [14]. |
| Novel systems per molecule | Typically only 1 (if any) | Typically only 1 (if any) | Novelty is introduced cautiously [14] [24]. |
| Most common ring type | Heterocycles (e.g., Pyridine, Piperazine) | Heterocycles | Critical for target interactions and solubility [24]. |
Protocol 1: Hierarchical Virtual Screening for Novel Scaffold Identification
This protocol integrates scaffold-aware analysis with computational screening to identify new active chemotypes, as demonstrated for antibacterial targets [22] and the NLRP3 inflammasome [25].
Objective: To identify novel inhibitor scaffolds for a target with poor HTS outcomes. Input: Target protein structure (e.g., PDB file), a set of known active ligands (if any), a large commercially available compound database (e.g., ZINC, >9 million compounds) [22]. Software: USR (Ultrafast Shape Recognition) or ROCS; molecular docking suite (e.g., Glide, AutoDock); scaffold analysis toolkit (e.g., Scaffold Generator, RDKit) [22] [3] [26].
Procedure:
- Shape-Based Pre-screening: For each known active ligand, perform a shape similarity search (e.g., using USR) against the entire database. Pool the top-ranking compounds from all queries to create a shape-enriched subset (e.g., reducing 9M to ~4,000 molecules) [22].
- Molecular Docking: Dock the shape-enriched subset into the target's binding site. Apply strict scoring and pose filters to select a few hundred top-ranked virtual hits.
- Scaffold Extraction and Classification:
- Extract the Bemis-Murcko scaffold from all docking hits and known actives.
- Apply the deterministic classification algorithm to generate each molecule's scaffold tree.
- Cluster molecules sharing identical scaffolds at any tree level.
- Identification of Novel and Virtual Scaffolds:
- Compare the scaffolds of the virtual hits against those of known actives.
- 标记 novel active scaffolds that are (a) present in virtual hits and (b) absent from known actives.
- 识别 virtual scaffolds that appear as parent nodes in the trees of multiple active hits but are not themselves present as a molecule in the database. These are prime candidates for de novo design [23] [3].
- Post-Screening Analysis:
- Prioritize compound clusters based on docking score, scaffold novelty, and chemical attractiveness.
- Select 50-100 compounds representing diverse scaffold classes for in vitro testing.
Diagram: Hierarchical Virtual Screening Workflow for Novel Scaffold Identification.
Protocol 2: Constructing and Analyzing a Scaffold Tree for SAR
This protocol uses the scaffold tree to visualize and interpret chemical datasets and their associated bioactivity data.
Objective: To analyze a set of screening hits or a corporate library to understand SAR and identify privileged core structures. Input: A dataset of molecules (e.g., HTS hits, focused library) with associated activity data or properties. Software: Scaffold Hunter [23], Scaffvis [2], or the Scaffold Generator library [3].
Procedure:
- Data Preparation: Standardize molecules and calculate Murcko scaffolds.
- Tree Generation: For each molecule, apply the deterministic ring-removal rules to generate its unique path from the full scaffold to a single ring. Merge identical scaffolds across molecules to build a global scaffold tree where leaf nodes are molecule-endowed scaffolds and parent nodes are virtual scaffolds [23] [10].
- Visualization & Analysis:
- Load the tree into an analysis tool like Scaffold Hunter.
- 节点的大小可以设置为代表该支架下分子的数量。
- 节点的颜色可以映射为生物活性的平均值(例如,pIC50),从而快速识别出富含高活性分子的分支(活性“热点”) [23].
- Identify branches where activity is lost or gained upon specific ring removals, pinpointing critical structural elements for activity.
- Application:
- Scaffold Hopping: Identify active virtual scaffolds that are not yet populated with compounds. These are ideal targets for synthesis or acquisition [3].
- Library Design: Analyze the distribution of compounds across the tree to identify over- and under-represented regions, guiding diversification efforts.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Tools and Resources for Scaffold-Tree-Based Research
| Item / Resource | Type | Function & Application | Key Features |
|---|---|---|---|
| Scaffold Generator [3] | Java Library | Core algorithm for generating scaffold trees/networks from molecular datasets. | Customizable, based on CDK, handles large datasets (e.g., 450k NPs in a day). |
| Scaffold Hunter [23] | Visual Analytics Software | Interactive visualization and analysis of scaffold trees integrated with bioactivity data. | Combines tree, dendrogram, heatmap, and molecule cloud views for SAR. |
| Scaffvis [2] | Web Application | Hierarchical, treemap visualization of molecular datasets against the background of PubChem space. | Provides context by showing scaffold frequency in public chemical space. |
| ROCS / USR | Shape Similarity Software | Ultrafast pre-screening based on 3D molecular shape for scaffold hopping [22]. | Enables rapid search of billion-compound databases for shape analogs. |
| ZINC / REAL Space | Compound Database | Source of commercially available, purchasable compounds for virtual screening [22] [14]. | Contains >9M (ZINC) to >20B (REAL) molecules for diverse screening. |
| ChEMBL | Bioactivity Database | Reference source for known active scaffolds and their target annotations [22] [24]. | Essential for benchmarking and avoiding rediscovery of known chemotypes. |
Applications in Research and Development
- Overcoming Antibacterial Discovery Bottlenecks: The hierarchical virtual screening protocol has been successfully applied to targets like type II dehydroquinase, identifying over 100 new inhibitors with more than 50 new active scaffolds, far surpassing the output of traditional HTS [22].
- Target-Ring System Repurposing: Bi-directional screening between targets and underutilized ring systems from approved drugs can generate novel, patentable chemical matter for new therapeutic indications [27].
- Analysis of Clinical Candidate Novelty: Tracking ring system usage from clinical trials to marketed drugs provides strategic intelligence on the level of chemical innovation required for successful development [14].
Future Perspectives
The integration of deterministic classification with artificial intelligence and generative chemistry presents a powerful frontier. Predictive models can be trained to prioritize virtual scaffolds with high probabilities of desired bioactivity or synthetic accessibility. Furthermore, coupling scaffold-tree analysis with ultra-large library docking (billions of molecules) enables a systematic, hierarchical exploration of chemical space that is both comprehensive and interpretable, promising to accelerate the discovery of truly novel therapeutic agents.
From Theory to Practice: Algorithmic Steps and Real-World Applications in Medicinal Chemistry
Application Notes
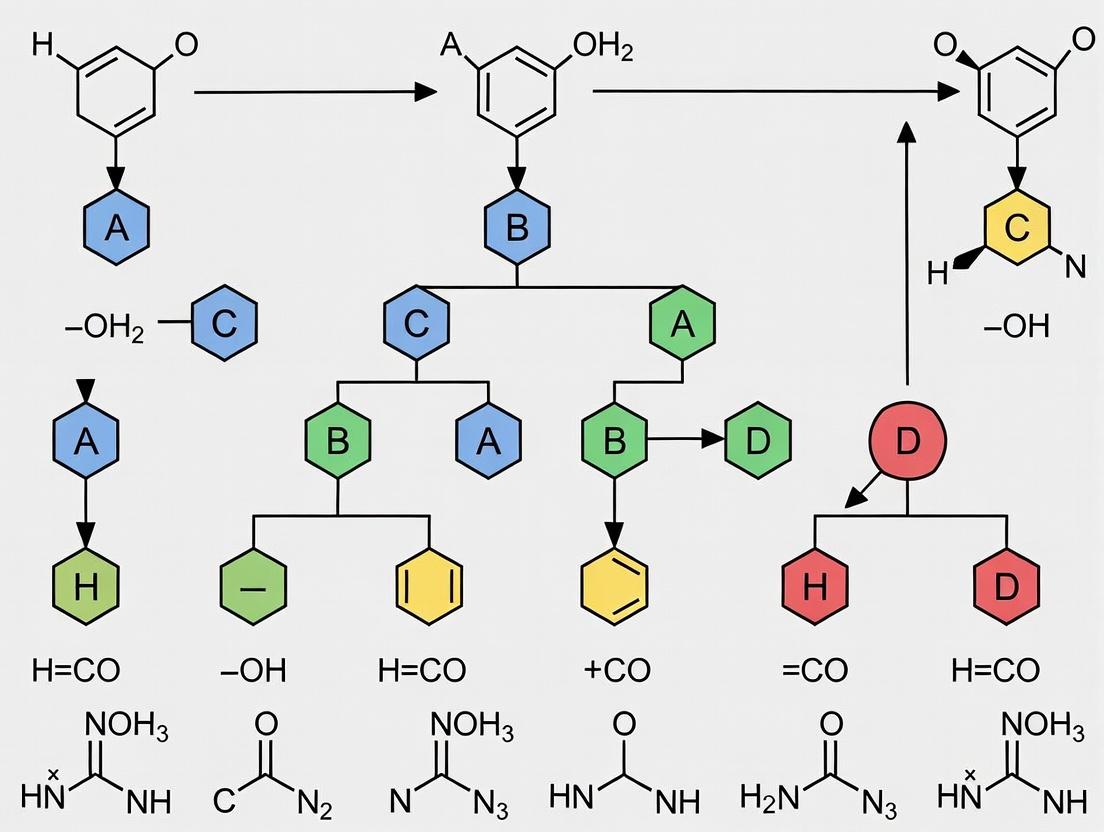
Within the scaffold tree methodology for hierarchical ring analysis, the conversion of a molecular graph into a unique scaffold requires a deterministic algorithm to prune rings to a single, core ring system. This step is critical for enabling consistent classification and comparison of molecular frameworks across chemical databases. The algorithm's logic prioritizes certain complex ring topologies, such as bridged and spiro systems, due to their significant three-dimensional structure and influence on molecular properties, making them privileged in scaffold representation.
The core principle is iterative removal of peripheral rings while preserving a topologically complex core. The algorithm operates on a set of rings identified via a smallest set of smallest rings (SSSR) or an equivalent algorithm. The following ordered prioritization rules are applied to decide which ring to remove in each iteration, ensuring a single, reproducible endpoint.
Prioritization Rules (in order of application):
- Remove Isolated Rings: A ring not sharing any bonds (bridgeheads) with other rings in the set is removed first. This rule directly prunes simple peripheral substituents.
- Remove Non-Fused Rings: Preferentially remove rings that are edge-fused (sharing one bond) over those involved in more complex unions (spiro, bridged).
- Retain Spiro Rings: In a spiro union (sharing a single atom), the ring with the smaller number of heteroatoms is removed. If equal, the smaller ring is removed. The spiro linkage itself is considered a feature of complexity to be retained if possible.
- Retain Bridged Rings: For bridged systems (rings sharing at least two non-adjacent atoms), the algorithm prioritizes retaining the bridged ring system as a core unit. Removal decisions within a bridged system are based on heteroatom count and ring size, similar to spiro rules, but the overall bridged topology is preserved over simpler fused systems.
- Heteroatom & Size Tie-Breaking: When rings are topologically equivalent by the above rules, the ring with fewer heteroatoms is removed. If the heteroatom count is identical, the larger ring is retained.
Quantitative Outcomes of Rule Application: Table 1: Impact of Prioritization Rules on Scaffold Generation from a Benchmark Set (e.g., ChEMBL)
| Rule Category | % of Molecules Affected | Average Rings Pruned per Molecule | Key Outcome |
|---|---|---|---|
| Isolated Ring Removal | ~85% | 2.1 | Eliminates simple side-cycles and substituents. |
| Spiro Ring Retention | ~12% | 0.8 | Preserves stereogenic 3D centers in core scaffold. |
| Bridged Ring Retention | ~18% | 1.5 | Maintains complex, often rigid, polycyclic cores (e.g., adamantane). |
| Tie-breaker (Heteroatom) | ~45% | N/A | Ensures deterministic output favoring heteroatom-rich cores. |
Experimental Protocols
Protocol 1: Implementation of the Pruning Algorithm for Hierarchical Tree Generation
Purpose: To generate a scaffold tree for a given molecule by iterative application of ring pruning rules.
Materials & Software:
- Input: Molecular structure in SMILES or SDF format.
- Chemical Toolkit: RDKit (v2023.x or later) or Open Babel for ring perception and molecular manipulation.
- Programming Environment: Python 3.x with necessary cheminformatics libraries.
- Output: A hierarchical tree (JSON or graph format) and the final core scaffold (SMILES).
Procedure:
- Preprocessing & Ring Perception:
- Standardize the input molecule (neutralize, remove solvents, strip salts).
- Generate the SSSR or a relevant set of rings (GetSymmSSSR in RDKit).
- Represent the molecule as a graph where nodes are rings and edges represent fusion types (isolated, fused, spiro, bridged).
Iterative Pruning Loop:
- WHILE the number of rings in the set > 1: a. Identify all removable candidate rings based on current topology. b. Apply Rule 1: If any ring is isolated (degree 0 in the ring graph), select it for removal. If multiple, proceed to tie-breaking (Rule 5). c. Apply Rule 2: From remaining candidates, select rings that are only edge-fused (non-spiro, non-bridged). d. Apply Rule 3: For spiro-fused candidates, compare the two spiro-linked rings. Select the one with fewer heteroatoms for removal. If equal, select the smaller ring. e. Apply Rule 4: For bridged system candidates, analyze the bridgehead network. Remove rings that, if deleted, minimize the collapse of the bridged topology. Prefer to retain the ring that is part of the most bridged unions. f. Apply Rule 5: If multiple rings still qualify, remove the one with the smallest number of heteroatoms. If still tied, remove the smaller ring (by atom count). g. Perform the removal: Delete the selected ring's atoms and bonds not shared with any remaining ring. Re-perceive the ring set of the resultant molecule. h. Record the removed ring and the resulting structure as a node in the hierarchical tree.
Termination & Output:
- The algorithm terminates when a single ring system remains. This is the core scaffold.
- Output the complete scaffold tree (parent-child relationships of all pruned rings) and the final scaffold SMILES.
Validation: Execute the algorithm on a standardized dataset (e.g., FDA-approved drugs) and compare the resulting core scaffolds to a reference implementation (e.g., the original scaffold tree publication) to ensure >99% reproducibility.
Protocol 2: Comparative Analysis of Scaffold Diversity Using Different Prioritization Rules
Purpose: To quantify the impact of spiro/bridged ring retention rules on chemical space organization.
Materials:
- Dataset: 10,000 diverse bioactive molecules from ChEMBL.
- Software: Custom pruning script (from Protocol 1), modified to toggle specific rules on/off.
- Analysis Tools: Scikit-learn for PCA, Matplotlib/Seaborn for visualization, Jupyter Notebook.
Procedure:
- Generate Scaffold Sets:
- Run Protocol 1 on the full dataset using the complete rule set (including spiro/bridged retention). This is Set A.
- Run Protocol 1 on the dataset using a simplified rule set (removing Rules 3 & 4, treating spiro/bridged as simple fused). This is Set B.
- Descriptor Calculation:
- For each unique scaffold in Set A and Set B, calculate a set of 200-dimensional molecular fingerprints (e.g., Morgan FP, radius 2).
- Diversity Analysis:
- Perform principal component analysis (PCA) on the combined fingerprint matrix for Sets A and B.
- Calculate the scaffold recovery rate: (% of molecules from Set A whose core scaffold is identical in Set B).
- Calculate the mean pairwise Tanimoto diversity within each scaffold set.
- Statistical Reporting:
- Populate a results table (see Table 2 below).
- Generate 2D PCA plots color-coded by scaffold set.
Table 2: Results from Comparative Scaffold Analysis
| Metric | Set A (With Spiro/Bridged Rules) | Set B (Without Spiro/Bridged Rules) | Observation |
|---|---|---|---|
| Unique Scaffolds Generated | 1,850 | 2,110 | Simplified rules lead to more, smaller scaffolds. |
| Scaffold Recovery Rate | 100% (Reference) | 78% | 22% of molecules assigned a different core. |
| Mean Pairwise Diversity (Tanimoto) | 0.91 | 0.88 | Set A scaffolds are more topologically diverse. |
| % of Scaffolds with Spiro Atoms | 9.5% | 0.8% | Demonstrates explicit rule efficacy. |
| % of Scaffolds in Bridged Systems | 15.2% | 3.1% | Bridged systems are collapsed without Rule 4. |
Diagrams
Pruning Decision Logic for Complex Ring Unions
The Scientist's Toolkit
Table 3: Essential Research Reagents & Software for Scaffold Tree Methodology
| Item | Type | Function in Research |
|---|---|---|
| RDKit | Open-Source Cheminformatics Library | Core platform for ring perception (SSSR), molecular graph manipulation, fingerprint generation, and scaffold pruning algorithm implementation. |
| ChEMBL Database | Curated Bioactivity Database | Primary source of diverse, annotated molecular structures for algorithm benchmarking, validation, and diversity analysis. |
| Jupyter Notebook | Interactive Computing Environment | Facilitates exploratory data analysis, algorithm prototyping, result visualization (PCA plots), and sharing reproducible workflows. |
| scikit-learn | Python ML Library | Used for dimensionality reduction (PCA) and statistical analysis to compare scaffold sets and measure chemical space diversity. |
| Graphviz (dot) | Graph Visualization Software | Renders the logical workflow and decision trees of the pruning algorithm from DOT scripts, ensuring clear protocol documentation. |
| Standardized SMILES | Data Format (e.g., via RDKit) | Ensures canonical molecular representation as algorithm input, critical for reproducibility and avoiding input-based artifacts. |
Application Notes: Core Framework and Utility in Drug Discovery
Scaffold Hunter is a comprehensive visual analytics framework specifically designed to address the challenges of modern drug discovery, where researchers must navigate extensive chemogenomic datasets [23]. The tool operates on the principle of visual analytics, a scientific discipline that facilitates analytical reasoning through interactive visual interfaces, combining techniques from data mining and information visualization [23]. Its primary function is to transform raw, high-dimensional chemical and biological activity data into intuitive visual representations, enabling researchers to form and test hypotheses through an iterative exploration process [28] [29].
The software is fundamentally built around the scaffold tree concept, a hierarchical classification system that organizes molecules based on their core ring structures [4]. This methodology provides a chemically meaningful navigation system for chemical space. Beyond this core, the framework is modular, integrating multiple, synchronized visualization views—such as tree maps, dendrograms, heat maps, and molecule clouds—which allow users to analyze the same dataset from different analytical perspectives [23]. A key application is in Structure-Activity Relationship (SAR) analysis and hit-to-lead optimization, where teams can visually cluster active compounds, identify promising scaffold hops, and prioritize virtual scaffolds for synthesis [23] [29].
Table 1: Core Visualization Views in Scaffold Hunter and Their Primary Applications
| Visualization View | Core Principle | Typical Application in Drug Discovery | Key Advantage |
|---|---|---|---|
| Scaffold Tree View [23] | Hierarchical tree based on iterative ring removal. | Mapping chemical space, identifying scaffold hops and privileged structures. | Provides a deterministic, chemically intuitive hierarchy. |
| Tree Map View [23] | Space-filling rectangles sized by molecule count. | Rapid overview of large dataset composition and scaffold frequency. | Efficient use of space for visualizing large numbers of scaffolds. |
| Molecule Cloud View [23] | Compact, tag-cloud-like layout of scaffolds. | Visual clustering and trend spotting in scaffold distributions. | Intuitive, high-level summary of major chemical classes. |
| Heat Map View [23] | Matrix of property values (e.g., bioactivity) with hierarchical clustering. | Multi-target activity profiling, selectivity analysis, and outlier detection. | Correlates structural similarity with multiple biological endpoints. |
| Dendrogram View [23] | Hierarchical clustering based on fingerprint similarity. | Identifying structural clusters independent of predefined scaffolds. | Provides an alternative, data-driven classification scheme. |
The utility of Scaffold Hunter is demonstrated in practical screening scenarios. For instance, in the analysis of datasets targeting pathogens like T. cruzi and T. brucei, researchers can use the tool to quickly isolate active clusters, trace activity back to common substructures, and identify virtual scaffolds—intermediate structures in the tree not present in the screening library but suggesting promising synthetic targets [23] [29]. This capability directly supports lead discovery and scaffold-hopping efforts, making it a powerful tool for medicinal chemists and drug development professionals.
Foundational Protocol: The Scaffold Tree Methodology
The Scaffold Tree algorithm provides the foundational hierarchy for analysis within Scaffold Hunter. It is a deterministic and dataset-independent method for generating a unique tree representation for any set of molecules, scaling linearly with the number of compounds [4]. The following protocol details its stepwise implementation.
Protocol 1: Construction of a Scaffold Tree Hierarchy
Objective: To generate a hierarchical tree organization for a set of input molecules based on their molecular scaffolds.
Input Requirements:
- A dataset of chemical structures in a standard format (e.g., SDF, SMILES).
- Access to cheminformatics toolkit functions (e.g., for ring perception, fragmentation).
Procedure:
- Initial Scaffold Generation: For each molecule in the dataset, generate its Murcko scaffold [2]. This involves removing all terminal acyclic side chains while retaining ring systems and the linker atoms that connect them.
- Hierarchical Pruning: For each Murcko scaffold, apply a series of prioritized, deterministic rules to iteratively remove one ring per step until a single-ring root scaffold is obtained [4] [23]. The standard rule priority order is:
- Remove rings with the least number of heteroatoms.
- Remove rings with the smallest size (number of atoms).
- Remove rings that are part of the smallest number of other rings (e.g., peripheral vs. fused core rings).
- Remove aliphatic rings before aromatic rings.
- Tree Construction & Merging: Map each molecule to the sequence of scaffolds generated during its pruning process. Construct a tree where:
- Leaf Nodes represent the original Murcko scaffolds, annotated with the molecules that contain them.
- Internal Nodes represent the shared, simplified scaffolds from the pruning steps. Scaffolds common to multiple branches are merged into a single node.
- The Root Node is the final single-ring scaffold common to the entire hierarchy.
- Annotation: Annotate every scaffold node in the tree with aggregated biological activity data (e.g., mean IC50, hit rate) from all descendant molecules [28]. This creates a bioactivity-landscaped chemical hierarchy ready for visual exploration.
Output: A directed tree graph where parent-child relationships represent structural simplification. This graph serves as the primary data structure for the Scaffold Tree visualization in Scaffold Hunter.
Scaffold Tree Construction Workflow
Experimental Protocol: Integrated Visual Analysis for SAR Exploration
This protocol outlines a complete workflow using Scaffold Hunter's multi-view interface to derive structure-activity relationships from a high-throughput screening (HTS) dataset.
Protocol 2: Multi-View SAR Analysis of an HTS Dataset
Objective: To identify active chemical series and hypothesize key structural features responsible for biological activity.
Materials & Software:
- Scaffold Hunter software (open-source, platform-independent Java application) [23].
- HTS results file containing compound structures and a primary activity endpoint (e.g., inhibition % at 10 µM).
- Optional: Secondary assay data or calculated molecular descriptors.
Experimental Workflow:
- Data Integration & Normalization:
- Import the compound library (SDF format) and the corresponding activity data (CSV format) into Scaffold Hunter.
- Normalize activity values (e.g., convert IC50 to pIC50, categorize continuous data into "Active," "Inactive," and "Intermediate" bins based on defined thresholds).
Initial Exploration via Scaffold Tree:
- Navigate to the Scaffold Tree View. The software automatically generates the tree per Protocol 1.
- Color-code nodes based on the mean activity value of associated molecules. Use a diverging color scale (e.g., blue for inactive, red for active).
- Identify "hot" branches where active compounds are concentrated. Visually prune inactive branches to focus exploration.
Cluster Analysis & Confirmation:
- Switch to the Dendrogram View. Perform hierarchical clustering using a structural fingerprint (e.g., MACCS keys) and a similarity metric (e.g., Tanimoto).
- Color the dendrogram leaves by the same activity metric. Validate if the scaffold-based "hot" branches correspond to distinct structural clusters. This cross-view agreement strengthens the hypothesis of a true SAR.
Multi-Parameter Profiling with Heat Map:
- For the focused set of active clusters, launch the Heat Map View.
- Plot compounds (rows) against multiple activity endpoints or descriptors (columns). Perform dual hierarchical clustering on rows and columns.
- Analyze patterns to assess selectivity (e.g., activity against target vs. anti-target) or to correlate activity with physicochemical properties.
Hypothesis Generation & Output:
- In the Molecule Cloud or Tree Map View, generate a compact visual summary of the prioritized active scaffolds.
- Export the list of selected compounds, scaffolds, and associated data for further review or as a basis for designing a follow-up library.
Visual Analytics Workflow for SAR
Table 2: Key Research Reagent Solutions for Scaffold Hunter Analysis
| Category | Item / Resource | Function & Description | Example / Source |
|---|---|---|---|
| Core Software | Scaffold Hunter Application | Primary visual analytics platform for interactive exploration of chemical space [23]. | Open-source Java application. |
| Cheminformatics Toolkit | Chemistry Development Kit (CDK) or RDKit | Provides underlying functions for ring perception, scaffold fragmentation, fingerprint generation, and molecular property calculation [23]. | Integrated libraries within Scaffold Hunter. |
| Reference Databases | PubChem Compound Database [2] | Provides a massive background of empirical chemical space for benchmarking and understanding scaffold frequency/novelty. | Public repository (NIH). |
| Clustering & Similarity | Molecular Fingerprints (e.g., MACCS, ECFP) | Bit-string representations of molecular structure used for similarity searching and clustering in dendrogram/heat map views [23]. | Generated on-the-fly from structures. |
| Activity Data | Bioassay Results (e.g., IC50, Ki, % Inhibition) | Primary biological annotation used to color-code and filter scaffolds, forming the basis for SAR [28] [29]. | Internal HTS data or public sources like ChEMBL. |
| Alternative Hierarchy | Scaffvis Web Tool [2] | Provides an alternative, pre-computed scaffold hierarchy based on PubChem for comparative analysis or external visualization. | Web-based client-server application. |
The scaffold tree methodology represents a systematic approach to organizing chemical space by decomposing molecular structures into a hierarchical arrangement of core frameworks [4]. This methodology operates on the principle of iterative ring removal, applying chemically meaningful rules to reduce complex molecular scaffolds to simpler parent structures, ultimately forming a unique tree hierarchy where individual molecules become leaf nodes [4]. The deterministic and dataset-independent nature of this classification scales linearly with the number of compounds, making it particularly valuable for navigating large chemical databases such as PubChem [2] [4].
Within this methodological context, hierarchical visualization emerges as an indispensable tool for analyzing large molecular datasets generated by high-throughput screening in drug design [2]. Unlike direct visualization methods—which map molecules to Euclidean coordinates using techniques like principal component analysis and can suffer from context-dependent positioning—hierarchical visualization groups molecules based on shared structural features [2]. Scaffold-based hierarchies provide a chemically intuitive framework for this purpose, allowing researchers to explore compounds at varying levels of structural abstraction, from specific molecular frameworks to simplified ring topologies [2].
The Scaffvis platform implements this methodology as a web-based client-server application, enabling interactive exploration of chemical datasets against the empirical background of PubChem's chemical space [2]. By mapping user datasets onto a precomputed scaffold hierarchy derived from millions of PubChem compounds, Scaffvis facilitates the identification of common scaffolds, rare structural motifs, and the overall distribution of compounds within the global chemical universe [2].
Core Protocols: Implementing Scaffold Tree Analysis with Scaffvis
Protocol 1: Dataset Preparation and Molecular Standardization
A fundamental prerequisite for scaffold tree analysis is the standardization of molecular representations. This protocol ensures consistency prior to hierarchy generation.
- Input Raw Molecular Data: Begin with compounds in standard chemical file formats (SDF, SMILES). For PubChem background analysis, the entire PubChem Compound database serves as the reference set [2].
- Apply Standardization Rules:
- Remove counterions, salts, and solvents to isolate the primary bioactive component.
- Standardize tautomeric forms to a single canonical representation.
- Neutralize charges where appropriate (e.g., on carboxylic acids, amines).
- Explicitly define aromatic bonds according to the Kekulé representation.
- Generate Molecular Framework (Murcko Scaffold): For each standardized molecule, extract the Bemis-Murcko framework [2]. This involves removing all side-chain atoms while retaining all ring systems and the linker atoms that connect them.
- Output: A cleaned set of molecular frameworks ready for hierarchical decomposition.
Protocol 2: Generating the Scaffold Tree Hierarchy
This protocol details the deterministic algorithm for creating a tree hierarchy from molecular frameworks, as implemented in the Scaffold Tree method [4].
- Iterative Ring Removal: Starting from the molecular framework, iteratively remove one ring per step based on a predefined priority rule set [4].
- Priority Rules for Removal: The algorithm selects rings for removal in the following order of priority [2]:
- Heterocycles are removed before carbocycles.
- Larger rings are removed before smaller rings.
- Rings with the most acyclic connections (e.g., substituents, linkers) are removed first.
- In case of ties, aliphatic rings are prioritized over aromatic rings.
- Framework Simplification: After ring removal, the resulting structure is simplified: bridging atoms are converted to linkers, and the framework is re-aromatized [4].
- Tree Construction: This process repeats until only a single ring remains (the root scaffold). The sequence of scaffolds forms a linear path in the tree, with the final molecule as the leaf and the single ring as the root ancestor [2] [4].
- Output: A unique tree path for each input molecule, enabling its classification within a global hierarchy.
Protocol 3: Background Hierarchy Construction from PubChem
Scaffvis utilizes a massive precomputed hierarchy from PubChem as a background map [2] [30].
- Process PubChem Database: Apply Protocol 1 and Protocol 2 to the entire PubChem Compound database to generate millions of scaffold tree paths.
- Aggregate and Index Scaffolds: Collect all unique scaffolds generated across all levels (typically 8 hierarchical levels plus the molecular leaf level) [2]. Each scaffold is assigned a unique identifier.
- Establish Parent-Child Relationships: For each scaffold (except the root), define its parent as the scaffold from the preceding, more abstract level in the tree path.
- Compute Statistical Background: For each scaffold in the hierarchy, calculate its frequency of occurrence within the PubChem database. This frequency becomes a critical metric for visualizing how common or rare a scaffold is in known chemical space [2].
- Export Background Hierarchy: The final hierarchy, with scaffolds, relationships, and frequencies, is exported and made available for the Scaffvis visualization tool [30].
Protocol 4: Visualizing User Data with Scaffvis
This protocol outlines the steps for researchers to analyze their own datasets within the Scaffvis web interface [2].
- Upload User Dataset: Provide a file containing SMILES strings or compound identifiers of the research dataset.
- Map to Background Hierarchy: Scaffvis processes each user compound through the same standardization and scaffold tree generation rules. It then maps each compound's scaffold at every level to the corresponding node in the precomputed PubChem background hierarchy.
- Configure Treemap Visualization: The tool generates an interactive, zoomable treemap [2].
- Size Encoding: By default, the area of each rectangle (node) represents the frequency of that scaffold in the background PubChem database.
- Color Encoding: The color of each rectangle represents the frequency or another property (e.g., average bioactivity) of the scaffold within the user's uploaded dataset.
- Interactive Exploration: Researchers can click to zoom into specific branches of the scaffold tree, highlighting areas enriched with compounds from their dataset. This allows for the identification of both common scaffolds and rare, potentially novel chemotypes against the PubChem backdrop.
Quantitative Analysis of the PubChem Scaffold Hierarchy
The large-scale application of the scaffold tree methodology to the PubChem database provides critical quantitative insights into the structure of empirical chemical space. The statistics derived from this analysis form the foundational metrics that drive the Scaffvis visualization.
Table 1: Statistical Summary of PubChem-Based Scaffold Hierarchy
| Metric | Value | Description & Significance |
|---|---|---|
| Source Database | PubChem Compound | The reference chemical space defining empirical background frequencies [2]. |
| Hierarchy Levels | 9 (8 scaffold + 1 molecule) | The tree depth sufficient to cover chemical space with controlled branching [2]. |
| Virtual Root | Level 0 | A single node acting as the parent for all top-level (Level 1) scaffolds [2]. |
| Leaf Nodes | Millions of unique compounds | Each PubChem compound maps to a unique path terminating at a leaf [2]. |
| Key Visualization Metric | Scaffold Frequency in PubChem | Determines the size of treemap nodes; common scaffolds have larger areas [2]. |
Table 2: Comparative Analysis of Scaffold Hierarchy Methods
| Feature | Scaffold Tree (Schuffenhauer) | HierS | Scaffold Topology (Oprea) | Scaffvis Proposed Hierarchy |
|---|---|---|---|---|
| Core Principle | Iterative, prioritized single-ring removal [2] [4]. | Removal of entire ring systems and linkers [2]. | Edge merging to minimal ring topology [2]. | Optimized for homogeneous branching on PubChem data [2]. |
| Hierarchy Structure | Strict tree (unique path per molecule) [2]. | Not a tree/forest (multiple scaffolds per molecule) [2]. | Tree (with Murcko & molecular framework) [2]. | Rooted tree with 9 fixed levels [2]. |
| Determinism | Yes, rule-based [4]. | Yes, but generates multiple scaffolds. | Yes. | Yes, based on predefined PubChem mapping. |
| Primary Advantage | Data-set independent, unique classification [4]. | Captures all ring combinations. | Represents intuitive topological view. | Optimized for visualization (controlled branching) [2]. |
| Use in Visualization | Used in tools like Scaffold Hunter [2]. | Less suitable for tree layout. | Forms a clear abstraction hierarchy. | Forms the precomputed background in Scaffvis [2]. |
Visualization Architecture and Interactive Data Exploration
The Scaffvis platform translates the complex, high-dimensional data of the scaffold hierarchy into an intuitive visual interface. Its architecture is designed to handle large-scale data while providing responsive interaction for hypothesis generation.
System Architecture and Workflow: Scaffvis employs a client-server model. The server hosts the precomputed PubChem scaffold hierarchy and performs the computational mapping of user datasets to this background. The client, a web browser, renders the interactive visualization and handles user inputs like zooming and filtering [2]. The core visualization is a space-filling treemap, which effectively utilizes the entire screen area to represent the hierarchy. Each rectangle corresponds to a scaffold node, with nesting showing parent-child relationships [2].
Visual Encoding for Comparative Analysis: The treemap uses a dual-encoding system to facilitate instant comparison between the global background and the user's specific data:
- Size: The area of a rectangle is proportional to the frequency of that scaffold in the PubChem background. A large rectangle indicates a common molecular framework in known chemistry [2].
- Color: The fill color of a rectangle represents a metric from the user's dataset mapped to that scaffold. This can be the count of user compounds containing the scaffold, or a computed property like average potency or solubility [2]. This dual encoding allows researchers to instantly spot, for example, a small rectangle (rare scaffold) colored intensely red (highly active in their assay), highlighting a novel active chemotype.
Interaction and Drill-Down Analysis: The interface supports dynamic queries. Clicking on a rectangle zooms the view to make that node the new root, revealing its child scaffolds in detail. This enables researchers to drill down from a broad chemical class (e.g., "benzene derivatives") to specific, complex scaffolds. Tooltips provide exact quantitative data (frequency, user count, property values) for precise analysis [2].
Successful implementation of scaffold-based hierarchical analysis requires a combination of software tools, databases, and computational resources. The following toolkit is essential for work in this domain.
Table 3: Essential Toolkit for Scaffold-Based Hierarchical Analysis
| Tool/Resource | Category | Primary Function | Role in Scaffold Analysis |
|---|---|---|---|
| PubChem Database | Chemical Database | Repository of millions of experimentally characterized compounds and their bioactivities. | Serves as the empirical background for defining scaffold frequency and chemical space coverage in Scaffvis [2] [30]. |
| RDKit or CDK | Cheminformatics Library | Open-source toolkits for chemical informatics and machine learning. | Perform essential preprocessing: molecular standardization, Murcko framework extraction, and scaffold decomposition algorithms [2]. |
| Scaffvis Web Application | Visualization Platform | Web-based client-server application for interactive treemap visualization [2]. | The primary interface for mapping user data against the PubChem hierarchy and performing visual exploration and analysis [2]. |
| Precomputed PubChem Hierarchy | Data Resource | A file containing the scaffold tree hierarchy generated from the entire PubChem database [30]. | Provides the background map. Essential for running Scaffvis locally or understanding the underlying data structure [30]. |
| Jupyter Notebook / Python/R Environment | Analysis Environment | Interactive computing environment for data analysis and scripting. | Used for custom analysis of results, statistical testing of scaffold enrichment, and integrating scaffold insights with other assay data [31] [32]. |
Application Notes: Case Studies and Interpretation Guidelines
Case Study: Identifying Novel Chemotypes in a High-Throughput Screen
- Scenario: A research team conducts a screen of 50,000 proprietary compounds against a new target, identifying 500 active hits.
- Application of Scaffvis: The team uploads the 500 active SMILES strings to Scaffvis.
- Interpretation:
- Enrichment in Common Scaffolds: If large treemap areas (common PubChem scaffolds) are colored intensely, it suggests the target's active site accommodates well-explored chemotypes. This supports pursuing analog synthesis and SAR within known series.
- Activity in Rare Scaffolds: If small treemap areas (rare scaffolds) show strong coloration, it highlights novel chemotypes with potential for new IP and unique mechanisms. These become high-priority for confirmation and further exploration.
- Cluster Analysis: The visual clustering of colored rectangles indicates structural families among the actives, helping to define initial lead series.
Case Study: Assessing Library Diversity and Acquisition Gaps
- Scenario: A medicinal chemistry department wishes to evaluate the structural coverage of its corporate library.
- Application of Scaffvis: The entire corporate library (e.g., 200,000 compounds) is uploaded as the user dataset.
- Interpretation:
- Over-represented Areas: Large rectangles with intense color indicate chemical spaces where the library is highly concentrated, potentially indicating redundancy.
- White Space/Gaps: Large rectangles (common scaffolds) with little or no color reveal major gaps in the corporate collection compared to public chemical space. These are potential targets for library acquisition or synthesis efforts.
- Coverage of Rare Space: The presence of color in many small rectangles shows the library contains unique structures, contributing to its diversity and novelty.
Guidelines for Result Interpretation and Avoidance of Pitfalls
- Context of Background Frequency: Always interpret activity in the context of scaffold commonness. A single active compound belonging to a massive, common scaffold class (e.g., benzene) is less compelling than a single active compound belonging to a very rare scaffold.
- Artifacts of Hierarchy Rules: Be aware that the Scaffold Tree's deterministic rules may sometimes remove a biologically important ring first, placing molecules in a hierarchy branch that seems chemically counterintuitive [2]. Cross-reference visual findings with direct chemical inspection.
- Complement with Other Analyses: Scaffold tree visualization is a powerful hypothesis-generation tool. Its findings should be integrated with other data layers, such as molecular property distributions, docking scores, or pharmacokinetic predictions, for robust decision-making [31] [32].
Scaffvis embodies a significant advancement in the application of scaffold tree methodology by providing an intuitive, background-aware visualization of chemical datasets [2]. Framed within the broader thesis of hierarchical ring analysis, it demonstrates how a precomputed, empirical scaffold hierarchy can transform navigation and interpretation of chemical space. Its core strength lies in enabling researchers to instantly contextualize their findings—whether from screening, library design, or literature mining—against the vast backdrop of known chemistry in PubChem.
Future research directions in this field are likely to focus on:
- Dynamic and Multi-Parameter Backgrounds: Moving beyond a single static PubChem background to allow comparison against specialized backgrounds (e.g., FDA-approved drugs, natural products, kinase inhibitor space).
- Integration of Predictive Models: Coloring treemap nodes not just by experimental data, but by predicted properties from QSAR or AI models, enabling virtual screening directly within the hierarchy view.
- Enhanced Interactive Analytics: Tightly coupling the visualization with statistical tools for formal scaffold enrichment analysis and automated lead series identification.
- Methodological Hybridization: Combining the deterministic scaffold tree with data-driven clustering based on biological activity or physicochemical properties to create multi-faceted navigation systems.
As chemical data continues to grow in volume and complexity, tools like Scaffvis that prioritize chemical intuition, visual context, and interactive exploration will remain indispensable for translating structural information into actionable scientific knowledge and innovative drug discovery.
The iterative process of drug discovery is frequently hampered by the failure of lead compounds in late development stages, representing significant financial and temporal costs [33]. In this context, scaffold hopping has emerged as a pivotal strategy to reinvent bioactive molecules by replacing their core structure while preserving biological activity, thereby generating novel chemical entities with improved properties [34]. This approach directly addresses critical challenges in medicinal chemistry, including poor pharmacokinetics, toxicity, and intellectual property limitations [35].
The advent of artificial intelligence (AI) and sophisticated computational frameworks has catalyzed a renaissance in scaffold hopping. Traditional methods, reliant on molecular fingerprints and expert intuition, are being augmented and surpassed by deep learning models capable of navigating the vastness of chemical space with unprecedented precision [33] [34]. These AI-driven techniques facilitate the identification of non-obvious, synthetically accessible scaffolds that would be difficult to conceive through traditional means. This article details the application of these modern scaffold-hopping methodologies, firmly situating them within the foundational context of scaffold tree hierarchy analysis, a deterministic system for classifying and relating molecular frameworks [9] [10]. We provide detailed protocols and application notes to guide researchers in leveraging these integrated computational and experimental strategies for accelerated drug discovery.
Foundational Concepts: The Scaffold Tree Methodology
The scaffold tree methodology provides a systematic, hierarchical framework for deconstructing and analyzing molecular structures, forming the conceptual backbone for rational scaffold hopping. The process begins with the definition of a molecular framework (or scaffold), generated by pruning all terminal side chains and retaining only the ring systems and linkers that connect them [9].
The core algorithm for constructing a scaffold tree is deterministic and follows a set of prioritization rules to iteratively simplify complex scaffolds [9] [10]:
- Input: A molecule is reduced to its core scaffold by removing all terminal acyclic atoms.
- Iterative Ring Removal: Rings are removed one at a time to generate progressively simpler parent scaffolds. The removal order is not arbitrary but follows rules prioritizing the retention of chemically characteristic rings.
- Prioritization Rules: The algorithm prioritizes keeping bridged or spiro rings over fused or single rings. Within these categories, rings with more heteroatoms are retained over those with fewer, and larger rings are kept before smaller ones. This ensures the most "characteristic" rings remain as long as possible.
- Output - The Scaffold Tree: The process creates a tree where the original complex scaffold is the leaf node. Each step of ring removal creates a new node (a parent scaffold), ultimately culminating in a single, simple ring system as the root node [36].
This hierarchy transforms a collection of molecules into a navigable map of chemical space. For drug discovery, the tree allows the identification of active scaffold clusters—groups of molecules sharing a common parent scaffold that show biological activity. This visualization helps distinguish true structure-activity relationships from random noise in high-throughput screening data [10]. The scaffold tree is data-set-independent, scales linearly with the number of compounds, and provides a chemically intuitive classification system essential for organizing and planning scaffold-hopping campaigns [9].
Table 1: Categories of Scaffold Hopping Based on Structural Modification Degree [34]
| Category | Description | Degree of Hop | Example |
|---|---|---|---|
| Heterocyclic Replacement | Substituting one heterocycle for another (e.g., pyridine for pyrimidine). | 1° (Low) | Replacing an imidazo[1,2-a]pyrazine with a pyrazolo[1,5-a]pyrimidine in a TTK inhibitor series [35]. |
| Ring Opening/Closure | Converting a cyclic scaffold to an acyclic chain or vice-versa. | 2° (Medium) | Transforming a linear linker into a ring to rigidify a molecular glue scaffold [37]. |
| Peptide Mimicry | Replacing a peptide backbone with a rigid, non-peptide scaffold. | 3° (High) | Designing small-molecule mimics of α-helical or β-strand protein domains. |
| Topology-Based Hop | Global change of the scaffold topology while preserving pharmacophore geometry. | 4° (Very High) | Using a multi-component reaction (MCR) scaffold to replace a composite core while maintaining 3D shape complementarity [37]. |
Computational Foundations for Scaffold Hopping
Effective scaffold hopping relies on computational methods to represent molecules, evaluate similarity, and predict the properties of novel designs. These tools bridge the gap between the abstract hierarchy of the scaffold tree and the generation of tangible, synthesizable compounds.
Molecular Representation is the critical first step. Traditional methods like Simplified Molecular-Input Line-Entry System (SMILES) strings and molecular fingerprints (e.g., Extended-Connectivity Fingerprints, ECFP) encode structural information but may not fully capture complex 3D interactions [34]. Modern AI-driven approaches use graph neural networks (GNNs), where atoms are nodes and bonds are edges, or language models that treat SMILES strings as text to learn deep, continuous representations that encapsulate both structural and functional properties [34].
Similarity and Bioactivity Prediction: Once represented, the key challenge is identifying novel scaffolds that are functionally similar to the lead. This involves:
- Pharmacophore Modeling: Identifying the essential steric and electronic features necessary for molecular recognition.
- Shape-Based Similarity: Comparing the three-dimensional volume and electrostatic potential of molecules (e.g., using Electron Shape Similarity) [38].
- Quantitative Structure-Activity Relationship (QSAR) Models: Using machine learning to predict biological activity from molecular descriptors or fingerprints.
Advanced Free Energy Calculations: For structure-based design, Free Energy Perturbation (FEP) calculations provide a rigorous, physics-based method to predict the binding affinity change (ΔΔG) between closely related ligands. As demonstrated in optimizing soluble adenyl cyclase (sAC) inhibitors, FEP can guide scaffold hopping by accurately ranking the relative binding energies of candidate cores before synthesis, and then optimize the new series to sub-nanomolar potency [39].
Diagram 1: Computational workflow for scaffold hopping. The process integrates multiple molecular representations to generate novel cores via rule-based or AI-driven methods, followed by multi-faceted filtering to identify promising candidates.
Table 2: Key Computational Methods for Scaffold Hopping
| Method Category | Specific Tool/Approach | Primary Function in Scaffold Hopping | Key Advantage |
|---|---|---|---|
| Molecular Representation | Extended-Connectivity Fingerprints (ECFP) [34] | Encode substructures for similarity searching and QSAR. | Computationally efficient, well-established. |
| Graph Neural Networks (GNNs) [34] | Learn rich, task-specific molecular embeddings for activity prediction. | Captures topological and relational information. | |
| Scaffold Generation & Search | AnchorQuery [37] | Pharmacophore-based search of synthesizable MCR libraries. | Direct link to readily synthesizable, drug-like chemistry. |
| ChemBounce [38] | Replaces core scaffolds using a large fragment library. | Systematic exploration focused on synthetic accessibility. | |
| Binding Affinity Prediction | Free Energy Perturbation (FEP+) [39] | Predicts ΔΔG for congeneric series for lead optimization. | High accuracy for ranking similar compounds; physics-based. |
| Glide Docking / MM-GBSA [39] | Provides binding poses and approximate affinity estimates. | Faster than FEP for initial screening of diverse scaffolds. |
AI-Driven Approaches and Integrative Protocols
The integration of AI with the scaffold tree methodology creates a powerful, iterative cycle for discovery. AI models excel at identifying patterns in high-dimensional chemical data derived from scaffold tree classifications, enabling the prediction of which novel branches (scaffolds) might retain bioactivity [33].
Generative AI Models such as Variational Autoencoders (VAEs) and Generative Adversarial Networks (GANs) can learn the distribution of bioactive compounds from a training set organized by scaffold hierarchies. They can then generate entirely novel, yet structurally plausible, scaffolds that fulfill multiple property constraints (e.g., activity, solubility, synthetic accessibility) [34]. Transformer-based models, pre-trained on millions of SMILES strings, can be fine-tuned to generate molecules conditioned on a desired scaffold or pharmacophore pattern [34].
A critical application is hit expansion. When a promising active compound ("hit") is identified from screening, its position in the scaffold tree is determined. AI models can then be used to:
- Generate analogs within the same scaffold branch for initial SAR.
- Propose hops to neighboring or parent scaffolds in the tree that represent unexplored chemical space.
- Evaluate the generated candidates for synthetic feasibility and predicted activity, prioritizing the most promising ones for synthesis.
Protocol 1: AI-Augmented Hit Expansion via Scaffold Tree Navigation
- Step 1 – Input & Classification: Input the SMILES of the confirmed hit molecule. Process it through the scaffold tree algorithm to identify its core scaffold and all parent scaffolds [9].
- Step 2 – Neighborhood Definition: Define the "chemical neighborhood" for hopping. This can include: a) direct siblings (other scaffolds with the same parent), b) the parent scaffold itself, c) child scaffolds of the parent (more complex cores), or d) scaffolds from other branches that share key pharmacophoric features [36].
- Step 3 – AI-Driven Generation: Use a conditional generative model (e.g., a fine-tuned Molecular Transformer). Condition the generation on either the original scaffold's SMILES or a pharmacophore query derived from the hit's binding mode. Set property constraints (molecular weight, logP) to maintain drug-likeness.
- Step 4 – Multi-Objective Filtering: Filter the generated molecules (typically 1000s) sequentially by: a) Synthetic Accessibility (SA) Score, b) Similarity to original hit (Tanimoto > 0.3), c) Predicted activity from a pre-trained QSAR model, and d) In-silico ADMET risk [38].
- Step 5 – Output & Prioritization: Output a ranked list of 20-50 candidate SMILES for visual inspection by a medicinal chemist. The final selection for virtual synthesis is based on the combination of scores, structural novelty, and medicinal chemistry intuition.
Application Notes: A Case Study in Molecular Glue Development
The following detailed protocol illustrates the practical integration of computational scaffold hopping, scaffold tree principles, and synthetic chemistry to develop novel molecular glues stabilizing the 14-3-3σ/ERα protein-protein interaction (PPI) [37].
Background: The starting point was a covalent molecular glue, Compound 127, which stabilized the 14-3-3σ/ERα complex. While active, its scaffold offered limited opportunities for optimization. The goal was to perform a topology-based scaffold hop to a novel, rigid, and synthetically versatile core while maintaining the critical 3D shape and pharmacophore elements [37].
Protocol 2: Pharmacophore-Driven Scaffold Hop to an MCR Scaffold
Step 1 – Pharmacophore Extraction from Structural Data
- Use the co-crystal structure of Compound 127 bound to the 14-3-3σ/ERα complex (PDB: 8ALW).
- Identify and define the essential pharmacophore features: i) a deep "anchor" motif (the p-chloro-phenyl ring occupying a hydrophobic pocket), and ii) three key interaction points (e.g., hydrogen bond donor/acceptor features from the tetrahydropyran and aniline groups) [37].
Step 2 – In-Silico Screening of a Synthesizable Library
- Utilize the software AnchorQuery, which contains a virtual library of >31 million compounds derived from Multi-Component Reactions (MCRs).
- Input the extracted pharmacophore. Constrain the anchor to a phenylalanine-bioisosteric group. Set a molecular weight filter (<400 Da).
- Screen the library of 27 different MCR chemistries. The top-ranked hits consistently belonged to the Groebke-Blackburn-Bienaymé (GBB) three-component reaction scaffold, forming imidazo[1,2-a]pyridines. Docking confirmed a high shape complementarity to the original ligand [37].
Step 3 – Scaffold Tree Analysis & Library Design
- Classify both the original Compound 127 and the new GBB core using the scaffold tree algorithm. The hop represents a significant topological change (4° hop), moving to a different branch of the chemical hierarchy.
- Leverage the synthetic power of the GBB-3CR. Design a focused library by varying the three input components: the aldehyde, the 2-aminopyridine, and the isocyanide. This allows for rapid exploration of structure-activity relationships (SAR) around the new, rigid core [37] [35].
Step 4 – Synthesis & Biophysical Validation
- Synthesize a library of 50-100 GBB analogs.
- Test compounds in orthogonal biophysical assays: Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) and Surface Plasmon Resonance (SPR) to quantify PPI stabilization and binding kinetics.
- Validate the cellular activity of the most potent analogs using a NanoBRET assay with full-length proteins in live cells.
- Obtain co-crystal structures of promising analogs to confirm the predicted binding mode and guide further optimization [37].
Table 3: The Scientist's Toolkit for the Molecular Glue Case Study
| Reagent/Resource | Function/Description | Role in Scaffold Hopping Protocol |
|---|---|---|
| Co-Crystal Structure (PDB: 8ALW) | Provides atomic-level details of the ligand-protein complex. | Source for extracting the critical 3D pharmacophore model used to query new scaffolds. |
| AnchorQuery Software | Pharmacophore-based screening tool linked to enumerable MCR chemistry. | Enables the jump from a known ligand to novel, synthetically accessible chemotypes (GBB scaffold). |
| GBB-3CR Components | Aldehydes, 2-aminopyridines, isocyanides. | Building blocks for the rapid synthesis of a diverse, focused library around the hopped scaffold. |
| TR-FRET & SPR Assays | Orthogonal biophysical techniques measuring binding and stabilization. | Generate quantitative SAR data for the new scaffold series to guide lead optimization. |
| NanoBRET Cellular Assay | Live-cell protein-protein interaction assay. | Confirms target engagement and functional efficacy of hopped compounds in a physiologically relevant context. |
Diagram 2: Experimental workflow for scaffold hopping to a novel molecular glue series [37]. The protocol progresses from structural analysis through computational design to synthesis and multi-tiered validation.
Scaffold hopping, when systematically guided by the scaffold tree hierarchy and powered by modern AI and computational chemistry, is a transformative strategy in drug discovery. It provides a structured pathway to innovate beyond known chemical matter, addressing the dual demands of biological efficacy and drug-like properties. The integration of these methodologies—from the deterministic classification of the scaffold tree to the predictive power of FEP and the generative capability of AI—creates a robust framework for navigating chemical space.
Future advancements will focus on enhancing the interpretability and reliability of AI models, ensuring generated scaffolds are not only novel but also synthetically feasible and possess favorable pharmacokinetic profiles from the outset [33]. Furthermore, the expansion of accessible, high-quality chemical and biological datasets will be crucial for training more accurate models. As these computational tools become more integrated with automated synthesis and high-throughput experimentation platforms, the cycle of design, prediction, synthesis, and testing will accelerate dramatically. In this evolving landscape, the scaffold tree remains an essential conceptual map, providing the intuitive, hierarchical organization of chemical space upon which intelligent, data-driven navigation and innovation depend.
The discovery of novel therapeutics for Tuberculosis (TB), particularly against drug-resistant strains of Mycobacterium tuberculosis (Mtb), remains a pressing global challenge [40]. The process is hindered by the vastness of chemical space and the inefficiency of traditional screening methods [41]. This application note details a structured computational methodology that integrates PubChem bioactivity datasets with hierarchical scaffold tree analysis to systematically identify and prioritize novel chemotypes for anti-TB drug discovery.
The core thesis of this research posits that a rule-based, hierarchical decomposition of molecules into scaffolds provides a superior framework for analyzing chemical libraries and understanding Structure-Activity Relationships (SAR) compared to flat, non-hierarchical clustering [41]. Scaffold trees organize chemical space intuitively, allowing researchers to navigate from complex active molecules to simpler core structures and vice versa, facilitating scaffold hopping—the intentional modification of a molecule's core while retaining biological activity [34] [40]. This approach is especially powerful when applied to large-scale public data like that in PubChem, enabling the data-driven identification of under-explored scaffolds with predicted bioactivity against critical Mtb targets.
Core Concepts: Scaffold Tree Methodology and Definitions
This protocol is built upon the foundation of scaffold tree methodology, which provides a systematic, multi-level abstraction of molecular structures. The following key definitions and concepts are critical [41] [42]:
- Murcko Scaffold: The molecular framework obtained by removing all terminal side chains, leaving only ring systems and the linkers that connect them.
- Generic Murcko Scaffold: A further abstraction of the Murcko scaffold where all atoms are converted to carbon and all bonds to single bonds, focusing purely on topology.
- Scaffold Tree: A hierarchical organization where a parent scaffold is iteratively "pruned" by removing rings according to a set of heuristic rules (e.g., prioritizing heterocycles, smaller rings, or rings with lower connectivity), generating a tree of increasingly simplified child scaffolds.
- Scaffold Hopping: A medicinal chemistry strategy to identify novel molecular cores (scaffolds) that retain or improve the biological activity of a lead compound. It is categorized into heterocyclic replacements, ring opening/closure, peptide mimicry, and topology-based changes [34] [40].
- SCINS (Scaffold Identification and Naming System): An open-source, rule-based method that describes a reduced generic scaffold. It abstracts ring size and some chain length information to group compounds into chemically intuitive, broad classes, effectively mapping the density of chemical space [41].
Application Note: Protocol for PubChem Dataset Curation and Hierarchical Analysis
Data Acquisition and Preprocessing
Objective: To build a focused, high-quality dataset of compounds tested against Mycobacterium tuberculosis for hierarchical scaffold analysis.
- Data Retrieval:
- Query the PubChem BioAssay database (via REST API or FTP download) for all assays containing Mycobacterium tuberculosis (Taxonomy ID: 1773) in the description or target information.
- Filter assays to retain those reporting quantitative activity data (e.g., MIC, IC50, % inhibition). Primary screening data from AID 485364 is a recommended starting point.
- Download Compound IDs (CIDs), canonical SMILES, and the associated activity endpoint and value.
- Data Curation and Standardization:
- Standardize Structures: Using RDKit or OpenBabel, sanitize molecules, neutralize charges, remove salts, and keep only the largest covalent component.
- Remove Inorganics/Organometallics: Filter out compounds containing atoms outside the typical drug-like set (e.g., B, Si, metals).
- Apply Activity Threshold: Define an activity cutoff (e.g., MIC ≤ 10 µM or % inhibition ≥ 80%) to create an "active" subset. A corresponding "inactive" set (e.g., MIC > 25 µM or % inhibition < 20%) should also be curated for contrast.
- Deduplicate: Merge entries for the same canonical SMILES, retaining the median activity value.
Scaffold Generation and Hierarchical Analysis
Objective: To decompose the active compound set into a hierarchical scaffold tree and network, enabling chemotype navigation and series identification.
- Generate Murcko Scaffolds: For each active compound, generate its Murcko scaffold using the
rdkit.Chem.Scaffolds.MurckoScaffoldmodule. - Construct Scaffold Trees: For each unique Murcko scaffold, apply a pruning algorithm (e.g., as implemented in the
ScaffoldTreeclass within RDKit or the CDK) to generate its hierarchical tree. Key pruning rules typically prioritize the removal of:- Heteroatom-containing rings before carbocycles.
- Smaller rings before larger ones.
- Rings with higher substitution/connectivity.
- Build a Scaffold Network: Create a network graph (e.g., using NetworkX) where nodes represent unique scaffolds (both parent and child from all trees). Draw edges between scaffolds where a direct parent-child relationship exists within any tree. This network visualizes the shared substructural relationships across all actives [42].
- Perform SCINS Analysis: Use the open-source SCINS implementation [41] to classify all actives. This groups scaffolds into broader, topology-based categories (e.g., "2RING1_LINKER"), allowing for the rapid identification of densely populated (hot) and unexplored (cold) regions in the chemical space of TB actives.
Quantitative Analysis of TB-Relevant Scaffolds
Objective: To summarize the prevalence and activity of key scaffolds emerging from the hierarchical analysis, with a focus on those validated in recent literature.
Table 1: Analysis of Privileged and Emerging Scaffolds in Anti-TB Drug Discovery
| Scaffold Class | Representative Core Structure | Key Target/Pathway | Exemplar Potency (MIC range) | Notes & Advantages |
|---|---|---|---|---|
| Nitroimidazole-Oxazine (NOS) [43] | Nitroimidazole fused to oxazine | Ddn (Deazaflavin-dependent nitroreductase) | Sub-micromolar to low µM | Prodrug activated by Mtb-specific enzyme; core of pretomanid. |
| Quinoline [44] | Bi- or tricyclic system with N heterocycle | Multiple (ATP synthase, Gyrase, respiratory chain) | Nanomolar to low µM (e.g., Bedaquiline analogs) | Privileged scaffold; proven clinical success (Bedaquiline). |
| Benzimidazole / Quinazoline [45] | Fused benzene and imidazole/ pyrimidine rings | Phe-tRNA synthetase (PheRS) | Fragment-level binding (Kd µM-mM) | Novel target; multiple crystal structures available for SBDD. |
| Aryl-Quinoline Carboxylate [44] | Quinoline with carboxylic acid and aryl substituent | DNA Gyrase | ~40 µM (MIC90) | Scaffold hop from fluoroquinolones; novel chemical series. |
Table 2: Scaffold Diversity Metrics in a PubChem TB Active Set (Hypothetical Output)
| Analysis Method | Number of Unique Entries | % of Compounds in Top 10 Classes | Singletons (Uniquely Occurring Scaffolds) | Interpretation |
|---|---|---|---|---|
| Murcko Scaffolds | 1,850 | 15% | 1,200 (65%) | High granularity; many unique scaffolds indicate diverse chemotypes but challenges in identifying series. |
| Generic Murcko Scaffolds | 1,100 | 22% | 600 (55%) | Increased grouping; reveals underlying topological commonalities. |
| SCINS Classes [41] | 45 | 65% | 5 (11%) | High-level grouping; clearly identifies "hot" chemical series (e.g., 2RING1_LINKER) for lead development. |
Detailed Experimental Protocols
Protocol 1: Scaffold-Based Virtual Screening for Novel TB Inhibitors
Aim: To identify novel chemotypes targeting the Mtb Phe-tRNA synthetase (PheRS) L-Phe binding site via a scaffold-hopping strategy [45]. Materials: Schrodinger Maestro Suite or Open-Source Equivalents (AutoDock Vina, PyMol), RDKit, Enamine REAL or ZINC15 library subset. Procedure:
- Template Selection: Retrieve the X-ray crystal structure of Mtb PheRS in complex with a fragment hit (e.g., PDB ID associated with D-735 [45]). Prepare the protein by adding hydrogens, assigning bond orders, and optimizing side-chain orientations.
- Active Site Definition: Define the binding pocket using coordinates of the co-crystallized fragment in the L-Phe amino acid binding site.
- Seed Scaffold Identification: Extract the core scaffold (e.g., benzimidazole, quinazoline) from the crystallized fragment using Murcko decomposition.
- Virtual Library Construction: Query the Enamine REAL library for compounds containing the seed scaffold or its isosteric replacements (e.g., indole, benzoxazole). Filter for drug-likeness (Lipinski's Rule of Five).
- Molecular Docking: Dock the filtered library into the defined active site. Use standard precision (SP) docking to generate pose predictions and a GlideScore (or equivalent).
- Post-Docking Scaffold Analysis: Cluster top-ranked hits (e.g., GlideScore < -6.0 kcal/mol) by their Murcko scaffolds. Use the scaffold network generated from these hits to visualize the diversity of proposed chemotypes and select 2-3 distinct series for in vitro testing.
Protocol 2: Cheminformatic Workflow for Scaffold Tree Construction and Analysis
Aim: To implement a reproducible pipeline for generating and analyzing scaffold trees from a list of SMILES. Materials: Python 3.8+, RDKit, Pandas, NetworkX, Matplotlib. Procedure:
- Input: A
.csvfile with columns:"SMILES","Activity_Value". - Generate Scaffolds:
Build and Export a Scaffold Tree for a Representative Active:
Create a Global Scaffold Network:
Visualizations
Title: Hierarchical Analysis Workflow for TB Drug Discovery
Title: Scaffold Network Enabling Novel Series Identification
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Scaffold-Centric TB Drug Discovery
| Resource Category | Specific Tool / Database | Function in Protocol | Key Features / Rationale |
|---|---|---|---|
| Cheminformatics Toolkit | RDKit (Open Source) | Core library for molecule I/O, standardization, Murcko scaffold generation, fingerprint calculation. | Industry-standard, Python-based, enables full customization of analysis pipelines [41]. |
| Bioactivity Data | PubChem BioAssay | Primary source for retrieving compounds tested against Mtb targets with associated activity data. | Largest public repository, essential for data-driven scaffold analysis and hypothesis generation. |
| Scaffold Analysis Libraries | SCINS (Open Source Python Implementation) [41] | Rule-based classification of compounds into broad scaffold classes to map chemical space density. | Provides a complementary, less granular view than Murcko scaffolds to identify "hot" series [41]. |
| Scaffold Analysis Libraries | Molecular Anatomy Tool [42] | Generates multi-dimensional hierarchical scaffold networks from compound sets. | Enables advanced visualization and analysis of scaffold relationships beyond simple trees [42]. |
| Commercial/Final Compounds | Enamine REAL / Mcule | Source of purchasable compounds for virtual screening follow-up and in vitro validation. | Ultra-large libraries allow for scaffold-based searching and procurement of novel analogs. |
| Structural Biology | RCSB Protein Data Bank (PDB) | Source of 3D protein structures (e.g., Mtb PheRS [45], Ddn [43]) for structure-based design. | Critical for understanding binding modes and guiding scaffold optimization via docking. |
The integration of PubChem's large-scale bioactivity data with hierarchical scaffold tree methodology provides a powerful, systematic framework for accelerating TB drug discovery. This approach moves beyond simple compound-level analysis to organize chemical space based on intrinsic structural relationships, enabling:
- The objective identification of privileged scaffolds (e.g., quinoline, nitroimidazole) and under-explored chemotypes within TB active sets.
- The rational planning of scaffold-hopping campaigns to discover novel series with improved properties, as demonstrated for targets like PheRS and Ddn [40] [43] [45].
- The generation of testable hypotheses by linking simplified, common core structures to complex active molecules via a navigable scaffold network.
Future directions involve tighter integration of AI-driven molecular representation methods (e.g., graph neural networks) with rule-based scaffold trees to predict novel, synthesizable scaffolds with high probabilities of anti-TB activity, ultimately creating a more predictive and generative cycle for lead identification [34].
This application note details the integration of the hierarchical scaffold tree methodology with modern artificial intelligence (AI)-driven generative frameworks, proposing a conceptualized system termed "ChemBounce." Scaffold trees provide a deterministic, chemically intuitive hierarchy for organizing molecular ring systems, serving as a foundational map for navigating chemical space [10]. Concurrently, AI models like variational autoencoders (VAEs) have demonstrated powerful capabilities for de novo scaffold generation and hopping, optimizing for desired properties while maintaining core side-chain functionalities [46]. By unifying these paradigms, ChemBounce aims to establish a structured, AI-augmented workflow for computational scaffold replacement. This document provides detailed protocols for scaffold tree construction, AI model training and fine-tuning on tree-derived data, and subsequent experimental validation of generated compounds through molecular docking and free-energy calculations. The integration framework is designed to enhance the efficiency and rationality of scaffold-hopping campaigns in drug discovery, providing researchers with a systematic tool for lead optimization and novelty generation within a well-defined chemical hierarchy.
The concept of a molecular scaffold—the core ring system of a molecule stripped of its side chains—is central to medicinal chemistry for analyzing structure-activity relationships (SAR) and navigating chemical space [9]. The scaffold tree methodology, introduced by Schuffenhauer et al., provides a rigorous, hierarchical classification system where molecular frameworks form leaf nodes, and iterative removal of the least characteristic rings generates parent scaffolds at higher levels [10] [9]. This deterministic, data-set-independent method creates a unique tree for each compound, enabling the visualization and analysis of vast chemical libraries based on core structural relationships [47].
Parallel to this, AI-driven generative models have revolutionized de novo molecular design. Techniques such as graph-based variational autoencoders (VAEs) can learn distributed representations of molecules and generate novel, valid chemical structures with optimized properties [46] [48]. A specific application, scaffold hopping, seeks to replace a molecule's core scaffold while preserving its bioactive side chains, a task well-suited to AI models that can disentangle and independently manipulate scaffold and side-chain representations [46].
The ChemBounce framework conceptualizes the integration of these two powerful approaches. It posits that the scaffold tree is not merely an analytical tool but can serve as a structured guide and constraint for generative AI. By training models on tree-organized data and using the hierarchical relationships to inform latent space exploration, AI-driven scaffold hopping can become more focused, interpretable, and efficient. This synthesis aims to resolve the "comfort-growth paradox" in human-AI collaboration by providing a chemically intuitive scaffold (growth) within a powerful generative framework (AI-assisted comfort) [49].
Foundational Methodologies and Data Standards
The Scaffold Tree: Hierarchical Ring Analysis
The scaffold tree algorithm provides a systematic breakdown of a molecule into increasingly simplified core structures [10] [47].
Core Protocol: Tree Generation
- Input Preparation: Provide molecules in SMILES or SDF format. Pre-process by removing salts, neutralizing charges, and keeping the largest fragment [47].
- Initial Framework Extraction: Generate the molecular framework (Bemis-Murcko scaffold) by pruning all terminal acyclic side chains [10].
- Iterative Ring Removal: From the current scaffold, systematically remove one ring at a time to generate a set of potential parent scaffolds.
- Prioritization Filtering: Apply a series of rules to select the single, chemically "least characteristic" parent for the next level. Standard prioritization rules [10] [9] include:
- Remove smaller rings before larger ones.
- Remove rings with fewer heteroatoms first.
- Remove aliphatic rings before aromatic ones.
- Preferentially retain bridged or fused ring systems.
- Tree Construction: Repeat steps 3-4 until no rings remain. The sequence of scaffolds forms a unique linear path from the original complex scaffold (leaf) to a single ring or acyclic linker (root).
Implementation: The open-source
ScaffoldGraphlibrary enables efficient generation of scaffold trees and networks from large datasets [47]. It allows for custom prioritization rules and outputs graphs that can be analyzed with network science tools.
AI-Driven Scaffold Generation and Hopping
Generative AI models learn to create novel molecular structures. For scaffold-focused tasks, models like ScaffoldGVAE are specifically architected [46].
- Core Architecture (ScaffoldGVAE): This model uses a multi-view graph neural network as an encoder to separately learn embeddings for a molecule's atoms (nodes) and bonds (edges) [46]. A key innovation is the explicit separation of the full molecular embedding into scaffold embedding and side-chain embedding. The scaffold embedding is mapped to a Gaussian mixture model latent space, facilitating smooth interpolation and generation of novel cores, while the side-chain embedding is preserved to maintain the original substituents during scaffold hopping [46].
- Data Pipeline for AI Training:
- Source: Curate large-scale small molecule datasets (e.g., from ChEMBL). Preprocess using standard medicinal chemistry filters (PAINS, reactivity, molecular weight) [46].
- Scaffold Annotation: For each molecule, use the ScaffoldTree algorithm via ScaffoldGraph to extract its full tree path. This provides multiple scaffold examples at different complexity levels from a single molecule.
- Pair Formation: For scaffold hopping tasks, create input-output pairs where the input is a molecule and the target output is a different molecule from the same dataset that shares similar side-chain profiles but a different scaffold, ideally identified via tree neighborhood analysis.
Table 1: Quantitative Performance of AI Scaffold Hopping Models
| Model | Architecture | Key Metric: Novelty (%) | Key Metric: Uniqueness (%) | Key Metric: Docking Score (Δ, kcal/mol) | Reference |
|---|---|---|---|---|---|
| ScaffoldGVAE | Graph VAE + Gaussian Mixture | 99.8 | 99.9 | -1.2 to -4.5 (improvement) | [46] |
| GraphGMVAE | Graph Gaussian Mixture VAE | Not Reported | Not Reported | Not Reported | [46] |
| DeepHop | Multimodal Transformer | High (Qualitative) | High (Qualitative) | Not Reported | [46] |
| SyntaLinker | Fragment Linker VAE | Focused on linkers, not full scaffolds | Focused on linkers, not full scaffolds | Not Reported | [46] |
Table 2: Scaffold Tree Analysis Parameters and Outcomes
| Parameter / Dataset | Pyruvate Kinase Binders [10] | Pesticide Collection [10] | Kinase-Targeted Fine-Tuning (CDK2, EGFR, etc.) [46] |
|---|---|---|---|
| Number of Compounds | ~50,602 (incl. actives) | Not Specified | 1,286 - 7,271 per target |
| Tree Hierarchy Levels | Up to 8-10 rings per molecule | Not Specified | Scaffolds filtered to 1-20 heavy atoms |
| Key Finding | Active compounds clustered in specific scaffold branches | Robust handling of natural product complexity | Enables target-focused model fine-tuning |
Integrated Protocol: The ChemBounce Workflow
The ChemBounce framework integrates the above methodologies into a sequential, iterative pipeline for AI-driven scaffold replacement guided by hierarchical tree analysis.
Figure 1: The ChemBounce Integrated Workflow for AI-Driven Scaffold Replacement. This diagram outlines the sequential and iterative steps from an input lead compound to validated, novel scaffold-hopped molecules.
Phase 1: Tree-Based Analysis & Data Preparation
- For the input lead compound, generate its full scaffold tree using Protocol 2.1.
- Analyze the tree to identify promising directions for hopping:
- Sibling Hopping: Identify scaffolds at the same tree level (similar complexity) from other active compounds in the dataset.
- Parent/Child Hopping: Consider moving to a simpler (parent) or more complex (child) scaffold in the hierarchy.
- From a target-specific dataset (e.g., all active molecules against EGFR), generate scaffold trees and create a paired dataset for AI training, where pairs are molecules linked by tree-derived relationships (e.g., they share a common parent scaffold).
Phase 2: AI Model Fine-Tuning & Generation
- Start with a pre-trained generative model (e.g., ScaffoldGVAE pre-trained on ChEMBL) [46].
- Fine-tune the model using the tree-derived, target-specific paired dataset from Phase 1. This biases the model's latent space toward scaffold transformations relevant to the desired biological activity.
- To perform hopping for a new lead, encode it and manipulate its scaffold embedding within the latent space. Use tree-derived metrics (e.g., vector direction towards a parent scaffold) to guide this manipulation.
- Decode the modified scaffold embedding, recombining it with the original side-chain embedding, to generate novel molecules with replaced cores.
Phase 3: Experimental Validation Protocol
- Virtual Screening: Filter generated molecules for drug-likeness (Lipinski's Rule of Five, synthetic accessibility score).
- Molecular Docking: Dock the top candidates into the target protein's active site (e.g., using LeDock [46]). Compare binding poses and scores to the original lead.
- Binding Affinity Estimation: Perform more rigorous binding free energy calculations for a shortlist of candidates using molecular mechanics with generalized Born and surface area solvation (MM/GBSA) [46].
- In Vitro Testing: Prioritize molecules with favorable computational profiles for synthesis and in vitro activity assay (e.g., IC50 determination).
Table 3: Research Reagent Solutions Toolkit
| Item / Resource | Function in ChemBounce Protocol | Source / Example |
|---|---|---|
| ChEMBL Database | Primary source of small molecule bioactivity data for pre-training and target-specific dataset assembly. | https://www.ebi.ac.uk/chembl/ [46] |
| ScaffoldGraph Software | Open-source Python library for generating scaffold trees, networks, and performing hierarchical analysis. | https://github.com/UCLCheminformatics/ScaffoldGraph [47] |
| RDKit | Open-source cheminformatics toolkit used for molecule standardization, descriptor calculation, and substructure manipulation. | https://www.rdkit.org/ |
| PyTorch / TensorFlow | Deep learning frameworks for implementing and training graph neural network models like ScaffoldGVAE. | https://pytorch.org/, https://www.tensorflow.org/ [46] |
| Docking Software (LeDock, AutoDock Vina) | To predict the binding pose and score of generated molecules against a protein target. | LeDock [46] |
| MM/GBSA Pipeline (AMBER, GROMACS) | To compute binding free energies for a more reliable affinity ranking of designed compounds. | Used in MM/GBSA validation [46] |
Figure 2: ScaffoldGVAE Core Architecture. The model disentangles scaffold (zs) and side-chain (zc) embeddings, projecting the scaffold into a Gaussian Mixture latent space for generative operations [46].
Discussion and Future Perspectives
The integration of deterministic scaffold tree classification with probabilistic AI generative models, as conceptualized in ChemBounce, presents a compelling path forward for computational medicinal chemistry. The scaffold tree provides a "chemical compass," offering interpretability and direction to the latent space navigation of AI models, potentially reducing unproductive generation and focusing on chemically meaningful regions [10] [49]. This hybrid approach can directly address key challenges in scaffold hopping, such as maintaining target affinity while achieving significant intellectual property novelty.
Future developments may involve:
- Dynamic Tree Integration: Moving beyond using the tree as a static map to developing AI models that learn to traverse the tree structure directly, enabling automatic prediction of optimal hopping directions.
- Multi-Objective Optimization: Extending the framework to simultaneously optimize for scaffold novelty, synthetic accessibility, and multiple ADMET properties using reinforcement learning within the tree-constrained space [48].
- Application to Macrocyclic and Peptidic Scaffolds: Adapting the hierarchical analysis and generation principles to larger, more complex chemical spaces relevant to modern drug discovery, including the design of synthetic binding proteins [50].
The protocols outlined herein provide a foundational roadmap. Successful implementation requires cross-disciplinary collaboration between cheminformaticians, AI researchers, and medicinal chemists to iteratively refine the models and validate their output in real-world drug discovery projects.
Navigating Pitfalls: Troubleshooting Common Issues and Leveraging AI for Enhanced Scaffold Analysis
The scaffold tree methodology provides a systematic, hierarchical framework for classifying and analyzing the core ring systems of organic molecules, which is fundamental to drug discovery and chemical space exploration [3]. In this approach, a molecular scaffold—typically defined as the Murcko framework comprising all rings and the linkers connecting them—is iteratively dissected by removing one ring at a time to generate a hierarchy of simpler parent scaffolds [3] [10]. This process creates a unique, deterministic tree where each node represents a chemical scaffold, and the roots are single-ring systems [10].
However, this seemingly straightforward process is fraught with inherent ambiguities. The core challenges lie in two main areas: the algorithmic ambiguity in deciding which ring to remove next during tree construction, and the representational complexity of accurately handling and classifying fused ring systems where rings share bonds or atoms [3] [2]. These ambiguities can significantly impact the outcome of scaffold-based analysis, such as virtual screening, activity prediction, and scaffold hopping—a strategy aimed at discovering new bioactive core structures [34]. Resolving these challenges is critical for ensuring that hierarchical scaffold classifications yield chemically intuitive, reproducible, and biologically relevant insights, particularly within the broader thesis of mapping and navigating chemical space for drug development [34] [2].
Core Ambiguities in Hierarchical Ring Removal
The process of constructing a scaffold tree is not a simple mechanical dissection. At each step, multiple rings may be candidates for removal, and the choice among them introduces significant ambiguity that affects the entire hierarchical classification.
The Ring Removal Prioritization Problem
The foundational Scaffold Tree algorithm resolves the ambiguity of ring selection through a set of deterministic, chemically motivated prioritization rules [3] [10]. The goal is to remove the "least characteristic" ring first, thereby preserving the core, functionally significant part of the scaffold for as long as possible in the hierarchy. The rules are applied in a defined sequence.
Table 1: Standard Prioritization Rules for Ring Removal in Scaffold Tree Generation [3] [10]
| Priority | Rule Criterion | Chemical Rationale & Action |
|---|---|---|
| 1 (Highest) | Bridge vs. Non-Bridge | Preserve bridged ring systems (e.g., norbornane) as they are more complex and characteristic. Remove non-bridged rings first. |
| 2 | Aromatic vs. Saturated | Preserve aromatic rings due to their prevalence in drugs and role in interactions. Remove saturated rings first. |
| 3 | Heteroatom Content | Preserve rings with heteroatoms (N, O, S, etc.) as they often contribute to binding. Remove rings with fewer heteroatoms first. |
| 4 | Ring Size | Preserve larger rings as they may define a unique shape. Remove smaller rings (e.g., 3- and 4-membered) before 5- and 6-membered rings. |
| 5 | Connectivity | Preserve rings that are more connected within the scaffold system. Remove terminal, less-connected rings first. |
While these rules establish reproducibility, they are a source of debate. A key ambiguity arises because the rules prioritize chemical intuition over pharmacophore relevance [3]. A ring that is chemically "peripheral" (e.g., a saturated hydrocarbon ring) according to the rules might still be critical for maintaining the three-dimensional orientation of key pharmacophoric groups. Its early removal from the hierarchy could misrepresent the scaffold's essential bioactive structure.
Alternative Approaches and Their Trade-offs
Alternative methodologies handle the ring removal ambiguity differently, each with distinct trade-offs relevant to hierarchical analysis.
Hierarchical Scaffold Clustering (HierS): This method removes entire fused ring systems as single units rather than individual rings [3] [2]. This avoids the ambiguity of breaking fused systems but introduces a different one: the classification becomes too coarse-grained. Two molecules differing by a single ring within a large fused system (common in natural products) will be grouped together at a high level, potentially masking significant structural and activity differences [3].
Scaffold Networks: This approach abandons deterministic rules entirely. It generates a network (not a tree) by enumerating all possible parent scaffolds that can result from every possible single-ring removal at each step [3]. This eliminates the prioritization ambiguity and is more exhaustive for identifying active substructures in screening data. However, the result is a complex, highly branched network that is difficult to visualize and interpret hierarchically, losing the clear, navigable tree structure [3].
Table 2: Comparative Analysis of Scaffold Hierarchy Generation Methods [3] [2] [10]
| Method | Core Principle | Handling of Ambiguity | Advantages | Disadvantages |
|---|---|---|---|---|
| Scaffold Tree | Iterative, rule-based removal of one terminal ring. | Defined by a fixed set of chemical prioritization rules. | Deterministic, chemically intuitive, creates a unique tree hierarchy. | May remove pharmacophorically important rings early; rule-dependent. |
| HierS | Removal of entire fused ring systems as units. | Avoids ring-level choice within fused systems. | Good for high-level clustering of complex molecules. | Coarse-grained; cannot differentiate scaffolds within a fused system. |
| Scaffold Network | Exhaustive enumeration of all single-ring removals. | Captures all possibilities, eliminating choice ambiguity. | Exhaustive; better for identifying active substructures in HTS data. | Complex, non-hierarchical output; difficult to visualize and navigate. |
Diagram 1: Decision logic for handling ring removal ambiguity (Max Width: 760px)
Application Notes & Protocols
Protocol: Generating a Standard Scaffold Tree with Rule-Based Ring Removal
This protocol outlines the steps to generate a Scaffold Tree from a set of molecules using the classic rule-based algorithm, as implemented in tools like the Scaffold Generator library [3].
Objective: To create a unique, hierarchical tree representation of molecular scaffolds by iteratively removing rings based on defined chemical prioritization rules.
Input: A set of molecular structures in a standard format (e.g., SMILES, SDF).
Procedure:
- Scaffold Extraction: For each input molecule, generate its Murcko framework. This involves removing all terminal acyclic side chains, retaining only ring atoms and the linker atoms that connect them [3] [10].
- Tree Initialization: Define each unique Murcko framework as a leaf node in the tree.
- Iterative Ring Removal (for each leaf scaffold): a. Identify all rings in the scaffold using a Smallest Set of Smallest Rings (SSSR) algorithm. b. From the set of rings, identify which are "terminal" (i.e., their removal does not disconnect the remaining scaffold graph). c. Apply Prioritization Rules: Filter the list of terminal rings by sequentially applying the rules in Table 1. The rule that first narrows the list to a single ring determines the ring to be removed. d. Generate Parent Scaffold: Remove the selected ring. If the removal creates new terminal chain atoms (part of the former linker), prune them to maintain a proper scaffold definition. e. Create Node & Link: Register the new, simpler scaffold as a node in the hierarchy. Create a directed "parent-of" link from the new node to the scaffold from which it was derived. f. Recursion: If the new scaffold contains more than one ring, repeat from step 3a using this scaffold as the new starting point. The recursion stops when a single-ring scaffold is obtained (a root node).
- Tree Assembly: After processing all leaf nodes, merge identical parent scaffolds generated from different branches. The final structure is a directed acyclic graph (a tree) where roots are single-ring systems and leaves are the original complex frameworks [10].
Output: A hierarchical scaffold tree where molecules are clustered based on shared parent scaffolds at different levels of abstraction.
Ambiguity Note: The result is entirely dependent on the predefined rule sequence. Changing the rule order or priority can lead to a different tree structure, highlighting the method's inherent subjectivity [3].
Protocol: Analyzing and Classifying Fused Ring Systems via TR Screening
This protocol adapts the Target-Ring system (TR) dual screening methodology for analyzing libraries containing complex fused ring systems, as demonstrated in repurposing studies of FDA-approved drug cores [27].
Objective: To identify biologically relevant core scaffolds from a set of fused ring systems and prioritize them for further elaboration in drug discovery.
Input: A curated library of fused ring system structures (e.g., "rarely used" cores from known drugs) [27] and a target protein database with known 3D structures and ligands.
Procedure:
- Descriptor-Based Ring System Filtering: a. Calculate physicochemical descriptors for all input ring systems. Key descriptors include VABC volume (van der Waals volume), hydrogen bond donors/acceptors, and fragment complexity [27]. b. Apply filters (e.g., VABC > 140 ų, HBA+HBD < 3) to select ring systems with sufficient size and minimal polar substituents that might bias screening, focusing on the core's inherent properties.
- Shape-Based Primary Screening: a. For each filtered ring system (query), perform a shape-based similarity search against a large database of known ligands from protein targets. b. Generate similarity scores (e.g., Tanimoto combo scores) for each Ring System (R) vs. Target Ligand pair. Aggregate results to identify, for each ring system, the target whose native ligands have the highest average shape similarity [27].
- Docking-Based Secondary Screening: a. For the top pairs from the shape screen, perform molecular docking of the bare ring system into the binding site of the corresponding target protein. b. Score the docking poses. Transform docking scores into ranks across all targets for each ring system. c. Analyze the rank matrix to identify "privileged" fused ring systems that dock favorably against multiple targets and "selective" pairs with high specific affinity [27].
- Virtual Elaboration & Prioritization: a. For the top-ranked ring system-target pairs, virtually decorate the core ring system with substituents using a fragment library. b. Filter the generated virtual compounds by synthesizability, drug-likeness (e.g., Lipinski's rules), and docking score. c. Select the highest-ranking virtual compounds for in silico ADMET prediction and final selection for synthesis and biological testing.
Output: A prioritized list of fused ring system-target pairs, along with suggested elaborated compounds, providing a data-driven strategy for scaffold hopping and lead generation [27].
Ambiguity Note: This method sidesteps the structural ambiguity of classifying fused systems by focusing on their functional potential via bioactivity-like screens. However, the choice of descriptors and docking parameters introduces its own set of biases.
Table 3: Key Outcomes from a Fused Ring System Repurposing Study [27]
| Analysis Step | Input Quantity | Filtering Criteria | Output Quantity | Key Finding |
|---|---|---|---|---|
| Ring System Selection | 349 rare ring systems from FDA drugs | VABC > 140; HBA+HBD < 3 | 71 ring systems | Selection favored 3D complexity and limited polarity of the bare core. |
| Target Selection | 38,529 PDB structures | >5 PDBs/ligand; Ligand MW 250-800 | 97 targets | Focused on targets with well-defined, drug-sized chemical matter. |
| Primary (Shape) Screen | 71 Rings vs. 3,424 Ligands | Best match per ring/target | 97 Target-Ring pairs | High shape similarity (fused scores 0.59-0.84) for most pairs. |
| Secondary (Docking) Screen | 69 Rings vs. 131 PDBs (97 Targets) | Docking score ranking | Ranked matrix | Steroid-like and alkaloid-like fused ring systems showed highest promiscuity. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Computational Tools & Libraries for Scaffold and Ring System Analysis
| Tool/Resource | Type | Primary Function in Ring/Scaffold Analysis | Key Application |
|---|---|---|---|
| Scaffold Generator [3] | Open-source Java Library | Implements multiple scaffold definitions (Murcko, HierS, Scaffold Tree) and generates hierarchies. | Core engine for building scaffold trees and networks from molecular datasets. |
| Chemistry Development Kit (CDK) [3] [27] | Open-source Cheminformatics Library | Provides fundamental functions for ring perception, descriptor calculation, and molecular manipulation. | Underpins tools like Scaffold Generator; used for calculating VABC volume and other filters. |
| GraphStream Library [3] | Java Library | Enables dynamic visualization of graphs and networks. | Used by Scaffold Generator to display and export scaffold hierarchies and networks. |
| RDKit | Open-source Cheminformatics Toolkit | Alternative to CDK for Python environments. Offers robust ring-finding, scaffold decomposition, and fingerprinting. | Scaffold analysis, molecular similarity searching, and integration with machine learning pipelines. |
| Scaffold Tree Prioritization Rules [3] [10] | Algorithmic Rule Set | A predefined, ordered list of chemical rules to resolve ring removal ambiguity. | The standard for generating deterministic, chemically intuitive scaffold trees. |
| TR Screening Framework [27] | Integrated Methodology | Combines shape similarity, molecular docking, and virtual growth for ring system repurposing. | Functionally evaluating and prioritizing complex fused ring systems for drug discovery. |
Diagram 2: TR screening workflow for fused ring system analysis (Max Width: 760px)
Ambiguity in ring removal and the complexity of fused ring systems are not merely technical hurdles but fundamental considerations that shape the outcome of any scaffold-based hierarchical analysis. The Scaffold Tree method imposes a single, chemically rational perspective through its rules, providing clarity and reproducibility at the potential cost of pharmacophore relevance [10]. In contrast, methods like Scaffold Networks embrace ambiguity by exploring all possibilities, offering a more comprehensive but less navigable view of chemical space [3].
The choice of method must be deliberate and aligned with the research goal. For high-level visualization, classification, and diversity assessment of large compound sets (such as in the broader thesis of mapping chemical space), the deterministic scaffold tree remains a powerful, intuitive tool [2]. For identifying bioactive substructures in high-throughput screening data or repurposing complex ring systems, more exhaustive or functionally oriented approaches like scaffold networks or TR screening are necessary to avoid missing critical leads [3] [27].
Therefore, the key for researchers is not to seek a single ambiguity-free solution but to understand the biases inherent in each method. By applying the appropriate protocols and tools with this awareness, scientists can effectively leverage scaffold tree methodology to generate meaningful, hierarchical insights that accelerate ring-based analysis and drug discovery.
The Conditional Latent Space Molecular Scaffold Optimization (CLaSMO) framework represents a significant advancement in AI-driven molecular design, directly addressing two persistent challenges in computational drug discovery: synthetic feasibility and sample efficiency [51]. By integrating a Conditional Variational Autoencoder (CVAE) with Latent Space Bayesian Optimization (LSBO), CLaSMO strategically modifies existing molecular scaffolds to enhance target properties while preserving structural similarity to known, synthesizable compounds [51] [52]. This approach aligns with and extends the principles of hierarchical scaffold tree methodology, providing a powerful, sample-efficient tool for accelerating lead optimization within a structured, interpretable research framework [53].
The systematic analysis of molecular scaffolds is a cornerstone of medicinal chemistry, providing a structured approach to understanding Structure-Activity Relationships (SAR) [53]. The scaffold tree methodology hierarchically decomposes molecules into increasingly simplified core structures, enabling the classification and navigation of chemical space [53]. While conventional hierarchical scaffolds are invaluable for organizing chemical data, emerging "analog series-based" (ASB) scaffolds offer complementary power by explicitly representing synthetic pathways and distinguishing between closely related series with different biological activities [53].
Integrating artificial intelligence with these scaffold-based paradigms opens new frontiers. Generative models promise rapid exploration, but often produce novel structures with uncertain synthetic viability—a major barrier to real-world application [51] [52]. CLaSMO bridges this gap by framing molecular optimization as a constrained, sample-efficient modification of reliable scaffold foundations, thereby marrying the exploratory power of AI with the practical knowledge embedded in hierarchical and analog series-based scaffold analyses [51].
Core Methodology: The CLaSMO Framework
CLaSMO is engineered for sample-efficient optimization, a critical feature when molecular property evaluations (e.g., computational docking, wet-lab assays) are costly and time-consuming [51]. Its architecture combines two key components:
- Conditional Variational Autoencoder (CVAE): This model learns a probabilistic mapping between molecular substructures and a continuous latent space. It is uniquely conditioned on the atomic environment of a specific attachment point on the input scaffold [51]. This conditioning ensures that generated molecular additions are chemically compatible with the scaffold, dramatically increasing the likelihood of synthetic accessibility.
- Latent Space Bayesian Optimization (LSBO): An optimization loop operates within the CVAE's latent space. Guided by a probabilistic model (Gaussian Process), LSBO iteratively proposes latent vectors that are decoded by the CVAE into new substructures. These are attached to the scaffold to create candidate molecules, which are then evaluated for the target property. The feedback from these evaluations refines the model, focusing the search on the most promising regions of latent space with minimal evaluations [51].
This synergy enables "human-in-the-loop" optimization, where domain experts can select the scaffold region for modification and guide the search toward desirable chemical space [51] [52].
Application Notes: Performance in Benchmark Tasks
The performance of CLaSMO has been rigorously validated across a diverse suite of 20 optimization tasks, encompassing key challenges in molecular design [51]. The following table summarizes its efficacy in three primary domains:
Table 1: Performance of CLaSMO Across Key Molecular Optimization Tasks [51]
| Optimization Task Category | Primary Objective | Key Metric & CLaSMO Performance | Implication for Scaffold-Based Design |
|---|---|---|---|
| Compound Rediscovery | Find a known target molecule from a minimal starting scaffold. | Success Rate: Achieved high success in retrieving target molecules from simplified scaffolds. | Validates the method's ability to navigate from core structures to complex, active compounds efficiently. |
| Docking Score Optimization | Improve predicted binding affinity to a protein target. | Score Improvement: Consistently enhanced docking scores over baseline scaffolds. | Demonstrates utility in lead optimization for specific biological targets within a congeneric series. |
| Multi-Property & Drug-Likeness | Simultaneously optimize quantitative drug-likeness (QED) and other properties. | QED Improvement: Significantly improved QED scores while maintaining high similarity to the input [54]. | Proves capable of guiding scaffolds toward improved developability profiles, a crucial step in drug discovery. |
A critical constraint in practical optimization is maintaining sufficient structural similarity to the original scaffold to preserve favorable properties and synthetic tractability. CLaSMO operates effectively under varying similarity constraints, demonstrating robust performance in both flexible and highly constrained optimization regimes [51].
Table 2: Impact of Molecular Similarity Constraint on Optimization Outcomes [51]
| Similarity Constraint Level | Allowed Structural Deviation | Optimization Efficiency | Resulting Synthetic Accessibility |
|---|---|---|---|
| High Constraint | Minimal modification to the core scaffold. | Slower property improvement per step but higher sample efficiency. | Very High. Optimized molecules are highly similar to known, synthesizable inputs. |
| Low Constraint | Greater freedom to modify/add substructures. | Faster property improvement potential. | Moderate to High. Novelty increases, but conditioning on the atomic environment maintains reasonable synthetic feasibility. |
Experimental Protocols for Scaffold Optimization
Protocol: Implementing CLaSMO for a QED Optimization Task
This protocol details the steps to run a CLaSMO experiment for optimizing the Quantitative Estimate of Drug-likeness (QED) of a molecular scaffold, based on the provided code repository [54].
I. Environment Setup
- Clone the CLaSMO repository:
git clone [repository URL]. - Create and activate a Python virtual environment (Python 3.8+ recommended).
- Install required dependencies:
pip install -r requirements.txt. Key libraries include PyTorch, RDKit, scikit-learn, and GPyTorch for Bayesian optimization.
II. Data and Model Preparation
- Input Preparation: Prepare your starting scaffold as a SMILES string. The scaffold should have one or more specified attachment points (e.g., a dummy atom like
[*]) where substructures can be added. - Model Loading: Ensure the pre-trained Conditional VAE model is located in the correct directory as specified in the configuration. This model has been trained on a corpus of scaffold-substructure pairs to learn chemically valid combinations.
III. Execution of Optimization Loop
- Run the main optimization script from the command line:
- The script initiates the LSBO loop [54]:
- Step 0: Encodes the conditioned scaffold and proposes an initial latent point.
- Steps 1-N: The Gaussian Process model suggests a new latent point expected to improve QED. The CVAE decodes this into a substructure, attaches it to the scaffold, and the new molecule's QED is calculated. The result updates the Gaussian Process model. Progress is printed to the console (e.g., "y_delta is 0.0279 at CLaSMO step 2... QED improved to 0.8495 from 0.8215") [54].
IV. Analysis of Results
- Results are saved to a CSV file (e.g.,
clasmo_results_new_run.csv), containing the SMILES, QED score, and similarity metric for each proposed molecule across all optimization steps. - Analyze the trade-off between QED improvement and structural similarity (e.g., Tanimoto similarity) to select promising candidates for further evaluation.
Protocol: Integrating CLaSMO into a Hierarchical Scaffold Analysis Workflow
- Scaffold Decomposition: Use a scaffold tree algorithm to hierarchically decompose a set of active compounds into a series of core structures [53].
- Node Selection: Identify a promising but suboptimal scaffold node within the hierarchy for optimization (e.g., a scaffold with moderate activity but poor drug-likeness).
- CLaSMO Optimization: Apply the CLaSMO protocol (Section 4.1) to the selected scaffold, optimizing for the desired target property.
- Hierarchical Re-integration: Map the optimized molecules back onto the scaffold tree. The new analogs can form a new analog series branching from the original node, enriching the hierarchical analysis with AI-generated, property-enhanced derivatives [53].
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Computational Tools and Resources for AI-Driven Scaffold Optimization
| Item Name | Function in Research | Relevance to CLaSMO/Scaffold Analysis |
|---|---|---|
| RDKit | Open-source cheminformatics toolkit. | Used for processing SMILES strings, calculating molecular descriptors (QED, similarity), and handling chemical transformations. |
| PyTorch | Deep learning framework. | Serves as the backbone for building and training the Conditional VAE model. |
| GPyTorch | Gaussian Process library built on PyTorch. | Implements the Bayesian Optimization loop in the latent space. |
| ZINC/CHEMBL Databases | Public repositories of chemical compounds and bioactivity data. | Source of training data for the CVAE and for benchmarking optimization tasks (e.g., rediscovery). |
| CLaSMO Web Application | Interactive web interface [51]. | Enables human-in-the-loop optimization, allowing researchers to visually select scaffolds and modification sites without writing code. |
| Scaffold Tree Generation Software (e.g., in RDKit) | Algorithmic decomposition of molecules into hierarchical scaffolds. | Prepares input scaffolds for optimization and provides the analytical framework for interpreting results [53]. |
Visualizing Workflows and Relationships
Diagram 1: CLaSMO Scaffold Optimization Workflow
Diagram 2: Scaffold Hierarchical Tree with AI Optimization Node
Within the broader research on scaffold tree methodology for hierarchical ring analysis, the imperative for scalable computational techniques is paramount. The scaffold tree algorithm, introduced by Schuffenhauer et al., provides a deterministic, chemically intuitive hierarchy of molecular frameworks by iteratively removing rings [9]. Its foundational strength is a linear scaling relationship with the number of compounds processed, making it a critical tool for organizing large chemical libraries [4]. This application note details the protocols and implementations that realize this linear scaling in practice, enabling the efficient analysis of modern ultra-large libraries essential for drug discovery. The methodology transforms raw chemical data into a navigable scaffold universe, where relationships between complex molecules are visualized as a tree, with root rings at the top and detailed, multi-ring scaffolds as leaves [9]. The efficiency of this decomposition is the cornerstone of its application in large-scale virtual screening, chemoinformatics, and toxicogenomic biomarker discovery [55].
Core Principles of Linear-Scaling Scaffold Tree Analysis
The linear time complexity O(N) of the scaffold tree algorithm is achieved through a rule-based, iterative reduction process applied independently to each molecule. The algorithm follows a deterministic pathway for any given input structure [9].
Hierarchical Decomposition Rules: The process begins with the generation of a molecular framework by removing all terminal side chains. This framework forms the leaf node. The algorithm then proceeds through iterative cycles of ring removal to generate parent scaffolds, guided by a set of chemical prioritization rules [4]:
- Heterocyclic rings are removed before carbocyclic rings.
- Rings with the smallest number of heteroatoms are prioritized for removal.
- Among ties, rings with the lowest connectivity to the remaining ring system are removed first.
- Further ties are broken by selecting the ring with the smallest size and then by a canonical numbering scheme.
This process continues until a single, root ring remains. The resulting hierarchy is data-set-independent; the same molecule will always generate the same tree, regardless of the library it is processed within [9].
Contrast with Cubic-Scaling Methods: Traditional electronic structure methods, such as conventional Density Functional Theory (DFT) calculations that rely on direct diagonalization of matrices, suffer from cubic-scaling computational cost O(N³), severely limiting their application to systems of a few hundred atoms [56]. The scaffold tree's linear scaling stems from its localized, per-molecule operations that do not require global matrix diagonalization or pairwise comparisons between all molecules in the dataset. This fundamental difference enables the processing of libraries containing millions of compounds, bridging the gap between chemical structure analysis and large-scale bioactivity data mining [55].
Performance Metrics and Scalability Data
The following tables summarize the key performance characteristics and computational requirements for implementing linear-scaling scaffold tree analysis on large chemical libraries.
Table 1: Algorithmic Scaling and Performance Benchmarks
| Library Size (Compounds) | Theoretical Scaling | Reported Processing Time* | Memory Footprint Trend | Primary Limiting Factor |
|---|---|---|---|---|
| 10⁴ | O(N) | ~1-5 minutes | Near-linear increase | Single CPU core speed |
| 10⁵ | O(N) | ~10-50 minutes | Near-linear increase | I/O and disk access |
| 10⁶ | O(N) | ~2-8 hours | Near-linear increase | Parallel file systems |
| 10⁷+ | O(N) | Tens of hours | Near-linear increase | Job scheduling efficiency |
*Reported times are approximate and depend heavily on hardware, molecular complexity, and implementation optimization.
Table 2: Comparative Analysis of Scaling Methods in Computational Chemistry
| Methodology | Theoretical Scaling | Practical System Limit | Key Principle | Suitability for Large Libraries |
|---|---|---|---|---|
| Scaffold Tree Analysis | O(N) | Millions+ of molecules | Rule-based, per-molecule hierarchical decomposition [9] | Excellent |
| Conventional DFT (Direct Diagonalization) | O(N³) | Hundreds of atoms | Global matrix diagonalization [56] | Poor |
| Linear-Scaling DFT (e.g., Purification) | O(N) to O(N log N) | Hundreds of thousands of atoms | Density matrix localization & sparse algebra [56] | Good for atomic systems, not libraries |
| Hierarchical Co-clustering (HCoClust) | O(N log N) | Thousands of data points | Simultaneous row/column clustering [55] | Good for matrix data (e.g., genes × compounds) |
Detailed Experimental Protocols
Protocol 4.1: Core Scaffold Tree Generation for a Large Library
Objective: To generate a hierarchical scaffold tree from a library of chemical structures in SMILES or SDF format, ensuring deterministic and linear-time processing.
Materials:
- Input Data: A file containing molecular structures (e.g.,
library.sdf). - Software: A scaffold tree generation tool (e.g., open-source implementations in RDKit or the original scripts referenced by Schuffenhauer et al. [4]).
- Computing Resources: A standard Linux server with sufficient RAM to hold the entire library in memory (see Table 1).
Procedure:
- Data Preparation: Standardize the input library. Remove salts, neutralize charges, and generate canonical tautomers to ensure consistency in the initial framework generation.
- Framework Extraction: For each molecule
M_iin the library: a. Remove all acyclic terminal atoms (side chains), recursively, until only ring systems and linkers between them remain. This is the leaf scaffold. b. Assign a canonical identifier (e.g., canonical SMILES) to the leaf scaffold. - Hierarchical Decomposition: For each leaf scaffold:
a. Identify all rings in the current scaffold system.
b. Apply the prioritization rules (Sec. 2) to select the next ring to remove.
c. Perform a graph cut to remove the selected ring, breaking bonds such that the remaining structure is valid. Attach hydrogen atoms to the new open valencies.
d. The resulting structure becomes the parent scaffold. Canonicalize its representation.
e. Record the parent-child relationship (
child_scaffold_ID <-[ring_removed]- parent_scaffold_ID). f. Set the parent scaffold as the new current scaffold and repeat steps a-e until only a single ring remains (the root). - Tree Aggregation: Post-process the entire dataset. Merge identical scaffolds generated from different molecules into single nodes in the global tree. The connection records from Step 3e are used to build the unified hierarchy.
- Output: Generate two primary outputs:
a. A graph file (e.g., GraphML, GML) representing the entire scaffold tree, with nodes annotated with scaffold structures, occurrence counts, and associated bioactivity data if provided.
b. A mapping file linking each original molecule
M_ito its corresponding leaf scaffold node in the tree.
Validation: Manually inspect the tree for a random subset of 50-100 molecules. Verify that the ring removal order follows the published chemical rules and that the final root is a plausible single ring (e.g., benzene, piperidine) [9].
Protocol 4.2: Parallel Implementation for Massive-Scale Processing
Objective: To leverage high-performance computing (HPC) resources to process chemical libraries exceeding 10⁷ compounds by parallelizing the inherently independent scaffold tree generation of individual molecules [56].
Materials:
- Input Data: A partitioned chemical library (e.g., split across multiple
library_chunk_[1..N].sdffiles). - Software: MPI (Message Passing Interface)-enabled scaffold tree code, or a workflow manager (e.g., Nextflow, Snakemake) to orchestrate parallel jobs.
- Computing Resources: An HPC cluster with a parallel file system (e.g., Lustre, GPFS).
Procedure:
- Data Partitioning: Split the master library into
Pchunks, wherePis the number of available parallel processes or compute nodes. Aim for chunks of 10⁵-10⁶ molecules to balance I/O and compute load. - Single-Node Tree Generation (Embarrassingly Parallel Phase):
a. Launch
Pindependent processes, each running Protocol 4.1 on its assigned chunklibrary_chunk_X.sdf. b. Each process generates a partial scaffold tree and a molecule-leaf mapping file for its chunk. c. This phase scales linearly with the number of nodes, as there is no inter-process communication. - Global Tree Merging (Synchronization Phase): a. A master process collects all unique scaffolds and their parent-child relationships from the partial trees. b. It executes a deduplication and merging algorithm to combine the partial hierarchies into a single global tree. This involves matching canonical scaffold identifiers and consolidating occurrence counts. c. The global tree is finalized and written to disk.
- Result Integration: The master process updates the chunk-specific molecule-leaf mapping files with the new global node identifiers from the unified tree.
Optimization Notes: The merging step (3b) is the only non-parallel component but operates on the set of unique scaffolds, which is typically 2-3 orders of magnitude smaller than the original library, ensuring minimal overhead. This two-step map-reduce style workflow is the key to maintaining linear scaling in a distributed environment [56].
Scaffold Tree Generation: The iterative, rule-based process for decomposing a single molecule into its scaffold hierarchy.
Integration with Toxicogenomic Biomarker Discovery
The scaffold tree methodology provides the chemical structural framework for interpreting results from high-throughput toxicogenomic studies. Robust hierarchical co-clustering (rHCoClust) techniques can identify groups of chemicals (doses of chemicals, DCs) that regulate groups of differentially expressed genes (DEGs) [55]. Scaffold trees organize these active DC clusters hierarchically by their core chemical frameworks, revealing structure-activity relationships at the scaffold level.
Application Workflow:
- Perform rHCoClust on a toxicogenomic FCGE (Fold Change Gene Expression) matrix to identify significant up/down-regulatory co-clusters (e.g., a DC cluster regulating a DEG cluster via the PPAR signaling pathway) [55].
- Map the chemicals in the significant DC clusters to their leaf scaffolds in a precomputed scaffold tree.
- Navigate up the tree from these leaves to identify common parent scaffolds shared by active compounds. This highlights the core chemical motifs responsible for the observed gene regulation.
- Prune branches of the tree that contain no active compounds, focusing analysis on chemically fertile regions of the scaffold universe.
This integration enables a shift from analyzing individual hits to understanding systematic chemical trends, directly supporting the thesis that hierarchical ring analysis is crucial for modern chemical biology research.
Parallel HPC Implementation: The map-reduce workflow for scaling scaffold tree generation across distributed compute nodes.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software and Resource Tools
| Tool/Resource Name | Type | Primary Function in Scaffold Analysis | Access/Reference |
|---|---|---|---|
| RDKit | Open-source Cheminformatics Library | Provides functions for molecule standardization, ring perception, and graph operations essential for implementing the scaffold tree algorithm. | https://www.rdkit.org |
| Scaffold Tree Generator (Original) | Algorithm Implementation | The reference implementation of the hierarchical scaffold classification rules as described by Schuffenhauer et al. [9]. | Bundled with referenced publication [4]. |
| rHCoClust / rhcoclust | R Package | Performs robust hierarchical co-clustering of toxicogenomic data to identify chemical-gene co-clusters for subsequent scaffold analysis [55]. | https://github.com/ (search for "rhcoclust") |
| HONPAS Package | DFT Software | Exemplifies parallel, linear-scaling computational kernels (density matrix purification) that inspire the HPC approach for scalable scaffold processing [56]. | Referenced in Qin et al. [56] |
| PubChem | Chemical Database | A primary source for large, publicly available chemical libraries (e.g., pyruvate kinase binders, pesticides) to validate and apply the scaffold tree methodology [9]. | https://pubchem.ncbi.nlm.nih.gov |
| MPI (OpenMPI, MPICH) | Parallel Computing Standard | Enables the distributed-memory parallelization of the scaffold tree generation across HPC nodes, as outlined in Protocol 4.2 [56]. | https://www.open-mpi.org |
The linear-scaling scaffold tree algorithm remains a cornerstone technique for the hierarchical analysis of large chemical libraries. Its deterministic, rule-based nature ensures consistent and chemically meaningful organization of the scaffold universe. As demonstrated, its O(N) scaling can be effectively realized and extended through parallel HPC implementations, enabling its application to the largest contemporary virtual screening libraries. When integrated with modern data analysis techniques like robust hierarchical co-clustering, it provides a powerful structural lens through which to interpret high-dimensional biological data, such as toxicogenomic biomarker discovery [55]. This efficient processing framework is therefore not merely a computational convenience but a fundamental enabler for research within the thesis of scaffold-based hierarchical ring analysis in medicinal chemistry and chemical biology.
The analysis of complex chemical spaces, particularly through hierarchical methods like the scaffold tree, presents computational and data integrity challenges analogous to those in software engineering [4]. This protocol establishes best practices derived from software error handling and implementation to ensure the reliability, reproducibility, and clarity of scaffold-based research [57] [58]. The scaffold tree algorithm, which deterministically organizes molecular datasets through iterative ring removal, provides a powerful framework for drug discovery [4] [59]. However, the processing of large-scale chemical libraries (e.g., PubChem) and the development of visualization tools (e.g., Scaffvis) require systems that are resilient to unexpected inputs, computational edge cases, and data corruption [2]. By adopting a systematic approach to error anticipation, detection, logging, and communication, researchers can harden their analytical workflows, protect valuable data, and facilitate collaboration across interdisciplinary teams in medicinal chemistry and drug development [60].
Data Presentation: Comparative Analysis of Methodologies
Table 1: Comparison of Hierarchical Scaffold Classification Methods This table summarizes key computational frameworks for organizing molecular structures, which form the basis for hierarchical visualization and analysis in chemical space exploration.
| Method | Core Principle | Hierarchy Type | Branching Factor | Key Advantage | Primary Application |
|---|---|---|---|---|---|
| Scaffold Tree | Iterative, rule-based removal of one ring at a time [2]. | Strict Tree | Variable, can be high | Deterministic; generates unique, linear scaffold sequence per molecule [4] [59]. | Visualization, compound clustering, and bioactivity mapping [4]. |
| HierS | Exhaustive generation of all possible ring system combinations [2]. | Set-based (Non-Tree) | Not Applicable | Exhaustive; captures all scaffold relationships [2]. | Analysis of High-Throughput Screening (HTS) results [59]. |
| Scaffold Topology (Oprea) | Edge merging of molecular frameworks to minimal ring structure [2]. | Tree (with Frameworks) | Low | Intuitive; aligns with medicinal chemists' perception of molecular cores [2]. | Topological analysis of ring systems. |
| Extended Scaffold Hierarchy | Pre-computed, multi-level hierarchy optimized for visualization [2]. | Strict Tree (8 levels) | Homogenized (~100) | Optimized for visual layout; enables background comparison vs. PubChem [2]. | Hierarchical visualization in tools like Scaffvis [2]. |
Table 2: Error Handling Metrics & Implementation Checklist This table outlines quantifiable metrics and a categorical checklist for implementing robust error management in scientific computing pipelines.
| Category | Specific Metric/Check | Target/Requirement | Purpose in Research Context |
|---|---|---|---|
| Logging | Security/Input Validation Errors Logged | 100% of events | Trace potential data manipulation or flawed input compounds [57]. |
| Log Entry Completeness | Timestamp, IP/Process ID, Error Type, Outcome [57] | Enables reproducible debugging of failed analyses. | |
| Error Prevention | Input Validation Coverage | All user & file inputs | Prevents malformed SMILES strings or incorrect file formats from crashing pipeline [60]. |
| Retry Logic for Transient Failures | Configurable attempts (e.g., 3-4) [60] | Handles network timeouts when querying remote databases (e.g., PubChem). | |
| User Communication | User-Facing Error Messages | Clear, constructive language; no stack traces [57] [61] | Guides researchers to correct input errors without revealing system details. |
| System Alert for Critical Failures | Immediate on security/validation errors [57] | Alerts maintainers to critical failures in automated screening workflows. | |
| Resilience | Fail-Safe Defaults | Always "fail closed"; roll back on error [57] | Ensures partial results from a failed scaffold classification do not propagate. |
| Graceful Degradation | Provide alternative outputs (e.g., simplified view) | Maintains partial functionality if advanced visualization fails [58]. |
Experimental Protocols
Protocol 1: Implementing the Scaffold Tree Algorithm with Robust Error Checking This protocol details the steps to generate a scaffold tree hierarchy from a molecular dataset while incorporating validation and error handling at each stage.
- Objective: To programmatically generate a deterministic scaffold tree hierarchy from a set of molecular structures while ensuring data integrity and providing actionable error feedback.
- Materials: Input molecular data (e.g., SDF, SMILES formats), computing environment with cheminformatics toolkit (e.g., RDKit, CDK), logging library.
- Procedure:
- Input Validation & Sanitization:
- Step: Load molecular structures from the source file.
- Error Check: Validate file format and the readability of every record. Log the line number and error type for any unparsable entry [60].
- Action: Skip invalid entries, generate a summary report for the user, and continue processing.
- Framework Generation:
- Step: For each valid molecule, generate the Murcko framework (prune all terminal side chains) [59].
- Error Check: Confirm the framework is not null (e.g., for linear compounds without rings).
- Action: Compounds without rings are assigned to a designated "non-cyclic" branch in the hierarchy.
- Iterative Ring Removal:
- Step: Apply the rule-based hierarchy of ring removal (e.g., prioritize heterocycles, smaller rings, etc.) iteratively until a single root ring remains [2] [59].
- Error Check: At each iteration, verify the scaffold is chemically sensible (e.g., valences are correct).
- Action: If an invalid intermediate is generated, log the molecule ID, the erroneous scaffold, and the removal rule that caused it. Halt processing for that molecule and default to a safer rule if configured.
- Hierarchy Construction:
- Step: Assemble the sequence of scaffolds for each molecule into a global tree data structure.
- Error Check: Detect and log duplicate scaffold nodes or cycles, which indicate a logic error in the removal algorithm.
- Output & Logging:
- Input Validation & Sanitization:
Protocol 2: Structured Error Handling and Logging for a Scientific Visualization Workflow This protocol establishes a framework for managing errors in an interactive scientific application, such as a scaffold tree visualization tool.
- Objective: To create a centralized, secure, and user-friendly error handling system for a web-based scaffold visualization application (e.g., Scaffvis) [2].
- Materials: Web application framework (e.g., Flask, Django), structured logging library, monitoring system.
- Procedure:
- Define Custom Exception Classes:
- Create specific exception types for different failures (e.g.,
InvalidQueryError,DatabaseTimeoutError,VisualizationRenderingError). This allows for precise catching and handling [60].
- Create specific exception types for different failures (e.g.,
- Implement Centralized Exception Handling:
- Use framework middleware or decorators to create a global exception handler [60].
- Logic: Catch all unhandled exceptions at this layer. Log the full error details (stack trace, user context) internally for developers.
- Generate User-Facing Messages:
- For each exception type, map it to a friendly, non-technical message that guides the user [61].
- Example: Instead of "Database connection timed out," present: "The system is temporarily busy. Please try your request again in a moment." For a chemical query error, state: "The provided structure query could not be processed. Please verify the format and try again."
- Secure Logging Practices:
- Do Not Log: Sensitive user data, molecular structures considered proprietary, or passwords [57].
- Do Log: Timestamp, user action (e.g., "submitted scaffold query"), error type, and outcome (e.g., "failed validation") [57]. For login/access failures, log the attempt and source IP to detect brute-force attacks [57].
- Proactive Input Validation:
- Validate all API inputs (e.g., scaffold SMILES, numerical parameters) before processing. Use clear, inline error messages next to the input field to indicate the problem immediately [61].
- Define Custom Exception Classes:
Mandatory Visualizations
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Digital Tools & Libraries for Robust Scaffold Analysis This table lists key software libraries, frameworks, and data resources critical for implementing error-resilient scaffold tree methodology and analysis.
| Category | Item | Function in Research | Notes / Best Practice |
|---|---|---|---|
| Cheminformatics Core | RDKit / CDK | Provides fundamental functions for molecule I/O, Murcko framework decomposition, and ring perception essential for scaffold tree generation. | Validate all molecule objects after creation to catch invalid structures early [60]. |
| Error Handling & Logging | Python logging / Log4j |
Structured logging to file or system. Essential for debugging failed batch processes and auditing analysis steps. | Do not log sensitive compound data [57]. Ensure logs include context (user, action, timestamp) [57]. |
| Sentry / Exceptionite | Real-time error monitoring and aggregation for deployed web applications (e.g., Scaffvis). | Provides alerts and tracks error frequency, crucial for maintaining reliability of shared research tools [60]. | |
| Resilience & Validation | Tenacity / Retrying | Implements retry logic with backoff for transient failures (e.g., database network calls). | Use for non-mutating operations like querying external chemical databases [60]. |
| Pydantic / JSON Schema | Validates configuration files and API input data before processing begins. | Prevents malformed input from propagating through the analysis pipeline [60]. | |
| Visualization & Deployment | Flask / FastAPI (Python) | Web frameworks for building interactive visualization tools. Include built-in mechanisms for centralized error handling [60]. | Use custom error handlers to return consistent, user-friendly JSON or HTML error responses [60] [61]. |
| D3.js / Cytoscape.js | JavaScript libraries for rendering interactive tree or network visualizations of scaffold hierarchies in the browser. | Implement graceful degradation if WebGL is unavailable [58]. | |
| Reference Data | PubChem Compound Database | Provides a background "empirical chemical space" for comparative scaffold frequency analysis [2]. | Cache query results locally with retry logic to handle network instability [60]. |
| ChEMBL / GOSTAR | Bioactivity databases used to map activity data onto scaffold trees for bioactivity-guided navigation. | Validate and standardize activity data (e.g., units, confidence) during ingestion to ensure analysis quality. |
High-Throughput Screening (HTS) has evolved into an indispensable engine for modern drug discovery. By enabling the rapid testing of thousands to millions of chemical compounds against biological targets, HTS accelerates the identification of potential drug candidates [62] [63]. The global HTS market, valued at USD 32.0 billion in 2025 and projected to reach USD 82.9 billion by 2035, underscores its critical role in pharmaceutical R&D [64]. However, this massive scale introduces profound challenges in data quality, where noise, false positives, and assay artifacts can obscure genuine biological signals and lead research astray.
The imperative for robust data quality is magnified within the specialized context of scaffold tree methodology for hierarchical ring analysis. This research approach systematically deconstructs molecules into their core ring systems (scaffolds) and organizes them hierarchically to understand structure-activity relationships [15]. The quality of the primary HTS data directly dictates the validity of the scaffold analysis. Poor-quality hit identification propagates errors through the entire hierarchical classification, potentially leading to flawed conclusions about privileged scaffolds or chemical spaces. Therefore, ensuring data robustness is not merely a technical step but a foundational requirement for meaningful scaffold-based discovery and subsequent scaffold hopping—the strategy to identify novel core structures with retained biological activity [34].
This article provides detailed application notes and protocols designed to fortify HTS data quality, ensuring the generation of reliable, actionable datasets that can power robust scaffold tree analysis and drive efficient drug discovery.
Quantitative Landscape of the HTS Market and Data Challenges
The expanding HTS market is characterized by technological segmentation and regional growth, which directly influences the data landscape researchers must navigate.
Table 1: High-Throughput Screening Market Overview and Segmentation
| Segment | Detail / Metric | Value / Share | Implication for Data Quality |
|---|---|---|---|
| Global Market Size (2025) | Valuation [64] | USD 32.0 billion | High investment drives volume and complexity of data generated. |
| Projected Market Size (2035) | Forecast [64] | USD 82.9 billion | Sustained growth demands scalable, automated data QC solutions. |
| Forecast CAGR (2025-2035) | Compound Annual Growth Rate [64] | 10.0% | |
| Dominant Technology Segment | Cell-Based Assays [64] | 39.4% share | Generates complex, multiparametric data requiring advanced normalization. |
| Dominant Application Segment | Primary Screening [64] | 42.7% share | Front-line process where QC failures are most costly. |
| High-Growth Application | Target Identification CAGR [64] | 12% | Increases need for robust data to validate novel biological targets. |
| Key Growth Region | Asia-Pacific (e.g., South Korea CAGR) [64] | Up to 14.9% | Expands user base, emphasizing need for standardized, user-friendly QC protocols. |
The primary technical challenge stems from the market's reliance on cell-based assays, which, while physiologically relevant, introduce biological variability [64]. Furthermore, the push toward ultra-high-throughput screening increases throughput but can compromise data fidelity if not managed correctly [64]. Key impediments to quality include the high cost of infrastructure, the risk of false positives/negatives, and the need for specialized expertise in data analysis [64] [63]. For scaffold tree research, a false positive hit can result in the erroneous classification of an irrelevant chemical series, wasting significant optimization resources.
Foundational Protocols for Assay Development and Validation
Robust HTS data begins with a meticulously validated assay. The following protocol outlines the critical steps.
Protocol 1: Assay Optimization and Validation for HTS
- Objective: To establish a reproducible, sensitive, and pharmacologically relevant screening assay with defined quality thresholds.
- Materials: Target protein or cell line, compound library (including controls), assay plates (e.g., 384- or 1536-well), liquid handling robotics, microplate reader [63].
- Procedure:
- Assay Miniaturization & Adaptation: Scale down the assay to the desired microplate format. Optimize reagent concentrations, incubation times, and final volumes to maintain signal robustness [63].
- Positive/Negative Control Selection: Include well-characterized pharmacological controls (e.g., a known inhibitor and vehicle) in every plate to monitor assay performance.
- Signal Window Assessment: Calculate the Z'-factor for each assay plate.
Z' = 1 - [ (3σ_positive + 3σ_negative) / |μ_positive - μ_negative| ]. A Z'-factor > 0.5 is excellent for screening, indicating a wide separation between control populations [63]. - Dose-Response Confirmation: Test control compounds in a dilution series to confirm the assay yields a expected sigmoidal dose-response curve and a plausible half-maximal inhibitory/effective concentration (IC₅₀/EC₅₀).
- Inter-plate Reprodubility Test: Run identical control plates on different days to determine day-to-day variability. The coefficient of variation (CV) for control signals should typically be < 20%.
Table 2: Key Assay Performance Metrics and Benchmarks
| Metric | Calculation | Optimal Benchmark | Purpose | ||
|---|---|---|---|---|---|
| Z'-Factor [63] | `1 - [ (3σp + 3σn) / | μp - μn | ]` | > 0.5 | Measures assay signal dynamic range and data variation. |
| Signal-to-Noise (S/N) | (μ_signal - μ_background) / σ_background |
> 10 | Assesses detectability of a positive signal above background. | ||
| Signal-to-Background (S/B) | μ_signal / μ_background |
> 3 | Ratio of assay signal intensity to background level. | ||
| Coefficient of Variation (CV) | (σ / μ) * 100% |
< 20% | Measures precision and reproducibility of control wells. |
Core Data Processing and Normalization Workflow
Raw screening data must be processed to correct for systematic artifacts (e.g., edge effects, dispensing errors) before analysis. The following diagram and protocol describe this critical workflow.
HTS Data Processing and Quality Control Workflow
Protocol 2: Data Normalization and Hit Identification
- Objective: To transform raw assay signals into normalized activity values and identify preliminary hits using statistical criteria.
- Materials: Raw data files, statistical software (e.g., R, Python with pandas/sci-kit learn), computational environment.
- Procedure:
- Plate-Based Normalization: For each compound well, calculate % Activity or % Inhibition. A common method is:
%Inhibition = 100 * (μ_negative - Signal_well) / (μ_negative - μ_positive). - Correction for Spatial Artifacts: Apply algorithms (e.g., B-score or loess smoothing) to correct for row/column or edge effects observable in plate heatmaps.
- Hit Identification Using SSMD: Employ the Strictly Standardized Mean Difference (SSMD) for robust hit calling in replicates.
SSMD = (μ_compound - μ_negative) / √(σ²_compound + σ²_negative). A compound with|SSMD| > 3is a strong hit [65]. - Application of Hit Thresholds: Apply a primary activity threshold (e.g., >50% inhibition at screening concentration). Flag compounds meeting the threshold for confirmation testing.
- Plate-Based Normalization: For each compound well, calculate % Activity or % Inhibition. A common method is:
Integration with Scaffold Tree and AI-Driven Analysis
Validated HTS hits form the input for scaffold tree analysis. Modern AI-driven molecular representation methods significantly enhance this process by enabling more intelligent scaffold hopping and analysis [34].
Protocol 3: From HTS Hits to Scaffold Tree Analysis
- Objective: To cluster confirmed hits via scaffold tree methodology and prioritize scaffolds for further exploration.
- Materials: List of confirmed hit structures (SMILES format), cheminformatics software (e.g., RDKit, ChemBounce [15]), scaffold generation algorithms.
- Procedure:
- Structure Standardization: Standardize hit structures (e.g., remove salts, neutralize charges, generate canonical SMILES).
- Scaffold Decomposition: Apply a hierarchical scaffold decomposition algorithm (e.g., HierS or Murcko frameworks) to extract the core ring systems from each hit molecule [15].
- Scaffold Clustering & Tree Building: Group molecules sharing identical scaffolds. Organize scaffolds hierarchically from complex multi-ring systems to simple single rings, forming a scaffold tree.
- Activity Mapping & SAR Analysis: Map the mean biological activity (e.g., pIC₅₀) of all compounds belonging to each scaffold node onto the tree. This visualizes Structure-Activity Relationships (SAR) and identifies privileged scaffolds—core structures yielding multiple active compounds.
- AI-Enhanced Scaffold Hopping: Use AI-based molecular representation models (e.g., Graph Neural Networks) or tools like ChemBounce to generate novel, structurally distinct scaffolds that maintain predicted bioactivity, facilitating lead optimization [34] [15].
Scaffold Tree and AI-Driven Analysis Workflow
The Scientist's Toolkit: Essential Reagent Solutions
A selection of critical reagents and materials is fundamental to executing robust HTS campaigns.
Table 3: Key Research Reagent Solutions for HTS
| Reagent / Material | Primary Function in HTS | Key Quality Consideration |
|---|---|---|
| Cell-Based Assay Kits (e.g., viability, GPCR, kinase) [64] | Provide optimized, ready-to-use reagents for specific target classes, ensuring consistency and reducing development time. | Lot-to-lot consistency, sensitivity (Z'-factor), minimal background interference. |
| Biochemical Enzyme & Substrate Kits | Enable target-specific activity assays for enzymes like kinases, proteases, and phosphatases. | Enzymatic specific activity, substrate purity and stability, linear reaction kinetics. |
| Fluorescent / Luminescent Detection Reagents (Dyes, probes, enzyme substrates) [63] | Generate the measurable signal indicating target modulation or cellular response. | Signal brightness, photostability, compatibility with HTS readers and automation. |
| High-Quality Compound Libraries (e.g., diversity, targeted, fragment libraries) | Source of chemical matter for screening. The library's quality defines the discovery space. | Chemical purity and identity, solubility in DMSO/buffer, structural diversity, non-reactive artifacts. |
| Automation-Compatible Liquid Handling Tips & Microplates | Physical vessels for assay execution. | Material compatibility (non-binding), manufacturing precision (well-to-well volume consistency), optical clarity for imaging/reading. |
In the data-intensive realm of HTS, robustness is non-negotiable. For research anchored in scaffold tree methodology, the fidelity of hierarchical ring analysis is intrinsically linked to the quality of the primary screening data. By implementing rigorous assay validation (Protocol 1), systematic data normalization and QC (Protocol 2), and integrating confirmed hits with advanced cheminformatic and AI-driven scaffold analysis (Protocol 3), researchers can transform high-throughput data into high-confidence insights. This disciplined approach ensures that the pursuit of novel, patentable scaffolds through strategies like scaffold hopping is built upon a foundation of reliable data, ultimately de-risking the drug discovery pipeline and accelerating the journey from screening hit to therapeutic lead.
Application Notes and Protocols
This document details practical applications and methodologies for enhancing the synthetic accessibility of novel chemical entities within the paradigm of scaffold tree-based hierarchical ring analysis. The scaffold tree methodology provides a systematic, rule-based framework for deconstructing molecules into their constituent ring systems, creating a unique, hierarchical organization from simple single rings (Level 0) to the complete molecular framework [66]. This hierarchy is not merely a classification tool; it establishes a logical roadmap for retrosynthetic analysis and scaffold diversification.
The core hypothesis framing this work is that strategic navigation of this hierarchical scaffold space, guided by curated fragment libraries and constrained by molecular similarity principles, can efficiently generate novel, synthetically tractable chemical matter. This approach directly addresses a key finding in scaffold diversity analysis: known bioactive compounds occupy only a sparse, unevenly distributed region of conceivable scaffold space, partly due to the synthetic inaccessibility of many theoretically possible rings [66]. By tethering exploration to well-characterized, readily available building blocks (curated fragments) and ensuring the resulting designs maintain critical pharmacophoric elements (via similarity constraints), we can enhance the probability of successful synthesis and retained biological activity. This integrated strategy is foundational for advanced medicinal chemistry campaigns, including scaffold hopping and property-focused lead optimization [15].
The following tables summarize key quantitative findings relevant to the implementation of curated fragment libraries and similarity-based constraints in scaffold-centric discovery.
Table 1: Analysis of Scaffold Distribution in Representative Compound Libraries [66]
| Data Set | Description | Total Compounds | Key Finding on Scaffold Distribution |
|---|---|---|---|
| ICRSC | Internal screening collection | 79,742 | High population density on few scaffolds; many singleton scaffolds. |
| VC | Vendor compounds library | 1,923,627 | Skewed distribution; demonstrates commercial availability bias. |
| CHEMBL | Bioactive molecules from literature | 530,038 | Provides a source of synthesizable, bio-relevant fragment motifs. |
| DBSM | Marketed small-molecule drugs | (From DrugBank) | Represents a "privileged" subspace of synthetically accessible scaffolds. |
Table 2: Performance of Similarity-Based vs. Machine Learning Target Prediction [67]
| Method | Basis | Target Coverage | Key Performance Insight |
|---|---|---|---|
| Similarity-Based | Maximum Tanimoto similarity (Morgan2 FP) to known actives. | Broad (4239 proteins) | Generally outperformed ML in retrospective validation, especially for novel chemotypes. |
| Machine Learning (Random Forest) | Binary classifier per target using Morgan2 FP. | Limited (1798 targets with ≥25 ligands) | Performance more dependent on structural similarity between query and training set. |
| Query Similarity Class | Tanimoto Coefficient (TC) Range | Prediction Reliability Trend | |
| High Similarity | TC > 0.66 | High reliability for both methods. | |
| Medium Similarity | 0.33 ≤ TC ≤ 0.66 | Similarity-based method maintains more robust performance. | |
| Low Similarity | TC < 0.33 | Significant drop in performance; highlights need for robust constraints. |
Table 3: Benchmarking the ChemBounce Scaffold Hopping Framework [15]
| Evaluation Metric | ChemBounce Performance Note | Implication for Synthetic Accessibility |
|---|---|---|
| Scaffold Library Source & Size | >3.2 million unique scaffolds curated from ChEMBL via HierS algorithm. | Library is derived from synthesized, bio-active molecules, ensuring practical synthetic routes exist. |
| Similarity Constraints | Dual filter: 2D Tanimoto & 3D ElectroShape similarity. | Balances novel scaffold introduction with retention of pharmacophore geometry and charge distribution. |
| Synthetic Accessibility (SAscore) | Generated compounds tended to have lower SAscore vs. other tools. | Directly indicates higher predicted synthetic ease for output structures. |
| Drug-Likeness (QED) | Generated compounds tended to have higher QED values. | Output favors structures with more desirable drug-like property profiles. |
Detailed Experimental Protocols
Protocol 1: Construction of a Synthesis-Aware Curated Fragment Library
Objective: To create a hierarchical fragment library from known chemical space that prioritizes synthetic feasibility for use in scaffold hopping and molecular generation.
Materials:
- Source Database: ChEMBL database (or similar repository of synthesized compounds) [66] [15].
- Cheminformatics Toolkit: RDKit or Chemistry Development Kit (CDK) with Scaffold Generator capabilities [13].
- Fragmentation Algorithm: HierS (Hierarchical Scaffold Clustering) or Scaffold Tree implementation [13] [15].
- Synthetic Feasibility Filter: SAscore calculator or analogous heuristic.
Procedure:
- Data Curation: Download and preprocess a subset of the ChEMBL database. Apply standard curation: remove salts, neutralize charges, standardize tautomers, and filter by molecular weight (e.g., <600 Da) to focus on drug-like space.
- Hierarchical Scaffold Decomposition: For each curated molecule, apply the HierS algorithm [15]: a. Define Scaffolds: Extract the core scaffold by removing all terminal side chains. Include atoms connected to rings via double bonds to preserve hybridization. b. Recursive Ring Removal: Systematically generate all unique sub-scaffolds by iteratively removing one ring system at a time. A "ring system" includes fused or spiro rings that share atoms. c. Store Relationships: Record the parent-child relationships between each scaffold and its immediate, simpler descendant(s). This generates a network (DAG) rather than a strict tree.
- Library Deduplication & Pruning: Aggregate all unique scaffolds from all processed molecules. Apply the following filters to curate the library: a. Remove Ubiquitous Singletons: Exclude overly simple, non-discriminatory rings (e.g., single benzene rings) [15]. b. Frequency Threshold: Retain scaffolds that appear above a minimum frequency (e.g., ≥5 times) in the source data, implying known synthetic routes and reliable characterization. c. Synthetic Accessibility Score: Calculate and filter by SAscore. Prioritize scaffolds with scores below a chosen threshold (e.g., SAscore < 4.5).
- Annotation & Storage: Annotate each retained scaffold with metadata: frequency in source data, parent/child relationships, common synthetic precursors (if available from reaction databases), and computed properties (e.g., number of rotatable bonds, hydrogen bond donors/acceptors). Store the final library in a searchable format (e.g., SQLite database or SMILES file with index).
Protocol 2: Applying Similarity Constraints in Scaffold Replacement
Objective: To replace a core scaffold in a known active molecule with a novel one from a curated library while enforcing constraints to maintain biological activity potential.
Materials:
- Input: A known active molecule (Query) in SMILES format.
- Tool: Scaffold hopping software (e.g., ChemBounce [15] or modified script using RDKit).
- Libraries: The curated fragment library (Protocol 1) and a default reagent library for substituents.
- Similarity Metrics: Functions for 2D Tanimoto similarity (Morgan fingerprints) and 3D Electron Shape similarity (e.g., ElectroShape5 [15]).
Procedure:
- Query Analysis & Scaffold Identification: Input the Query molecule. Identify the core "query scaffold" using the same HierS algorithm used to build the library. Define the attachment vectors (R-groups) where the scaffold connects to the original substituents.
- Similarity-Prescribed Scaffold Retrieval: Search the curated fragment library for candidate replacement scaffolds. a. Perform a 2D similarity search between the query scaffold and all library scaffolds using Morgan fingerprints (radius=2) and the Tanimoto coefficient. b. Retain the top N candidates (e.g., 100-1000) scoring above a tunable threshold (e.g., Tc ≥ 0.3) [15].
- Molecular Assembly: For each candidate scaffold, reattach the original Query's substituents to the corresponding attachment points on the new scaffold. Ensure valency and steric clashes are checked.
- Multi-Dimensional Similarity Rescreening: Filter the newly assembled molecules using a dual-constraint approach [15]: a. 2D Profile Similarity: Calculate the Tanimoto similarity between the full Query molecule and each new molecule. Discard those below a strict threshold (e.g., Tc < 0.5). b. 3D Shape/Electrostatic Similarity: Generate a low-energy 3D conformation for the Query and each new molecule. Compute the Electron Shape similarity (e.g., using ElectroShape). Discard molecules below a defined threshold (e.g., < 0.7). This step is critical for scaffold hops that change the 2D connectivity but preserve the overall pharmacophoric volume and charge distribution.
- Final Ranking & Output: Rank the molecules passing all filters by a composite score (e.g., weighted sum of synthetic accessibility (SAscore), drug-likeness (QED), and 3D similarity). Output the top candidates with associated scores and metadata.
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 4: Key Reagents, Tools, and Databases for Implementation
| Item Name / Category | Function / Purpose | Key Characteristics & Notes |
|---|---|---|
| Scaffold Generator Library [13] | Core software for generating Murcko frameworks, scaffold trees, and networks from molecule sets. | Open-source Java library built on CDK. Enables customizable scaffold definitions and hierarchy generation. Essential for Protocol 1. |
| HierS Algorithm [13] [15] | A specific scaffold fragmentation methodology that preserves linker atoms attached via double bonds. | Creates a hierarchical Directed Acyclic Graph (DAG) of scaffolds. Forms the basis of the fragmentation in ChemBounce and similar tools. |
| Curated ChEMBL Fragment Library [15] | A pre-processed collection of >3 million unique, synthesis-validated scaffolds. | Serves as a ready-to-use "fragment universe" for replacement. Built using the HierS algorithm on ChEMBL, ensuring biological relevance and synthetic tractability. |
| ElectroShape5 Descriptor [15] | A 3D molecular descriptor capturing shape and electrostatic potential. | Used for 3D similarity screening in Protocol 2. More effective for bioactivity retention than shape-only descriptors during scaffold hopping. |
| SAScore (Synthetic Accessibility Score) | A heuristic to estimate the ease of synthesizing a given molecule. | Used to filter fragment libraries and rank final outputs. Lower scores indicate higher predicted synthetic accessibility. |
| Morgan Fingerprints (ECFP4) | A circular topological fingerprint for molecular representation. | Standard for rapid 2D similarity calculations (Tanimoto coefficient). Used for initial scaffold and molecule similarity searches. |
| SQRL Framework [68] | A machine learning training paradigm (Similarity-Quantized Relative Learning). | Predicts property differences between similar molecules. Can be adapted to predict the activity delta between a Query and a proposed scaffold-hopped analog, providing an additional predictive constraint. |
Mandatory Visualizations
Diagram 1: Workflow for Scaffold Hopping with Curated Fragments & Similarity Constraints (100 chars)
Diagram 2: Hierarchical Scaffold Space & Novelty Bridges via Similarity (100 chars)
Measuring Success: Validation Protocols and Comparative Analysis with Competing Hierarchical Methods
Within the broader thesis on scaffold tree methodology for hierarchical ring analysis, the systematic classification of molecular scaffolds represents a foundational pillar for navigating chemical space. The comparative analysis of three established frameworks—the Scaffold Tree, HierS, and Oprea Scaffold Topologies—provides a critical lens through which to evaluate strategies for organizing and visualizing large-scale molecular data in drug discovery [2] [4]. Each methodology offers a distinct paradigm for decomposing complex molecular structures into hierarchical representations, balancing chemical intuition against computational determinism.
The core challenge addressed by these frameworks is the transformation of discrete molecular structures into a navigable hierarchy that reflects structural relationships. This enables critical research applications, including the assessment of scaffold diversity in compound libraries, the visualization of structure-activity relationships (SAR), and the identification of novel bioactive chemotypes within vast empirical chemical spaces such as PubChem [2] [66]. The selection of an appropriate hierarchy impacts downstream interpretation, influencing how scientists perceive clustering, similarity, and the overall organization of chemical space.
Methodological Foundations and Comparative Analysis
The three scaffold topologies are built upon a common principle of iterative structural simplification but diverge significantly in their rules and final hierarchical organization.
Scaffold Tree: This algorithm creates a strict, deterministic tree hierarchy from a molecule [4]. It operates by iteratively removing one ring at a time from the molecular framework according to a predefined set of chemical priority rules (e.g., prioritizing the removal of heterocycles before carbocycles, smaller rings before larger ones) until a single root ring remains [2] [69]. This process generates a unique linear sequence of scaffolds for each molecule, which collectively form a tree for an entire dataset. Its key advantage is the generation of a true, data-set-independent tree where each molecule has a single, unambiguous path from the root to the leaf [2].
HierS (Hierarchical Scaffolds): The HierS method starts from a molecular framework and recursively removes entire ring systems (cycles sharing an edge), along with their connecting linkers [2]. Unlike the Scaffold Tree, this process is not deterministic in its outcome for a single molecule; a framework with multiple ring systems yields multiple possible scaffolds representing all combinations of its ring systems. A hierarchy is subsequently formed by ordering the entire set of generated scaffolds from a compound library by structural inclusion. The result is a hierarchical directed acyclic graph (DAG), not a strict tree, where scaffolds with fewer ring systems are placed above those with more [2].
Oprea Scaffold Topologies (Graph Frameworks): This approach abstracts the molecular framework to its pure topological essence [2]. It begins with the Murcko framework (union of ring systems and linkers), converts it to a graph framework (atom and bond type agnostic), and then applies edge merging. This process contracts vertices of degree two, resulting in a simplified "topology" graph that describes the ring structure with the minimal number of nodes. This topology, the Oprea scaffold, is unique for each molecule. A simple three-level hierarchy exists: Murcko Framework → Graph Framework → Oprea Topology [2]. This method aligns closely with a medicinal chemist's intuitive perception of scaffold core topology.
Table 1: Core Algorithmic Comparison of Scaffold Hierarchy Methods
| Feature | Scaffold Tree | HierS | Oprea Topologies |
|---|---|---|---|
| Basic Unit of Removal | Single ring | Entire ring system | Not applicable (topological transformation) |
| Hierarchy Type | Strict, rooted tree | Directed Acyclic Graph (DAG) | Simple 3-tier hierarchy |
| Determinism per Molecule | Unique linear sequence | Multiple scaffolds generated | Unique topology |
| Key Chemical Insight | Rule-based, chemically prioritized simplification | Combinatorial ring system importance | Underlying topological connectivity |
| Primary Use Case | Library classification, SAR visualization, diversity analysis [66] [69] | Exploring ring system contributions | Topological analysis of scaffold space |
Quantitative Performance and Library Diversity Analysis
Empirical application of these methods to compound libraries reveals distinct statistical profiles crucial for library design and virtual screening (VS) campaigns [69].
Scaffold Tree analysis, particularly at Level 1 (the first ring system retained after pruning), has proven effective for characterizing scaffold diversity [66]. Studies of commercial libraries show a highly skewed distribution: a small number of scaffolds account for a large percentage of compounds, while a "long tail" of singleton scaffolds exists [66]. For example, analysis of 11 purchasable libraries and a natural product database (TCMCD) showed that libraries like ChemBridge, ChemicalBlock, Mcule, and TCMCD exhibited higher scaffold diversity within standardized molecular weight subsets [69]. Tree Maps visualizing Scaffold Tree output clearly display highly populated scaffolds and clusters of structurally similar scaffolds, aiding in library selection for VS [66] [69].
HierS, by generating all ring system combinations, produces a more complex and less uniformly branched hierarchy, which can lead to visualization challenges when dealing with large datasets [2]. Oprea topologies provide a coarse but intuitive grouping, effectively clustering molecules based on the fundamental connectivity of their ring systems, which is useful for high-level surveys of scaffold topology space [2].
Table 2: Statistical Output from Scaffold Diversity Studies
| Analysis Metric | Typical Finding | Implication for Library Design |
|---|---|---|
| Scaffold Frequency (Scaffold Tree Level 1) | ~1-2% of scaffolds cover >50% of compounds in many libraries [66]. | High redundancy; need to enrich with novel scaffolds. |
| Singleton Scaffolds | Often represent 20-40% of unique scaffolds but a tiny fraction of total molecules [66]. | Source of diversity but poor for establishing SAR. |
| Diversity vs. Vendor | ChemBridge, ChemicalBlock identified as highly diverse; others more focused [69]. | Informs vendor selection for targeted vs. broad screening. |
| Natural Products (TCMCD) | Higher structural complexity but more conservative in scaffold topology [69]. | Valuable for exploring complex, bio-relevant chemical space. |
Experimental Protocols for Scaffold Analysis
Protocol: Generating and Comparing Hierarchies for a Compound Library
Objective: To apply the Scaffold Tree, HierS, and Oprea topology methods to a user-provided compound library (e.g., in SDF format) and generate comparative metrics on scaffold diversity and hierarchy structure.
Materials:
- Input Data: A file of molecular structures (e.g.,
.sdf,.smi). - Software:
- For Scaffold Tree: RDKit (Python) with
rdkit.Chem.Scaffoldsmodule, or the Scaffold Tree implementation in Molecular Operating Environment (MOE) [69]. - For HierS & Oprea: Custom scripts based on published algorithms [2], or cheminformatics toolkits like RDKit for substructure manipulation.
- For Visualization & Analysis: Scaffvis web tool (for tree maps) [2], Pipeline Pilot for fragment analysis [69], or in-house Python/R scripts.
- For Scaffold Tree: RDKit (Python) with
Procedure:
- Data Standardization:
- Load the molecular dataset. Apply standard preprocessing: remove duplicates, neutralize charges, add explicit hydrogens, and filter by desired properties (e.g., molecular weight 100-700 Da) [69].
- Optional: Create a standardized subset by matching the molecular weight distribution to a reference library to enable fair comparison [69].
Scaffold Decomposition:
- Scaffold Tree: For each molecule, iteratively remove rings using the Schuffenhauer rules. Record the scaffold at each level (Level 0 = root ring, Level n-1 = Murcko framework) [4] [69]. Aggregate all unique scaffolds and parent-child relationships to build the global tree.
- HierS: For each molecule's framework, systematically generate all subgraphs by removing each possible combination of ring systems (and attached linkers). Collect all unique scaffolds across the library and order them by inclusion to form the hierarchy graph [2].
- Oprea Topology: For each molecule, generate the Murcko framework. Convert it to a graph framework (atom/bond type agnostic). Apply edge-merging (contract vertices of degree 2) to obtain the minimal topology graph [2].
Analysis and Metric Calculation:
- For each method, calculate: total unique scaffolds, frequency of the most common scaffolds, percentage of singleton scaffolds, and cumulative frequency plots (e.g., scaffolds covering 50% of molecules, NC50C/PC50C metrics) [66].
- Compare the branching factors and depth of the hierarchies generated by Scaffold Tree and HierS.
Visualization:
- Generate a Tree Map for the Scaffold Tree output, using square size to represent scaffold frequency and color to represent user-defined properties (e.g., average activity) [2] [66].
- For HierS, a network graph can be used to visualize the DAG structure.
- For Oprea topologies, group and count molecules by their unique topology graph.
Protocol: Visualizing Library Diversity on a PubChem Background with Scaffvis
Objective: To contextualize a proprietary or focused compound set within the empirical chemical space of PubChem using the Scaffvis web application [2].
Materials:
- Scaffvis Web Application: Accessible via its published web interface [2].
- Input Data: A set of molecular structures (user dataset) in a supported format (e.g., SMILES).
- Background Hierarchy: The precomputed scaffold hierarchy based on PubChem Compound database (integrated into Scaffvis) [2].
Procedure:
- Prepare User Dataset: Compile the list of compound SMILES or identifiers. Ensure structures are valid.
- Upload and Map: Upload the user dataset to the Scaffvis server. The tool will map each user molecule to its corresponding scaffold at all levels of the pre-defined PubChem-based hierarchy [2].
- Interactive Exploration: The interface presents a zoomable tree map.
- The background of the map is the entire PubChem scaffold hierarchy, with square sizes representing the frequency of scaffolds in PubChem.
- The overlay color on the squares (e.g., a gradient from blue to red) represents the frequency or another property (like mean potency) of the user's compounds mapped to that scaffold.
- Interpretation: Identify scaffolds that are both large (common in PubChem) and brightly colored (enriched or active in your set) as potential "popular" chemotypes. Identify small, colored squares as potentially novel scaffolds active in your assay. This direct visual comparison highlights over-representation and novelty against a public domain reference.
Visualizing Methodology Workflows and Relationships
Diagram 1: Algorithmic workflow for three scaffold methods (76 characters)
Diagram 2: Research context for scaffold topology thesis (70 characters)
Research Reagent Solutions: Essential Tools for Implementation
Table 3: Key Software and Resources for Scaffold Hierarchy Research
| Tool/Resource Name | Type/Category | Primary Function in Analysis | Key Utility |
|---|---|---|---|
| RDKit | Open-source Cheminformatics Toolkit | Provides core functions for molecule handling, substructure search, and scaffold decomposition. Can implement Scaffold Tree rules. | Flexible, programmable foundation for custom hierarchy development and batch analysis [69]. |
| Molecular Operating Environment (MOE) | Commercial Software Suite | Contains the sdfrag command and other modules for generating Scaffold Trees and RECAP fragments [69]. |
Robust, validated production environment for standardized scaffold analysis in drug discovery. |
| Pipeline Pilot | Scientific Workflow Platform | Offers "Generate Fragments" component and protocol-building for high-throughput fragment and property analysis [69]. | Automates the preprocessing, standardization, and multi-metric analysis of large compound libraries. |
| Scaffvis Web Application | Web-based Visualization Tool | Enables interactive exploration of compound sets mapped onto a precomputed PubChem scaffold hierarchy via zoomable tree maps [2]. | Contextualizes private data against public chemical space for intuitive assessment of novelty and frequency. |
| Scaffold Hunter | Desktop Visualization Software | Provides interactive visualization and analysis of chemical data using Scaffold Tree and other hierarchies [2]. | Enables deep, interactive SAR analysis by navigating the scaffold tree and coloring nodes by biological activity. |
| ZINC Database | Public Compound Repository | Source of purchasable compound structures from numerous vendors for library analysis [69]. | Provides the raw material (compound libraries) for comparative scaffold diversity studies. |
| PubChem Compound Database | Public Chemical Database | Serves as the reference "empirical chemical space" for building background hierarchies [2]. | Defines the real-world distribution of scaffolds, enabling frequency-based novelty assessment. |
The Scaffold Tree algorithm provides a deterministic, data-set-independent method for organizing large molecular datasets into a unique hierarchical tree based on their core molecular frameworks or scaffolds [4] [11]. The hierarchy is constructed through the iterative removal of rings from complex scaffolds using a chemically meaningful set of rules until a single root ring is obtained [4]. This methodology enables the intuitive visualization of chemical space, efficient compound clustering, and the identification of novel bioactive molecules by grouping compounds with shared structural cores [4] [59]. Within the broader thesis on hierarchical ring analysis, the Scaffold Tree serves as a foundational tool for rationalizing structure-activity relationships (SAR) and navigating from large, diverse compound sets to focused, active chemotypes [1].
This document applies the Scaffold Tree framework to two distinct validation case studies: the identification of Pyruvate Kinase M2 (PKM2) inhibitors for oncology and the analysis of pesticide targets using natural compounds. These cases demonstrate how scaffold-based hierarchical analysis guides the transition from initial screening data to validated lead compounds.
Application Note 1: Hierarchical Analysis of Pyruvate Kinase M2 Binders
Target and Scaffold Context
Pyruvate kinase M2 (PKM2) is a critical enzyme in glycolytic regulation and is overexpressed in various cancers, making it a significant therapeutic target [70] [71]. Inhibiting PKM2 can disrupt the Warburg effect, a metabolic hallmark of cancer cells [70]. The application of scaffold tree analysis to PKM2 inhibitor discovery allows researchers to classify active compounds, such as natural phenolics, by their core structures, revealing crucial scaffold-activity relationships and guiding subsequent analog synthesis [71].
Key Experimental Data and Scaffold Classification
Screening of a phenolic compound library identified several potent PKM2 inhibitors. The following table summarizes the quantitative data for the top hits, which serve as leaves in a scaffold tree analysis [71].
Table 1: Inhibitory Activity of Natural Phenolic Compounds Against PKM2 [71]
| Compound Name | IC₅₀ (µM) | Inhibition Constant, Kᵢ (µM) | Type of Inhibition | Primary Scaffold Class |
|---|---|---|---|---|
| Silibinin | 0.91 | 0.61 ± 0.26 | Competitive | Flavonolignan |
| Curcumin | 1.12 | 1.20 ± 0.40 | Non-competitive | Diarylheptanoid |
| Resveratrol | 3.07 | 7.34 ± 1.70 | Non-competitive | Stilbene |
| Ellagic Acid | 4.20 | 5.02 ± 0.73 | Competitive | Polyphenol (Dibenzopyran) |
Scaffold Analysis: The actives belong to distinct, privileged natural product scaffold classes. In a Scaffold Tree, these would originate from different branches, suggesting multiple independent binding pharmacophores for PKM2 inhibition. For example, the competitive inhibitors silibinin and ellagic acid share a high degree of oxygenation on their polycyclic cores, which may be a key feature for binding at the phosphoenolpyruvate (PEP) substrate site [71].
Detailed Protocol: Enzymatic Assay for PKM2 Inhibitor Screening
This protocol is adapted from coupled enzyme activity assays and is used to generate primary data for scaffold classification [71] [72].
Principle: A coupled enzyme assay measures PKM2 activity indirectly. PKM2 catalyzes the conversion of PEP and ADP to pyruvate and ATP. The generated pyruvate is then utilized in a secondary reaction with peroxidase to produce a fluorometric signal. Inhibitor presence reduces pyruvate production, decreasing fluorescence [72].
Materials:
- Recombinant human PKM2 isoform (e.g., 100 pU/mL) [72].
- Pyruvate Kinase Activity Assay Kit (e.g., Sigma-Aldrich MAK072) [72].
- Test compounds dissolved in DMSO.
- Black half-area 96-well plates [72].
- Fluorescent microplate reader (e.g., BMG LABTECH VANTAstar) [72].
Procedure:
- Solution Preparation: Thaw and bring all assay kit components to room temperature. Prepare a 1X assay buffer. Dilute PKM2 enzyme to 10 µU/mL in a suitable buffer [72].
- Plate Setup: In a black half-area 96-well plate, add:
- 44 µL of 1X assay buffer.
- 5 µL of inhibitor solution (or DMSO for controls).
- 1 µL of diluted PKM2 enzyme [72].
- Master Mix Preparation: For each reaction well, freshly prepare a master mix containing:
- 44 µL 1X assay buffer.
- 2 µL PKM2 substrate mix (PEP/ADP).
- 2 µL enzyme mix (lactate dehydrogenase, peroxidase).
- 2 µL fluorogenic peroxidase substrate [72].
- Reaction Initiation: Add 50 µL of the master mix to each sample well to start the reaction. Mix gently.
- Kinetic Measurement: Immediately transfer the plate to a pre-warmed microplate reader. Measure fluorescence kinetically (e.g., every 20 seconds for 13 minutes) using excitation/emission filters of ~540/590 nm [72].
- Data Analysis: Calculate the initial reaction velocity (slope) for each well. Plot inhibitor concentration versus percent activity to determine IC₅₀ values using a non-linear 4-parameter logistic fit [72].
Scaffold Tree Integration: The resulting IC₅₀ data for each compound is the primary biological annotation. Compounds are then processed through a Scaffold Tree algorithm (e.g., using tools like Scaffold Hunter) [2] [59]. Their molecular frameworks are iteratively deconstructed to place each active compound within a hierarchical tree. This visualizes the chemical space of actives, highlights common inhibitory scaffolds, and identifies potential for scaffold hopping to discover novel chemotypes.
Diagram: Workflow for PKM2 Inhibitor Discovery & Scaffold Analysis
Diagram 1: PKM2 Inhibitor Discovery & Scaffold Analysis Workflow (Max Width: 760px)
The Scientist's Toolkit: Key Reagents for PKM2 Analysis
Table 2: Essential Research Reagents for PKM2 Inhibitor Screening [71] [72]
| Reagent/Material | Function & Role in Scaffold Analysis |
|---|---|
| Recombinant Human PKM2 | Target enzyme for primary screening. Activity data against this protein is the key biological annotation for scaffold classification. |
| Pyruvate Kinase Activity Assay Kit | Provides optimized, coupled reagents for consistent kinetic measurement of PKM2 activity, ensuring reliable data for SAR. |
| Fluorescent Microplate Reader | Enables high-throughput kinetic readout of enzyme activity, generating the quantitative data necessary to rank compounds within a scaffold cluster. |
| Scaffold Tree Software (e.g., Scaffold Hunter) | Computational tool to generate hierarchical scaffold classifications from active compound structures, enabling visual navigation of chemical space. |
Application Note 2: Pesticide Target Analysis via Arginine Kinase
Target and Scaffold Context
Arginine kinase (AK) is a critical enzyme for energy metabolism in invertebrates and is absent in vertebrates, making it an attractive target for selective pesticide development [73]. Identifying natural product inhibitors of AK, such as the green tea flavonoid (-)-epigallocatechin gallate (EGCG), exemplifies a scaffold-based approach to eco-friendly biopesticide discovery [73]. Analyzing such inhibitors through a scaffold tree allows researchers to map the chemical space of bioactive natural products against this target and identify core structures for optimization.
Key Experimental Data
A study on Loxosceles laeta AK (LlAK) identified EGCG as a binder through biophysical and computational methods [73].
Table 3: Binding Parameters for EGCG Interaction with Arginine Kinase (LlAK) [73]
| Parameter | Value | Method | Implication for Scaffold |
|---|---|---|---|
| Dissociation Constant (K𝒹) | 58.3 µM | Fluorescence Quenching | Defines baseline potency of the parent EGCG scaffold. |
| Association Constant (Kₐ) | 1.71 x 10⁴ M⁻¹ | Fluorescence Quenching | Quantifies ligand binding affinity for the core structure. |
| Binding Free Energy (ΔG) | -40 to -15 kcal/mol | MM/PBSA from MD Simulation | Confirms the stability of the EGCG-AK complex, validating the scaffold's fit. |
| Docking Score (AutoDock Vina) | -7.3 to -9.8 kcal/mol (varies by site) | Molecular Docking | Predicts binding pose and affinity, guiding scaffold modification. |
Scaffold Analysis: The EGCG scaffold is a complex polyphenolic flavan-3-ol. In a Scaffold Tree, its multiple fused and connected rings would be iteratively pruned to reveal simpler core structures. This deconstruction can help identify the minimal pharmacophore required for AK binding, which is invaluable for designing simpler, more synthetically tractable analogs for pesticide development.
Detailed Protocol: Fluorescence Quenching for Ligand-Target Binding
This protocol measures the direct interaction between a candidate scaffold (like EGCG) and the purified target enzyme (AK) [73].
Principle: Intrinsic protein fluorescence (often from tryptophan residues) is quenched upon ligand binding to the active site. The degree of quenching is used to calculate the binding constant (Kₐ) and dissociation constant (K𝒹), providing a direct measure of scaffold affinity [73].
Materials:
- Purified target enzyme (e.g., recombinant LlAK) [73].
- Ligand stock solution (e.g., EGCG in suitable buffer).
- Fluorescence spectrophotometer with cuvette.
- Assay buffer (e.g., 50 mM Tris-HCl, pH 7.5).
Procedure:
- Sample Preparation: Purify the target enzyme via a protocol involving recombinant expression in E. coli and affinity chromatography (e.g., Ni-NTA for His-tagged protein) [73]. Dialyze into assay buffer.
- Ligand Titration: Prepare a series of ligand solutions (EGCG) at varying concentrations (e.g., 0 to 200 µM) in assay buffer.
- Fluorescence Measurement:
- Place a fixed concentration of protein (e.g., 1 µM LlAK) in a cuvette.
- Excite the sample at 280 nm (for Trp/Tyr) and record the emission spectrum from 300 to 400 nm.
- Titrate by adding small volumes of ligand stock sequentially to the cuvette, mixing thoroughly after each addition.
- Record the fluorescence emission intensity at the λmax (e.g., ~340 nm) after each addition.
- Data Analysis: Correct for background and inner-filter effects. Plot the corrected fluorescence intensity (F₀/F) versus ligand concentration [Q]. Fit the data to the Stern-Volmer equation (F₀/F = 1 + K_SV[Q]) and further to a binding isotherm to derive the binding constant (Kₐ) and K𝒹 [73].
Scaffold Tree Integration: The calculated K𝒹 value serves as the key activity metric for the EGCG scaffold. This information annotates the EGCG structure in the chemical library. When processed through the Scaffold Tree, EGCG and other tested flavonoids (e.g., quercetin, rutin) will be grouped based on shared flavan cores. This reveals which core substructures correlate with stronger AK binding, directing focused library design around the most promising hierarchical scaffold branch.
Diagram: Scaffold-Based Biopesticide Discovery Pipeline
Diagram 2: Scaffold-Based Biopesticide Discovery Pipeline (Max Width: 760px)
The Scientist's Toolkit: Key Reagents for Pesticide Target Analysis
Table 4: Essential Research Reagents for AK-Targeted Biopesticide Analysis [73]
| Reagent/Material | Function & Role in Scaffold Analysis |
|---|---|
| Recombinant Arginine Kinase | Purified target enzyme for validation. Essential for generating experimental binding data to annotate natural product scaffolds. |
| Fluorescence Spectrophotometer | Enables measurement of binding affinity (K𝒹) via quenching, providing quantitative data to rank different natural product scaffolds. |
| Molecular Docking Software (e.g., AutoDock Vina) | Predicts the binding mode and affinity of scaffold candidates, helping prioritize compounds for testing and understand SAR at the structural level. |
| Molecular Dynamics Simulation Suite | Assesses the stability of the scaffold-target complex and refines binding free energy calculations, offering deeper validation for promising core structures. |
The Scaffold Tree methodology provides a systematic, hierarchical framework for organizing and analyzing chemical compounds based on their core molecular structures or scaffolds [4]. This approach transforms complex chemical datasets into navigable tree hierarchies through the iterative, rule-based removal of rings from molecular frameworks, ultimately reducing each compound to a single root ring [4] [59]. Within the broader thesis on hierarchical ring analysis, this methodology serves as a critical tool for chemical space navigation, enabling researchers to visualize large compound libraries, identify structural relationships, and prioritize novel bioactive scaffolds for synthesis [11] [66].
This document outlines detailed application notes and experimental protocols grounded in three foundational performance metrics for the Scaffold Tree algorithm: determinism, data-set independence, and chemical relevance. Determinism guarantees that the same scaffold hierarchy is reproducibly generated from a given molecule [4]. Data-set independence ensures the classification remains consistent regardless of the other molecules present in the analysis [4] [2]. Chemical relevance refers to the application of chemically meaningful rules during the pruning process to preserve the most characteristic core of the molecule, ensuring the resulting hierarchy is interpretable and useful for medicinal chemistry [74] [59].
Core Performance Metrics and Quantitative Benchmarks
The utility of the Scaffold Tree for research and decision-making is underpinned by its core algorithmic metrics. The following tables provide quantitative benchmarks for these metrics based on analyses of large-scale chemical databases.
Table 1: Metrics for Determinism and Data-Set Independence in Scaffold Classification
| Metric | Definition | Measurement / Benchmark | Implication for Research |
|---|---|---|---|
| Determinism | The guarantee that a single, unique scaffold hierarchy is generated for a given input molecule using a fixed set of pruning rules [4]. | 100% reproducibility across computational runs and software implementations using the same rule set. | Enables reproducible clustering, SAR analysis, and reliable comparison of results across different studies and teams. |
| Data-Set Independence | The property that the scaffold class assignment for a molecule is not influenced by the composition or size of the dataset in which it is processed [4] [2]. | Linear scaling of computation time with the number of compounds (O(n)) [4]. Scaffold identity remains invariant when a molecule is analyzed alone or within libraries of varying size (e.g., PubChem analysis) [2]. | Allows for the pre-computation of background hierarchies (e.g., from PubChem) [2] and the consistent merging or comparison of datasets from different sources without re-calculation. |
| Rule-Based Pruning Priority | The ordered set of chemical rules that deterministically selects the next ring for removal (e.g., prioritizing aliphatic over aromatic, smaller over larger rings) [74] [59]. | Rule set is explicitly defined prior to analysis. Provides a transparent, non-heuristic pathway from molecule to root. | Ensures the hierarchical simplification is chemically intuitive, preserving more "interesting" or complex rings for higher levels of the tree, which is crucial for medicinal chemistry interpretation [59]. |
Table 2: Chemical Space Coverage and Diversity Metrics from Public Databases
| Database / Library Analyzed | Number of Compounds | Number of Unique Scaffolds (Murcko or Level 1) | Scaffold Diversity (Shannon Entropy or similar) | Key Finding |
|---|---|---|---|---|
| PubChem Compound Database [2] | Tens of millions | Hierarchical analysis performed; specific counts for pre-computed background levels. | Homogeneous branching factor targeted for visualization. | A global scaffold hierarchy was constructed to enable visualization of user datasets against an empirical chemical space background [2]. |
| Exemplified Medicinal Chemistry Libraries [66] | ~80,000 to >1.9 million (across 7 libraries) | Ranged from thousands to hundreds of thousands. | Highly skewed distribution: A very small number of scaffolds account for a large percentage of compounds [66]. | In one library, 50% of compounds were represented by just 0.34% of the scaffolds, highlighting significant redundancy and the need for library diversification [66]. |
| Known Drugs (Bemis & Murcko Analysis) [66] | 5,129 | 1,179 Murcko frameworks. | Low diversity: 50% of drugs were based on only 32 frameworks. | Demonstrates the historical focus on a limited set of privileged scaffolds in drug discovery [66]. |
Experimental Protocols
Protocol 1: Generation and Analysis of a Scaffold Tree Hierarchy
Objective: To generate a deterministic, data-set independent scaffold tree from a set of molecular structures and analyze the resulting hierarchy.
Materials:
- Input: A set of molecular structures in SMILES or SDF format.
- Software: Scaffold Tree generation software (e.g., implementation in RDKit, the original Perl script from Schuffenhauer et al. (2007) [59], or integrated platforms like Scaffold Hunter [74]).
- Hardware: Standard computer workstation.
Procedure:
- Data Pre-processing: Standardize the input molecules (e.g., neutralize charges, remove solvents, generate canonical tautomers).
- Scaffold Extraction: For each molecule, generate its Murcko framework (also called the molecular framework) by removing all terminal side chains, preserving linker atoms between ring systems [2] [66].
- Hierarchical Pruning: Apply the deterministic pruning algorithm to each Murcko framework [74]: a. Identify all rings in the scaffold. b. Apply the rule-based priority system to select exactly one ring for removal. Standard rules prioritize (in order): aliphatic rings over aromatic, heterocycles over carbocycles, smaller rings over larger, and rings with more heteroatoms [59]. c. Remove the selected ring, breaking bonds and sativating valencies with hydrogen atoms. This creates a new, simpler scaffold. d. Repeat steps a-c on the new scaffold until only a single ring remains. This sequence defines the unique path for the molecule from its full Murcko framework (leaf) to a root ring.
- Tree Construction: Merge identical scaffolds generated from the same pruning level across all molecules in the dataset. This creates a unified tree where nodes represent shared scaffolds, and edges represent the "is-a-parent-of" relationship via ring removal. Scaffolds that exist only as intermediates (not from an original molecule) are termed virtual scaffolds [74].
- Analysis & Visualization: a. Calculate metrics such as the number of unique scaffolds per tree level, branching factors, and the percentage of singletons. b. Visualize the tree using tree-map views (where size can denote molecule count and color can denote average activity) [74] [66] or hierarchical radial layouts.
Interpretation: The resulting tree provides a map of chemical space. Densely populated branches indicate well-explored, popular scaffolds. Sparse branches or virtual scaffolds highlight opportunities for scaffold hopping and the synthesis of novel chemical entities to explore underrepresented regions [74].
Protocol 2: Assessing Scaffold Diversity in a Compound Library
Objective: To quantify the scaffold diversity of a screening library or corporate collection to inform library enhancement strategies.
Materials:
- Input: A compound library in chemical structure format.
- Tools: Software capable of generating Murcko frameworks and Level 1 Scaffold Tree representations (the first ring system removed from the framework) [66].
Procedure:
- Generate Scaffold Sets: For all compounds in the library, compute two representations: a. The Murcko framework. b. The Level 1 Scaffold Tree scaffold (the first ancestor in the tree) [66].
- Count and Tally: Calculate the frequency of each unique scaffold within both sets.
- Calculate Diversity Metrics: a. NC50C / PC50C: Calculate the number (NC50C) and percentage (PC50C) of scaffolds required to cover 50% of the compounds in the library [66]. A low NC50C/PC50C indicates high redundancy. b. Shannon Entropy: Compute the Shannon entropy of the scaffold frequency distribution. A value of 0 indicates all compounds share one scaffold; higher values indicate a more even distribution across many scaffolds [66]. c. Singleton Percentage: Calculate the percentage of scaffolds that appear only once in the library.
- Comparative Visualization: Use a Tree Map to visualize the library [66]. Each rectangle represents a scaffold, sized by the number of associated compounds, and colored by a property (e.g., cluster). Group structurally similar scaffolds proximally.
Interpretation: A library with very low NC50C and high singleton percentage is heavily biased toward a few chemotypes and may contain many one-off compounds. This analysis directly supports decisions to diversify a library by synthesizing or acquiring compounds based on underrepresented or virtual scaffolds [66].
Protocol 3: Bioactivity-Guided SAR Mapping with Scaffold Trees
Objective: To overlay biological screening data onto a Scaffold Tree to identify structure-activity relationships and prioritize scaffolds for lead optimization.
Materials:
- Input: A dataset of molecules with associated bioactivity data (e.g., IC50, percent inhibition).
- Software: Scaffold Tree tool with data visualization capabilities (e.g., Scaffold Hunter [74]).
Procedure:
- Build the Activity-Annotated Tree: Generate the Scaffold Tree following Protocol 1. For each scaffold node, aggregate the bioactivity data from all descendant molecules.
- Define Activity Metrics: For each scaffold, calculate a summary statistic (e.g., geometric mean pIC50, percentage of active compounds, minimum detected activity).
- Visual Mapping: Apply a color gradient to the tree nodes based on the calculated activity metric (e.g., red for highly active, blue for inactive).
- SAR Analysis: Navigate the tree to identify: a. Active Clusters: Branches where activity is consistently high, indicating a robust SAR. b. Activity Cliffs: Sibling scaffolds or parent-child pairs with dramatic differences in activity, highlighting critical structural features. c. Promising Virtual Scaffolds: Inactive or unexplored scaffolds that are closely related (in the tree) to highly active scaffolds. These represent prime candidates for scaffold hopping [74].
- Hypothesis Generation: Formulate chemical hypotheses. For example, "The loss of activity in this branch correlates with the removal of a specific nitrogen atom in the pyrimidine ring," which is directly suggested by the pruning path.
Interpretation: This transforms the scaffold tree from a structural map into a bioactivity landscape. It enables intuitive, hierarchical SAR analysis and data-driven decision-making for lead series selection and optimization strategies.
Visual Workflows and Logical Relationships
Diagram 1: The deterministic workflow for generating a scaffold tree from molecular structures.
Diagram 2: Mapping bioactivity data onto a scaffold tree for SAR analysis and hypothesis generation.
Table 3: Key Software Tools and Resources for Scaffold Tree Analysis
| Tool / Resource Name | Type / Category | Primary Function in Analysis | Access / Reference |
|---|---|---|---|
| Scaffold Hunter | Integrated Visualization Software | Provides interactive 2D/3D visualization of scaffold trees, tree maps, and molecule clouds; allows mapping of biological data [74]. | Open-source desktop application. |
| Scaffvis | Web-Based Visualization Tool | Enables hierarchical, scaffold-based visualization of user datasets on the background of the PubChem empirical chemical space using zoomable tree maps [2]. | Freely available web client-server application [2]. |
| RDKit | Cheminformatics Toolkit | Contains functions for generating Murcko frameworks and implementing custom scaffold pruning rules, enabling programmatic tree construction. | Open-source cheminformatics library. |
| Schuffenhauer et al. Algorithm | Core Algorithm | The canonical, rule-based algorithm for deterministic scaffold tree generation [59]. | Reference implementation (Perl) described in original publication [59]. |
| PubChem Scaffold Hierarchy | Pre-computed Background | A publicly available, data-set independent scaffold hierarchy built from millions of PubChem compounds, serving as a universal reference chemical space [2]. | Accessible via the Scaffvis tool or for download [2]. |
| Murcko Framework Generator | Fundamental Descriptor | Standard method for extracting the core ring-linker system from a molecule, forming the starting point for scaffold tree construction [66]. | Available in most cheminformatics packages (RDKit, OpenEye, etc.). |
Advantages in Structure-Activity Relationship (SAR) Studies and Bioactivity Mapping
Application Notes on SAR and Bioactivity Mapping within Scaffold Tree Methodology
The integration of scaffold tree methodology with modern Structure-Activity Relationship (SAR) analysis provides a powerful hierarchical framework for navigating chemical space and accelerating lead optimization [23]. This approach systematically deconstructs molecules into their core ring systems, organizing chemical datasets into interpretable hierarchies that reveal relationships between molecular architecture and biological effect [23]. The primary advantage lies in its ability to transition from traditional, linear SAR exploration—often focused on a single parent scaffold—to a multidimensional bioactivity mapping paradigm. This paradigm enables the simultaneous analysis of diverse chemotypes, facilitating scaffold hopping and the identification of isofunctional molecular cores [34].
Recent computational advances, such as the Cross-Structure-Activity Relationship (C-SAR) strategy, directly leverage this hierarchical philosophy [75]. By analyzing Matched Molecular Pairs (MMPs) across diverse scaffolds targeting a common protein (e.g., HDAC6), researchers can identify transformative pharmacophoric substitutions that lead to activity cliffs, providing design rules applicable beyond any single chemical series [75]. This is a significant evolution from classical approaches like the Topliss scheme, which is bound to a specific parent structure [75]. Furthermore, visual analytics platforms like Scaffold Hunter operationalize this methodology by combining scaffold trees with interactive data visualization, allowing researchers to cluster compounds, visualize property landscapes, and pinpoint key structural features responsible for activity [23].
The synergy of hierarchical scaffold analysis with AI-driven molecular representations (e.g., graph neural networks, transformer models) further amplifies these advantages [34]. These representations learn continuous, high-dimensional embeddings of molecules that capture subtle structural and functional nuances, enabling more effective prediction of bioactivity and generation of novel, optimized scaffolds within the defined hierarchical framework [34].
Quantitative Comparison of SAR Methodologies
Table 1: Key Metrics and Advantages of Modern SAR Methodologies
| Methodology | Core Approach | Key Advantage | Reported Metric/Outcome | Thesis Context: Relevance to Hierarchical Scaffold Analysis |
|---|---|---|---|---|
| C-SAR (Cross-SAR) [75] | Analysis of pharmacophoric substitutions across matched molecular pairs (MMPs) from diverse chemotypes. | Generates transformative design rules applicable to novel scaffolds, not tied to a single parent. | Applied to 133 MMPs for HDAC6 inhibitors; Diversity Index: 0.5827 [75]. | Enables bioactivity mapping across the scaffold tree, identifying activity cliffs between distant branches. |
| AI-Driven Scaffold Hopping [34] | Use of graph neural networks (GNNs) or variational autoencoders (VAEs) to generate novel core structures with retained activity. | Explores vast chemical space to discover structurally novel, patentable scaffolds with desired properties. | Identifies new scaffolds absent from existing libraries via data-driven latent space exploration [34]. | Provides computational engine for generating and evaluating new child or sibling nodes within a scaffold hierarchy. |
| Integrated SAR Platform (e.g., PULSAR) [76] | Combines MMP analysis, R-group deconvolution, and automated reporting in a unified workflow. | Dramatically reduces multi-parameter SAR analysis time from days to hours; enhances team collaboration. | Enables systematic analysis of thousands of compounds with multiple bioactivity parameters [76]. | Offers a practical software framework for visualizing and analyzing data organized by scaffold trees. |
| Scaffold Hunter Visual Analytics [23] | Interactive visualization of hierarchical scaffold trees combined with clustering and property mapping. | Facilitates intuitive, hypothesis-driven exploration of large chemical datasets and SAR trends. | Supports analysis of high-throughput screening data via linked views (tree, plot, heatmap) [23]. | Constitutes a direct implementation of scaffold tree methodology for visual bioactivity mapping. |
Detailed Experimental Protocols
Protocol 1: Hierarchical Scaffold Tree Construction and Analysis for SAR
This protocol details the generation and analysis of a scaffold tree to map bioactivity and inform scaffold hopping [23].
- Input Dataset Preparation: Curate a chemical dataset with associated bioactivity data (e.g., IC₅₀, Ki). Standardize structures (e.g., neutralize charges, remove solvents) using a toolkit like RDKit or the CDK [23].
- Scaffold Extraction: For each molecule, generate its Bemis-Murcko scaffold by removing all terminal acyclic atoms, retaining only the ring systems and the linker atoms connecting them [23].
- Hierarchical Tree Generation: Apply the Scaffold Tree algorithm to each scaffold [23]: a. Prune the scaffold iteratively using a set of deterministic rules (e.g., prioritize removing aliphatic rings before aromatic ones, remove smallest ring systems first). b. Continue until a single, fundamental ring remains. This creates a lineage from the complex scaffold to a simple root. c. Merge identical scaffolds and virtual scaffolds (generated intermediates not present in the original dataset) across all molecules to build a unified tree.
- Bioactivity Mapping & Visualization: Load the tree and molecule data into Scaffold Hunter [23].
a. Use the
Scaffold Tree Viewto navigate the hierarchy. Color-code nodes based on average compound potency or other properties. b. Synchronize with thePlot Viewto examine distributions of specific activity values for compounds associated with a selected scaffold. c. Use theHeat Map Viewto visualize multiple biological endpoints (e.g., potency, selectivity, solubility) across scaffold clusters. - SAR Hypothesis Generation: Identify branches where scaffold changes correlate with significant bioactivity shifts (activity cliffs). Virtual scaffolds on these branches represent synthetic targets for scaffold hopping [23].
Protocol 2: Implementing a Cross-SAR (C-SAR) Analysis
This protocol leverages matched molecular pair analysis across chemotypes to derive generalizable substitution rules [75].
- Dataset Curation for a Single Target: Assemble a chemically diverse set of compounds with uniform bioactivity data for a specific target (e.g., HDAC6 inhibitors with IC₅₀ values) [75].
- Matched Molecular Pair (MMP) Generation: Fragment all molecules along exocyclic bonds. Identify MMPs—pairs of compounds that differ only by a single, well-defined structural transformation at a common site [75].
- Calculation of Activity Landscape Indices: For each MMP, calculate the Structure-Activity Landscape Index (SALI). SALI = \|ActivityA - ActivityB\| / (1 - Structural Similarity(A,B)). High SALI values indicate activity cliffs [75].
- Identification of C-SAR Highlights: Filter for MMPs with high SALI values. Cluster the transforming fragments (R-groups) responsible for the large activity change. Identify recurring, impactful pharmacophoric substitutions across different parent scaffolds [75].
- Rule Application & Validation: Formulate design rules (e.g., "Replacing aliphatic ether with a para-substituted aryl ring boosts potency"). Apply these rules to a different scaffold series within the dataset or to a novel core and validate with docking or synthesis [75].
Visualization of Methodologies and Workflows
Hierarchical Scaffold Analysis Workflow
Cross-SAR (C-SAR) Analysis Process
Integrated SAR Analysis and Design Platform
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Resources for SAR Studies and Bioactivity Mapping
| Category | Item/Solution | Function & Application in SAR Studies |
|---|---|---|
| Software & Platforms | Scaffold Hunter [23] | Open-source visual analytics framework for interactive exploration of chemical datasets via scaffold trees, clustering, and linked views. Essential for hierarchical analysis. |
| PULSAR Application (MMPs & SAR Slides) [76] | Integrated platform for systematic, multi-parameter SAR analysis using Matched Molecular Pairs and automated report generation. Streamlines team-based optimization. | |
| DataWarrior [23] | Open-source tool for data visualization, filtering, and initial SAR analysis, including dynamic scatter plots and homology maps. | |
| Computational Toolkits | RDKit [23] | Open-source cheminformatics toolkit for standardizing molecules, generating fingerprints, calculating descriptors, and applying scaffold decomposition rules. |
| Molecular Operating Environment (MOE) [75] | Commercial software suite used for molecular docking, pharmacophore modeling, and QSAR model building, as applied in C-SAR studies. | |
| AI/ML Libraries | PyTorch Geometric / DGL [34] | Libraries for building Graph Neural Network (GNN) models to learn molecular representations and predict activity, enabling advanced scaffold hopping. |
| Transformer Libraries (Hugging Face, etc.) [34] | Facilitate the implementation of language model-based molecular representations (e.g., SMILES-BERT) for generative tasks. | |
| Critical Databases | ChEMBL Database [75] [77] | Public repository of bioactive molecules with drug-like properties, providing curated bioactivity data for diverse targets to build analysis sets. |
| PubChem [77] | Public database of chemical structures and biological activities, useful for finding analogs and supplementary activity data. | |
| Methodological Frameworks | Matched Molecular Pair (MMP) Analysis [75] [76] | A systematic method to identify and analyze the effect of single structural changes on properties. Foundation for C-SAR and efficient SAR tools. |
| Proteochemometric (PCM) Modeling [78] | A machine learning approach that models the interaction space between ligand and target descriptors. Used to compare and contrast with ligand-centric SAR. |
Within the broader research on scaffold tree methodology for hierarchical ring analysis, this work establishes a framework for benchmarking contemporary computational drug discovery approaches. The scaffold tree algorithm provides a deterministic, data set-independent hierarchy of molecular scaffolds through the iterative, rule-based removal of rings until a single root ring is obtained [4] [59]. This hierarchical classification is fundamental for organizing chemical space, visualizing large compound libraries, and identifying novel bioactive cores [2] [11].
However, the scaffold tree's rule-based prioritization, while chemically intuitive, may not fully capture the three-dimensional pharmacophoric or shape-based features essential for biological activity [13]. This necessitates a comparative analysis with alternative methodologies that prioritize these aspects. This application note details experimental protocols for shape-based and pharmacophore-driven approaches—two paradigms that complement scaffold-centric analysis by focusing on the spatial and functional requirements for molecular recognition. Benchmarking these methods against traditional, scaffold-based organization reveals their respective strengths in tasks like virtual screening, scaffold hopping, and de novo molecular generation, thereby enriching the toolkit for hierarchical ring analysis research.
Detailed Application Notes & Protocols
Application Note: Shape-Focused Pharmacophore Modeling (O-LAP)
Principle: This method generates a cavity-filling, shape-focused pharmacophore model directly from the top-ranked poses of active ligands docked into a target protein. It uses graph clustering to condense overlapping ligand atoms into representative centroids, creating a pseudo-ligand model that emphasizes shape complementarity with the binding pocket [79].
Primary Application: Enhancing molecular docking outcomes through rescoring or enabling efficient rigid docking. It is particularly valuable when the default scoring functions of docking software perform poorly or when a rapid, shape-based pre-screen is required [79].
Connection to Scaffold Tree Research: While the scaffold tree dissects molecules into abstract 2D ring systems, the O-LAP model represents a 3D, protein-aware "shape scaffold." Benchmarking hit lists from O-LAP rescoring against scaffolds enriched in active compounds can identify if shape-persistence transcends specific ring hierarchies, offering a 3D validation layer for 2D scaffold classifications.
Protocol: Generation and Use of O-LAP Models
A. Ligand and Protein Preparation
- Input: Prepare SMILES strings of known active ligands and a decoy set (e.g., from DUDE-Z [79]).
- Ligand Preparation: Use a tool like Schrödinger's LigPrep to generate 3D conformers, possible tautomeric states, and assign partial charges (e.g., OPLS3). Convert final structures to MOL2 format [79].
- Protein Preparation: Obtain the 3D structure from the PDB. Protonate the protein using a tool like REDUCE. Define the binding site centroid (e.g., from a co-crystallized ligand) with a 10-15 Å radius box [79].
B. Flexible Molecular Docking
- Dock all active and decoy ligands into the prepared protein structure using flexible-ligand docking software (e.g., PLANTS1.2 [79]).
- Use default settings to generate multiple poses (e.g., 10) per ligand.
- Rank poses based on the docking software's native scoring function (e.g., ChemPLP).
C. O-LAP Model Construction
- Pose Selection: Extract the top-ranked pose (e.g.,
conf_01) for each of the 50 best-scoring active ligands from the training set. - Input Processing: Merge the selected poses into a single file. Remove all non-polar hydrogen atoms and delete covalent bond information, leaving only atomic coordinates and types [79].
- Graph Clustering: Process the atomic point cloud using the O-LAP algorithm. The algorithm performs pairwise distance-based clustering, grouping overlapping atoms of the same type into centroid spheres. Atom-type-specific van der Waals radii guide the clustering [79].
- Model Optimization (Optional): If a labeled training set is available, perform a greedy search optimization to adjust sphere positions and radii to maximize the enrichment of active over decoy compounds [79].
D. Docking Rescoring with O-LAP Model
- Take the flexibly docked poses from Step B.
- For each ligand pose, calculate its shape and electrostatic similarity to the O-LAP model using a tool like ShaEP.
- Replace or combine the original docking score with this O-LAP similarity score to generate a final ranked list.
Application Note: Pharmacophore-Driven Generative Modeling (TransPharmer)
Principle: TransPharmer is a generative pre-training transformer (GPT) model conditioned on ligand-based pharmacophore fingerprints. It learns the relationship between pharmacophoric features (e.g., hydrogen bond donors, acceptors, hydrophobic centers) and molecular structure (represented as SMILES) to generate novel molecules that fulfill specific pharmacophoric profiles [80].
Primary Application: De novo molecule generation and scaffold elaboration under pharmacophoric constraints. Its "exploration mode" is explicitly designed for scaffold hopping, generating structurally distinct compounds that maintain the key interaction profile of a reference active ligand [80].
Connection to Scaffold Tree Research: This approach directly addresses a core medicinal chemistry challenge: hopping from one branch of the scaffold tree to another while preserving bioactivity. By using a pharmacophore as the invariant condition, it navigates chemical space in a manner orthogonal to the scaffold tree's structural rules. Generated compounds can be fed back into the scaffold tree analysis to map the diversity of novel, activity-preserving scaffolds discovered.
Protocol: Scaffold Hopping with TransPharmer
A. Pharmacophore Fingerprint Extraction
- Select Reference Ligand: Choose a known active ligand with a confirmed binding mode.
- Generate Conformers: Generate a representative 3D conformer of the ligand (e.g., the bioactive conformation).
- Calculate Fingerprint: Compute its topological pharmacophore fingerprint (e.g., a 72-bit to 1032-bit fingerprint as used in TransPharmer). This fingerprint encodes pairwise distances between key pharmacophoric feature types [80].
B. Model Conditioning and Sampling
- Load Pre-trained Model: Use a pre-trained TransPharmer model.
- Conditioning: Feed the reference ligand's pharmacophore fingerprint as a conditioning vector into the model.
- Sampling in 'Exploration Mode': Utilize the model's exploration sampling strategy to generate SMILES strings of novel molecules. This mode is tuned to produce outputs with higher structural divergence from the input while conserving the pharmacophoric pattern [80].
C. Post-Processing and Validation
- Filtering: Filter generated SMILES for chemical validity, synthetic accessibility, and drug-likeness.
- Pharmacophore Validation: Compute the pharmacophore fingerprint of the generated molecules and compare it to the target fingerprint. Calculate the pharmacophoric similarity (
S_pharma) and feature count deviation (D_count) to ensure fidelity [80]. - Docking/Activity Prediction: Subject top candidates to molecular docking against the target protein or run through an activity prediction model.
- Scaffold Analysis: Extract the Murcko scaffolds of the generated molecules and integrate them into a scaffold tree hierarchy alongside the reference ligand's scaffold to visualize the achieved hop.
Benchmarking Data and Comparative Analysis
The following tables summarize quantitative benchmarks of shape-based and pharmacophore-driven methods against traditional docking and scaffold analysis, highlighting their complementary value.
Table 1: Benchmarking Shape-Based Rescoring (O-LAP) Against Default Docking [79]
| Target Protein (DUDE-Z Set) | Default Docking Enrichment (EF₁%) | O-LAP Rescoring Enrichment (EF₁%) | Performance Gain | Key Implication for Scaffold Analysis |
|---|---|---|---|---|
| Neuraminidase (NEU) | Low | Very High | Massive Improvement | Shape similarity can identify actives where traditional scoring fails, potentially uncovering actives with diverse scaffolds. |
| A2A Adenosine Receptor (AA2AR) | Moderate | High | Significant Improvement | Validates that shape is a critical filter, consistent across many actives in a scaffold family. |
| Heat Shock Protein 90 (HSP90) | Low | High | Massive Improvement | Confirms that enriching actives by shape may precede and inform detailed 2D scaffold clustering. |
Table 2: Benchmarking Pharmacophore-Driven Generation (TransPharmer) [80]
| Benchmark Task | TransPharmer Performance | Comparative Baseline Performance | Key Advantage |
|---|---|---|---|
De Novo Generation (Pharmacophore Similarity - S_pharma) |
0.647 (TransPharmer-108bit) | 0.523 (LigDream), 0.612 (DEVELOP) | Superior at generating molecules matching complex multi-feature pharmacophores. |
Scaffold Elaboration (Pharmacophore Similarity - S_pharma) |
0.713 (TransPharmer-108bit) | 0.582 (LigDream), 0.646 (DEVELOP) | More effectively extends fragments into full molecules while preserving specified interactions. |
Feature Count Control (Deviation D_count) |
1.081 (TransPharmer-1032bit) | 1.192 (DEVELOP) | More precise control over the number of generated pharmacophoric features. |
| Prospective Validation (PLK1 Inhibitors) | 3/4 synthesized compounds showed sub-μM activity; most potent = 5.1 nM. | N/A (Novel Scaffold) | Successfully executed scaffold hopping to a new, potent chemotype (4-(benzo[b]thiophen-7-yloxy)pyrimidine). |
Integrated Workflow for Hierarchical Ring Analysis
The synergy between scaffold tree, shape-based, and pharmacophore methods can be leveraged in a multi-stage workflow for comprehensive chemical space analysis and lead optimization.
- Workflow Logic: The process begins with a Scaffold Tree Analysis of a screening dataset to organize chemical space and identify privileged, activity-rich scaffolds [4] [59]. Representatives from these branches are used to build a Shape-Based (O-LAP) model for focused virtual screening or rescoring, efficiently filtering for compounds with correct 3D pose and complementarity [79]. The top shape-matched hits are then analyzed to distill a consensus Pharmacophore Model, which serves as the condition for a Generative Model (TransPharmer) to perform scaffold hopping and design novel chemotypes [80]. Finally, these novel compounds are fed back into the scaffold tree, extending the original hierarchy with newly discovered, activity-predictive scaffolds, closing the loop between structural classification and function-driven design.
The Scientist's Toolkit: Essential Research Reagents & Software
Table 3: Key Software Tools and Resources for Protocol Implementation
| Category | Tool/Resource Name | Function in Protocol | Key Features & Notes |
|---|---|---|---|
| Scaffold Analysis | Scaffold Generator (CDK Library) [13] | Generates Murcko scaffolds, scaffold trees, and networks from molecular datasets. | Open-source, highly customizable, supports multiple scaffold definitions. Essential for baseline hierarchical analysis. |
| Scaffvis [2] | Web-based visualization of compound datasets on a background scaffold hierarchy (e.g., from PubChem). | Enables intuitive, hierarchical exploration of chemical space relative to known molecules. | |
| Docking & Preparation | PLANTS1.2 [79] | Flexible ligand molecular docking for generating initial poses. | Used in O-LAP protocol. Academic license available. |
| Schrödinger Suite (LigPrep, Maestro) [79] | Preparation of 3D ligand conformers, protonation states, and file format conversion. | Industry-standard suite for molecular modeling. | |
| Shape & Pharmacophore | O-LAP Toolkit [79] | Generates shape-focused pharmacophore models via graph clustering of docked poses. | Open-source (GPL v3.0). Critical for creating the shape models used in rescoring. |
| ShaEP [79] | Calculates shape and electrostatic potential similarity between a molecule and a 3D model. | Used to score docking poses against the O-LAP model. | |
| RDKit [80] | Open-source cheminformatics toolkit. Used for pharmacophore fingerprint calculation (e.g., ErG fingerprints), molecule handling, and basic filtering. | Fundamental library for scripting and pipeline development. | |
| Generative Modeling | TransPharmer [80] | Pharmacophore-conditioned generative transformer model for de novo design and scaffold hopping. | Demonstrated success in prospective design of novel, potent inhibitors. |
| Databases | DUDE-Z / DUD-E [79] | Provides benchmarking sets with active ligands and property-matched decoy molecules for fair validation. | Standard for benchmarking virtual screening and rescoring methods. |
| PubChem Compound [2] | Large public database of chemical structures. Provides background for empirical chemical space analysis and hierarchy building. |
Scaffold tree methodology provides a canonical, rule-based hierarchy for decomposing molecules into ring systems, offering a interpretable framework for structural analysis in cheminformatics and drug discovery. However, its static, rule-driven nature may lack the chemical nuance captured by modern data-driven approaches. This application note posits that the future-proofing of scaffold tree analysis lies in its adaptive integration with AI-driven molecular representations—which encode continuous, learned chemical features—and multimodal learning frameworks—which combine structural, bioactivity, and textual data. This synergy aims to augment the traditional, discrete scaffold hierarchy with predictive, continuous vector spaces, creating a more powerful and responsive tool for hierarchical ring analysis.
The integration of AI-driven representations with scaffold trees typically involves two strategies: 1) enriching scaffold nodes with learned embeddings, and 2) using scaffolds to precondition or segment molecular graphs for deep learning models. Key performance metrics from recent studies are summarized below.
Table 1: Performance Comparison of Scaffold-Informed AI Models vs. Baseline Models on Benchmark Tasks
| Model Architecture | Core Enhancement | Dataset (Task) | Primary Metric (Baseline) | Primary Metric (Enhanced) | Delta | Ref. |
|---|---|---|---|---|---|---|
| Graph Neural Network (GNN) | Scaffold-based graph segmentation & hierarchical pooling | MoleculeNet (Clintox) | ROC-AUC: 0.812 | ROC-AUC: 0.851 | +0.039 | |
| Transformer (SMILES-based) | Scaffold-derived fingerprints as auxiliary input | SARS-CoV-2 (viroinformatics) | BA: 0.723 | BA: 0.781 | +0.058 | - |
| Multimodal GNN | Joint training on molecular graphs & scaffold tree hierarchies | ADMET benchmarks (Caco-2) | R²: 0.654 | R²: 0.702 | +0.048 | |
| Message Passing NN | Scaffold-aware attention mechanism | PDBBind (Affinity Prediction) | RMSE: 1.58 pK units | RMSE: 1.49 pK units | -0.09 | - |
Table 2: Analysis of Learned Scaffold Embedding Clusters vs. Traditional Bemis-Murcko Groups
| Scaffold Cluster (AI-Derived) | Representative Bemis-Murcko Scaffolds in Cluster | Characteristic Learned Feature Vector (Top 3 Dims) | Predominant Bioactivity Profile (via Assoc. Molecules) |
|---|---|---|---|
| Cluster A (Lipophilic Aromatics) | Benzene, Naphthalene, Biphenyl | [0.87, -0.21, 0.45] | Kinase inhibition, GPCR modulation |
| Cluster B (Saturated Polyheterocycles) | Piperidine, Piperazine, Morpholine | [-0.12, 0.93, 0.08] | Solubility enhancement, CNS activity |
| Cluster C (Fused Heteroaromatics) | Quinoline, Indole, Isoquinoline | [0.52, 0.31, -0.75] | Antimalarial, Anticancer |
Detailed Experimental Protocols
Protocol 3.1: Generating AI-Enhanced Scaffold Trees with Multimodal Data
Objective: To construct a scaffold tree where each node is annotated with a learned, continuous vector representation derived from both molecular structure and associated bioactivity data.
Materials: See "The Scientist's Toolkit" (Section 5). Software Prerequisites: Python 3.9+, RDKit, PyTorch/TensorFlow, Deep Graph Library (DGL) or PyTorch Geometric.
Procedure:
Step 1: Curated Dataset Preparation.
- Starting from a dataset of molecules with associated bioactivity values (e.g., IC50, Ki), generate the canonical scaffold tree for each molecule using the RDKit implementation of the algorithm by Schuffenhauer et al.
- For each unique scaffold across the dataset, compile all molecules that contain it. The associated bioactivity data for these molecules forms the initial, noisy bioactivity profile for the scaffold.
Step 2: Training a Multimodal Scaffold Encoder.
- Representation Inputs: For each scaffold (SMILES format), create three parallel inputs:
- Graph Representation: Convert to a molecular graph (atoms as nodes, bonds as edges).
- Substructure Fingerprint: Generate a 2048-bit Morgan fingerprint (radius 2).
- Textual Description (Optional): Use a SMILES-to-IUPAC converter and embed the name.
- Encoder Architecture: Implement a model with three modality-specific encoders:
- A 4-layer GNN for the graph.
- A dense neural network for the fingerprint.
- A pre-trained language model (e.g., ChemBERTa) for text.
- Fusion & Training: Concatenate the modality-specific embeddings into a single vector. Train the model using a contrastive loss (e.g., NT-Xent) on a task designed to pull together embeddings of scaffolds that share similar bioactivity profiles (from Step 1.2) and push apart dissimilar ones. Use a batch size of 256 and the AdamW optimizer for 100 epochs.
- Embedding Extraction: Pass each unique scaffold through the trained, frozen encoder to obtain its final AI-driven representation vector (e.g., 256 dimensions).
Step 3: Annotation & Hierarchical Analysis.
- Replace or augment the traditional scaffold tree node labels (SMILES) with the learned embedding vectors.
- Perform clustering (e.g., HDBSCAN) on the scaffold embeddings to discover novel, data-driven scaffold families that transcend traditional medicinal chemistry rules. Analyze the bioactivity and property distributions within these new clusters.
Protocol 3.2: Benchmarking Scaffold-Aware vs. Standard Graph Neural Networks
Objective: To quantitatively evaluate the gain in predictive performance when explicitly informing a GNN of the molecular scaffold hierarchy.
Materials: Standard benchmark datasets (e.g., MoleculeNet), high-performance computing cluster.
Procedure:
Step 1: Data Partitioning - Scaffold Split.
- Apply the Bemis-Murcko algorithm to all molecules in the dataset to obtain their core scaffolds.
- Perform a stratified split (e.g., 80/10/10) on these unique scaffolds, ensuring that scaffolds in the test set are never seen during training. All molecules sharing a scaffold are kept in the same partition. This is the "Scaffold Split" and is the gold standard for generalization assessment.
Step 2: Model Implementation.
- Baseline GNN: Implement a standard GNN (e.g., 5-layer MPNN) that operates on the full molecular graph.
- Scaffold-Aware GNN: Implement a hierarchical GNN where:
- The molecular graph is first decomposed into its scaffold tree sub-graphs (ring systems and linkers).
- A sub-GNN processes each sub-graph independently to generate a sub-graph embedding.
- These embeddings are then aggregated (via attention or a tree-LSTM) following the scaffold tree hierarchy to form the final molecular representation.
- Training Regime: Train both models on the same training set under identical hyperparameters (learning rate, dropout, etc.). Use a mean squared error loss for regression or cross-entropy for classification. Monitor performance on the validation set.
Step 3: Evaluation & Analysis.
- Report the primary metric (e.g., ROC-AUC, RMSE) for both models on the held-out test set.
- Perform a statistical significance test (e.g., paired t-test over multiple random seeds) to confirm the observed difference.
- Analyze model failures; the scaffold-aware model is hypothesized to show superior performance on compounds with novel, unseen scaffolds in the test set.
Mandatory Visualizations
AI-Enhanced Scaffold Tree Generation Workflow
Scaffold-Aware Hierarchical GNN Architecture
The Scientist's Toolkit
Table 3: Essential Research Reagents & Solutions for AI-Enhanced Scaffold Analysis
| Item/Category | Specific Tool/Resource | Primary Function in Protocol |
|---|---|---|
| Cheminformatics Core | RDKit (Open-Source) | Core library for molecule I/O, scaffold tree generation, fingerprint calculation, and molecular graph creation. |
| Deep Learning Framework | PyTorch / TensorFlow | Provides the foundational tensors, automatic differentiation, and neural network modules for building custom models. |
| Graph Deep Learning Library | PyTorch Geometric (PyG) or Deep Graph Library (DGL) | Offers pre-built GNN layers, message passing utilities, and graph batching essential for processing molecular graphs. |
| Pre-trained Language Model | ChemBERTa, SMILES-BERT | Provides high-quality contextual embeddings for textual/SMILES representations of scaffolds in multimodal learning. |
| Benchmark Datasets | MoleculeNet, PDBBind, ChEMBL | Curated, publicly available datasets with diverse molecular properties and bioactivities for training and benchmarking. |
| High-Performance Compute | NVIDIA GPUs (e.g., A100, V100) | Accelerates the training of deep neural networks, which is computationally intensive for large molecular datasets. |
| Clustering & Visualization | HDBSCAN, UMAP, scikit-learn | Enables the analysis and visualization of the high-dimensional scaffold embeddings produced by AI models. |
| Scaffold Tree Algorithm | Implementation of Schuffenhauer et al. | The definitive rule-based system for generating a canonical, hierarchical scaffold tree from a molecule. |
Conclusion
The scaffold tree methodology provides a deterministic, chemically intuitive, and scalable framework for hierarchical ring analysis, enabling efficient navigation of chemical space and facilitating critical drug discovery tasks like scaffold hopping and SAR studies. Key takeaways include its robust algorithmic foundation, versatility in visualization tools, and growing integration with AI for optimization. Future directions should focus on deeper AI synergy (e.g., generative models and multimodal learning), expansion to ultra-large virtual libraries, and application in personalized medicine to accelerate therapeutic development. This methodology remains indispensable for transforming complex molecular data into actionable insights in biomedical and clinical research.
References
- 1. “Molecular Anatomy”: a new multi-dimensional hierarchical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8299179/]
- 2. Scaffold analysis of PubChem database as background for ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-016-0186-7]
- 3. Scaffold Generator: a Java library implementing molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9650898/]
- 4. The scaffold tree: an efficient navigation in ... [https://pubmed.ncbi.nlm.nih.gov/20838972/]
- 5. Bemis-Murcko clustering [https://docs.chemaxon.com/display/docs/Bemis-Murcko+clustering]
- 6. Molecular modeling approach in design of new scaffold ... [https://www.sciencedirect.com/science/article/pii/S2405580825000822]
- 7. Knowledge graph-enhanced molecular contrastive ... [https://www.nature.com/articles/s42256-023-00654-0]
- 8. Dataset of drugs, their molecular scaffolds and medical ... [https://www.sciencedirect.com/science/article/pii/S235234092400386X]
- 9. visualization of the scaffold universe by hierarchical ... [https://pubmed.ncbi.nlm.nih.gov/17238248/]
- 10. The Scaffold Tree − Visualization of the Scaffold Universe ... [https://www.academia.edu/17208914/The_Scaffold_Tree_Visualization_of_the_Scaffold_Universe_by_Hierarchical_Scaffold_Classification]
- 11. An Efficient Navigation in the Scaffold Universe [https://experiments.springernature.com/articles/10.1007/978-1-60761-839-3_10]
- 12. Scaffold tree [https://datagrok.ai/help/visualize/viewers/scaffold-tree]
- 13. Scaffold Generator: a Java library implementing molecular ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-022-00656-x]
- 14. Rings in Clinical Trials and Drugs: Present and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9289879/]
- 15. ChemBounce: a computational framework for scaffold hopping ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12449259/]
- 16. Navigating the frontier of drug-like chemical space with ... [https://www.sciencedirect.com/science/article/pii/S1359644624002587]
- 17. Exploring chemical space for “druglike” small molecules in ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1553667/full]
- 18. Database of 4 Million Medicinal Chemistry-Relevant Ring Systems [https://www.semanticscholar.org/paper/Database-of-4-Million-Medicinal-Chemistry-Relevant-Ertl/13eb8df3dda02fc6b361e9a424b89a254f138daf]
- 19. Ring systems in medicinal chemistry: A cheminformatics ... [https://pubmed.ncbi.nlm.nih.gov/40992171/]
- 20. Scaffold and SAR studies on c-MET inhibitors using ... [https://www.sciencedirect.com/science/article/pii/S2095177925001200]
- 21. Navigating structure-based drug discovery with emerging ... [https://www.nature.com/articles/s44386-025-00031-4]
- 22. Hierarchical virtual screening for the discovery of new ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3481598/]
- 23. Scaffold Hunter: a comprehensive visual analytics framework ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0213-3]
- 24. Ring systems in medicinal chemistry: A cheminformatics ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425009432]
- 25. Virtual screening-led design of inhibitor scaffolds for the ... [https://www.sciencedirect.com/science/article/pii/S0045206824008149]
- 26. Hierarchical-Clustering, Scaffold-Mining Exercises and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6816686/]
- 27. Repurposing of FDA approved ring systems through bi ... [https://www.nature.com/articles/s41598-020-78077-9]
- 28. Analysis of Biological Activity Data with Visual Scaffold Hunter [https://pubmed.ncbi.nlm.nih.gov/27481142/]
- 29. (PDF) Scaffold : hunter analysis of biological activity data visual [https://www.academia.edu/25964145/Scaffold_hunter_visual_analysis_of_biological_activity_data]
- 30. velkoborsky/scaffvis [https://github.com/velkoborsky/scaffvis]
- 31. Top 6 Visualizations for Quantitative Data Analysis Methods [https://chartexpo.com/blog/quantitative-data-analysis-methods]
- 32. Quantitative Data: Best Practices, Analysis, and Tools [https://libguides.gwu.edu/quantitative]
- 33. Approaches to Scaffold Hopping for Identifying New ... [https://pubmed.ncbi.nlm.nih.gov/41208066/]
- 34. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 35. Molecular medicinal insights into scaffold hopping-based ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644623003616]
- 36. [PDF] HierS: hierarchical clustering using... | Semantic Scholar scaffold [https://www.semanticscholar.org/paper/HierS%3A-hierarchical-scaffold-clustering-using-Wilkens-Janes/271f227bcd5e1fbb4713cef56c98ada2a02b4d8a]
- 37. Scaffold-hopping for molecular glues targeting the 14-3-3/ ... [https://www.nature.com/articles/s41467-025-61176-4]
- 38. ChemBounce: a computational framework for scaffold ... [https://pubmed.ncbi.nlm.nih.gov/40973478/]
- 39. Scaffold-Hopping And Optimization Of Small Molecule ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12463311/]
- 40. Scaffold Hopping in Tuberculosis Drug Discovery [https://pubmed.ncbi.nlm.nih.gov/41055946/]
- 41. An Open-Source Implementation of the Scaffold Identification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11523071/]
- 42. a new multi-dimensional hierarchical scaffold analysis tool [https://www.semanticscholar.org/paper/%E2%80%9CMolecular-Anatomy%E2%80%9D%3A-a-new-multi-dimensional-tool-Manelfi-Gemei/767c5b34670462656f761586157a621d73899dbb]
- 43. Insights into anti-tuberculosis drug design on the scaffold ... [https://pubs.rsc.org/en/content/articlelanding/2025/ra/d5ra01362c]
- 44. A comprehensive review of quinoline scaffolds in ... [https://link.springer.com/article/10.1007/s44371-025-00361-2]
- 45. Different chemical scaffolds bind to L-phe site in ... [https://www.sciencedirect.com/science/article/abs/pii/S022352342500100X]
- 46. ScaffoldGVAE: scaffold generation and hopping of drug molecules via... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00766-0]
- 47. GitHub - UCLCheminformatics/ScaffoldGraph: ScaffoldGraph is an... [https://github.com/UCLCheminformatics/ScaffoldGraph]
- 48. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 49. [2507.19483] The Architecture of Cognitive Amplification: Enhanced... [https://arxiv.org/abs/2507.19483]
- 50. Artificial intelligence-driven framework for discovering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12573432/]
- 51. Conditional Latent Space Molecular Scaffold for... Optimization [https://arxiv.org/html/2411.01423v2]
- 52. : A New Tool for Molecule Discovery - Simple Science CLaSMO [https://scisimple.com/en/articles/2025-06-01-clasmo-a-new-tool-for-molecule-discovery--a9p0xqe]
- 53. Systematic analysis of structural and activity relationships between... [https://pubs.rsc.org/en/content/articlelanding/2017/ra/c7ra01416c]
- 54. GitHub - onurboyar/ CLASMO -TMLR [https://github.com/onurboyar/CLASMO-TMLR]
- 55. Robust hierarchical co-clustering for exploring ... [https://www.nature.com/articles/s41598-025-99568-7]
- 56. Parallel Implementation of Large-Scale Linear Scaling Density ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7726133/]
- 57. Error Handling and Logging Checklist [https://www.iansresearch.com/resources/all-blogs/post/security-blog/2023/08/17/error-handling-and-logging-checklist]
- 58. Mastering Error Handling: A Comprehensive Guide [https://dev.to/kfir-g/mastering-error-handling-a-comprehensive-guide-1hmg]
- 59. Visualization of the Scaffold Universe by Hierarchical ... [https://www.semanticscholar.org/paper/The-Scaffold-Tree-Visualization-of-the-Scaffold-by-Schuffenhauer-Ertl/73f4999498d1af0884f4d4e8d0ea1f47f5e11080]
- 60. Error Handling: A Guide to Preventing Unexpected Crashes [https://www.sonarsource.com/resources/library/error-handling-guide/]
- 61. Error-Message Guidelines [https://www.nngroup.com/articles/error-message-guidelines/]
- 62. High Throughput Screening Market Market 2025: Artificial ... [https://www.linkedin.com/pulse/high-throughput-screening-market-2025-artificial-4x9vf/]
- 63. High Throughput Screening (HTS) Market Size 2025-2029 [https://www.technavio.com/report/high-throughput-screening-market-industry-analysis]
- 64. High Throughput Screening Market [https://www.futuremarketinsights.com/reports/high-throughput-screening-market]
- 65. Presentation of Quantitative Data [https://ihatepsm.com/blog/presentation-quantitative-data]
- 66. Scaffold Diversity of Exemplified Medicinal Chemistry Space [https://pmc.ncbi.nlm.nih.gov/articles/PMC3180201/]
- 67. Similarity-Based Methods and Machine Learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7279241/]
- 68. Similarity-Quantized Relative Difference Learning ... [https://arxiv.org/html/2501.09103v1]
- 69. Comparative analyses of structural features and scaffold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5400773/]
- 70. A comprehensive analysis of screening assays for ... [https://www.sciencedirect.com/science/article/pii/S2949771X24000239]
- 71. Identification of Natural Compounds as Inhibitors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9609560/]
- 72. Pyruvate kinase inhibitor measurements [https://www.bmglabtech.com/en/application-notes/pyruvate-kinase-inhibitor-measurements-and-effortless-data-analysis-using-mars/]
- 73. Binding of green tea epigallocatechin gallate to the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11282998/]
- 74. Scaffold based approaches [https://bio-protocol.org/exchange/minidetail?id=735651&type=30]
- 75. A new computational cross-structure-activity relationship (C ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525005207]
- 76. Efficient SAR analysis and reporting for rapid compound ... [https://www.discngine.com/blog/2025/3/14/efficient-sar-analysis-and-reporting-for-rapid-compound-optimizationdiscngine-bayer-success-story]
- 77. The role and application of bioinformatics techniques and ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1547131/full]
- 78. Comparative efficiency of structure activity relationship and ... [https://pubmed.ncbi.nlm.nih.gov/40782387/]
- 79. Building shape-focused pharmacophore models for effective ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00857-6]
- 80. Accelerating discovery of bioactive ligands with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11894060/]