Cross-Validation Accelerates Discovery: Integrating Genome Mining with Dereplication for Novel Natural Products
This article provides a comprehensive guide for researchers and drug development professionals on the strategic integration of genome mining and dereplication to accelerate the discovery of novel bioactive natural products.
Cross-Validation Accelerates Discovery: Integrating Genome Mining with Dereplication for Novel Natural Products
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the strategic integration of genome mining and dereplication to accelerate the discovery of novel bioactive natural products. It explores the foundational concepts of biosynthetic gene clusters (BGCs) and mass-spectral dereplication, details step-by-step methodologies for their combined application, addresses common technical challenges and optimization strategies, and presents frameworks for rigorous cross-validation. By synthesizing these approaches, the article outlines a robust pipeline that minimizes the re-discovery of known compounds, prioritizes promising leads, and enhances the efficiency of translating genomic potential into new therapeutic candidates.
The Synergy of Prediction and Identification: Core Concepts in Genome Mining and Dereplication
The discovery of novel bioactive natural products, a critical source for new pharmaceuticals and agrochemicals, has been transformed by two complementary computational and analytical pillars: genome mining and dereplication. Both strategies aim to solve the central problem of rediscovery in natural product research but operate from opposite directions [1] [2].
Genome mining is a forward, in-silico prediction strategy. It involves bioinformatically analyzing microbial (meta)genomes to identify Biosynthetic Gene Clusters (BGCs)—groups of co-localized genes that encode the enzymatic machinery for producing a specialized metabolite [3] [2]. The core premise is that the genetic blueprint precedes and predicts chemical output. In contrast, dereplication is a reverse, analytical chemistry strategy. It involves the rapid chemical screening of extracts—from microbial fermentations or plant materials—to identify known compounds early in the discovery pipeline. This prevents wasted effort on re-isolating and re-characterizing known entities [1] [4].
Within the broader thesis of cross-validating genome mining predictions with dereplication results, these pillars form a synergistic validation cycle. Genome mining offers a hypothesis (a predicted BGC and its putative product), while dereplication provides an empirical test (detection and identification of molecules from a cultured organism). Their integration is essential for efficiently navigating the vast landscape of microbial and plant chemical diversity to prioritize truly novel leads for drug development [1].
Pillar One: Genome Mining - From Genetic Blueprint to Chemical Prediction
Core Principles and Methodologies
Genome mining operates on the fundamental biosynthetic principle that genes for natural product synthesis are clustered in microbial genomes [3]. The workflow begins with the identification of conserved "backbone" or "signature" enzymes—such as non-ribosomal peptide synthetases (NRPS), polyketide synthases (PKS), or terpene synthases—which serve as baits for BGC detection [2].
Modern tools like antiSMASH (the Antibiotics and Secondary Metabolite Analysis Shell) use libraries of profile hidden Markov models (pHMMs) to detect these signature domains and define the boundaries of BGCs [5] [2]. The methodology extends beyond simple detection to functional prediction. For example, advanced implementations can predict specific metabolite classes, such as non-ribosomal peptide (NRP) metallophores (metal-chelating compounds), by searching for genes encoding distinctive chelator biosynthesis pathways (e.g., for catechol or hydroxamate groups) within NRPS clusters [5].
A key experimental protocol for validation involves heterologous expression: cloning the predicted BGC into a surrogate host (like Streptomyces coelicolor) to induce production and isolate the compound [2]. Alternatively, gene knockout experiments, where core biosynthetic genes are deleted, are performed to link the cluster to the observed metabolite, followed by comparative metabolomics (e.g., LC-MS) of wild-type and mutant strains to confirm the absence of the target compound [6].
Performance and Applications
Genome mining has proven exceptionally powerful for large-scale, taxonomic analyses of biosynthetic potential. A 2025 study mining 187 fungal genomes from the Alternaria genus and related taxa identified 6,323 BGCs, with an average of 34 BGCs per genome [3]. This reveals a much greater hidden biosynthetic capacity than observable through traditional cultivation. The performance of automated prediction tools continues to improve. For instance, a specialized algorithm for detecting NRP metallophore BGCs in antiSMASH achieved a 97% precision and 78% recall against manual expert curation [5].
The applications are vast:
- Taxonomic and Evolutionary Insights: Mapping BGC distribution across a phylogeny can reveal horizontal transfer events and evolutionary relationships. The study of Alternaria showed BGC patterns generally correlated with phylogeny, identifying unique clusters in specific sections like Infectoriae [3].
- Targeted Discovery of Specific Compound Classes: Tools can be tuned to find clusters for desired activities. The metallophore detector enabled a survey of 69,929 bacterial genomes, predicting that 25% of all bacterial NRPS clusters encode metallophore production [5].
- Uncovering Novel Regulatory Loci: Innovative approaches combine genome mining with transcriptomic data. Research on Streptomyces coelicolor used the regulon of an iron master regulator to co-discover a novel operon (desJGH) essential for the biosynthesis of the siderophore desferrioxamine, a cluster missed by standard BGC detection tools [6].
Table 1: Key Performance Metrics of Genome Mining Tools and Studies
| Tool / Study Focus | Core Methodology | Dataset Scale | Key Performance Metric | Primary Application |
|---|---|---|---|---|
| antiSMASH (general BGC detection) [5] [2] | Profile HMMs for signature domains | Virtually unlimited genomes | Identifies core biosynthetic enzymes and cluster boundaries | Broad-spectrum BGC discovery |
| antiSMASH NRP Metallophore Detector [5] | pHMMs for chelator biosynthesis genes | 69,929 bacterial genomes | 97% precision, 78% recall vs. manual curation | Targeted discovery of metallophores |
| Fungal BGC Mining in Alternaria [3] | antiSMASH-based pipeline | 187 fungal genomes | Avg. 34 BGCs/genome; identified 548 Gene Cluster Families (GCFs) | Taxonomic distribution & mycotoxin risk assessment |
| Regulation-Guided Mining (e.g., DmdR1 regulon) [6] | Integration of TF binding site prediction & transcriptomics | Genome of S. coelicolor | Discovered novel essential operon (desJGH) for a known metabolite | Prioritizing BGCs with shared regulatory logic |
Pillar Two: Dereplication - From Chemical Analysis to Identity Verification
Core Principles and Methodologies
Dereplication functions as the quality-control checkpoint of natural product discovery. Its goal is to rapidly identify known compounds within a complex mixture before engaging in lengthy isolation processes [7] [4]. The standard experimental protocol is centered on Liquid Chromatography coupled with tandem Mass Spectrometry (LC-MS/MS).
A typical dereplication workflow involves [7] [4]:
- Sample Preparation: Often includes a cleanup step (e.g., Solid-Phase Extraction with C-18 cartridges) to remove interfering sugars and salts, enhancing chromatographic resolution and MS signal [7].
- LC-MS/MS Analysis: The extract is separated by liquid chromatography, and eluting compounds are ionized (commonly by Electrospray Ionization - ESI) and analyzed by a high-resolution mass spectrometer. Key data collected include: precursor ion mass (with high mass accuracy, often < 5 ppm error), isotopic pattern, retention time (RT), and fragmentation pattern (MS/MS spectrum) [4].
- Database Matching: The acquired MS/MS spectra are queried against reference spectral libraries. These can be public databases like GNPS (Global Natural Products Social Molecular Networking), MassBank, or mzCloud, or curated in-house libraries built from authentic standards [4].
- Annotation & Validation: Compounds are annotated based on spectral matches. Confidence is increased by cross-referencing with other data (e.g., UV spectra) or, definitively, by comparison with an authentic analytical standard [7].
Performance and Applications
Modern dereplication is highly effective at parsing complexity. A 2025 study of a polyherbal liquid formulation (PLF) containing ten plant extracts used LC-MS/MS to identify 70 compounds (44 unique and 26 shared) in a single analysis, successfully attributing them to specific plant contributors [7]. The efficiency gains are substantial; developing a targeted in-house MS/MS library for 31 common phytochemicals enabled their rapid dereplication in 15 different food and plant samples, drastically reducing the time needed for compound identification [4].
Primary applications include:
- Prioritization of Novelty: The foremost role is to flag known molecules, allowing researchers to focus resources on unknown chromatographic features with novel MS signatures [4].
- Standardization of Complex Formulations: Dereplication provides the chemical fingerprints necessary for quality control of herbal medicines, ensuring batch-to-batch consistency and validating ingredient sources [7].
- Accelerating Lead Discovery: By quickly identifying bioactive compounds in screening hits, the downstream isolation pipeline is streamlined. This integrates seamlessly with genome mining: the molecule detected via dereplication can be linked back to a predicted BGC [1].
Table 2: Representative Dereplication Workflows and Outcomes
| Study / Application | Sample Type | Core Analytical Platform | Key Outcome / Performance | Strategic Purpose |
|---|---|---|---|---|
| Polyherbal Formulation (PLF) Analysis [7] | Liquid syrup with 10 plant extracts | LC-MS/MS with SPE C-18 cleanup | Identified 70 compounds; attributed 44 to specific plants. | Standardization and quality control of complex mixtures. |
| In-house Phytochemical Library [4] | 15 diverse food and plant extracts | LC-HR-ESI-MS/MS | Rapid dereplication of 31 target compounds across all samples. | Accelerated screening and validation of common bioactive metabolites. |
| Peptidic Natural Product Discovery [1] | Microbial fermentation extracts | LC-MS/MS integrated with genomic data (peptidogenomics) | Connects detected peptides to biosynthetic gene clusters. | Bridging analytical chemistry with genomic predictions. |
Comparative Analysis: Strengths, Limitations, and Synergies
Genome mining and dereplication are not competing but complementary. Their direct comparison highlights the rationale for an integrated approach.
Table 3: Comparative Analysis of Genome Mining vs. Dereplication
| Aspect | Genome Mining | Dereplication |
|---|---|---|
| Primary Input | DNA sequence (genome/metagenome) | Chemical extract (crude or partially purified) |
| Core Objective | Predict biosynthetic potential and novel chemical scaffolds. | Identify existing chemical entities to avoid rediscovery. |
| Key Strength | Reveals vast, hidden biosynthetic capacity (e.g., 34 BGCs/genome in fungi) [3]. Unbiased by cultivation conditions. | Provides direct, empirical chemical evidence. Fast and high-throughput for known compounds. |
| Major Limitation | Predicts potential, not actual production. Many BGCs are "silent" under lab conditions. Prediction of exact chemical structure can be error-prone [2]. | Blind to compounds not in reference libraries. Cannot predict novel scaffolds de novo. Requires the organism to produce the compound under test conditions. |
| Typical Output | Catalog of predicted BGCs and putative compound classes (e.g., NRPS-derived metallophore) [5]. | List of identified compounds with confidence levels (e.g., 70 compounds in an herbal syrup) [7]. |
| Computational vs. Analytical Load | High computational load for sequence analysis and prediction. | High analytical load for chromatography and mass spectrometry. |
The Synergy for Cross-Validation: The limitations of one pillar are addressed by the strengths of the other. A genome mining prediction (e.g., a novel NRPS cluster) guides targeted cultivation and analysis. Subsequent dereplication of the organism's extract can either: a) identify the predicted compound class, validating the in-silico hypothesis, or b) reveal a novel molecule, prompting the re-interpretation of the BGC's function. Conversely, a novel molecule found via dereplication can trigger a targeted genome mining effort to find its BGC, enabling genetic engineering and yield optimization [1]. This iterative loop of prediction and validation is the essence of a robust natural product discovery pipeline.
Integrated Workflow and Visualization
The most effective discovery pipelines interweave genome mining and dereplication into a single workflow. This integrated approach is foundational to the thesis of cross-validation.
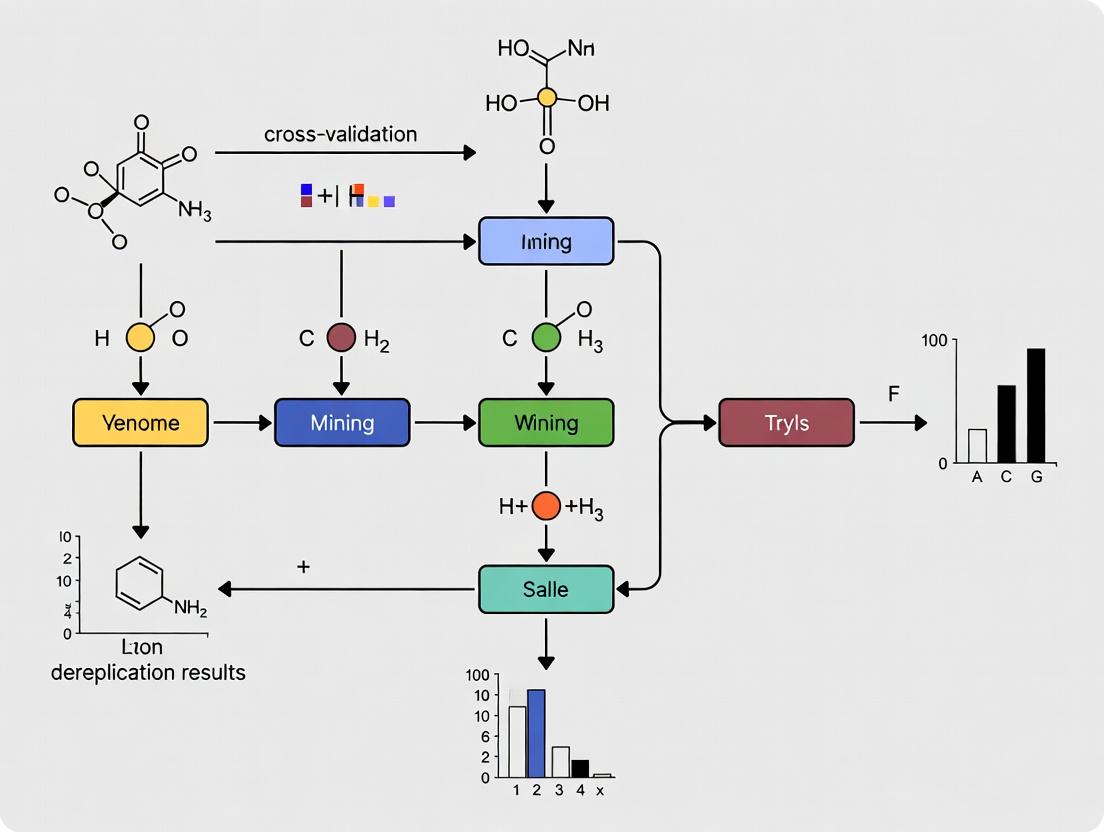
Diagram 1: Integrated Genome Mining & Dereplication Workflow. This diagram illustrates the synergistic, cyclical relationship between the two pillars, forming a cross-validation loop.
This integrated process can be formalized into a structured cross-validation framework.
Diagram 2: Cross-Validation Framework for BGC-Metabolite Linking. This diagram formalizes the parallel analysis and comparison steps that constitute the core of a cross-validation thesis.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Implementing an integrated genome mining and dereplication strategy requires a suite of specialized computational tools and analytical resources.
Table 4: Essential Research Toolkit for Integrated Discovery
| Tool / Resource Name | Category | Primary Function | Key Application in Workflow |
|---|---|---|---|
| antiSMASH [5] [3] [2] | Genome Mining Software | Identifies and annotates biosynthetic gene clusters in genomic sequences. | The primary engine for BGC prediction and initial functional annotation (e.g., NRPS, PKS, metallophore). |
| MIBiG (Minimum Information about a BGC) [3] | Reference Database | A curated repository of experimentally characterized BGCs. | Used as a reference for comparing predicted BGCs to known ones for in-silico dereplication. |
| GNPS (Global Natural Products Social Molecular Networking) [1] [4] | Mass Spectrometry Platform | A web-based platform for storing, sharing, and analyzing mass spectrometry data, especially MS/MS. | Core platform for experimental dereplication via spectral matching and molecular networking to find related compounds. |
| LC-HR-MS/MS System (e.g., Q-TOF, Orbitrap) [7] [4] | Analytical Instrumentation | Provides high-resolution precursor and fragment ion masses for accurate compound identification. | Generates the empirical metabolomic data (retention time, accurate mass, MS/MS spectra) for dereplication. |
| C-18 Solid Phase Extraction (SPE) Cartridges [7] | Sample Preparation Reagent | Removes salts, sugars, and other polar interferents from complex biological extracts. | Critical cleanup step prior to LC-MS to reduce ion suppression and improve chromatographic resolution for dereplication. |
| Authentic Chemical Standards [7] [4] | Research Reagents | Pure compounds used as analytical references. | Provides definitive, highest-confidence identification during dereplication and is used to build in-house MS/MS libraries. |
The discovery of novel natural products (NPs) for drug development is at a critical juncture. While NPs have contributed to 60% of marketed small-molecule drugs, the path from gene cluster to validated lead compound remains fraught with inefficiencies and high attrition rates [8]. The central bottleneck is no longer a lack of data but an overabundance of unvalidated predictions. Modern genome mining can scan tens of thousands of bacterial genomes to predict biosynthetic potential, and AI models can generate plausible 3D structures. However, without rigorous, multi-layered cross-validation, these computational hits remain mere hypotheses, wasting valuable resources in downstream experimental validation [5]. This guide compares the core methodologies defining the current landscape—genome mining, structure prediction, and high-throughput screening—within the essential framework of cross-validation. By objectively evaluating their performance data and experimental protocols, we provide researchers with a clear roadmap for integrating validation at every step to accelerate the translation of genetic blueprints into tangible therapeutic candidates.
Performance Comparison of Core Discovery and Validation Methodologies
The following tables summarize the quantitative performance and key characteristics of the primary technologies discussed, providing a basis for objective comparison.
Table 1: Performance Metrics of Genome Mining & Structure Prediction Tools
| Methodology | Tool/Approach | Primary Function | Reported Performance | Key Advantage for Cross-Validation |
|---|---|---|---|---|
| Automated Genome Mining | antiSMASH with NRP metallophore rules [5] | Detects biosynthetic gene clusters (BGCs) for non-ribosomal peptide metallophores. | 97% precision, 78% recall against manual curation. | High-precision rule set reduces false positives, providing a reliable starting point for experimental validation. |
| 3D Structure Prediction | NatGen (Deep Learning Framework) [8] | Predicts chiral configurations and 3D conformations of NPs from 2D structures. | 96.87% accuracy on benchmark; 100% in a prospective study of 17 plant NPs; Avg. RMSD <1 Å. | Generates testable structural hypotheses for unknown NPs, enabling computational docking and property prediction. |
| Metagenome Analysis | Co-assembly & Binning (e.g., for CRC microbiomes) [9] | Recovers genomes, including uncultivated species, from complex metagenomic samples. | Enabled CRC prediction with 0.90-0.98 AUROC using selected genomes. | Uncovers "microbial dark matter," expanding the search space for novel BGCs beyond cultured organisms. |
Table 2: Comparison of Screening & Validation Paradigms
| Paradigm | Typical Throughput | Data Output | Key Cross-Validation Requirement | Common Pitfalls (False Signals) |
|---|---|---|---|---|
| High-Throughput Screening (HTS) [10] | 10,000 – 100,000 compounds/day | Hit compounds with activity readout (e.g., IC50). | Orthogonal assays to confirm target engagement; cheminformatic triage. | Assay interference from chemical reactivity, aggregation, autofluorescence [10]. |
| Pharmacotranscriptomics (PTDS) [11] | Moderate (depends on sequencing scale) | Genome-wide expression profiles; pathway modulation signatures. | Independent cohort validation; connection to phenotypic endpoints. | Confounding by off-target cellular effects; requires careful model training. |
| Structure-Based Virtual Screening (SBVS) [12] | Millions of compounds in silico | Ranked list of predicted binders; binding poses. | Experimental affinity testing (e.g., SPR, ITC); benchmark on diverse "Core Sets" [12]. | Scoring function biases; overfitting on benchmark datasets; poor synthesizability of hits [12]. |
Detailed Experimental Protocols for Critical Validation Steps
Protocol for Genome Mining-Driven Discovery and Validation of Metallophores
This protocol, based on the automated detection of non-ribosomal peptide (NRP) metallophore biosynthetic gene clusters (BGCs), outlines a complete cycle from in silico prediction to chemical and functional validation [5].
- Step 1: Automated BGC Detection. Input bacterial genome assemblies (e.g., FASTA files) into the antiSMASH software (version 7.1+) with the integrated NRP metallophore detection module enabled [5]. The algorithm scans for specific chelator biosynthesis genes (e.g., for catechols, hydroxamates, salicylates) within NRPS clusters.
- Step 2: In Silico Prioritization. Filter results based on BGC novelty, cluster completeness, and taxonomic origin. Prioritize BGCs from understudied bacterial lineages (e.g., certain myxobacteria or cyanobacteria) to maximize discovery potential [5].
- Step 3: Cultivation and Compound Induction. Culture the source bacterium under trace metal-limiting conditions (e.g., using iron-depleted media) to induce metallophore production. Perform small-scale (e.g., 50 mL) and large-scale (e.g., 10 L) fermentations.
- Step 4: Chemical Dereplication and Isolation. Concentrate culture supernatants via solid-phase extraction. Employ liquid chromatography-high-resolution mass spectrometry (LC-HRMS) to dereplicate by comparing observed masses and fragmentation patterns to databases (e.g., GNPS). Ispute pure compounds using guided preparative HPLC.
- Step 5: Structural Elucidation & Cross-Validation. Determine the 2D structure of the isolated metallophore using nuclear magnetic resonance (NMR) spectroscopy (¹H, ¹³C, 2D experiments). This is the critical cross-validation step: Compare the elucidated structure to the one predicted from the BGC analysis (e.g., predicted amino acid sequence, chelator type). Discrepancies require re-examination of gene function annotations.
- Step 6: Functional Validation. Confirm metal-chelating activity and specificity using a chrome azurol S (CAS) assay and growth promotion assays in metal-deficient media supplemented with the purified compound.
Protocol for Cross-Validating Predicted 3D Structures of Natural Products
This protocol validates the output of AI-based 3D structure predictors like NatGen, which is essential for downstream structure-based design [8].
- Step 1: Input Preparation. Compile a set of natural product 2D structures (SMILES strings) with unknown or uncertain stereochemistry. These can be sourced from repositories like COCONUT.
- Step 2: 3D Conformation Generation. Process the 2D inputs using the NatGen framework. The model will generate predictions for chiral centers and output low-energy 3D conformations in a standard format (e.g., SDF).
- Step 3: In Silico Benchmarking (For Known Structures). For NPs with experimentally solved crystal structures (e.g., from the Cambridge Structural Database), calculate the root-mean-square deviation (RMSD) between the predicted and experimental atomic coordinates to quantify accuracy.
- Step 4: Experimental Cross-Validation (For Unknowns). For novel NPs, the predicted 3D structure must guide experimental validation. Use the predicted conformation for computational studies like molecular docking to a putative target. The critical step is to use the prediction to inform the design of a synthetic route to the proposed stereoisomer or to analyze spectroscopic data (e.g., computational NMR chemical shift prediction vs. experimental data, or prediction of optical rotation).
- Step 5: Prospective Validation Loop. The ultimate validation is the successful total synthesis of the predicted stereostructure and confirmation of its identity and bioactivity, closing the loop between genome-based prediction, AI-based structural hypothesis, and chemical reality [8].
Protocol for Metagenomic BGC Discovery with Cross-Cohort Validation
This protocol validates the disease relevance of BGCs recovered from uncultivated microbes, as demonstrated in colorectal cancer (CRC) microbiome studies [9].
- Step 1: Cohort Selection and Metagenomic Co-assembly. Select metagenomic sequencing datasets from case and control cohorts (e.g., CRC patients vs. healthy individuals). Perform de novo co-assembly of reads within each cohort using assemblers like MEGAHIT or metaSPAdes to create a pooled set of longer scaffolds [9].
- Step 2: Genome Binning and Quality Control. Cluster scaffolds into metagenome-assembled genomes (MAGs) using binning tools (e.g., MetaBAT2, MaxBin2). Retain only medium- to high-quality MAGs (completeness >50%, contamination <10%) as per MIMAG standards [9].
- Step 3: BGC Mining and Abundance Profiling. Run antiSMASH on the recovered MAGs to identify BGCs. Map raw sequencing reads from each individual sample back to the BGC-containing scaffolds to generate abundance profiles.
- Step 4: Statistical Association & Model Building. Use machine learning (e.g., Random Forest) to identify BGCs whose abundances are most important for classifying cases vs. controls. Calculate feature importance scores.
- Step 5: Cross-Cohort Validation. This is the essential validation step. Take the top predictive BGCs identified in one cohort (e.g., Asian cohort) and test their predictive power on a completely independent cohort (e.g., Caucasian cohort). A lack of overlap in specific "important" BGCs may indicate population-specific signatures, while validated overlap strengthens general biological relevance [9].
- Step 6: In Vitro Functional Testing. Heterologously express high-priority, novel BGCs in a model host (e.g., Streptomyces coelicolor) to isolate the encoded compound and test its bioactivity in relevant disease models.
Visualizing Workflows and Validation Frameworks
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for NP Discovery and Validation
| Category | Item/Reagent | Primary Function in Validation | Key Consideration |
|---|---|---|---|
| Bioinformatics & Genomics | antiSMASH Software Suite [5] | Standardized detection & annotation of BGCs; enables reproducible mining. | Must be used with latest rule sets (e.g., for metallophores) and updated databases. |
| Genome Taxonomy Database Toolkit (GTDB-tk) [9] | Consistent taxonomic classification of MAGs; essential for comparative ecology. | Critical for identifying novel taxa harboring uncharacterized BGCs. | |
| Analytical Chemistry | Chrome Azurol S (CAS) Assay Solution | Universal, colorimetric detection of siderophore and metallophore activity. | Serves as a rapid functional validation for iron-chelating BGC predictions [5]. |
| NMR Solvents (e.g., DMSO-d⁶, CDCl₃) & Internal Standards (TMS) | Solubilize NPs and provide a reference for structural elucidation via NMR. | Purity and isotopic enrichment are critical for obtaining high-resolution spectra. | |
| Molecular Biology | Heterologous Expression Kits (e.g., for S. coelicolor or E. coli) | Express BGCs from uncultivable hosts to isolate and characterize the encoded compound. | Choice of host and vector must be compatible with BGC size and genetic requirements. |
| Screening & Assays | Validated Target Protein & Biochemical Assay Kit | Confirm target engagement for hits from virtual or HTS campaigns. | Use orthogonal assay formats (e.g., fluorescence + SPR) to rule out artifactural inhibition [10] [12]. |
| Cell-based Phenotypic Assay Reagents | Confirm biological activity in a more physiologically relevant context. | Links target-based screening to cellular function; essential for mechanistic studies [11]. |
The contemporary paradigm of natural product discovery has shifted from traditional activity-guided isolation to a data-driven hypothesis-generating approach. This transition is anchored in the cross-validation of genomic potential with chemical evidence, forming the core thesis of modern research. Genome mining predicts the biosynthetic capacity of an organism, while mass spectrometry-based dereplication identifies the actual molecules produced. The convergence of these lines of evidence—verifying that predicted gene clusters (BGCs) yield detected metabolites—is critical for prioritizing novel bioactive compounds and accelerating drug discovery. This guide objectively compares the key enablers of this workflow: the foundational databases MIBiG and GNPS, and the essential bioinformatics tools antiSMASH and DEREPLICATOR+.
Comparative Analysis of Core Databases and Tools
The efficacy of the genome mining-dereplication cycle depends on the performance and integration of specialized resources. The following tables provide a quantitative and functional comparison of these core enablers.
Table 1: Comparison of Foundational Databases for Cross-Validation
| Feature | MIBiG (Minimum Information about a Biosynthetic Gene cluster) | GNPS (Global Natural Products Social Molecular Networking) |
|---|---|---|
| Primary Purpose | Repository of experimentally validated Biosynthetic Gene Clusters (BGCs) for genome mining reference and training [13]. | Public repository and ecosystem for organizing, sharing, and analyzing tandem mass spectrometry (MS/MS) data [14]. |
| Key Content | Curated BGC entries with gene annotations, compound structures, and bioactivities. Version 3.0 contains 2,692 entries [13]. | Crowd-sourced mass spectral libraries and raw data from thousands of studies, encompassing billions of mass spectra [14] [15]. |
| Role in Cross-Validation | Provides the "genomic blueprint" standard for comparing newly identified BGCs from antiSMASH, helping prioritize novel clusters [13] [16]. | Provides the "chemical evidence" for dereplication. Serves as the primary data source for tools like DEREPLICATOR+ to identify known compounds [14]. |
| Critical Metrics | - 2,692 curated BGC entries (v3.0) [13]. - 1,188 entries with cross-linked chemical structures [13]. - 1,002 entries with annotated bioactivities [13]. | - Billions of mass spectra archived [15]. - >98% of spectra represent "dark matter" (unknown compounds) [15]. - Enables identification of five times more molecules than previous approaches with DEREPLICATOR+ [14]. |
Table 2: Comparison of Essential Bioinformatics Tools
| Feature | antiSMASH (antibiotics & Secondary Metabolite Analysis Shell) | DEREPLICATOR+ |
|---|---|---|
| Primary Function | Detects and annotates Biosynthetic Gene Clusters (BGCs) in genomic data [16]. | Identifies known natural products from tandem mass spectrometry data by searching against structure databases [14]. |
| Core Algorithm | Rule-based system using profile Hidden Markov Models (pHMMs) to identify signature biosynthetic enzymes [16]. | Fragmentation graph algorithm that matches experimental spectra to in-silico fragmented chemical structures [14]. |
| Scope & Coverage | Detects 81 different types of BGCs (as of v7.0) in bacterial, fungal, and plant genomes [16]. | Dereplicates peptides, polyketides, terpenes, benzenoids, alkaloids, flavonoids, and more [14]. |
| Key Performance | Identified an average of 34 BGCs per genome in a study of 187 Alternaria fungi genomes [17]. | Identified 488 unique compounds (at 1% FDR) in Actinobacterial spectra, a twofold increase over its predecessor and with more spectra per compound [14]. |
| Integration Role | Input for MIBiG: Newly characterized BGCs can be submitted to MIBiG [13]. Input for Dereplication: Predicts potential product structures for targeted MS analysis. | Input from GNPS: Searches GNPS's massive spectral repository [14]. Validation for Mining: Confirms the production of metabolites from predicted BGCs. |
Detailed Experimental Methodologies
The validation of integrated workflows relies on standardized experimental protocols. Below are detailed methodologies for key experiments that generate data for tools like antiSMASH and DEREPLICATOR+.
Protocol for Genome Sequencing and BGC Mining (as applied inAlternariastudies)
This protocol outlines the steps for obtaining genomic data and mining it for biosynthetic potential, as described in large-scale fungal studies [17].
- DNA Extraction & Sequencing: Extract high-quality genomic DNA from pure microbial or fungal culture. Utilize Illumina short-read sequencing platforms (e.g., NextSeq500). Process raw reads with Trimmomatic (v0.38) to remove adapters and low-quality bases [17].
- Genome Assembly: Perform de novo assembly of quality-filtered reads using SPAdes (v3.12) or a similar assembler [17].
- Gene Prediction: Employ a standardized pipeline (e.g., funannotate v1.8.7) on all assemblies to ensure consistent gene model prediction and functional annotation [17].
- BGC Detection with antiSMASH: Submit the assembled genome sequence (FASTA format) to the antiSMASH web server (v7.0) or run the standalone tool. Use default parameters to detect known cluster types (e.g., polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS)) [16].
- BGC Analysis & Prioritization: Compare antiSMASH-predicted BGCs against the MIBiG reference database using the built-in KnownClusterBlast function. Group BGCs into Gene Cluster Families (GCFs) using tools like BiG-SCAPE to visualize biosynthetic potential across strains [17].
Protocol for Metabolite Profiling and Dereplication via GNPS/DEREPLICATOR+
This protocol describes the generation and analysis of mass spectrometry data for dereplication, forming the chemical validation pillar [14] [15].
- Sample Preparation & LC-MS/MS: Extract metabolites from microbial culture or environmental sample using appropriate solvents (e.g., methanol/ethyl acetate). Perform reversed-phase liquid chromatography (LC) coupled to a high-resolution tandem mass spectrometer (HR-MS/MS).
- Data Preprocessing: Convert raw MS files to open formats (e.g., .mzML). Use computational tools (e.g., MZmine) for peak picking, alignment, and deconvolution to create a list of precursor ions and associated MS/MS spectra.
- Data Submission to GNPS: Upload the processed MS/MS data file to the GNPS platform. Annotate the dataset with critical metadata (strain, growth conditions, etc.) [14].
- Molecular Networking: Use the GNPS molecular networking workflow to cluster similar MS/MS spectra. This visualizes chemical relatedness and groups analogs of the same molecular family [15].
- Dereplication with DEREPLICATOR+: Within the GNPS environment, select the DEREPLICATOR+ workflow for dereplication. The tool will automatically search all spectra against its integrated databases (e.g., AntiMarin, DNP) and report metabolite-spectrum matches (MSMs) with a statistical score and False Discovery Rate (FDR) estimation [14].
- Result Validation: Manually inspect high-scoring matches, checking for consistency of fragmentation patterns. Use the molecular network to propagate identifications to related, unknown spectra within the same cluster.
Visualizing the Integrated Workflow
The cross-validation of genome mining and dereplication is a multi-step, iterative process. The following diagram illustrates the logical workflow and data flow between the key enablers.
Cross-Validation of Genome Mining and Dereplication Workflow
Detailed Algorithmic Pathways
Understanding the internal logic of the core bioinformatics tools is key to interpreting their results. The following diagrams detail the primary algorithmic pathways for antiSMASH and DEREPLICATOR+.
Table 3: The Scientist's Toolkit: Essential Research Reagents & Resources
| Item Category | Specific Item/Resource | Function in Cross-Validation Workflow |
|---|---|---|
| Sequencing & Genomics | Illumina NextSeq500 / NovaSeq Platforms | Provides high-throughput, short-read genomic DNA sequencing for BGC discovery [17]. |
| SPAdes Assembler | Performs de novo genome assembly from short reads, constructing contiguous sequences (contigs) for mining [17]. | |
| Funannotate Pipeline | Standardizes gene prediction and functional annotation across diverse genomes, ensuring consistent input for antiSMASH [17]. | |
| Mass Spectrometry | High-Resolution LC-MS/MS System (e.g., Q-TOF, Orbitrap) | Generates high-quality tandem mass spectra with accurate mass measurements, essential for database matching [14]. |
| Solvent Systems (e.g., Methanol, Ethyl Acetate) | Used for comprehensive extraction of secondary metabolites from microbial cultures or environmental samples. | |
| Software & Databases | antiSMASH (v7.0) | The primary tool for detecting and annotating biosynthetic gene clusters in genomic data [16]. |
| DEREPLICATOR+ | Advanced algorithm for identifying known natural products from MS/MS spectra against chemical structure databases [14]. | |
| MIBiG Database (v3.0) | Curated reference database of known BGCs used to assess novelty and predict function [13]. | |
| GNPS Platform | Central repository and analysis suite for mass spectrometry data, enabling dereplication and molecular networking [14] [15]. | |
| Specialized Tools | BiG-SCAPE / BiG-SLiCE | Tools for comparing and networking BGCs, identifying gene cluster families across genomes [17] [15]. |
| HypoRiPPAtlas | Database of hypothetical RiPP structures predicted from genomes, used as a custom target for DEREPLICATOR+ searches [15]. |
antiSMASH BGC Detection Algorithm Pathway
DEREPLICATOR+ Dereplication Algorithm Pathway
The discovery of microbial natural products has transitioned from a serendipitous, phenotype-driven endeavor to a data-driven, targeted deep-mining operation [18]. This paradigm shift is central to a broader thesis on the cross-validation of genome mining with dereplication results, a process essential for linking predicted biosynthetic potential with actual chemical output. Historically, only a fraction of a microbe's biosynthetic gene clusters (BGCs) are expressed under standard conditions, leaving a vast reservoir of "silent" or "cryptic" clusters undiscovered [18]. Modern discovery pipelines now integrate genomics, metabolomics, and advanced bioinformatics to systematically bridge this gap. These integrated strategies have led to the discovery of 185 novel microbial natural products between 2018 and 2024, demonstrating the efficacy of moving from genomic prediction to metabolomic confirmation [18]. This guide objectively compares the core technologies and methodologies underpinning this modern pipeline, providing researchers with a framework for validating genomic predictions with experimental metabolomic data.
Comparative Analysis of Core Discovery Platforms and Performance
The contemporary discovery landscape is defined by synergistic platforms that combine genomic prediction, metabolomic analysis, and intelligent prioritization. The following table compares the key technological approaches, their primary functions, and their role in the cross-validation workflow.
Table 1: Comparison of Core Technologies in the Integrated Discovery Pipeline
| Technology Category | Representative Tools/Platforms | Primary Function | Role in Cross-Validation |
|---|---|---|---|
| Genome Mining & BGC Prediction | antiSMASH 7.0, DeepBGC, PRISM 4, RIPP | Predicts and annotates biosynthetic gene clusters from genomic data. | Generates hypotheses about chemical potential; identifies targets for metabolomic search. |
| Metabolomics & Dereplication | GNPS (Global Natural Products Social), SIRIUS, MS-DIAL | Analyzes mass spectrometry data to identify known compounds and highlight novel features. | Provides experimental evidence to confirm or refute genomic predictions; prevents rediscovery. |
| Multi-Omics Integration | Feature-Based Molecular Networking (FBMN), SPECO, MSSN | Correlates genomic clusters with metabolomic features through data integration. | Directly links a predicted BGC to its observable metabolic product, closing the discovery loop. |
| AI & Machine Learning Platforms | Exscientia (Generative Chemistry), Insilico Medicine (Target Discovery) | Accelerates compound design and prioritization using predictive models. | Enhances prediction accuracy for BGC products and properties, informing validation strategies [19]. |
The performance of these platforms is quantified by their output and efficiency. A landmark study utilizing an integrated bioinformatics pipeline—combining multilayer sequence similarity network (MSSN), short peptide and enzyme co-localization (SPECO) analysis, and AlphaFold-Multimer—successfully identified 1,057 P450-modified RiPPs gene clusters from 20,399 actinomycete genomes [18]. This led to the heterologous expression and characterization of nine new macrocyclic peptides, validating the predictive power of the integrated approach [18]. Compared to traditional single-tool analyses, strategies combining tools like PRISM and ClusterFinder have increased structural diversity coverage by 40% [18].
Table 2: Quantitative Performance Metrics of Discovery Strategies (2018-2024)
| Performance Metric | Traditional Isolation | Genome Mining Only | Integrated Multi-Omics Pipeline | Data Source |
|---|---|---|---|---|
| Novel Compounds Discovered | Low (High Rediscovery) | Medium (Theoretical) | High (185 compounds reported) | [18] |
| BGC Product Linkage Rate | Not Applicable | Low (~25%) | High (Validated by design) | [18] |
| Annotation Accuracy for Unknowns | N/A | N/A | Up to 65% higher than database-only | [18] |
| Discovery Timeline (Target to Validation) | 3-5 years | 1-2 years (for expression) | <1 year (streamlined workflow) | [19] [18] |
Experimental Protocols for Cross-Validation
The core thesis of cross-validating genome mining with dereplication is operationalized through specific experimental protocols. These methodologies ensure that a predicted "silent cluster" is conclusively linked to a "known spectrum" or a novel compound.
Protocol 1: Integrated Genomics-Metabolomics for Novel RiPP Discovery
This protocol details the workflow for discovering novel ribosomally synthesized and post-translationally modified peptides (RiPPs) [18].
- Genome Sequencing & Assembly: Obtain high-quality genome sequences using long-read technologies (e.g., PacBio HiFi) for complete BGC assembly [18].
- In silico BGC Prediction: Use RiPP-specific tools (e.g., RiPPER, antiSMASH) to identify precursor peptides and associated modification enzymes (e.g., P450s) [18].
- Bioinformatic Prioritization:
- Perform BlastP and EFI-EST analysis to build sequence similarity networks (SSNs) of target enzyme families [18].
- Apply SPECO (short peptide and enzyme co-localization) to identify genomic loci where precursor peptides and modifying enzymes are co-localized [18].
- Use AlphaFold-Multimer to predict physical interactions between precursor peptides and enzyme pockets, filtering for genuine biosynthetic pairs [18].
- Metabolomic Analysis:
- Culture the native or a heterologous expression host (e.g., S. albus).
- Acquire high-resolution LC-MS/MS data (using Orbitrap or FT-ICR systems).
- Process data with GNPS and SIRIUS to create molecular networks and predict molecular formulas for metabolites.
- Cross-Validation & Identification:
- Compare the accurate mass and fragmentation pattern of observed metabolites with the in silico predicted mass of the RiPP product from the prioritized BGC.
- Isolate the target compound and use advanced NMR (e.g., cryogenic probes with 2D experiments like COSY, HSQC) for full structural elucidation [18].
- Confirm bioactivity through targeted assays.
Protocol 2: AI-Enhanced Prioritization & Validation
This protocol incorporates AI platforms to accelerate the prioritization of BGCs or compound designs for experimental validation [19].
- Target/BGC Identification: Use AI-driven knowledge graphs (e.g., BenevolentAI) or generative models to identify novel disease targets or prioritize cryptic BGCs based on predicted bioactivity or chemical novelty [19].
- In silico Compound Design: For a selected target, employ generative chemistry platforms (e.g., Exscientia's Centaur Chemist) to design novel small molecules with optimized properties. These platforms can compress design cycles by ~70% and require 10x fewer synthesized compounds [19].
- Predictive Metabolomics: Apply machine learning tools (e.g., DeepMass) to predict MS/MS spectra of AI-designed compounds or predicted natural products [18].
- Experimental Testing & Loop Closure:
- Synthesize top-ranking compounds or heterologously express the prioritized BGC.
- Acquire experimental LC-MS/MS data and compare it to the AI-predicted spectra.
- Use the experimental results (bioactivity, ADME data) to retrain and refine the AI models, creating a closed Design-Make-Test-Analyze loop [19].
Critical Methodological Note: Evaluation Metrics for Predictive Models
When developing or using machine learning models for BGC prediction or spectrum forecasting, the choice of evaluation metric is critical. For binary classification tasks (e.g., BGC vs. non-BGC, active vs. inactive), researchers must avoid misleading metrics like accuracy and F1 score, which perform poorly on imbalanced datasets common in biological discovery [20]. Instead, the Matthews Correlation Coefficient (MCC) should be employed, as it provides a more reliable and informative measure of model quality by considering all four confusion matrix categories (true positives, false positives, true negatives, false negatives) [20]. Furthermore, for model validation, repeated hold-out validation (e.g., performing 1000 random 80/20 train/test splits) is recommended over simple k-fold cross-validation. This approach provides more universal and generalizable performance estimates than a single arbitrary data partition [20].
The Integrated Discovery Pipeline: A Systems View
The following diagram illustrates the complete workflow for moving from a silent genomic cluster to a known metabolomic spectrum, integrating the technologies and protocols described above.
Diagram 1: The Integrated Genome-to-Metabolome Discovery Pipeline. This workflow visualizes the systematic process from genomic prediction to experimental cross-validation, culminating in a confirmed link between a biosynthetic gene cluster and its metabolic product.
The Scientist's Toolkit: Essential Research Reagents & Platforms
Successful execution of the cross-validation pipeline depends on access to specific computational tools, databases, and experimental resources.
Table 3: Essential Research Toolkit for Genome Mining & Dereplication
| Tool/Resource Name | Category | Primary Function & Role in Cross-Validation | Key Feature |
|---|---|---|---|
| antiSMASH 7.0+ | Genome Mining | Identifies and annotates BGCs in microbial genomes. The starting point for generating genomic hypotheses. | Integrates HMMs & AI; >40 annotatable BGC types [18]. |
| GNPS (Global Natural Products Social) | Metabolomics/Dereplication | Community MS/MS data repository and analysis platform for dereplication and molecular networking. | Enables feature-based molecular networking (FBMN) to find novel metabolites [18]. |
| SIRIUS | Metabolomics | Predicts molecular formulas and structures from MS/MS data using fragmentation trees. | Crucial for annotating unknowns not found in libraries [18]. |
| AlphaFold-Multimer | Bioinformatics | Predicts 3D structures of protein complexes (e.g., enzyme-precursor peptide). | Validates physical interaction in BGCs, prioritizing clusters for expression [18]. |
| EFI-EST & EFI-GNT | Bioinformatics | Generates Sequence Similarity Networks (SSNs) and Genome Neighborhood Networks. | Visualizes relationships within enzyme families to identify novel variants [18]. |
| Cryogenic NMR Probes (600 MHz+) | Structure Elucidation | Provides high-sensitivity NMR data for structural determination of trace novel compounds. | Sensitivity increased by ~30%, enabling stereochemistry solving of microgram quantities [18]. |
| PacBio HiFi Sequencing | Genomics | Produces highly accurate long reads for complete, gap-free genome assemblies. | Essential for capturing entire, often large, BGCs in single contigs [18]. |
The Critical Cross-Validation Workflow
The final and most critical conceptual diagram details the decision logic of the cross-validation process itself, where genomic prediction and metabolomic evidence converge.
Diagram 2: The Cross-Validation Decision Logic for BGC-Metabolite Linking. This flowchart outlines the critical decision points in experimentally validating whether a predicted biosynthetic gene cluster produces a known or novel metabolite, ensuring rigorous and efficient discovery.
Building the Integrated Pipeline: A Step-by-Step Workflow for Combined Analysis
The quest for novel bioactive natural products has entered a transformative phase, moving beyond random screening to precision-guided discovery. Genome mining represents the cornerstone of this shift, enabling researchers to decipher the genetic blueprints—Biosynthetic Gene Clusters (BGCs)—that encode for specialized metabolites directly from microbial genomes [3]. However, the sheer scale of genomic data presents a new challenge: predicting which of the thousands of detected BGCs are both novel and capable of producing bioactive compounds [21]. This is where the principle of cross-validation with dereplication becomes critical. By integrating genomic predictions with experimental metabolomic data, researchers can prioritize BGCs most likely to yield novel chemistry, thereby accelerating the discovery pipeline and mitigating the high rate of compound rediscovery [22].
This guide provides a comparative analysis of current methodologies for targeted genome mining, situating them within a broader research thesis that emphasizes the validation of in silico predictions with high-resolution mass spectrometry and dereplication strategies. We evaluate the performance of integrated approaches against standalone techniques, presenting experimental data and protocols to inform the strategies of researchers and drug development professionals [23].
Comparative Landscape of Genome Mining and Dereplication Tools
The efficacy of a discovery pipeline hinges on the selection and integration of computational and experimental tools. The table below provides a comparative overview of core methodologies, highlighting their primary functions, strengths, and suitability for cross-validation workflows.
Table 1: Comparison of Core Methodologies for Targeted Genome Mining and Dereplication
| Methodology Category | Representative Tool/Approach | Primary Function | Key Strength | Limitation for Cross-Validation |
|---|---|---|---|---|
| BGC Prediction & Analysis | antiSMASH [24] [21] [25] | Identifies and annotates BGCs in genomic data. | Comprehensive; supports multiple BGC classes; user-friendly. | Predicts potential, not expressed metabolites; high false-positive rate for novelty. |
| Comparative Genomics | EDGAR, BPGA Pan-genome Analysis [24] [25] | Identifies unique genomic regions (e.g., BGCs) by comparing multiple genomes. | Pattern-independent; highlights strain-specific adaptations. | Requires multiple high-quality genomes; does not confirm bioactive production. |
| Spectral Dereplication | DEREPLICATOR+ [23], GNPS Molecular Networking [22] | Identifies known metabolites in MS/MS data by searching spectral libraries. | Rapidly filters out known compounds; high-throughput. | Limited to known compounds in libraries; struggles with novel scaffold families. |
| Integrated Genomic & Metabolomic Validation | Peptidogenomics/Genome-Guided Discovery [22] [25] | Links MS/MS spectra to predicted BGCs via in silico spectrum prediction. | Directly connects genotype to chemotype; validates BGC activity. | Computationally intensive; requires high-quality genome and metabolome. |
| Generative AI for Bioactive Design | TransPharmer (Pharmacophore-aware GPT) [26] | De novo generation of novel molecular structures constrained by bioactive features. | Enables scaffold hopping; designs novel structures beyond natural templates. | Generated structures require de novo synthesis and functional validation. |
Cross-Validation Strategies: Integrating Genomics with Metabolomics
The most promising strategies for novel discovery involve converging evidence from independent genomic and metabolomic analyses. The following experimental protocols detail two high-yield approaches.
Protocol 1: Integrated Genome Mining and Comparative Genomics
This protocol uses a subtractive, pattern-independent strategy to pinpoint BGCs uniquely associated with a bioactive strain [25].
- Genome Sequencing and Assembly: For the bioactive producer strain and closely related, non-producing strains, perform whole-genome sequencing. Assemble reads using a tool like SPAdes [24]. Assess quality with QUAST [3].
- Uniform Gene Prediction & Annotation: Process all assemblies through a unified pipeline (e.g., funannotate for fungi [3] or PGAP/RAST for bacteria [24]) to ensure comparable gene calls.
- Primary BGC Candidate List: Analyze the producer strain's genome with antiSMASH to generate a list of all predicted BGCs [21] [25].
- Unique Region Identification: Conduct a pan-genome or comparative genomic analysis (using tools like EDGAR or BPGA) to identify all genes and genomic regions present in the producer but absent in the non-producers [24] [25].
- Candidate Intersection: Cross-reference the antiSMASH candidate list with the list of unique genomic regions. BGCs appearing on both lists represent high-priority targets likely responsible for the unique bioactivity [25].
- Functional Genetic Validation: Use site-directed mutagenesis (e.g., gene knockout) on key genes within the prioritized BGC. A significant reduction or loss of the original bioactivity in the mutant confirms the BGC's role [25].
Protocol 2: Genome-Guided Dereplication and Peptidogenomics
This protocol directly links mass spectrometry data to genomic predictions, validating BGC expression and identifying novel metabolites [22] [23].
- Metabolite Extraction and MS/MS Analysis: Culture the producing organism under conditions that elicit bioactivity. Prepare crude extracts and analyze using LC-MS/MS with data-dependent acquisition [23].
- Dereplication via Spectral Networking: Process MS/MS data through the Global Natural Products Social (GNPS) platform. Use molecular networking to cluster related spectra and DEREPLICATOR+ to automatically annotate nodes corresponding to known natural products [22] [23].
- In Silico Spectrum Prediction from BGCs: Using the sequenced genome, predict BGCs with antiSMASH. For RiPP or NRP BGCs, use tools like NRPSpredictor2 or RiPP prediction modules to predict the core peptide structure [22]. Generate theoretical MS/MS fragmentation spectra for predicted structures.
- Cross-Validation Match: Search the unannotated, putatively novel nodes in the molecular network against the database of in silico predicted spectra. A high-confidence match validates that the BGC is expressed and pinpoints its specific chemical product [23].
- Isolation and Structure Elucidation: Target the fermentation and purification of the metabolite linked to the validated BGC for full structural characterization via NMR.
Performance Analysis: Quantitative Outcomes of Integrated Approaches
The success of integrated strategies is evidenced by quantitative improvements in discovery rates and prioritization efficiency, as shown in the following data from recent studies.
Table 2: Experimental Output and Efficiency of Discovery Workflows
| Study & Organism | Methodology | Key Quantitative Outcome | Impact on Novelty & Prioritization |
|---|---|---|---|
| Alternaria spp. (123 genomes) [3] | Large-scale antiSMASH mining & GCF analysis. | Identified 6,323 BGCs, grouped into 548 Gene Cluster Families (GCFs). 9 unique GCFs in divergent sections identified as ideal diagnostic markers. | Enabled taxonomic prioritization; revealed that the alternariol mycotoxin GCF is restricted to specific sections, guiding food safety monitoring. |
| Xenorhabdus/Photorhabdus spp. (13 genomes) [21] | antiSMASH + BiG-SCAPE similarity networking. | Identified 178 putative BGCs; network analysis showed 146 similar to known BGCs and 22 orphan clusters. | Clearly differentiated known from potential novelty; orphan clusters (e.g., novel NRPS/T1PKS) are prime targets for heterologous expression. |
| Actinomyces Spectra Analysis [23] | Dereplication with DEREPLICATOR+. | At 0% FDR, identified 154 compounds (8194 MS matches), a 2-fold increase over prior tools. Uncovered 10 metabolites (PKs, terpenes) missed by peptide-specific tools. | Dramatically improved dereplication throughput and accuracy, efficiently clearing known compounds to reveal novel chemical space. |
| Pantoea agglomerans [25] | Integrated antiSMASH + Comparative Genomics (EDGAR) + Mutagenesis. | antiSMASH listed 24 candidates; comparative genomics narrowed to a single 14-kb unique BGC. Knockout confirmed its role in antibiotic production. | Reduced candidate list from 24 to 1, demonstrating extreme prioritization efficiency and direct functional validation. |
Visualizing the Integrated Discovery Workflow
The following diagram illustrates the logical flow and decision points of a cross-validated genome mining and dereplication pipeline, integrating the protocols and concepts described above.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful execution of the described workflows relies on a suite of specialized bioinformatics tools and experimental resources.
Table 3: Essential Research Toolkit for Targeted Genome Mining and Dereplication
| Tool/Resource Name | Category | Primary Function in Workflow | Key Application Note |
|---|---|---|---|
| antiSMASH | Bioinformatics | Core BGC detection and annotation from genome assemblies [24] [21] [25]. | The standard first-pass tool; configure to run "known cluster blast" for initial dereplication. |
| BiG-SCAPE/CORASON | Bioinformatics | Constructs similarity networks of BGCs to group them into families (GCFs) [21]. | Critical for assessing BGC novelty at a sequence level and prioritizing orphan clusters. |
| funannotate | Bioinformatics | Unified pipeline for fungal genome annotation, essential for consistent gene calls [3]. | Use to re-annotate public genomes for fair comparative analysis. |
| GNPS & DEREPLICATOR+ | Mass Spectrometry | Cloud platform for MS/MS data analysis, molecular networking, and automated dereplication [22] [23]. | DEREPLICATOR+ significantly expands identifiable compound classes compared to earlier tools. |
| NRPSpredictor2 / RiPP modules | Bioinformatics | Predicts substrate specificity of NRPS adenylation domains or core peptide sequences for RiPPs [22]. | Generates predicted chemical structures for in silico spectrum matching in peptidogenomics. |
| SPAdes | Bioinformatics | Genome assembly from Illumina and other NGS reads [3] [24]. | Use in careful combination with quality assessment tools (QUAST) to ensure assembly fidelity. |
| TransPharmer | Generative AI | Generates novel molecular structures guided by pharmacophore fingerprints [26]. | Useful for scaffold hopping and designing synthetic analogs inspired by natural product hits. |
In the field of natural product discovery, the critical challenge of dereplication—the rapid identification of known compounds within complex extracts—has been transformed by computational mass spectrometry. This process is essential for avoiding the costly re-isolation of known molecules and for prioritizing novel chemical entities for drug development [27]. The integration of dereplication results with genome-mining predictions forms a powerful cross-validation framework. This synergy allows researchers to verify the functional output of biosynthetic gene clusters (BGCs) identified in microbial genomes with actual metabolite production, thereby bridging genomic potential with chemical reality [23]. Modern dereplication engines, particularly algorithmic approaches like DEREPLICATOR+, are central to this integrative strategy, enabling the high-throughput annotation of tandem mass spectrometry (MS/MS) data against vast databases of known natural products [23].
Comparative Performance Analysis of Dereplication Tools
The landscape of computational tools for annotating MS/MS data is diverse, ranging from spectral library search engines to in silico fragmentation algorithms. The following analysis compares the performance and scope of key tools, with a focus on DEREPLICATOR+ and its predecessors.
Table 1: Core Algorithmic Comparison of Dereplication Tools
| Tool | Primary Approach | Compound Classes Covered | Key Innovation | Reported Identification Increase vs. Predecessors |
|---|---|---|---|---|
| DEREPLICATOR+ [23] | Fragmentation graph matching & molecular networking | Peptides, polyketides, terpenes, benzenoids, alkaloids, flavonoids, lipids | Extended fragmentation model beyond peptides; integrated spectral networking | 5x more unique compounds than previous dereplication efforts in GNPS data [23] |
| DEREPLICATOR [27] | Theoretical spectrum generation for peptides via bond disconnection | Peptidic Natural Products (PNPs: NRPs & RiPPs) | First high-throughput PNP dereplicator with statistical validation (p-values, FDR) | Order of magnitude more PNPs identified in GNPS than prior efforts [27] |
| Classical Molecular Networking (GNPS) [28] | Cosine similarity-based clustering of MS/MS spectra | All, but requires library matches for annotation | Visual organization of related spectra into molecular families | Foundation for network-based discovery; enables variant discovery |
| SIRIUS [28] | Combinatorial fragmentation & isotope pattern analysis | Small molecules (typically < 500 Da) | CSI:FingerID for database searching using fragmentation trees | Increased metabolite identification rates fivefold over earlier approaches [23] |
The expansion from DEREPLICATOR to DEREPLICATOR+ represents a quantum leap in scope. While DEREPLICATOR was highly effective for peptidic natural products (PNPs), it was limited to this class [27]. DEREPLICATOR+ generalizes the underlying algorithm, enabling the identification of a much broader spectrum of natural product classes, including polyketides and terpenes, which are major sources of therapeutic agents [23]. This is evidenced by experimental data: when analyzing Actinomyces spectra at a stringent 0% False Discovery Rate (FDR), DEREPLICATOR+ identified 154 unique compounds, compared to 66 identified by DEREPLICATOR—a 2.3-fold increase [23]. Notably, among these identifications were critical compound classes that the original tool missed, including polyketides and terpenes [23].
Table 2: Experimental Performance Benchmark on Real-World Datasets
| Dataset (Source) | Number of Spectra | DEREPLICATOR+ Identifications (1% FDR) | Key Findings and Comparative Advantage |
|---|---|---|---|
| SpectraActiSeq (Actinomyces strains) [23] | 651,770 | 488 unique compounds (8,194 MSMs) | Identified chalcomycin and its variants; found 2.2x more spectra per compound on average than DEREPLICATOR. |
| SpectraGNPS (Global repository) [23] | ~248 million | Not explicitly totaled (applied to all spectra) | Enabled searching of the entire GNPS infrastructure; cornerstone for large-scale, crowd-sourced dereplication. |
| SpectraCyan (Cyanobacteria) [23] | ~11.9 million | Applied for cross-validation with genomes of 4 Moorea strains. | Directly linked MS/MS identifications to genomic potential in strains with sequenced genomes. |
Beyond pure identification counts, a critical metric is the biological verifiability of the results. In the SpectraActiSeq study, DEREPLICATOR+ identified 24 high-confidence metabolites (score threshold ≥15, 0% FDR). Strikingly, 17 out of these 24 (71%) were independently confirmed as being produced by Actinomyces species according to the AntiMarin database, demonstrating the tool's high precision and biological relevance [23].
Experimental Validation: Protocols for Cross-Validation with Genome Mining
The true power of dereplication is realized when its results are integrated with genomic data. The following protocol, derived from the validation of DEREPLICATOR+, outlines a robust framework for cross-validation.
Experimental Workflow for Integrated Genome Mining and Dereplication:
Genome Sequencing and BGC Prediction:
- Isolate genomic DNA from the microbial strain of interest.
- Perform whole-genome sequencing (e.g., Illumina, PacBio).
- Annotate the genome using tools like antiSMASH to identify and predict the classes of Biosynthetic Gene Clusters (BGCs) present (e.g., NRPS, PKS, terpene synthase) [23].
Metabolite Profiling via LC-MS/MS:
- Culture the microbe under various conditions to stimulate secondary metabolism.
- Extract metabolites using appropriate solvents (e.g., ethyl acetate for non-polar compounds, methanol/water for polar compounds).
- Analyze extracts using reversed-phase Liquid Chromatography coupled to high-resolution tandem Mass Spectrometry (LC-HRMS/MS) in Data-Dependent Acquisition (DDA) mode.
Computational Dereplication:
- Convert raw MS data to open formats (.mzML, .mzXML).
- Submit the MS/MS spectra to the DEREPLICATOR+ tool via the GNPS platform or standalone version.
- Search against comprehensive natural product databases (e.g., AntiMarin, Dictionary of Natural Products).
- Apply stringent statistical filters (e.g., 1% FDR, p-value < 10⁻⁷) to generate a list of high-confidence compound identifications [23].
Cross-Validation Analysis:
- Compare: Map the classes of compounds identified by DEREPLICATOR+ (e.g., non-ribosomal peptide, polyketide) against the classes of BGCs predicted by genome mining (e.g., NRPS, PKS).
- Corroborate: A strong correlation (e.g., identification of an NRPS-derived peptide when an NRPS cluster is present) validates both the genomic prediction and the MS/MS annotation.
- Discover: Compounds identified without a corresponding known BGC may indicate a novel or silent cluster. Conversely, a predicted BGC with no detected metabolite may be inactive under the tested conditions, guiding future cultivation experiments.
This workflow was successfully applied to cyanobacterial strains (Moorea spp.), where DEREPLICATOR+ annotations from the SpectraCyan dataset were directly cross-referenced with the genomes of four cultured strains, functionally validating the genomic potential of these organisms [23].
Visualizing the Integrated Workflow
The following diagram illustrates the logical workflow for cross-validating genome mining predictions with dereplication results, a core thesis of modern natural product discovery.
Diagram 1: Integrated Genome Mining & Dereplication Workflow
The dereplication process itself, as implemented by algorithms like DEREPLICATOR+, involves a sophisticated computational pipeline. The following diagram details its key steps from data input to statistically validated identifications.
Diagram 2: DEREPLICATOR+ Algorithmic Pipeline
Successful dereplication and cross-validation studies rely on a suite of databases, software platforms, and analytical standards. The following table details the essential components of this research toolkit.
Table 3: Research Toolkit for Dereplication and Cross-Validation Studies
| Tool/Resource | Type | Primary Function in Dereplication | Key Feature/Note |
|---|---|---|---|
| Global Natural Products Social (GNPS) [28] | Web Platform / Repository | Crowdsourced repository of MS/MS spectra; hosts dereplication tools (DEREPLICATOR+) and enables molecular networking. | Central hub for public MS/MS data analysis and community standards. |
| AntiMarin Database [23] [27] | Chemical Structure Database | Curated database of known microbial metabolites. Serves as a primary target database for dereplication searches. | Contains ~60,908 compounds; flags Actinomyces-origin compounds [23]. |
| Dictionary of Natural Products [23] | Chemical Structure Database | Comprehensive database of characterized natural products. Used to expand search space beyond microbial metabolites. | Contains over 250,000 compounds; provides broad chemical coverage [23]. |
| Molecular Networking [28] | Data Analysis Technique | Groups related MS/MS spectra based on similarity, enabling discovery of structural variants and propagation of annotations. | Foundational to the variable dereplication of novel variants of known compounds [27]. |
| High-Resolution LC-MS/MS System | Instrumentation | Generates the primary experimental data (MS/MS spectra) for dereplication. High mass accuracy is critical. | Required for data acquisition in DDA or DIA mode. |
| antiSMASH | Bioinformatics Software | Predicts Biosynthetic Gene Clusters (BGCs) from genomic data, providing the "genomic potential" for cross-validation. | Generates hypotheses about the types of compounds (NRPS, PKS, etc.) a strain can produce. |
| ClassyFire [23] | Bioinformatics Tool | Automatically classifies identified compounds into chemical ontology classes (e.g., benzenoid, lipid). | Used post-dereplication to analyze the chemical diversity of identified compounds [23]. |
The accelerating discovery of microbial biosynthetic potential through genome sequencing has created a critical bottleneck: the efficient prioritization of truly novel bioactive compounds from a sea of known entities and redundant genetic information. This challenge sits at the intersection of two complementary fields: genomic prediction, which uses statistical and machine learning models to forecast phenotypes or biosynthetic potential from genetic data, and dereplication, the process of rapidly identifying known compounds or genetic elements to focus resources on novelty. Framed within a broader thesis on the cross-validation of genome mining with dereplication results, this guide argues that strategic, bidirectional integration of these disciplines is not merely beneficial but essential for modern natural product discovery and microbial genomics. Isolating novel antibiotics from soil bacteria, for instance, requires integrating cultivation, bioactivity screening, mass spectrometry (MS) dereplication, and genomic analysis to confirm discoveries and uncover molecules missed by single methods [29].
This comparison guide objectively evaluates the tools, methodologies, and data frameworks that enable this integration. We provide experimental data and protocols to compare the performance of leading genomic prediction models and dereplication algorithms, demonstrating how their combined application validates findings, reduces false leads, and accelerates the path from genetic sequence to novel therapeutic agent.
Core Concepts and Definitions
- Genomic Prediction: A suite of statistical methods used to predict complex phenotypic traits, breeding values, or functional potential (e.g., antibiotic production) from dense genetic marker data or whole-genome sequences. Models range from parametric methods like GBLUP to non-parametric machine learning algorithms such as Random Forest and XGBoost [30] [31].
- Dereplication: In natural product discovery, dereplication uses analytical techniques (e.g., MS, NMR) and database matching to identify known compounds in bioactive extracts early in the pipeline [32] [33]. In genomics, it refers to computational methods for selecting a non-redundant, representative subset of genomes from a larger collection to avoid bias and reduce computational burden [34].
- Cross-Validation: A fundamental resampling technique used to assess the predictive performance and generalizability of genomic models. It involves partitioning data into training and validation sets iteratively to obtain robust accuracy estimates, which is crucial for model selection and tuning [35] [36].
- Strategic Integration: The bidirectional flow of information where genomic predictions guide dereplication priorities (e.g., prioritizing extracts from strains with unique biosynthetic gene clusters), and dereplication results validate and refine genomic models (e.g., using known compound identification to improve BGC product prediction algorithms).
Methodological Comparisons
Genomic Prediction Models: Performance and Applications
The choice of genomic prediction model significantly impacts the accuracy of trait forecasting. Performance varies based on trait heritability, genetic architecture, and dataset size.
Table 1: Comparison of Genomic Prediction Model Performance
| Model Category | Specific Model | Typical Use Case | Key Strength | Reported Accuracy (Range/Notes) | Computational Demand |
|---|---|---|---|---|---|
| Parametric | GBLUP / rrBLUP | Polygenic traits, additive genetic effects [35]. | Robust, simple, no hyperparameter tuning needed [31]. | Competitive across diverse traits [35] [31]. | Low to Moderate |
| Parametric (Bayesian) | BayesA, BayesB, BayesC | Traits with major loci or non-normal effect distributions [35]. | Flexible priors can model complex architectures. | Similar to GBLUP on many traits; excels with specific architectures [35]. | High |
| Semi-Parametric | RKHS (Reproducing Kernel Hilbert Spaces) | Modeling non-additive genetic effects [30]. | Captures complex, non-linear relationships. | Can outperform linear models for non-additive traits [30]. | Moderate to High |
| Non-Parametric (ML) | Random Forest (RF) | Complex traits, interaction effects [30] [31]. | Handles high-dimensional data, models interactions. | +0.014 mean accuracy gain over GBLUP in one benchmark [30]. | Moderate |
| Non-Parametric (ML) | XGBoost (XGB) | Large datasets with complex patterns [31]. | High predictive accuracy, efficient computation. | +0.025 mean accuracy gain over GBLUP [30]; fast fitting. | Low to Moderate (fitting) |
| Non-Parametric (ML) | Support Vector Machine (SVM) | Binary classification tasks (e.g., disease presence) [31]. | Effective in high-dimensional spaces. | Similar performance to GBLUP for binary traits in canines [31]. | High (large datasets) |
Note: Accuracy gains are context-dependent. Studies like [31] found no significant difference between GBLUP and ML models for several canine health traits, highlighting the importance of dataset-specific evaluation.
Dereplication Tools: Genomic and Metabolomic Approaches
Dereplication tools address redundancy at both the genetic and chemical levels.
Table 2: Comparison of Dereplication Tools and Strategies
| Tool/Strategy | Primary Domain | Core Methodology | Key Function | Advantage | Reference |
|---|---|---|---|---|---|
| skDER | Genomic Dereplication | Uses skani for efficient Average Nucleotide Identity (ANI) calculation, offers dynamic & greedy clustering [34]. | Selects representative genome subset from thousands based on ANI. | Scalable, reduces computational bias in downstream analyses. | [34] |
| CiDDER | Genomic Dereplication | Protein-cluster saturation; iteratively picks genomes covering unique protein space [34]. | Maximizes pangenome diversity with minimal genomes. | Protein-centric view ideal for functional diversity studies. | [34] |
| DAS Tool | Metagenomic Binning | Dereplication, aggregation, and scoring of bins from multiple algorithms [37]. | Integrates outputs of various binning tools to produce optimal genome set. | Recovers more high-quality genomes than any single tool. | [37] |
| MS/MS with GNPS | Metabolomic Dereplication | Tandem mass spectrometry data matched against spectral libraries [29] [32]. | Identifies known metabolites in complex extracts. | Rapid annotation, prioritizes extracts with novel spectra. | [29] |
| Regulation-Guided Mining | Functional Prioritization | Links Biosynthetic Gene Clusters (BGCs) to regulatory networks and co-expression data [6]. | Predicts BGC function and ecological role for prioritization. | Provides a third dimension (regulation) beyond sequence and chemistry. | [6] |
Integrated Workflows for Cross-Validation
The most powerful discovery pipelines create a closed loop where genomic and metabolomic data cross-validate each other.
Workflow Diagram: Integrated Genome Mining and Dereplication Pipeline
The following diagram illustrates the strategic, bidirectional integration of genomic prediction and dereplication within a discovery pipeline.
Protocol for Paired Cross-Validation in Integrated Studies
A robust cross-validation protocol is essential for testing the integrated model's ability to predict bioactivity from genomic data.
Table 3: Protocol for k-Fold Cross-Validation of an Integrated Genomic Prediction Model
| Step | Action | Purpose | Key Parameters & Notes |
|---|---|---|---|
| 1. Dataset Preparation | Compile data: Genomes (or BGC features), paired bioactivity outcomes (e.g., active/inactive, compound identity from dereplication). | Create linked genomic-phenotypic dataset. | Ensure each strain has both genomic data and a validated dereplication/activity label [29]. |
| 2. Stratified Partitioning | Randomly split strain dataset into k equal folds (e.g., k=5 or 10), maintaining class balance (active/inactive ratio). | Ensure each fold is representative of the whole dataset. | Prevents folds with no active examples. Use paired sampling as in [35]. |
| 3. Iterative Training & Validation | For each fold i: Use folds {1...k} except i as training set; fold i as validation set. | Assess model generalizability to unseen data. | Train integrated model (e.g., ML classifier on genomic features) on training set. |
| 4. Prediction & Comparison | Use trained model to predict bioactivity/compound class for validation strains. Compare predictions to dereplication-confirmed labels. | Measure predictive accuracy. | Metrics: Accuracy, Precision, Recall, AUC-ROC. Compare to a null model. |
| 5. Aggregate Results | Calculate average performance metrics across all k iterations. | Obtain robust estimate of model performance. | Provides mean and variance of accuracy, indicating stability [35] [36]. |
| 6. Model Refinement | Use results to adjust feature selection (e.g., BGC types), model architecture, or hyperparameters. | Optimize the final model. | Prevents overfitting to specific dataset partitions. |
The following diagram details this iterative validation cycle, which is central to refining the integrated system.
Experimental Protocols for Key Integration Steps
Protocol 1: Integrated Discovery from Environmental Samples
This protocol, adapted from a study recovering antibiotics from soil, exemplifies the physical workflow [29].
- In situ Cultivation: Use microbial diffusion chambers with semi-permeable membranes incubated in native soil to recover diverse, often uncultivable bacteria [29].
- High-Throughput Screening: Screen crude extracts from recovered isolates against target pathogens (e.g., MRSA, VRE).
- Mass Spectrometry Dereplication: Analyze active extracts via LC-MS/MS. Process data through the Global Natural Products Social Molecular Networking (GNPS) platform to match spectra against known compound libraries [29].
- Genomic Sequencing & Mining: Sequence genomes of active, dereplicated strains. Use tools like antiSMASH to identify Biosynthetic Gene Clusters (BGCs).
- Cross-Validation: For extracts with no GNPS match but confirmed bioactivity, examine BGCs for novelty. For extracts with a GNPS match, verify the presence of the corresponding known BGC. This step confirms the pipeline's accuracy and highlights cases where genomics detects clusters for compounds missed by MS (e.g., streptothricin in [29]).
- Model Building: Use data from steps 3-5 to train a classifier (e.g., Random Forest) that predicts bioactivity or compound class from genomic features (BGC types, regulatory genes).
Protocol 2: Genomic Dereplication for Representative Sampling
This protocol ensures downstream genomic analyses are efficient and unbiased [34].
- Input: A large collection (>1000) of genome assemblies for a target genus/species.
- ANI-based Dereplication (skDER): Calculate pairwise ANI using
skani. Run theskDER dynamicalgorithm with thresholds (e.g., ANI >99.5%, AF >90%) to select a representative set that minimizes redundancy while maintaining genetic breadth. - Protein-Cluster Dereplication (CiDDER): Alternatively, or in parallel, run
CiDDERto select the minimal set of genomes that achieve a user-defined saturation (e.g., 95%) of the total protein cluster diversity. - Analysis & Validation: Perform comparative genomics (e.g., phylogeny, pangenome) on the dereplicated set. Validate that key functional diversity (e.g., known BGC types) is retained by comparing to the full set.
- Feedback to Prediction: Use the representative genomes and their associated metadata (e.g., isolation source, phenotype) as a refined, unbiased training set for building more generalizable genomic prediction models.
Results and Validation Through Case Studies
Case Study 1 - Multi-Omic Dereplication [29]: Screening of 1,218 soil bacterial isolates yielded 120 active against multidrug-resistant pathogens. MS dereplication via GNPS identified known antibiotics (e.g., actinomycin D) in 33% of active strains. Genomic analysis confirmed the corresponding BGCs and, critically, uncovered the production of additional antibiotics like streptothricin and nigericin in some strains that were not initially detected by MS. This demonstrates how genomics can feed back into and expand upon metabolomic dereplication results.
Case Study 2 - Regulation-Guided Mining [6]: A novel strategy integrated transcriptional regulatory network analysis with co-expression data in Streptomyces coelicolor. By identifying genes co-regulated with the iron-responsive regulator DmdR1, researchers discovered a novel operon (desJGH) involved in desferrioxamine biosynthesis, which had been missed by standard BGC prediction tools. This "regulation-based prioritization" is a form of in silico functional dereplication that feeds genomic predictions into a prioritization schema.
Case Study 3 - Benchmarking Prediction Models [31]: A comparison of GBLUP, Random Forest, SVM, XGBoost, and MLP for predicting health and behavior traits in guide dogs found no statistically significant difference in model performance for the tested traits. This underscores that simpler, more interpretable models like GBLUP can be sufficient, especially when dataset size is limited, and highlights the importance of empirical cross-validation within one's specific system.
The Researcher's Toolkit
Table 4: Essential Research Reagent Solutions and Tools for Integrated Studies
| Category | Item / Software / Database | Primary Function in Integration | Key Features / Notes |
|---|---|---|---|
| Cultivation & Screening | Microbial Diffusion Chambers [29] | Recovers diverse, hard-to-cultivate microbes from environmental samples. | Enables in situ cultivation; key for accessing novel chemical diversity. |
| Reasoner's 2A (R2A) & SMS Agar [29] | Culture media for isolation and growth of soil bacteria. | Low-nutrient media often preferred for environmental isolates. | |
| Dereplication (Metabolomic) | LC-MS/MS System | Generates high-resolution spectral data for compounds in extracts. | Essential for metabolomic profiling. |
| GNPS (Global Natural Products Social) [29] | Public platform for MS/MS spectral library matching and molecular networking. | Core tool for rapid metabolomic dereplication; community-driven. | |
| Dereplication (Genomic) | skDER & CiDDER [34] | Selects non-redundant genome subsets based on ANI or protein-cluster saturation. | Prevents bias, reduces compute time for downstream pangenome/BGC analysis. |
| DAS Tool [37] | Integrates bins from multiple metagenomic binning algorithms. | Recovers more high-quality metagenome-assembled genomes (MAGs). | |
| Genome Mining & Prediction | antiSMASH | Predicts Biosynthetic Gene Clusters (BGCs) from genomic data. | Standard tool for initial genomic potential assessment. |
| EasyGeSe [30] | Curated resource of datasets for benchmarking genomic prediction methods. | Enables fair comparison of new models across diverse species/traits. | |
| R/pyR, scikit-learn, EMMREML [36] | Software environments for implementing GBLUP, Bayesian, and ML models. | Flexible environments for building custom genomic prediction pipelines. | |
| Cross-Validation Framework | Custom scripts for k-fold partitioning | Implements paired, stratified cross-validation schemes. | Critical for obtaining unbiased performance estimates (see Protocol 3.2). |
The strategic, bidirectional integration of genomic prediction and dereplication creates a powerful, self-validating discovery engine. Genomic predictions prioritize strains and BGCs for costly experimental dereplication, while dereplication results provide the essential ground truth to train, test, and refine genomic models via rigorous cross-validation. As evidenced by the tools and case studies presented, there is no single best model or tool; the optimal pipeline depends on the specific biological question, data type, and scale. Success lies in consciously designing workflows that allow these two streams of information to converse, ensuring that computational predictions are grounded in experimental chemistry and that laboratory efforts are focused on the most promising targets for novel discovery. This integrated approach is paramount for efficiently navigating the vast chemical and genetic landscapes towards new therapeutic breakthroughs.
The discovery of novel bioactive natural products from actinomycetes is increasingly guided by computational genome mining, which identifies Biosynthetic Gene Clusters (BGCs) encoding these compounds. However, a persistent challenge is the high rate of BGC rediscovery and the difficulty in linking predicted gene clusters to expressed metabolites, a problem known as the "genome-metabolome gap" [38]. This underscores the critical need for a robust cross-validation framework, where in silico genome mining predictions are systematically validated with experimental metabolomics and dereplication data. This integrated approach is essential to move from speculative genetic potential to confirmed novel chemistry.
The FK-family of metabolites, which includes commercially significant immunosuppressants like FK506 (tacrolimus) and rapamycin (sirolimus), serves as an exemplary case [39]. These complex polyketides are produced by modular polyketide synthases (PKS), and their BGCs exhibit high sequence similarity yet direct the production of distinct molecular scaffolds. Accurately differentiating these closely related BGCs and linking them to their specific chemical products is a definitive test for modern genome mining and dereplication pipelines. This case study examines the tools and methodologies enabling this cross-validation, focusing on the journey from predicting an FK-family BGC to identifying its final metabolite within actinomycete strains.
Comparative Analysis of Genome Mining & Dereplication Platforms
The initial step in natural product discovery is the comprehensive identification and prioritization of BGCs. Researchers can select from a suite of tools, each with distinct strengths in detection, comparison, and analysis.
Table 1: Comparison of Major Genome Mining Tools for BGC Analysis
| Tool | Primary Approach | Key Strength | Limitation for Cross-Validation | FK-Family Application Example |
|---|---|---|---|---|
| antiSMASH [39] | Rule-based, HMM profiles | Excellent for BGC detection & initial classification within a single genome. Industry standard. | Not designed for all-vs-all comparisons across large genome sets; cannot mark query genes as optional. | Identifies PKS Type I clusters characteristic of FK-family but may miss evolutionary variants. |
| GATOR-GC [39] | Targeted, proximity-weighted similarity | Flexible (required/optional queries), performs all-vs-all comparisons, computes GATOR Focal Scores (GFS) for evolutionary insight. Automates deduplication. | Newer tool with a less extensive user base than antiSMASH. | Successfully differentiated FK-family BGCs (e.g., rapamycin vs. FK506) by chemistry using GFS [39]. |
| BiG-SCAPE/CORASON [39] | Comparative genomics, phylogeny | Groups BGCs into Gene Cluster Families (GCFs); useful for evolutionary analysis. | Typically used downstream of antiSMASH; not for initial detection. | Can cluster known FK-family BGCs to understand genomic relationships post-prediction. |
| PRISM | Deep learning & rule-based | Predicts chemical structures directly from genomic sequence. | Predictions are probabilistic and require strong experimental validation. | Can propose a core scaffold for a novel FK-family-like BGC. |
Following genomic prediction, the chemical space of cultivated strains must be analyzed to link BGCs to their products. Dereplication platforms are critical for this step.
Table 2: Comparison of Dereplication and Metabolomics Platforms
| Platform/Method | Core Technology | Key Strength | Role in Cross-Validation |
|---|---|---|---|
| GNPS Molecular Networking [40] [41] | LC-MS/MS data visualization and database matching | Maps metabolite relationships within and across samples; identifies knowns and clusters unknowns. | Central hub for cross-validation. Links MS/MS spectra from extracts to BGC predictions, highlighting novel metabolites. |
| Metabolomics (LC-MS/MS) [41] [42] | High-resolution mass spectrometry | Detects and relatively quantifies thousands of metabolites in a single extract. | Provides the experimental chemical profile to validate the expressed potential of a BGC. |
| Cytotoxicity/Activity Screening [43] [42] | Cell-based or biochemical assays (e.g., BSLA [40]) | Identifies extracts/fractions with desired bioactivity for prioritization. | Guides the isolation process towards bioactive metabolites predicted from certain BGC classes (e.g., cytotoxic compounds from PKS clusters). |
| Database Integration (MIBiG, NP Atlas) [39] [40] | Curated repositories of known BGCs and metabolites | Provides ground truth data for comparing predictions and spectral matches. | Essential for ruling out rediscovery. A novel FK-family BGC should not match known MIBiG entries closely. |
Experimental Protocols for Cross-Validation
The integration of genomic and metabolomic data requires standardized, detailed protocols. Below are generalized methodologies adapted from recent studies for each key stage.
Table 3: Summary of Key Experimental Protocols for Integrated Discovery
| Protocol Stage | Detailed Methodology | Purpose in Cross-Validation |
|---|---|---|
| 1. Genome Sequencing & BGC Prediction | • DNA Extraction: Use kits for high-GC content bacteria (e.g., TIANamp kit) [44].• Sequencing: Perform long-read sequencing (PacBio/Nanopore) for complete BGC assembly [41].• Assembly & Annotation: Use Unicycler/SPAdes, annotate with Prokka [41] [44].• BGC Mining: Run antiSMASH on the complete genome. Use GATOR-GC with queries for key FK-family PKS domains for targeted analysis [39] [41]. | Obtains the complete genetic blueprint. Identifies and classifies all BGCs, specifically targeting FK-family-like architecture for further study. |
| 2. Metabolite Profiling & Dereplication | • Cultivation & Extraction: Grow strain on solid media (e.g., V8 agar), extract metabolites with ethyl acetate via sonication [41].• LC-MS/MS Analysis: Use reversed-phase chromatography coupled to a high-resolution tandem mass spectrometer.• Molecular Networking: Process raw MS/MS data with MZmine, upload to GNPS for analysis. Annotate nodes using spectral library matches [41] [42]. | Generates the experimental chemical profile of the strain. Dereplicates known compounds and organizes unknown metabolites into families, creating a map for novel chemistry. |
| 3. Comparative Multi-Omics Analysis | • Strain Grouping: Cluster phylogenetically related strains with differential bioactivity (e.g., strong vs. weak inhibition of a target pathogen) [41].• Correlation Analysis: Statistically link the presence/absence of specific BGCs and MS/MS spectral features (metabolites) across the strain groups.• Isolation & Structure Elucidation: Use bioactivity and molecular networking to guide the purification of target metabolites via HPLC. Elucidate structures using NMR and MS [40]. | Directly connects a genomic feature (BGC) to an expressed metabolite and a phenotypic outcome, providing strong evidence for function. |
Case Studies in Integrated Discovery
Differentiating FK-Family BGCs with GATOR-GC
A 2025 study demonstrated the utility of the GATOR-GC tool for targeted mining. When applied to differentiate BGCs for the chemically distinct but genetically similar FK-family metabolites (rapamycin and FK506), GATOR-GC used its proximity-weighted similarity scoring (GFS) to successfully cluster them separately according to their specific chemistries [39]. This precise discrimination, which may be challenging for broader tools, is crucial for accurate prediction before chemical analysis begins.
From Anti-PhytophthoraActivity to Borrelidin Identification
A 2025 study on actinomycetes inhibiting the plant pathogen Phytophthora infestans provides a textbook example of cross-validation [41]. Researchers began with 63 actinomycete strains pre-characterized for differential inhibition levels. They then:
- Sequenced Genomes and identified BGCs using antiSMASH.
- Profiled Metabolomes using LC-MS/MS and GNPS molecular networking.
- Performed Comparative Analysis, correlating BGCs and spectral features present only in highly active strains but absent in inactive ones.
This workflow pinpointed the known metabolite borrelidin as the major active compound and putatively identified over 75 other compounds associated with activity, directly linking genotype (a specific PKS BGC) to chemical phenotype (borrelidin production) and biological activity [41].
Discovering Cytotoxic Potential in a NovelMicrobacteriumSpecies
A study on Microbacterium alkaliflavum sp. nov., isolated from mangrove sediments, combined taxonomy, genome mining, and metabolomics [42]. Genome analysis revealed 8 BGCs, including one for desferrioxamines. Concurrent LC-MS/MS metabolomics and molecular networking identified 10 cytotoxic compounds in the extracts, which showed activity against nasopharyngeal carcinoma cell lines. This holistic approach confirmed the strain's novel taxonomic status and simultaneously validated its genome-predicted biosynthetic potential with actual cytotoxic metabolite production.
Diagram 1: Cross-validation workflow integrating genome mining and metabolomics.
Diagram 2: Differentiating closely related FK-family BGCs by chemical output.
The Scientist's Toolkit: Essential Reagents & Materials
Table 4: Research Reagent Solutions for Integrated Discovery Workflows
| Item/Category | Function & Application | Example/Notes |
|---|---|---|
| Specialized Growth Media | Activates silent BGCs and supports secondary metabolism in diverse actinomycetes. | Gauze's Agar [44], V8 Agar [41], 2216E Marine Agar [44]. Using a variety is key (OSMAC approach). |
| DNA Extraction Kits (High-GC) | Efficient lysis and purification of genomic DNA from actinomycetes for sequencing. | Spin-column based kits like TIANamp Bacteria DNA Kit [44]. |
| LC-MS/MS Grade Solvents | High-purity solvents for metabolite extraction and chromatography to prevent interference. | Ethyl Acetate, Methanol, Acetonitrile, Water. Used for solid-liquid extraction [41]. |
| Metabolomics Standards | Internal standards for instrument calibration and quality control in mass spectrometry. | Includes a range of known compounds to ensure analytical reproducibility. |
| Bioassay Components | Enables phenotypic screening to guide the discovery process towards a biological target. | Bioluminescent Reporter Strains (for BSLA assay) [40], pathogen spores, cell lines (e.g., NPC lines TW03, 5-8F) [42], culture media. |
| Reference Databases | Essential for annotating genomes and dereplicating metabolites. | MIBiG (BGCs) [39], GNPS Spectral Libraries [40], GTDB (taxonomy) [44]. |
Overcoming Hurdles: Solving Data, Technical, and Interpretation Challenges
The discovery of novel natural products, a critical source for new drug leads, has been fundamentally transformed by genome mining—the bioinformatic analysis of genomes to identify biosynthetic gene clusters (BGCs) [45]. However, a persistent challenge lies in the cross-validation of in silico genome mining predictions with experimental analytical results, a process known as dereplication. This gap is primarily driven by two technical bottlenecks: the prevalence of incomplete genome assemblies, which obscure full BGCs, and poor-quality tandem mass spectra, which hinder confident compound identification [4] [46].
Incomplete genomes, especially those derived from short-read sequencing, fail to capture repetitive regions, complex structural variants, and full gene clusters, leading to a significant underestimation of an organism's biosynthetic potential [47] [48]. Concurrently, poor spectral fragmentation in mass spectrometry generates uninterpretable data, wasting computational resources and obscuring the detection of novel compounds [46]. This guide provides a comparative analysis of modern strategies and tools designed to address these gaps, facilitating a more robust and predictive workflow for researchers aiming to validate genomic predictions with metabolomic evidence.
Comparative Analysis of Solutions for Incomplete Genomes
The quality of genome mining is intrinsically linked to the completeness and continuity of the input genomic data. Incomplete assemblies fragment BGCs across multiple contigs, preventing their identification or leading to erroneous predictions.
Genome Sequencing and Assembly Strategies
Advanced sequencing and assembly strategies are paramount for overcoming incompleteness. The table below compares the predominant approaches.
Table 1: Comparison of Genomic Sequencing & Assembly Approaches for BGC Recovery
| Approach | Key Technology | Advantages for BGC Mining | Limitations | Typical Outcome for BGCs |
|---|---|---|---|---|
| Short-Read (Illumina) | High-accuracy reads (150-300 bp) | Low cost per base; high accuracy for SNPs; well-established pipelines [49]. | Very short reads cannot resolve repeats; BGCs frequently fragmented [48]. | Highly fragmented clusters; missed repetitive regions of PKS/NRPS. |
| Long-Read (PacBio HiFi, ONT) | Reads of 10 kb to >100 kb | Spans repetitive regions and full operons; enables complete microbial genomes [48]. | Higher cost; historical lower base accuracy (improved with HiFi). | Complete, contiguous BGCs on single contigs; accurate representation of gene order. |
| Hybrid Assembly | Combination of short and long reads | Leverages short-read accuracy and long-read continuity; cost-effective compromise [48]. | Computational complexity in merging datasets. | High-quality, complete assemblies; improved accuracy in repetitive domains. |
| Telomere-to-Telomere (T2T) | Ultra-long reads + advanced phasing | Closes nearly all gaps; resolves centromeres and complex structural loci [47] [48]. | Resource-intensive; currently applied to select, high-value genomes. | Gold-standard completeness; reveals variation in complex, clinically relevant loci (e.g., MHC) [47]. |
Bioinformatics Tools for Genome Mining and Binning
Given the cost of generating complete genomes for all samples, sophisticated binning tools are used to reconstruct genomes from metagenomic data. A recent benchmark evaluated 13 binning tools across different data types (short-read, long-read, hybrid) and binning modes (single-sample, multi-sample, co-assembly) [50].
Table 2: Performance of Top Metagenomic Binning Tools for Recovering High-Quality MAGs
| Tool | Recommended Data/Binning Mode | Key Algorithm | Performance Highlight | Utility for BGC Mining |
|---|---|---|---|---|
| COMEBin [50] | Multi-sample binning (all data types) | Contrastive learning & data augmentation for contig embeddings. | Ranked 1st in 4 out of 7 data-binning combinations. | Recovers most near-complete MAGs, maximizing BGC discovery potential. |
| MetaBinner [50] | Single & multi-sample binning | Ensemble algorithm using multiple feature types. | Ranked 1st in 2 data-binning combinations; good scalability. | Reliable for diverse projects; balances performance and speed. |
| MetaBAT 2 [50] | General-purpose, efficient | Tetranucleotide frequency & coverage with Expectation-Maximization. | Highlighted as an efficient binner with excellent scalability. | Practical first-pass analysis for large-scale metagenomic surveys. |
| VAMB [50] | Multi-sample short-read binning | Deep variational autoencoder (VAE) for feature integration. | Efficient and performant with multi-sample short-read data. | Effective for large cohort studies (e.g., human gut, marine). |
Key Finding from Benchmarks: Multi-sample binning, which uses coverage information across multiple related samples, consistently outperforms single-sample and co-assembly modes. It recovered 54% to 194% more near-complete MAGs from marine datasets, directly translating to a greater capacity for discovering complete BGCs [50].
Specialized Genome Mining Algorithms
Beyond assembly, the algorithms used to identify BGCs vary in sensitivity and specificity.
Table 3: Comparison of Genome Mining Algorithm Approaches
| Algorithm Type | Representative Tools/Strategies | Detection Principle | Strengths | Weaknesses & Data Gaps |
|---|---|---|---|---|
| Rule-based / pHMM | antiSMASH [5], PRISM | Profile Hidden Markov Models (pHMMs) for conserved biosynthetic domains. | High precision for canonical pathways (PKS, NRPS); well-curated. | Misses novel, non-canonical BGCs lacking known domain signatures [45]. |
| Machine Learning | DeepBGC, | Trained on known BGCs to recognize genomic features. | Can identify BGCs with weak or unknown domain signatures. | Performance depends on training data; risk of overfitting. |
| Regulation-based | Strategy from [6] | Identifies BGCs via shared transcriptional regulators/co-expression. | Reveals silent/clustered BGCs; predicts ecological function. | Requires high-quality regulon data and expression datasets. |
| Metallophore-specific | antiSMASH metallophore module [5] | pHMMs for chelator biosynthesis genes (e.g., EntA, SalSyn). | 97% precision, 78% recall for NRP metallophore BGCs. | Limited to known chelator types; new pathways require manual curation. |
Experimental Protocol for Regulation-Based Mining (as in [6]):
- Target a Master Regulator: Select a transcription factor (TF) linked to a physiological response (e.g., iron starvation regulator DmdR1).
- Predict Regulon: Bioinformatically predict all TF binding sites (TFBS) across the genome to map the regulon.
- Integrate Co-expression: Analyze RNA-seq data to identify genes within the regulon that are co-expressed under the TF-inducing condition.
- Prioritize Unknown BGCs: Identify co-expressed, predicted operons that are not annotated as known BGCs by standard tools (e.g., antiSMASH).
- Experimental Validation: Genetically delete the candidate BGC and use comparative metabolomics (LC-MS) to identify the lost compound.
Comparative Analysis of Solutions for Poor Spectral Fragmentation
Dereplication relies on high-quality tandem mass spectra (MS/MS) to compare experimental data against reference libraries. Poor fragmentation leads to ambiguous or failed identifications.
Spectral Quality Assessment and Filtering
A critical first step is pre-filtering spectra to remove uninterpretable data. A method using Fisher Linear Discriminant Analysis (FLDA) on multiple spectral features was shown to effectively eliminate poor-quality spectra [46].
Table 4: Features for Assessing Tandem Mass Spectral Quality
| Feature Category | Specific Features | Rationale | Impact of Poor Quality |
|---|---|---|---|
| Signal Intensity | Total ion current (TIC), Signal-to-noise ratio [46] | High-quality spectra have sufficient signal. | Low intensity peaks are indistinguishable from noise. |
| Peak Distribution | Number of peaks, Average peak distance [46] | Good fragmentation yields regularly spaced peaks. | Random or too few peaks prevent sequence inference. |
| Fragment Patterns | Presence of water/ammonia losses, complementary ion pairs [46] | Indicative of predictable peptide fragmentation. | Absence of expected patterns suggests non-peptide ions or interference. |
| Charge State | Precursor charge determination [46] | Informs fragmentation patterns and database search parameters. | Misassignment leads to incorrect interpretation. |
Experimental Protocol for Spectral Quality Filtering:
- Feature Extraction: For each MS/MS spectrum, compute a vector of quality features (e.g., peak count, TIC, complementarity score).
- Model Application: Process the feature vector through a pre-trained classifier (e.g., FLDA model) to assign a quality score or binary label (high/low quality).
- Filtering: Remove all spectra labeled as low quality prior to database searching or molecular networking.
- Benefit: This can reportedly reduce unnecessary database search time by over 50% by removing 20-30% of poor-quality spectra [46].
Dereplication and Spectral Matching Strategies
Once quality spectra are obtained, the choice of dereplication strategy significantly impacts success rates.
Table 5: Comparison of Dereplication and Spectral Analysis Approaches
| Approach | Description | Advantages | Limitations & Data Gaps |
|---|---|---|---|
| Public MS/MS Library Search | Matching against open databases (GNPS, MassBank) [4]. | Broad coverage; essential for known compound identification. | Limited spectral coverage for rare/novel NPs; variable spectral quality. |
| In-house Library Construction | Creating a custom library from analyzed standards [4]. | High confidence annotations for targeted compounds; control over QC. | Resource-intensive to build and maintain; limited to available standards. |
| Molecular Networking (GNPS) | Clustering MS/MS spectra by similarity to explore chemical space [4]. | Can annotate unknowns via analogy to knowns in cluster; reveals novel variants. | Requires good spectral quality for meaningful clustering; analogs may be unknown. |
| Database Search with Degeneracy | Searching with relaxed accuracy (e.g., >10 ppm mass error). | Increases putative identifications. | Dramatically increases false positives; requires manual validation. |
Experimental Protocol for Building an In-House Dereplication Library (adapted from [4]):
- Standard Pooling: Combine analytical standards into pools based on logP values and exact masses to minimize co-elution.
- LC-MS/MS Data Acquisition: Analyze each pool using a uniformly optimized LC-ESI-MS/MS method in positive and/or negative mode.
- Collision Energy Ramping: Acquire MS/MS spectra at multiple collision energies (e.g., 10, 20, 30, 40 eV) to capture comprehensive fragmentation patterns.
- Metadata Compilation: For each compound, document its name, formula, observed exact mass (<5 ppm error), retention time, and all adduct ions ([M+H]⁺, [M+Na]⁺).
- Library Deployment: Use the curated library to screen experimental samples, matching retention time, precursor mass, and MS/MS spectral patterns.
Integrated Cross-Validation Workflow
The synergy between improved genomic data and high-quality spectral analysis forms the basis of robust cross-validation. The following diagram synthesizes this integrated approach, highlighting how addressing initial data gaps enhances the final validation step.
Diagram 1: Integrated workflow showing how addressing data gaps enables robust cross-validation between genome mining and dereplication.
Table 6: Essential Research Reagents and Resources for Cross-Validation Studies
| Item / Resource | Function / Purpose | Example / Specification | Source/Reference |
|---|---|---|---|
| Long-Read Sequencing Kit | Generate long sequencing reads for complete genome/BGC assembly. | PacBio HiFi or Oxford Nanopore Ultra-Long DNA library prep kits. | [47] [48] |
| Metagenomic Binning Software | Reconstruct metagenome-assembled genomes (MAGs) from complex samples. | COMEBin, MetaBinner for multi-sample binning (highest quality). | [50] |
| Specialized Genome Mining Tool | Identify specific classes of BGCs beyond standard tools. | antiSMASH with metallophore HMMs (for siderophores). | [5] |
| LC-MS/MS Grade Solvents | Mobile phase for high-resolution metabolomics to minimize background noise. | Methanol, acetonitrile, water, formic acid (Optima LC/MS grade). | [4] |
| Authentic Analytical Standards | Build in-house spectral libraries for targeted, high-confidence dereplication. | Compounds like quercetin, catechin, betulinic acid (purity >97%). | [4] |
| Spectral Quality Assessment Tool | Pre-filter poor-quality MS/MS spectra before time-consuming database searches. | Software implementing FLDA or similar classifier on spectral features. | [46] |
| Regulation Network Data | Enable regulation-based genome mining to find silent or co-regulated BGCs. | RNA-seq datasets under specific stimuli; TF binding site predictions. | [6] |
Within the paradigm of data-driven natural product discovery, a central thesis is the cross-validation of genome mining predictions with experimental metabolomics data. This integration is crucial for de-orphaning biosynthetic gene clusters (BGCs) but is persistently challenged by false positives—both in silico BGC predictions that do not yield detectable metabolites and spectral matches that incorrectly annotate compounds. This guide objectively compares the performance of contemporary computational and integrative strategies designed to refine these predictions and enhance the fidelity of discovery pipelines.
Performance Comparison of Refinement Strategies
The following tables quantify the effectiveness of different approaches to reduce false positives in BGC prediction and metabolomic annotation, based on recent experimental studies.
Table 1: Performance of Correlation-Based Metabologenomics Scoring Methods [51] This study on 110 fungi evaluated three scoring methods for linking Gene Cluster Families (GCFs) to mass spectrometry signals.
| Scoring Method | Input Data (GCF/Metabolite) | Core Calculation | Performance on 25 Known Links | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| Pattern Matching | Binary / Binary | Pearson’s chi-squared test (p-value) | Statistically significant for 21/25 known pairs [51] | Easy statistical interpretation; robust to noise. | Misses linkages with low metabolite expression. |
| Correlation Scoring | Binary / Binary | Weighted scoring matrix (Presence/Presence: +10; Absence/Presence: -10) | Effective for high-confidence linkage ranking [51] | Penalizes contradictory data (GCF absent but ion present). | Requires optimized threshold for significance. |
| Intensity Ratio Analysis | Binary / Quantitative | Ratio of avg. ion abundance (GCF+ vs. GCF- strains) | Identifies strong, abundant metabolite signals [51] | Uses quantitative data; mitigates MS column bleed artifacts. | Biased toward highly abundant metabolites. |
Table 2: Precision of Automated BGC Prediction and Spectral Annotation Tools These data summarize the precision of specialized tools for predicting specific BGC types and annotating metabolomic data.
| Tool / Strategy | Target | Reported Precision & Recall | Key Outcome | Experimental Validation |
|---|---|---|---|---|
| antiSMASH NRP Metallophore Detection [5] | Automated detection of NRP metallophore (e.g., siderophore) BGCs | 97% precision, 78% recall (vs. manual curation) [5] | First automated census predicted 25% of bacterial NRPS clusters encode metallophores [5]. | Characterization of novel metallophores from Pseudomonas and Streptomyces matched genomic predictions [5]. |
| GNPS Feature-Based Molecular Networking (FBMN) with AI Tools [18] | Annotation of unknown metabolites in extracts | Up to 65% higher accuracy than database-dependent methods alone [18] | Enables annotation in non-model strains by leveraging community MS/MS data and in silico predictions. | Used in discovery of >185 novel microbial NPs (2018-2024) [18]. |
| Regulation-Guided Genome Mining [6] | Prioritizing BGCs with shared regulatory context (e.g., iron regulation) | Identified a novel DFO biosynthesis locus missed by standard BGC miners [6] | Links BGCs to physiological conditions, reducing prioritization of silent clusters. | Deletion of predicted locus (desJGH) in S. coelicolor altered DFO B/E precursor balance [6]. |
Detailed Experimental Protocols
The effectiveness of the strategies in Table 1 relies on rigorous, standardized experimental workflows. Below are detailed protocols for key cited experiments.
This protocol outlines the process for generating and correlating genomic and metabolomic data from fungal strains.
1. Genomic Data Processing & GCF Network Generation:
- Genome Sequencing & Assembly: Sequence 110 Ascomycete fungi (combining public and newly assembled genomes). Assemble using appropriate long- or short-read assemblers.
- BGC Prediction: Analyze all genomes with antiSMASH (v6.0 or later) using standard parameters to identify BGCs [51].
- Create Protein Domain Arrays: Convert each BGC into an array of its constituent protein domains.
- Calculate Similarity & Network GCFs: Calculate pairwise similarity between all BGCs based on shared domains and sequence identity. Group BGCs into Gene Cluster Families (GCFs) using a defined similarity threshold (e.g., 50% to 90%). The choice of threshold dramatically affects GCF number and size [51].
2. Metabolomic Data Acquisition & Dereplication:
- Fungal Cultivation & Extraction: Grow each of the 110 strains in three different culture conditions to stimulate diverse secondary metabolism. Perform liquid-liquid extraction of metabolites from each culture [51].
- LC-MS/MS Analysis: Analyze all 330 extracts via high-resolution Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). Use consistent chromatography and data-dependent MS/MS acquisition.
- Data Processing: Process raw MS data (e.g., using MZmine, MS-DIAL). Perform peak picking, alignment, and background subtraction. The result is a data matrix of MS1 ions (e.g., 9,301 ions) [51].
- Dereplication: Annotate ions by searching MS1 and MS/MS data against in-house and public spectral libraries (e.g., GNPS). Use in silico prediction tools (e.g., SIRIUS) for molecular formula and structure suggestions [51] [18].
3. Correlation-Based Scoring:
- Create a binary GCF presence/absence matrix across all strains.
- For Pattern Matching and Correlation Scoring, create a binary metabolite ion presence/absence matrix. For Intensity Ratio Analysis, use a quantitative ion abundance matrix [51].
- For each GCF-ion pair, calculate the linkage score using the chosen method(s) from Table 1.
- Validate the Model: Use a set of known BGC-metabolite pairs (e.g., 25 validated linkages from the MIBiG repository) to assess which scoring method and GCF networking parameters yield the highest true positive rate [51].
This protocol describes a strategy to uncover novel, co-regulated BGCs that may be missed by sequence-based mining.
1. Identify a Master Regulator and its Regulon:
- Select a model organism with well-characterized physiology (e.g., Streptomyces coelicolor for iron metabolism).
- Focus on a global transcriptional regulator (e.g., the iron-dependent regulator DmdR1).
- Bioinformatically predict the DmdR1 regulon: Identify the conserved binding site motif for DmdR1. Scan the genome to predict all transcription factor binding sites (TFBS). Define the set of genes (the regulon) likely controlled by DmdR1 [6].
2. Integrate Regulon Data with Co-Expression Networks:
- Analyze public or newly generated global gene expression datasets (e.g., RNA-seq under iron-limited vs. iron-replete conditions).
- Calculate gene co-expression networks (e.g., using Pearson Correlation Coefficients).
- Integrate datasets: Identify genes that are both part of the predicted DmdR1 regulon and co-expressed with known iron-related biosynthesis genes (e.g., the core desferrioxamine BGC) [6].
3. Discover and Validate a Novel Locus:
- The integrated analysis revealed the desJGH operon, located distantly from the core DFO BGC, as both DmdR1-regulated and co-expressed with known DFO genes [6].
- Genetic Validation: Construct in-frame deletion mutants of target genes (desG, desH) in S. coelicolor.
- Metabolic Profiling: Perform LC-MS analysis of wild-type and mutant strains under iron-limiting conditions. Use extracted ion chromatograms for known DFOs (B and E).
- Characterize Phenotype: The mutants showed a strong reduction in DFO B and an increase in DFO E, confirming the locus's role in tailoring the DFO precursor pool [6].
Visualizing Key Workflows
The following diagrams illustrate the logical relationships and experimental workflows for the core strategies discussed.
Integrated Metabologenomics for BGC-Metabolite Linking
Regulation-Guided BGC Prioritization Workflow
The Scientist's Toolkit: Research Reagent Solutions
This table details essential materials and tools for implementing the cross-validation workflows described.
| Item / Reagent | Function in Cross-Validation | Application Notes |
|---|---|---|
| antiSMASH Software Suite [51] [5] | The standard for automated BGC prediction in genomic sequences. Identifies clusters based on profile HMMs of core biosynthetic enzymes. | Essential for the genomics arm. Version 6.0+ includes specialized detection modules (e.g., for NRP metallophores with 97% precision) [5]. |
| GNPS Platform & Spectral Libraries [18] | A crowdsourced platform for mass spectral data sharing and molecular networking. Enounces feature-based molecular networking (FBMN) for metabolomic dereplication. | Critical for the metabolomics arm. Compares experimental MS/MS spectra to reference libraries to annotate known compounds and cluster unknowns [18]. |
| MIBiG Repository (Minimum Information about a BGC) [51] | A curated genomic and chemical database of experimentally validated BGCs and their metabolites. | Serves as the essential ground-truth dataset for training and validating new correlation or prediction algorithms (e.g., validating 25 known pairs) [51]. |
| Iron-Depleted Culture Media | Used to experimentally induce the expression of iron-scavenging BGCs (e.g., siderophores) in microbial cultures. | Key reagent for the functional validation of regulation-guided mining. Creating iron-limiting conditions activates the DmdR1 regulon, allowing detection of associated metabolites like desferrioxamines [6]. |
| Reference Metabolite Standards | Authentic chemical standards for known natural products (e.g., desferrioxamine B, lovastatin). | Used to confirm LC-MS/MS-based annotations by matching retention time and fragmentation pattern. Vital for converting spectral "hits" into verified identifications and reducing annotation false positives. |
The pursuit of novel bioactive compounds from microbial communities hinges on two parallel tracks: genome mining of biosynthetic gene clusters (BGCs) and the mass spectrometry-based dereplication of metabolites. A critical thesis in modern natural product discovery is the need for cross-validation between these tracks to confirm the link between a predicted genetic potential and an expressed chemical product [23]. This process is computationally monumental, requiring the analysis of thousands of metagenomes to recover high-quality genomes and correlate them with spectral data. Success depends on optimizing throughput—maximizing the scale, speed, and cost-efficiency of analysis. This guide compares how next-generation, AI-enhanced cloud workflows like the Metagenomics-Toolkit meet this challenge against traditional and alternative modern methods [52] [53].
Performance Comparison: Metagenomics-Toolkit vs. Alternative Workflows
The following tables provide a quantitative comparison of the Metagenomics-Toolkit's capabilities against other common strategies, focusing on scalability, accuracy, and resource efficiency.
Table 1: Comparison of Workflow Strategies for Large-Scale Metagenome Analysis
| Feature / Strategy | Traditional Local HPC | Standard Cloud Pipeline | Metagenomics-Toolkit (AI/Cloud-Optimized) |
|---|---|---|---|
| Core Optimization | Maximizes use of fixed, local hardware. | Elastic scaling of compute nodes. | ML-predicted resource allocation & elastic cloud scaling [52] [53]. |
| Scalability | Limited by local cluster size & queue times. | High, but can incur cost from over-provisioning. | Very High. Efficient scaling for 100s-1000s of samples (e.g., 757 sewage samples) [52] [53]. |
| Cost Efficiency | High capital expenditure; low variable cost. | Variable; risk of wasted spend on unused resources. | Higher. ML reduces CPU/RAM waste, lowering compute costs [52]. |
| Key Advantage | Full control, no data transfer costs. | Flexibility, access to latest hardware. | Balanced efficiency & scalability. Automated, reproducible, and cost-effective for massive studies [53]. |
| Best For | Single projects with stable, predictable needs. | Projects with fluctuating or urgent compute needs. | Large-scale, reproducible projects like global microbiome surveys or cross-validation studies [52]. |
Table 2: Benchmarking Results for Key Analytical Steps in Metagenomics
| Analytical Step | Tool / Method | Reported Performance Metric | Context & Comparison |
|---|---|---|---|
| Taxonomic Profiling / Pathogen Detection | Kraken2/Bracken | Highest F1-score; detects pathogens at 0.01% abundance [54]. | Outperformed MetaPhlAn4 (limited at 0.01%) and Centrifuge in food safety benchmarking [54]. |
| Metagenomic Binning | COMEBin | Ranked 1st in 4 of 7 data-type/binning-mode combinations [50]. | Leading modern tool using contrastive learning; excels in multi-sample binning [50]. |
| Metagenomic Binning | Multi-sample Binning | Recovered 54-125% more near-complete MAGs vs. single-sample [50]. | Consistently superior strategy across short-read, long-read, and hybrid data types [50]. |
| Dereplication | DEREPLICATOR+ | Identified 5x more molecules vs. prior approaches in GNPS data [23]. | Enables cross-validation by linking mass spectra to diverse natural product classes [23]. |
Experimental Protocols for Cross-Validation Studies
The integration of genome-centric metagenomics and dereplication requires rigorous experimental design. Below is a detailed methodology based on current best practices and cited studies.
Protocol 1: Co-assembly and Binning for Low-Abundance Genome Recovery This protocol is designed to recover metagenome-assembled genomes (MAGs), including uncultivated species, for association with phenotypes like disease or metabolite production [9].
- Sample Collection & Sequencing: Collect multiple metagenomic samples from a cohort (e.g., patient groups, environmental gradients). Perform whole-metagenome shotgun sequencing using either Illumina (for depth) or PacBio/ONT (for contiguity) [55] [9].
- Co-assembly: Perform a de novo co-assembly of all reads from multiple samples within a cohort using a assembler like metaSPAdes (for short reads) or metaFlye (for long reads). This pools sequencing depth to aid in assembling genomes of low-abundance organisms [9].
- Binning: Apply multi-sample binning to the co-assembled contigs. Use coverage information across all samples with a high-performance tool like COMEBin or MetaBinner [50]. This step groups contigs into putative MAGs.
- Quality Filtering & Dereplication: Assess MAG quality using CheckM2. Filter for medium-quality (completeness >50%, contamination <10%) or near-complete MAGs. Dereplicate the MAGs across cohorts using tools like dRep to create a non-redundant genome catalog [50].
- Annotation & Correlation: Annotate MAGs for Biosynthetic Gene Clusters (BGCs) (using antiSMASH) and other functions. Statistically correlate MAG abundance (from read mapping) with target phenotypes (e.g., disease status) to identify "important" genomes [9].
Protocol 2: Metabolite Extraction and Dereplication for Cross-Validation This protocol details the complementary mass spectrometry workflow to identify known metabolites and enable connection to genomic data [23].
- Metabolite Extraction: From the same or biologically linked samples, perform solvent-based extraction of metabolites.
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Analyze the extracts using LC-MS/MS to generate tandem mass spectra for thousands of metabolites.
- Spectral Dereplication: Process the raw spectra. Use a dereplication tool like DEREPLICATOR+ to search spectra against natural product databases (e.g., AntiMarin, GNPS). This identifies known metabolites and their structural variants without re-isolation [23].
- Molecular Networking: Upload data to the Global Natural Products Social Molecular Networking (GNPS) platform. Create a molecular network to visualize spectral similarities and identify clusters of related metabolites, expanding the discovery of variants [23].
- Cross-Validation: Compare results from Protocol 1 and Protocol 2. The key step is to attempt to link BGCs from important MAGs to the dereplicated metabolites or molecular families, based on predicted biosynthetic class and co-occurrence, validating that genomic potential translates to chemical expression.
Workflow Architecture and Cross-Validation Logic
The following diagrams illustrate the optimized workflow of the Metagenomics-Toolkit and the logical framework for cross-validation.
Diagram 1: AI-Optimized Cloud Workflow of the Metagenomics-Toolkit. This architecture shows how machine learning dynamically manages computational resources within a scalable cloud environment to process metagenomic data from raw reads to annotated results [52] [53].
Diagram 2: Framework for Cross-Validating Genome Mining with Dereplication. This logic flow illustrates the parallel bioinformatic and chemical analysis tracks that converge to validate connections between biosynthetic gene clusters and expressed metabolites [23] [9].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents, Kits, and Databases for Integrated Metagenomics & Dereplication
| Item | Category | Primary Function in Research |
|---|---|---|
| High-Fidelity (HiFi) Long-Read Sequencing Kits (PacBio) | Sequencing Reagent | Generate long reads (~10-25 kb) with very high accuracy (Q30+), enabling complete assembly of BGCs and repeat regions [55]. |
| DNA Extraction Kits for Complex Matrices (e.g., soil, stool) | Sample Prep | Lyse diverse microbial cells and isolate high-quality, inhibitor-free genomic DNA suitable for long-read sequencing [56]. |
| Metagenomic Sequencing Library Prep Kits | Sample Prep | Fragment DNA and attach platform-specific adapters for next-generation sequencing on Illumina, Nanopore, or PacBio platforms [56]. |
| GTDB (Genome Taxonomy Database) | Bioinformatics Database | Provides a standardized bacterial and archaeal taxonomy for consistent classification of newly recovered MAGs [9]. |
| MIBiG (Minimum Information about a Biosynthetic Gene Cluster) | Bioinformatics Database | A curated repository of known BGCs and their metabolites, used as a reference for annotating and prioritizing novel BGCs from MAGs. |
| AntiMarin / GNPS Spectral Libraries | Chemistry Database | Curated repositories of mass spectra for known natural products, essential for dereplicating metabolites and avoiding rediscovery [23]. |
| LC-MS Grade Solvents & Columns | Chemistry Reagent | Essential for high-resolution metabolite separation and mass spectrometry analysis to generate high-quality fragmentation spectra. |
The discovery of novel bioactive natural products has entered a transformative "deep-mining era," driven by advances in genomics and metabolomics [18]. High-throughput sequencing reveals that a vast reservoir of biosynthetic gene clusters (BGCs) exists in microbial genomes, with estimates suggesting that in model genera like Streptomyces, only approximately 10% of BGCs are expressed under standard laboratory conditions [18]. This discrepancy between genomic potential and observed chemical output defines the critical "genome-metabolome gap." The majority of BGCs remain "silent" or "cryptic," not yielding detectable quantities of their encoded compounds under typical cultivation, representing a major untapped resource for drug discovery [18] [57].
Bridging this gap requires a synergistic, cross-validated strategy. On one side, genome mining provides a predictive roadmap, identifying the genetic potential for natural product synthesis through tools like antiSMASH and DeepBGC [5] [58]. On the other, dereplication through analytical chemistry, primarily mass spectrometry, rapidly identifies known compounds in complex extracts, preventing redundant rediscovery and highlighting novel chemistry [7] [23]. The convergence of these fields—where genomic predictions are validated by metabolomic detection, and unknown metabolomic features are traced back to genetic origins—forms the core of a modern paradigm for unlocking silent clusters. This guide provides a comparative analysis of the leading cultivation, elicitation, genome mining, and dereplication strategies, focusing on their integrated application to efficiently discover novel microbial metabolites.
Comparative Analysis of Genome Mining and Dereplication Platforms
The efficacy of a silent cluster discovery pipeline hinges on the initial in silico prediction and subsequent analytical validation. The table below compares the core functionality, strengths, and limitations of leading genome mining and dereplication tools.
Table 1: Comparison of Genome Mining and Dereplication Platforms
| Tool/Strategy | Primary Function | Key Strength | Major Limitation | Typical Use Case in Cross-Validation |
|---|---|---|---|---|
| antiSMASH [5] [58] | Broad-spectrum BGC detection & classification. | Gold standard; wide BGC class coverage (40+ types); integrates new modules (e.g., metallophore detection). | Rule-based; may miss atypical/hybrid clusters; predicts potential, not expression. | Initial genome survey to catalog silent BGCs and prioritize targets. |
| DeepBGC [18] [58] | BGC detection using deep learning (BiLSTM). | Better generalization for novel architectures; context-aware. | Can yield false positives on diverse genomes; performance depends on training data. | Complementary tool to antiSMASH for identifying clusters with weak sequence homology. |
| RFBGCpred [58] | Machine-learning classifier for 5 major BGC classes. | High accuracy (98.02%) for focused classes (PKS, NRPS, etc.); handles hybrid clusters well. | Limited to predefined classes; not a full detection pipeline. | High-confidence classification of specific, prioritized BGC types. |
| LC-MS/MS Dereplication [7] [4] | Compound identification via spectral matching. | High sensitivity; can quantify compounds; direct chemical evidence. | Requires reference standards or libraries; blind to compounds not in database. | Rapid identification of known metabolites in extracts to highlight novel peaks. |
| Molecular Networking (GNPS) [23] [59] | Organizes MS/MS spectra by similarity into networks. | Identifies compound families and analogs; can annotate unknowns by relation to knowns. | Computational intensity; annotation confidence depends on network topology and libraries. | Visualizing chemical diversity of an extract and connecting novel features to known scaffolds. |
| DEREPLICATOR+ [23] | Algorithm for dereplicating spectra against diverse metabolite databases. | Identifies multiple compound classes (PKs, terpenes, etc.); high-throughput. | Dependent on quality and scope of underlying structural databases. | Automated, large-scale dereplication of mass spectrometry datasets from multiple strains. |
Experimental Protocols for Cultivation and Metabolite Analysis
The first experimental step is to coax the expression of silent BGCs. The One Strain Many Compounds (OSMAC) approach is fundamental, systematically varying cultivation parameters [18]. Key protocols include:
- Media Variation: Cultivating the strain in 2-3 chemically distinct media (e.g., complex ISP2, defined R2A, seawater-based). Co-cultivation with other microbes or the addition of signaling molecules (e.g., N-acetylglucosamine) can also mimic ecological competition and trigger defense responses [5].
- Elicitor Addition: Adding sub-inhibitory concentrations of antibiotics, heavy metals (e.g., iron, zinc), or enzyme inhibitors (e.g., histone deacetylase inhibitors for fungi) at mid-log phase can stress the organism and activate cryptic pathways [5].
- Genetic Elicitation: For genetically tractable strains, CRISPRi-mediated knockdown of repressor genes or overexpression of pathway-specific transcriptional activators can directly switch on silent clusters [18].
Metabolite Extraction and LC-MS/MS Analysis
Following cultivation, metabolites are extracted and prepared for analysis. A critical dereplication protocol involves specialized sample preparation for complex matrices, as demonstrated in the analysis of polyherbal formulations [7].
- Sample Cleanup Protocol (for complex broths or extracts): Use Solid Phase Extraction (SPE) with C-18 reversed-phase cartridges. Condition cartridges with methanol and water. Load sample, wash with 5-15% methanol to remove polar interferents like sugars, then elute target metabolites with 80-100% methanol. This step significantly reduces matrix effects and enhances chromatographic resolution and MS ionization efficiency [7].
- LC-MS/MS Analysis for Dereplication: Perform analysis using a UHPLC system coupled to a high-resolution tandem mass spectrometer (e.g., Q-TOF) [4] [59].
- Chromatography: Use a C-18 column. Employ a gradient from 5% to 98% acetonitrile (with 0.1% formic acid) over 20-25 minutes [59].
- Mass Spectrometry: Acquire data in both Data-Dependent Acquisition (DDA) and Data-Independent Acquisition (DIA, e.g., SWATH) modes. DDA provides clean MS/MS spectra for library matching, while DIA captures fragmentation data for all ions, ensuring no minor metabolites are missed [59].
- Dereplication Workflow: Convert raw data to .mzML format. For DIA data, use MS-DIAL to deconvolute and create pseudo-MS/MS spectra. Submit both DDA spectra and deconvoluted DIA data to the GNPS platform for molecular networking and library search against public spectral repositories (MassBank, HMDB, NIST) and in-house libraries [4] [59].
Integrated Genomic-Metabolomic Cross-Validation Workflow
The most powerful strategy directly links genomic prediction to metabolomic observation.
- Genome Sequencing and Mining: Sequence the strain using a long-read platform (e.g., PacBio HiFi) for a complete genome. Annotate BGCs using antiSMASH and DeepBGC [18] [58].
- Targeted Metabolite Prediction: For high-priority, silent BGCs (e.g., those with unusual architecture), use tools like PRISM or antiSMASH to predict the core scaffold of the encoded metabolite (e.g., a non-ribosomal peptide sequence) [18].
- Extract Analysis and Correlation: Analyze crude extracts from OSMAC experiments using high-resolution LC-MS/MS. Process the data with molecular networking on GNPS.
- Cross-Validation: Search for the exact mass of the predicted metabolite core scaffold or its plausible derivatives in the LC-MS data. Investigate the molecular network neighborhood of any matching mass for related analogs. If a potential match is found, use tandem MS to compare the experimental fragmentation pattern with in silico predictions of the hypothesized structure [23]. This creates a feedback loop where genomics guides metabolomics, and metabolomics validates genomics.
Diagram Title: Cross-validation workflow linking genomic prediction with metabolomic analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of the described strategies relies on a suite of specialized reagents, materials, and bioinformatics resources.
Table 2: Key Research Reagent Solutions for Silent Cluster Discovery
| Item | Function / Purpose | Example / Specification |
|---|---|---|
| Solid Phase Extraction (SPE) C-18 Cartridges [7] | Sample cleanup to remove interfering sugars, salts, and polar matrix components from complex culture extracts prior to LC-MS, enhancing signal clarity. | 1 g/6 mL capacity; used with methanol/water gradients for washing and elution. |
| LC-MS Grade Solvents | Mobile phase for ultra-high-performance liquid chromatography (UHPLC) to ensure minimal background noise and ion suppression in mass spectrometry. | Methanol, Acetonitrile, Water (ISO 3696 Grade I), with additives like Formic Acid (0.1%). |
| Chemical Elicitors | To induce stress responses and activate silent biosynthetic pathways during cultivation. | Sub-inhibitory antibiotics, heavy metal salts (FeCl₃, ZnCl₂), N-acetylglucosamine. |
| High-Resolution Tandem Mass Spectrometer | The core analytical instrument for dereplication, providing accurate mass and fragmentation data for compound identification. | Q-TOF (Quadrupole-Time of Flight) or Orbitrap-based systems. |
| Spectral Reference Libraries | Databases for matching experimental MS/MS spectra to identify known compounds. | GNPS libraries, NIST, MassBank, HMDB, and custom in-house libraries [4] [23]. |
| Genome Mining Software (Local/Web) | Bioinformatics tools for the in silico identification and analysis of BGCs. | antiSMASH (web or CLI), DeepBGC, RFBGCpred [5] [18] [58]. |
| Molecular Networking Platform | Web-based platform to analyze LC-MS/MS data, visualize chemical relationships, and dereplicate compounds via community tools. | The Global Natural Products Social Molecular Networking (GNPS) platform [23] [59]. |
Case Studies in Strategy Application
Dereplication of a Complex Polyherbal Formulation
A study on a polyherbal liquid formulation (Linkus syrup) containing ten plant extracts demonstrated a robust dereplication protocol [7]. Researchers used SPE C-18 cleanup followed by LC-MS/MS analysis, identifying 70 compounds. By correlating these compounds with analyses of individual plant extracts, they attributed 44 compounds uniquely to specific species and found 26 shared compounds. This peak intensity-based correlation served as a dereplication and standardization method, ensuring quality control. While applied to plants, this precise analytical workflow is directly transferable to microbial fermentations, where it can distinguish strain-specific metabolites from media components or co-culture exchange products.
Genome-Guided Discovery of Novel RiPPs
Advanced genome mining integrated with metabolomics was showcased in the discovery of novel ribosomally synthesized and post-translationally modified peptides (RiPPs) [18]. Researchers used a bioinformatics pipeline combining sequence similarity networks (SSNs) and AlphaFold-Multimer structure prediction to identify genes encoding novel P450 enzymes linked to RiPP biosynthesis. After heterologous expression of the predicted BGCs in a tractable host (E. coli or S. albus), LC-MS analysis detected new macrocyclic peptides (e.g., kitasatides, micitides), whose structures were confirmed by NMR. This case exemplifies the direct pathway from in silico prediction of a silent cluster in a native genome to heterologous expression and final compound discovery.
Diagram Title: Dereplication workflow using molecular networking and database search.
Unlocking the chemical potential of silent BGCs is no longer reliant on serendipity but is a rational, data-driven process. The most effective strategy is an iterative, cross-validated cycle that leverages the complementary strengths of genome mining and metabolomic dereplication. Genome mining offers a hypothesis ("this strain can make a compound like this"), while advanced dereplication through high-resolution LC-MS/MS and molecular networking provides the testable evidence.
Future directions will deepen this integration. Tools like DEREPLICATOR+, which can dereplicate diverse compound classes and connect to genomic data, will become more central [23]. Machine learning models, already used for BGC prediction (e.g., DeepBGC, RFBGCpred) and resource optimization in workflows like the Metagenomics-Toolkit, will increasingly predict optimal elicitation conditions or link spectral features directly to BGC types [58] [60]. For researchers, the key is to build a pipeline that flexibly incorporates these evolving tools, always using one line of evidence to inform and validate the other, thereby systematically transforming silent genetic potential into novel chemical discoveries.
Establishing Confidence: Frameworks for Cross-Validation and Method Assessment
The contemporary discovery of microbial natural products has evolved into a data-driven deep-mining era, pivoting from serendipitous isolation to a targeted, hypothesis-driven process [18]. This paradigm is built on a core validation loop, where in silico genome mining predictions are experimentally tested and refined through advanced analytical dereplication. Genome mining tools systematically unearth hidden biosynthetic gene clusters (BGCs) from genomic data, predicting their chemical potential [18]. Dereplication strategies, primarily leveraging high-resolution mass spectrometry (HRMS) and sophisticated algorithms, then analyze the organism's actual metabolome to rapidly identify known compounds and highlight novel ones [40] [23]. The convergence of these two streams—genomic potential and metabolomic reality—creates a powerful feedback cycle for confirming discoveries, minimizing rediscovery, and accelerating the path to novel therapeutics. This guide compares the leading tools and platforms that enable this integrated workflow, providing researchers with a framework for selecting optimal strategies to validate their genome mining predictions.
Comparative Analysis of Genome Mining & Dereplication Platforms
The initial stage of the validation loop relies on computational tools to predict biosynthetic potential. The following table compares the core algorithms, outputs, and validation utilities of major genome mining platforms.
Table 1: Comparison of Major Genome Mining Platforms for BGC Prediction
| Tool / Platform | Core Algorithm & Approach | Primary Output & Strengths | Key Validation Utility | Reported Performance/Notes |
|---|---|---|---|---|
| antiSMASH 7.0 | Rule-based, using Hidden Markov Models (HMMs) to detect known BGC core biosynthetic enzymes [18]. | Identifies and annotates >40 types of known BGCs; provides detailed modular architecture for NRPS/PKS clusters [18]. | Excellent for generating testable hypotheses on clusters with known biosynthetic logic; output guides targeted LC-MS analysis. | Industry standard; improved precision/recall for specific classes like NRP-metallophores when using curated HMM modules [61]. |
| DeepBGC | Deep learning using Bi-directional Long Short-Term Memory (BiLSTM) and Random Forest classifiers [18]. | Detects both known and "orphan" BGCs with novel architectures; effective in under-explored phylogenetic groups [18]. | Uncovers cryptic clusters missed by rule-based methods, expanding the search space for novel chemistry. | Useful for identifying novel BGC families in non-model organisms (e.g., Verrucomicrobia) [18]. |
| PRISM 4 | Combinatorial logic to predict chemical structures from NRPS/PKS gene clusters. | Generates predicted chemical structures for non-ribosomal peptides and polyketides. | Provides a concrete, testable chemical formula and mass for dereplication; direct link to m/z search. | Prediction accuracy depends on cluster annotation quality; ideal for cross-referencing with HRMS data. |
| RiPPer & RODEO | Heuristic and SVM-based analysis focused on ribosomally synthesized and post-translationally modified peptides (RiPPs) [18]. | Identifies precursor peptides and predicts RiPP core structures based on enzyme families. | Targets a specific, diverse class of natural products; predictions are often small peptides amenable to MS/MS dereplication. | Specialized for RiPP discovery; can be integrated with genomics to find P450-modified RiPPs [18]. |
The subsequent analytical phase employs dereplication to test these predictions. The table below contrasts leading dereplication methodologies and platforms.
Table 2: Comparison of Dereplication Platforms & Methodologies
| Platform / Method | Core Technology | Strengths & Application | Limitations | Reported Performance |
|---|---|---|---|---|
| GNPS Molecular Networking | Tandem MS (MS/MS) spectral similarity networking via the Global Natural Products Social platform [18] [23]. | Visualizes related metabolites in extracts; clusters known and unknown compounds; enables community-wide data sharing and annotation. | Requires high-quality MS/MS data; annotations rely on available spectral libraries. | Central to modern workflows; used to analyze hundreds of millions of spectra [23]. |
| DEREPLICATOR+ | Algorithm that searches MS/MS spectra against structure databases by modeling fragmentations [23]. | Dereplicates peptides, polyketides, terpenes, alkaloids, etc.; identifies structural variants. | Performance depends on database coverage and fragmentation model accuracy. | Identified 5x more molecules from GNPS data than previous approaches; found 488 compounds (1% FDR) in a test Actinomyces dataset [23]. |
| Feature-Based Molecular Networking (FBMN) | LC-MS data alignment coupled with GNPS, integrating chromatographic and spectral data [18]. | Improves network accuracy by aligning features across samples; better for quantitative studies. | More complex data processing pipeline. | Coupled with AI tools (SIRIUS), can annotate unknowns in extracts with ~65% higher accuracy than database-dependent methods alone [18]. |
| ISDB & NAP | In silico fragmentation databases and network annotation propagation. | Predicts MS/MS spectra for putative structures from genomics (e.g., from PRISM) for comparison with experimental data. | Computational heavy; predictions may contain false positives. | Directly links genome mining (PRISM) and dereplication (GNPS) in a validated workflow. |
Experimental Protocols for Cross-Validation
A robust validation loop requires standardized protocols to connect genomic predictions with metabolomic analysis. Below are detailed methodologies for two key integrative approaches.
Protocol: Integrated Genomic-Dereplication Workflow for Novel Compound Discovery
This protocol outlines the steps from sequencing to validated compound identification [18] [23].
- Genome Sequencing & Assembly: Sequence the microbial strain using a high-accuracy platform (e.g., PacBio HiFi). Assemble reads into a complete or draft genome.
- In silico Genome Mining:
- Process the genome through multiple mining tools (e.g., antiSMASH, DeepBGC).
- Prioritize BGCs of interest based on novelty scores, phylogenetic context, or suspected product class.
- For prioritized NRPS/PKS/RiPP clusters, use tools like PRISM or RiPPer to predict a putative chemical structure or core mass.
- Metabolite Extraction & LC-HRMS/MS Analysis:
- Culture the strain under various conditions (OSMAC approach).
- Prepare crude organic extracts.
- Analyze by reversed-phase LC coupled to a high-resolution tandem mass spectrometer (e.g., Q-TOF, Orbitrap).
- Acquire data-dependent MS/MS spectra for ions above a threshold.
- Dereplication & Validation:
- Process raw data (feature detection, alignment).
- Submit MS/MS data to GNPS for FBMN analysis.
- Use DEREPLICATOR+ to screen features against natural product databases.
- Cross-validate: Search for the exact mass or predicted MS/MS spectrum of the compound(s) predicted by genome mining (Step 2).
- Identify novelty: Compounds not matched by dereplication, but associated with a predicted BGC, are high-priority novel candidates.
- Isolate and elucidate the structure of the candidate using targeted purification and NMR.
Protocol: Targeted MS/MS Validation of P450-Modified RiPP Predictions
This specialized protocol validates genome mining predictions for a specific enzyme-modified class [18].
- Bioinformatic Prediction:
- Use tools like RiPPer and sequence similarity networks (SSNs) to scan genomes for precursor peptides adjacent to P450 oxidase genes.
- Apply AlphaFold-Multimer to model precursor peptide-P450 enzyme interactions, identifying true biosynthetic pairs.
- Heterologous Expression:
- Clone the predicted RiPP BGC (including precursor and P450 genes) into an expression host like S. albus or E. coli.
- Targeted Metabolomic Analysis:
- Analyze culture extracts of the heterologous host and a control strain lacking the BGC via LC-HRMS/MS.
- Use ion chromatogram extraction for the exact mass of the predicted mature RiPP.
- Structural Confirmation:
- Acquire high-quality MS/MS spectra of the target ion unique to the expressing strain.
- Compare fragmentation patterns to the predicted macrocyclic or cross-linked structure.
- Confirm production by isolating the compound and determining its planar and stereochemical structure via NMR.
Workflow & Methodology Visualization
Validation Loop for Genome Mining & Dereplication Workflow
Dereplication Methodologies for Metabolite Identification
Table 3: Key Research Reagents & Computational Tools for the Validation Loop
| Tool/Reagent Category | Specific Example(s) | Primary Function in Validation Workflow |
|---|---|---|
| Genome Sequencing Service | PacBio HiFi, Oxford Nanopore MinION [18] | Provides high-quality, contiguous genomic data as the foundational input for all in silico mining. |
| Genome Mining Software | antiSMASH 7.0, DeepBGC, PRISM 4 [18] | Converts raw genome sequence into prioritized, interpretable BGC predictions and putative chemical structures. |
| Mass Spectrometry Platform | Orbitrap, Q TOF, FT ICR MS with UHPLC [18] | Generates the high-resolution mass and fragmentation spectral data required for accurate dereplication. |
| Dereplication Platform | GNPS, DEREPLICATOR+, SIRIUS/CSI:FingerID [18] [23] | Analyzes MS/MS data to filter out known compounds and propose identities for unknowns via spectral matching or AI. |
| Natural Product Database | MIBiG, AntiMarin, Dictionary of Natural Products [23] | Curated repositories of known compounds and their associated spectra, essential as a reference for dereplication. |
| Heterologous Expression Host | Streptomyces albus J1074, E. coli BL21(DE3) [18] | Enables controlled expression of silent or cryptic BGCs predicted by mining, linking genes directly to metabolites. |
| Structure Elucidation Instrument | NMR with Cryoprobes (600 MHz+), Microcrystal Electron Diffraction [18] | Provides definitive structural and stereochemical proof for novel compounds flagged by the validation loop. |
The discovery of fungal secondary metabolites (SMs), which include vital pharmaceuticals, agrochemicals, and concerning mycotoxins, has been revolutionized by genome mining. This approach identifies biosynthetic gene clusters (BGCs) that encode the enzymatic machinery for SM production [3]. In fungi like Alternaria—a genus of significant agricultural and food safety importance due to its prolific production of phytotoxins and mycotoxins—genome mining reveals a vast, untapped metabolic potential [3] [62]. However, the presence of a BGC is only a prediction of chemical capacity; it does not confirm the actual production of the metabolite under laboratory or natural conditions [51].
This gap between genetic potential and chemical reality defines the "cold start" problem in natural product discovery and necessitates robust cross-validation strategies. Dereplication, the process of identifying known compounds in a mixture using techniques like mass spectrometry, prevents redundant rediscovery [23]. The integration of genomics (predicting what could be made) and metabolomics (detecting what is made) into a "metabologenomics" framework is therefore critical [51]. This guide objectively compares the methodologies, tools, and analytical strategies for validating genome mining predictions with dereplication results, using recent Alternaria research as a primary case study to highlight best practices and remaining challenges.
Core Concepts and Workflow for Cross-Validation
The fundamental goal is to establish a confirmed link between a Biosynthetic Gene Cluster (BGC) and its metabolic product. This process involves two parallel streams of data generation and analysis that must converge.
Genomics Stream: This begins with high-quality genome sequencing and assembly. For fungi, tools like funannotate are used for consistent gene prediction and annotation [3]. BGCs are then identified using specialized algorithms such as antiSMASH [5] [51]. To manage the thousands of BGCs discovered in large-scale studies, they are grouped into Gene Cluster Families (GCFs) based on protein domain similarity. GCFs cluster BGCs predicted to produce the same or structurally related molecules, enabling comparative analysis across strains [3] [51].
Metabolomics Stream: This involves culturing organisms under various conditions to elicit SM production, followed by metabolite extraction and analysis via Liquid Chromatography-High Resolution Mass Spectrometry (LC-HRMS/MS) [63]. The resulting mass spectral data is processed to detect "mass features." Dereplication tools like DEREPLICATOR+ or GNPS Molecular Networking compare these features to spectral libraries of known compounds to provide putative identifications [23].
Cross-Validation Logic: The core integrative analysis tests for a statistical association between the presence (or absence) of a specific GCF in a set of genomes and the detection (or intensity) of a specific metabolite in the corresponding metabolomic profiles of those strains [51]. A strong, statistically significant association provides evidence that the GCF encodes the biosynthesis of that metabolite.
Diagram 1: Integrated Metabologenomics Workflow for BGC-Metabolite Cross-Validation. The workflow illustrates the parallel genomics and metabolomics streams that converge in a statistical correlation analysis to generate testable hypotheses linking specific Gene Cluster Families (GCFs) to metabolite production [3] [51].
Comparison of Primary Methodological Approaches
Three main computational strategies exist for correlating GCF and metabolomics data, each with distinct strengths, weaknesses, and optimal use cases [51].
Diagram 2: Scoring Method Comparison for Metabologenomics. This diagram contrasts three primary algorithms for linking GCFs to metabolites, highlighting their different data inputs, core calculations, and inherent analytical trade-offs [51].
1. Pattern Matching (Binary Co-occurrence)
- Principle: Uses a simple chi-squared test on binary presence/absence matrices for both GCFs and metabolite ions [51].
- Best For: Initial, high-throughput screening for strong, unambiguous links where metabolite production is consistent across strains with the GCF.
- Limitation: It fails to detect linkages for metabolites that are produced at low levels or under specific conditions not captured in the assay, leading to false negatives [51].
2. Correlation Scoring (Weighted Binary)
- Principle: Applies a weighted scoring matrix that rewards co-presence, strongly penalizes metabolite presence without the GCF (a likely false positive), and mildly rewards co-absence [51].
- Best For: The recommended general-purpose method. It effectively prioritizes robust correlations while filtering out noise, making it suitable for large, noisy datasets.
- Limitation: The final score is not a probabilistic measure, requiring careful interpretation and threshold setting.
3. Intensity Ratio Analysis (Quantitative)
- Principle: Uses quantitative metabolite intensity (peak height) data. The score is the ratio of the average metabolite abundance in strains with the GCF versus strains without it [51].
- Best For: Identifying the genetic basis of highly expressed, core metabolites. It can overcome technical artifacts like "column bleed" in LC-MS.
- Limitation: Heavily biased toward high-abundance metabolites and will miss clusters encoding low-yield or tightly regulated compounds.
Table 1: Comparison of Correlation Methods for Metabologenomics
| Method | Primary Input Data | Core Calculation | Key Advantage | Key Disadvantage | Optimal Use Case |
|---|---|---|---|---|---|
| Pattern Matching [51] | Binary (Presence/Absence) for GCFs and Ions | Pearson's Chi-squared Test | Provides a clear, statistically significant p-value. | Insensitive to low-abundance or conditionally expressed metabolites. | Initial screening for strong, constitutive links. |
| Correlation Scoring [51] | Binary (Presence/Absence) for GCFs and Ions | Weighted Scoring Matrix | Effectively penalizes false positives (ion without GCF). | Score requires empirical threshold setting; not a direct probability. | General-purpose analysis of large, complex datasets. |
| Intensity Ratio Analysis [51] | Binary GCF data & Quantitative Ion Intensity | Mean(Ion w/ GCF) ÷ Mean(Ion w/o GCF) | Accounts for variation in metabolite expression levels. | Inherently biased toward high-abundance metabolites. | Finding clusters for dominant, core metabolic products. |
Case Study: BGC and Metabolite Distribution inAlternaria
Recent large-scale studies on the fungal family Pleosporaceae, which includes Alternaria, provide a concrete example of applying comparative genomics and the challenges of cross-validation [3] [63].
Genomic Potential: A 2025 study mining 187 genomes (123 Alternaria, 64 related genera) identified 6,323 BGCs, averaging 34 BGCs per genome (29 for Alternaria) [3] [64]. These were classified into 548 Gene Cluster Families (GCFs). Key findings include:
- The GCF for the mycotoxin alternariol (AOH) was restricted to sections Alternaria and Porri, informing targeted food safety monitoring [3].
- The sections Infectoriae and Pseudoalternaria possessed unique GCF profiles, offering candidates for diagnostic markers, though none were linked to known compounds [3] [64].
- Phylogenetic analysis showed that GCF presence/absence patterns generally correlated with evolutionary relationships, indicating vertical transmission plays a key role in BGC distribution [3].
Metabolomic Reality & Integration: A complementary 2022 metabolomics study on Alternaria section Alternaria used untargeted LC-HRMS to profile 36 isolates [63]. It successfully detected a unique chemical phenotype for the dehydrocurvularin family of toxins in three strains. Subsequent genomic examination confirmed the associated BGC was located in a subtelomeric accessory region, a genomic location often associated with strain-specific and horizontally transferred traits [63]. This exemplifies a targeted cross-validation where a metabolic signature guided the genomic search.
Table 2: Key Genomic and Metabolomic Statistics from Recent *Alternaria Studies*
| Analysis Type | Metric | Result | Interpretation & Implication |
|---|---|---|---|
| Comparative Genomics [3] [64] | Total Genomes Analyzed | 187 (123 Alternaria, 64 other Pleosporaceae) | Unprecedented scale for this taxon. |
| Total BGCs Predicted | 6,323 | Vast, untapped biosynthetic potential. | |
| Average BGCs per Genome | 34 (29 for Alternaria) | Confirms Alternaria as metabolically prolific. | |
| BGCs Grouped into GCFs | 548 Gene Cluster Families | Enables pattern analysis and prioritization. | |
| Metabolomics & Cross-Validation [63] | Strains Profiled Metabolically | 36 (Section Alternaria) | Phenotypic diversity exists within a section. |
| Unique Chemical Phenotype Detected | Dehydrocurvularin toxin family in 3 strains | Metabolomics can pinpoint rare chemotypes. | |
| Genomic Location of Correlated BGC | Subtelomeric accessory region | Links metabolite specificity to flexible genomic regions prone to horizontal transfer. |
Diagram 3: Phylogenetic Distribution of Key BGC Traits in Alternaria. This diagram synthesizes findings from large-scale genomics, showing how different BGCs and GCFs map onto the phylogeny of Alternaria, with direct implications for food safety, diagnostics, and regulation [3].
Experimental Protocols for Key Analyses
1. Genome Sequencing, BGC Prediction & GCF Networking
- Genome Assembly: Use a hybrid sequencing approach (e.g., Illumina for accuracy, Oxford Nanopore for long reads) for high-quality genomes [62]. Assemble with tools like SPAdes or Unicycler and assess completeness with BUSCO [3] [62].
- Gene Prediction: Annotate all genomes with a consistent pipeline (e.g., funannotate for fungi) to avoid technical bias [3].
- BGC Detection: Run antiSMASH with appropriate settings for fungi to identify BGCs [5] [51].
- GCF Networking: Calculate pairwise similarity between all BGCs based on shared protein domains and sequence identity. Use a tool like BiG-SCAPE to cluster BGCs into GCFs at a defined similarity threshold (e.g., 50% permissive to 90% strict). The threshold choice significantly impacts results: stricter thresholds create many small GCFs (likely identical products), while permissive thresholds create fewer, larger GCFs (structurally related products) [51].
2. Untargeted Metabolomics & Dereplication
- Culture & Extraction: Grow fungal strains on multiple media types (e.g., CYS80, DRYES, MMK2) to stimulate diverse SM production. Extract agar plugs with ethyl acetate [63].
- LC-HRMS/MS Analysis: Analyze extracts using reversed-phase UPLC coupled to a high-resolution mass spectrometer (e.g., Thermo Orbitrap). Use a gradient of acetonitrile/water with formic acid over a 10-20 minute method [63].
- Data Processing: Convert raw files, perform peak picking, alignment, and background subtraction using software like MS-DIAL or MZmine.
- Dereplication: Export MS/MS spectra and analyze via the Global Natural Products Social Molecular Networking (GNPS) platform. Use the DEREPLICATOR+ tool to search spectra against natural product libraries (e.g., AntiMarin, DNP) for putative identifications [23].
3. Correlation-Based Cross-Validation
- Data Matrix Preparation: Create a binary matrix (1/0) for GCF presence across all strains. Create a corresponding matrix for metabolite presence (binary) or peak intensity (quantitative) [51].
- Statistical Correlation: Apply one or more of the methods from Table 1. For the weighted Correlation Scoring method [51], use a scoring system such as:
- GCF present, Ion present: +10
- GCF absent, Ion present: -10
- GCF present, Ion absent: 0
- GCF absent, Ion absent: +1
- Validation: Prioritize high-scoring pairs for downstream genetic validation (e.g., gene knockout) or chemical isolation.
Table 3: Key Research Reagents and Computational Tools for Metabologenomics
| Category | Item / Tool Name | Primary Function in Workflow | Key Consideration / Note |
|---|---|---|---|
| Wet-Lab & Sequencing | Potato Dextrose Agar (PDA) / CYSA80 Media [63] [62] | Standardized fungal culturing to elicit secondary metabolism. | Using multiple media types is crucial for metabolic diversity. |
| Illumina NextSeq / HiSeq Platforms [3] [62] | High-throughput short-read sequencing for accurate genome assembly. | Often used in hybrid strategies with long-read tech. | |
| Oxford Nanopore MinION [65] | Long-read sequencing to resolve repetitive regions and complete genomes. | Essential for assembling BGCs often found in complex regions. | |
| Bioinformatics - Genomics | funannotate Pipeline [3] | Unified gene prediction and functional annotation of fungal genomes. | Reduces bias from using different annotation pipelines. |
| antiSMASH [5] [51] | The standard tool for genome-wide identification and annotation of BGCs. | Continuously updated; version choice affects results. | |
| BiG-SCAPE / CORASON | Clusters predicted BGCs into Gene Cluster Families (GCFs) based on similarity. | The similarity cutoff parameter is critical and must be reported. | |
| Bioinformatics - Metabolomics | MZmine / MS-DIAL | Open-source software for processing raw LC-MS data (peak picking, alignment). | Generates the quantitative feature tables for analysis. |
| Global Natural Products Social (GNPS) [23] | Web-based platform for mass spectral data sharing, dereplication, and molecular networking. | Central repository for community data and tools. | |
| DEREPLICATOR+ [23] | Algorithm for dereplicating MS/MS spectra against databases of known natural products. | Extends beyond peptides to polyketides, terpenes, etc. | |
| Integrated Analysis | In-house Python/R Scripts | Implementing correlation scoring (e.g., weighted matrix) and statistical tests. | Custom code is often required for specific study designs. |
| Paired Omics Data Platform | A public repository specifically for linked genomic and metabolomic datasets. | Facilitates meta-analysis and sharing of integrated data. |
The cross-validation of genome mining predictions with metabolomic dereplication results has matured from a conceptual goal to a practicable, high-throughput framework. As demonstrated in Alternaria, integrating these approaches transforms a static list of predicted BGCs into a dynamic map of expressed chemical diversity, directly linking taxonomy, genetics, and phenotype. The correlation-based methods, particularly weighted correlation scoring, provide a robust statistical framework to prioritize the most promising BGC-metabolite pairs for further experimental characterization [51].
Future advancements will stem from addressing current limitations: improving the detection and expression of "silent" BGCs, expanding high-quality spectral libraries for dereplication, and developing more sophisticated, possibly AI-driven, algorithms that can predict chemical structures directly from BGC sequences [66]. Furthermore, standardizing workflows and depositing paired datasets in public repositories will be essential for the community to build upon these integrative analyses, accelerating the discovery of novel fungal natural products for application in drug development, agriculture, and food safety.
The systematic discovery of novel natural products and biosynthetic pathways hinges on two interdependent computational processes: genome mining and dereplication. Genome mining involves scanning microbial genomes to identify biosynthetic gene clusters (BGCs) responsible for producing specialized metabolites, such as antibiotics and siderophores [5]. Dereplication is the subsequent step of efficiently identifying and filtering out known compounds or genetic elements to prioritize novelty [67]. The core thesis of modern discovery pipelines posits that the robustness of findings is significantly enhanced through the cross-validation of results from these two domains. This article provides a comparative guide, grounded in recent experimental data, to evaluate the performance of leading software tools in both fields. By objectively comparing benchmarks on speed, accuracy, and scalability, we aim to equip researchers and drug development professionals with the evidence needed to select optimal tools, thereby accelerating the translation of genomic potential into novel therapeutic leads.
Performance Benchmarks: Quantitative Comparisons
Benchmarking Metagenomic Binning and Dereplication Tools
The performance of tools varies significantly based on data type, algorithm, and specific use case. The following tables consolidate key quantitative findings from recent benchmarking studies.
Table 1: Performance of Metagenomic Binning Tools Across Data Types [50] This table summarizes the performance of top-ranked binning tools in recovering Moderate or higher Quality (MQ), Near-Complete (NC), and High-Quality (HQ) Metagenome-Assembled Genomes (MAGs) from a marine dataset (30 samples). Multi-sample binning consistently outperforms single-sample modes.
| Data Type | Binning Mode | Top-Performing Binner | MQ MAGs (Median) | NC MAGs (Median) | HQ MAGs (Median) | Key Strength |
|---|---|---|---|---|---|---|
| Short-Read (mNGS) | Multi-sample | COMEBin [50] | 1101 | 306 | 62 | Best overall recovery |
| Short-Read (mNGS) | Single-sample | MetaBinner [50] | 550 | 104 | 34 | Efficient for per-sample analysis |
| Long-Read (HiFi/Nanopore) | Multi-sample | COMEBin [50] | 1196 | 191 | 163 | Superior with long-read data |
| Hybrid (Short+Long) | Multi-sample | MetaBinner [50] | 1334 | 219 | 176 | Best for hybrid data integration |
Table 2: Performance of Sequence Search and Dereplication Tools [67] This table compares the speed, accuracy, and resource utilization of lightweight dereplication tools using a large bacterial contig dataset (~10 GB, 934k contigs).
| Tool | Algorithm Basis | Search Speed (100k queries) | Clustering Adjusted Rand Index (ARI) | Max Memory Footprint | Primary Use Case |
|---|---|---|---|---|---|
| Blini [67] | Fractional MinHash, Mash Distance | 25 seconds | 0.997 - 1.000 | 38 - 462 MB | Rapid large-scale dereplication |
| MMseqs2 [67] | K-mer matching & alignment | >30 min (for 1 query) | 1.000 | 3 - 6 GB | Accurate, alignment-based clustering |
| Sourmash [67] | Fractional MinHashing | ~36 days (est. for 100k) | N/A | Variable | General-purpose similarity search |
Table 3: Accuracy of Automated Genome Mining Predictions [5] Specialized modules within genome mining platforms can achieve high accuracy in predicting specific metabolite types, such as non-ribosomal peptide (NRP) metallophores.
| Tool / Module | Target BGC Type | Precision | Recall | Key Detection Metric |
|---|---|---|---|---|
| antiSMASH NRP Metallophore Detector [5] | NRP Siderophores & Metallophores | 97% | 78% | Presence of chelator biosynthesis genes |
| Regulation-based Mining [68] | Iron-regulated Siderophores (e.g., Desferrioxamine) | Functional Association | N/A | Co-expression with regulator (DmdR1) binding sites |
Experimental Protocols and Methodologies
Protocol for Benchmarking Metagenomic Binning Tools
A comprehensive benchmark of 13 binning tools was conducted using five real-world datasets (human gut, marine, cheese, activated sludge) under seven data-binning combinations [50].
- Datasets & Preprocessing: The study used short-read (mNGS), long-read (PacBio HiFi, Oxford Nanopore), and hybrid sequencing data. Reads were assembled using metaSPAdes (short-read) and metaFlye (long-read).
- Binning Modes: Three modes were evaluated: (1) Co-assembly binning: all samples assembled together before binning; (2) Single-sample binning: each sample assembled and binned independently; (3) Multi-sample binning: samples assembled independently but coverage profiles calculated across all samples during binning.
- Evaluation Metrics: Reconstructed MAGs were evaluated with CheckM2. MAGs were classified as: Moderate or higher quality (MQ): >50% completeness, <10% contamination; Near-complete (NC): >90% completeness, <5% contamination; High-quality (HQ): NC criteria plus presence of rRNA and tRNA genes [50].
- Key Finding: Multi-sample binning recovered significantly more HQ MAGs than single-sample binning (e.g., 233% more in a 30-sample human gut dataset) [50].
Protocol for Evaluating Dereplication Speed and Accuracy
The performance of the dereplication tool Blini was evaluated against Sourmash and MMseqs2 using simulated and real sequence data [67].
- Search Function Test:
- Small Dataset: 100 viral genomes were queried against the RefSeq viral database. Blini completed the search in 0.5 seconds, compared to 126 seconds (Sourmash) and 151 seconds (MMseqs2).
- Large Dataset: 100,000 random 10kbp fragments (with 0.1% SNPs) were queried against a 10 GB bacterial contig dataset. Blini processed all queries in 25 seconds, achieving a throughput of 5100 queries per second.
- Clustering Function Test:
- Two simulated datasets were used: one with sequences containing 1% random SNPs, and another with random fragments of original sequences.
- Clustering accuracy was measured using the Adjusted Rand Index (ARI). Blini achieved an ARI between 0.989 and 1.0, comparable to MMseqs2, while using substantially less memory (e.g., 38 MB vs. 3 GB at a high scale setting) [67].
Protocol for Genome Mining and Cross-Validation with Dereplication
A study on Sophora flavescens demonstrates an integrated workflow for metabolite discovery that cross-validates different analytical techniques [59].
- Sample Analysis: Root extracts were analyzed via LC-MS/MS using both Data-Dependent Acquisition (DDA) and Data-Independent Acquisition (DIA) modes.
- Dereplication Pipeline:
- Molecular Networking: DIA data was processed to create a molecular network on the GNPS platform, clustering compounds by MS/MS spectral similarity.
- Database Matching: DDA data was simultaneously matched against public spectral libraries.
- Annotation & Isomer Discrimination: Results from both pathways were combined, and isomers were resolved by analyzing their extracted ion chromatograms (EICs).
- Outcome: This complementary strategy annotated 51 compounds, demonstrating that DIA-based networking is powerful for detecting trace components, while DDA provides cleaner spectra for direct matching [59].
Protocol for Regulation-Based Genome Mining
An innovative strategy used regulatory network analysis to predict and prioritize BGC function in Streptomyces coelicolor [68].
- Regulon Prediction: Genome-wide Transcription Factor Binding Sites (TFBS) for 17 regulators were predicted using position weight matrices from the LogoMotif database.
- Co-expression Integration: RNA-seq co-expression data was integrated with TFBS predictions to expand regulons and identify co-regulated operons.
- Functional Discovery: This approach identified the novel operon desJGH as part of the iron-regulated DmdR1 regulon. Experimental deletion confirmed its essential role in modulating the production of the siderophore desferrioxamine B [68].
- Significance: This method provides a functional filter for prioritizing BGCs that may be missed by sequence-based mining alone, adding a regulatory layer for cross-validation.
Visualizing Workflows and Logical Relationships
Diagram 1: Cross-Validation Framework for Genome Mining & Dereplication
This diagram outlines the integrated workflow where genomic and metabolomic pipelines inform and validate each other.
Diagram 2: Integrated Dereplication Strategy for Metabolites
This diagram details the multi-pronged LC-MS/MS strategy for comprehensive metabolite annotation [59].
Table 4: Key Reagents, Software, and Databases for Integrated Studies This table lists critical resources for executing the experimental protocols and analyses described in this guide.
| Category | Item Name | Function in Research | Example Use / Note |
|---|---|---|---|
| Sequencing & Assembly | PacBio HiFi / Oxford Nanopore | Generates long-read sequences for improved genome assembly and binning [50]. | Essential for resolving repetitive BGC regions. |
| metaSPAdes / metaFlye | Assemblers for short-read and long-read metagenomic data, respectively [50]. | Produces contigs for subsequent binning. | |
| Binning Software | COMEBin [50] | High-performance binner using contrastive learning; top-ranked in multi-sample benchmarks. | Recommended for short/long/hybrid data in multi-sample mode. |
| MetaBinner [50] | Stand-alone ensemble binner; excels with hybrid data. | Efficient for generating initial component results. | |
| Genome Mining | antiSMASH [5] | Predicts BGCs in genomic data; includes specialized detectors (e.g., for NRP metallophores). | Core platform for BGC discovery; 97% precision for metallophores [5]. |
| StreptoBase / LogoMotif DB | Provides curated genome and regulatory data for model organisms like S. coelicolor [68]. | Used for regulation-based mining predictions. | |
| Dereplication | Blini [67] | Lightweight tool for rapid nucleotide sequence search and clustering. | Processes 100k queries in 25 sec; minimal RAM footprint [67]. |
| GNPS Platform [59] | Web-based ecosystem for mass spectrometry data analysis and molecular networking. | Central for metabolomic dereplication and annotation. | |
| Evaluation & QC | CheckM2 [50] | Assesses the quality (completeness, contamination) of MAGs. | Defines MQ, NC, and HQ MAG standards for benchmarking. |
| MS-DIAL / MZmine [59] | Software for processing LC-MS/MS data, especially from DIA scans. | Converts raw DIA data into formats suitable for GNPS. | |
| Reference Data | RefSeq / GTDB | Curated genomic reference databases for taxonomy and annotation. | Used for search index construction and phylogenetic mapping [67] [5]. |
| MassBank / NIST / mzCloud | Tandem mass spectral libraries for metabolite identification. | Targets for direct spectral matching in dereplication [59]. |
The discovery of novel bioactive natural products, a critical source for new therapeutics, has been revitalized by the convergence of genome mining and advanced mass spectrometry (MS)-based dereplication. This integrated pipeline addresses a central challenge: efficiently distinguishing novel metabolites from known compounds within complex biological extracts. Genome mining predicts the biosynthetic potential of a microbial strain by identifying gene clusters, such as those for non-ribosomal peptide synthetases (NRPS) or polyketide synthases (PKS). However, the correlation between the presence of a biosynthetic gene cluster (BGC) and the actual production of the corresponding compound is imperfect due to silent or poorly expressed clusters [69].
Conversely, tandem mass spectrometry (MS/MS) analysis of an extract provides direct evidence of produced metabolites. Dereplication algorithms analyze these spectra against databases of known compounds to prevent redundant rediscovery [27]. The core thesis of modern discovery is the cross-validation of these two data streams. A prioritized target for costly heterologous expression and isolation emerges not from a genomic correlation alone, but from a causative link established when a detected MS/MS signal cannot be explained by known compounds in databases yet is plausibly linked to a predicted BGC. This guide provides a comparative analysis of the computational and experimental frameworks that enable this transition from correlation to causation, focusing on performance metrics, experimental validation, and practical workflow integration for researchers and drug development professionals.
Comparative Analysis of Genome Mining and Dereplication Platforms
The initial prioritization of clusters relies on computational tools for genome analysis and spectral interpretation. The table below compares the key functionalities, performance, and integration capabilities of major approaches.
Table 1: Comparison of Bioinformatics Tools for Genome Mining and Dereplication
| Tool / Approach | Primary Function | Key Metrics/Performance | Advantages | Limitations | Integration with Experimental Validation |
|---|---|---|---|---|---|
| Genome Mining (e.g., antiSMASH, PRISM) | Identifies & predicts BGCs from genomic data. | Predicts cluster type, core structure, potential bioactivity. | Provides hypothesis for compound structure; essential for elucidating biosynthesis. | Does not confirm compound production; high false-positive rate for novel compounds. | Target for heterologous expression; guides MS/MS spectral interpretation. |
| DEREPLICATOR [27] | Dereplicates peptidic natural products (PNPs) via MS/MS database search. | Identified 37 unique PNPs at p<10⁻¹¹ in benchmark; ~7.3% FDR at peptide level. | Specialized for NRPs/RiPPs; enables identification of variants via spectral networks. | Restricted to peptide-based compounds only. | Directly validates production of known PNPs; flags clusters producing known compounds. |
| DEREPLICATOR+ [23] | Dereplicates diverse natural product classes (PNPs, polyketides, terpenes, etc.). | Identified 5x more molecules than previous tools; 154 compounds at 0% FDR in Actinomyces dataset. | Broad coverage; identifies variants; suitable for large-scale GNPS data analysis. | Computational complexity higher than class-specific tools. | Core tool for cross-validation; identifies novelty gaps in MS/MS data linked to BGCs. |
| MINE Framework [70] | Prioritizes the most informative next experiment in a p>>n setting. | Model-guided adaptive design; maximizes discovery efficiency from limited samples. | Optimizes resource allocation in exploration phase; integrates prior omics data. | Requires initial dataset and ensemble modeling; not a direct identification tool. | Guides sequential experimental strategy (e.g., which strain to express next). |
| Predictive Metabolite Modeling [69] | Predicts community metabolite dynamics from genomic content. | Linear regression maps gene content to metabolite dynamics; applicable to denitrification. | Demonstrates principle of genotype-to-phenotype prediction for metabolism. | Currently demonstrated for specific, well-defined pathways in communities. | Provides ecological context for cluster expression and metabolite detection. |
The performance of dereplication tools is critically evaluated by their false discovery rate (FDR) and identification scope. For instance, DEREPLICATOR+ dramatically expanded discovery, identifying 488 unique compounds at a 1% FDR in Actinomyces spectra, compared to 73 by its predecessor [23]. This high-throughput identification is fundamental for filtering out known compounds and highlighting unknown spectral features that become candidates for novel cluster expression.
Cross-Validation Workflow: From Spectral Networks to Cluster Prioritization
The decisive step in prioritization is the cross-validation of genomic and spectroscopic evidence. The following workflow diagram outlines this integrated process.
Diagram 1: Integrated workflow for prioritizing heterologous expression targets. The workflow initiates with parallel genomic and metabolomic profiling of a microbial strain. Genome mining identifies all potential BGCs, while LC-MS/MS captures the actual metabolome. Dereplication against databases like AntiMarin or GNPS libraries filters the spectra into "known" and "unknown" groups [23]. The crucial integration occurs via molecular networking, which clusters MS/MS spectra based on similarity, often grouping structurally related molecules [27]. A high-priority target is generated when an "unknown" spectral cluster can be plausibly linked to a predicted BGC—for example, through a shared physicochemical property, a predicted molecular family, or a co-occurrence pattern across multiple strains [69]. This correlation, refined by bioinformatic filters, provides a causative hypothesis strong enough to justify the investment in heterologous expression.
Experimental Protocols for Validation and Isolation
Once a target cluster is prioritized, it must be experimentally validated. The core methods involve heterologous expression of the BGC and subsequent purification of the metabolite.
Heterologous Expression in a Model Host
The primary goal is to express the silent or poorly expressed BGC in a genetically tractable host like Pichia pastoris (for proteins) or Streptomyces coelicolor (for bacterial natural products).
Protocol: Heterologous Expression of a Biosynthetic Gene Cluster
- Cluster Isolation & Vector Construction: Isolate the target BGC from genomic DNA via PCR, cosmid library screening, or direct synthesis. Clone it into an appropriate expression vector with a strong, inducible promoter (e.g., PAOX1 for P. pastoris) and selectable markers.
- Host Transformation: Introduce the recombinant vector into the expression host. For P. pastoris, this is typically done via electroporation, followed by selection on media lacking histidine for integration into the genome [71].
- Screening & Expression: Screen transformants for vector integration via PCR. Inoculate positive clones into expression media. Induce expression at optimal cell density (e.g., with methanol for P. pastoris). Culture for a defined period (e.g., 72-96 hours).
- Harvest & Preliminary Analysis: Pellet cells via centrifugation. Prepare crude extracts from the supernatant (secreted proteins) or cell lysate (intracellular compounds). Analyze extracts via LC-MS/MS and compare to the original "unknown" spectrum from the native strain to confirm production.
Protein Purification and Functional Assay
For expressed bioactive proteins or peptides, functional validation requires purification.
Protocol: Multi-step Purification of a Recombinant Protein [71] This protocol, based on the purification of the human RANK extracellular domain from P. pastoris, exemplifies a standard approach.
- Concentration & Initial Clean-up: Concentrate the protein-containing culture supernatant using ultrafiltration (UF) with an appropriate molecular weight cutoff membrane.
- Size-Exclusion Chromatography (SEC): Apply the concentrated sample to a Sephadex G-50 column equilibrated with a suitable buffer (e.g., PBS). This step separates the target protein from other molecules based on hydrodynamic size.
- Ion-Exchange Chromatography (IEX): Pool the fractions containing the target protein and apply them to a Q-Sepharose Fast Flow anion-exchange column. Elute the protein using a linear gradient of increasing ionic strength (e.g., 0 to 1 M NaCl). This step separates proteins based on charge.
- Assessment & Storage: Analyze the purity of the final eluate via SDS-PAGE and Western blotting. Determine protein concentration. A purity of >95% is typically achieved for functional studies [71]. Aliquot and store at -80°C.
Table 2: Validation Metrics for Heterologous Expression and Purification
| Experimental Stage | Key Performance Metrics | Typical Target / Outcome | Purpose in Validation Pipeline |
|---|---|---|---|
| Heterologous Expression | - MS/MS spectral match to native unknown.- Titer of target compound (mg/L). | Detection of target ion; correlation of fragmentation pattern. | Confirms the BGC is responsible for producing the detected metabolite. |
| Protein Purification [71] | - Purity (% by SDS-PAGE).- Total yield (mg of protein).- Specific activity (if applicable). | >95% purity; sufficient yield for in vitro/in vivo assays. | Enables direct functional testing of the isolated compound. |
| Functional Assay [71] | - IC₅₀ in cell-based assay.- In vivo efficacy (e.g., tumor growth inhibition). | Statistically significant bioactivity vs. control. | Establishes the biological relevance and therapeutic potential of the novel metabolite. |
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of this pipeline depends on specific research reagents and platforms.
Table 3: Key Research Reagent Solutions for the Discovery Pipeline
| Item | Function in the Pipeline | Example / Specification | Role in Establishing Causation |
|---|---|---|---|
| Pichia pastoris Expression System | Heterologous host for protein & sometimes peptide expression. | Strains like GS115; vector pPIC9K for secretion [71]. | Provides a clean background to confirm the BGC's sole responsibility for metabolite production. |
| Sephadex G-50 & Q-Sepharose FF | Chromatography media for protein purification [71]. | For size-exclusion and anion-exchange chromatography, respectively. | Enables isolation of pure compound for unambiguous structural and functional validation. |
| Global Natural Products Social (GNPS) | Public mass spectrometry data repository and analysis platform [27] [23]. | Infrastructure for molecular networking and spectral library search. | Provides the reference database for dereplication and the network context to link unknown spectra. |
| AntiMarin / DNP Database | Curated chemical databases of natural products. | AntiMarin (~60k compounds); Dictionary of Natural Products (~255k compounds) [23]. | Essential reference for dereplication algorithms to define the "known" and thus highlight the "unknown". |
| PacBio HiFi Reads | Long-read sequencing technology for high-quality genome assembly [72]. | Provides reads of 15-20 kbp with Q30+ accuracy. | Produces contiguous genome assemblies essential for accurately identifying intact, complete BGCs. |
| Hi-C Sequencing Kit | Determines chromosomal conformation for scaffolding. | Proximity ligation assay (e.g., Dovetail AssemblyLink) [72]. | Places assembled BGC contigs within a chromosomal context, informing regulatory potential. |
The path from genomic correlation to causative validation in natural product discovery is now a structured, high-throughput pipeline. The comparative advantage lies not in any single tool but in their strategic integration. DEREPLICATOR+ and similar algorithms efficiently clear the field of known compounds [23]. Genome mining provides the genetic blueprint. Their cross-validation within molecular networks identifies high-probability novelty [27]. Finally, model-guided experimental design frameworks like MINE optimize the sequence of costly heterologous expression experiments in the face of complex data [70].
The resulting prioritized clusters move beyond correlation—they represent testable hypotheses with strong supporting evidence from orthogonal data types. This rational, integrated approach directly addresses the historical bottleneck of rediscovery, systematically guiding researchers and drug developers toward the most promising novel bioactive compounds for isolation and development.
Conclusion
The systematic cross-validation of genome mining with dereplication results represents a paradigm shift in natural product discovery, moving from serendipitous finding to a predictable, hypothesis-driven workflow. This integrated approach directly addresses the major bottlenecks of re-discovery and silent gene clusters by creating a self-informing cycle where genomic predictions guide analytical chemistry, and experimental data validates genomic hypotheses. For biomedical and clinical research, this translates to a more efficient pipeline for uncovering novel chemical scaffolds with bioactive potential. Future directions will be dominated by the deeper integration of artificial intelligence for predictive modeling, the expansion of unified multi-omics platforms, and the application of these strategies to complex microbiomes and metagenomic data, ultimately accelerating the delivery of new leads for drug-resistant infections, oncology, and other areas of unmet medical need.
References
- 1. , sequencing and identification of peptidic natural... Dereplication [https://pubs.rsc.org/en/content/articlehtml/2016/np/c5np00050e]
- 2. BJOC - Navigating and expanding the roadmap of natural product... [https://www.beilstein-journals.org/bjoc/articles/18/178]
- 3. Genome mining reveals the distribution of biosynthetic gene ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11754-z]
- 4. Rapid Dereplication of Bioactive Compounds in Plant and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380011/]
- 5. Automated genome mining predicts structural diversity and ... [https://elifesciences.org/reviewed-preprints/109154]
- 6. Genome mining based on transcriptional regulatory networks ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003183]
- 7. An effective dereplication strategy for analyzing a ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25018867]
- 8. Accurate Structure Prediction of Natural Products with ... [https://chemrxiv.org/engage/chemrxiv/article-details/6820654c927d1c2e660f6511]
- 9. Highly-accurate prediction of colorectal cancer through low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12449802/]
- 10. High-Throughput Screening - Drug Discovery [https://www.technologynetworks.com/drug-discovery/articles/high-throughput-screening-principles-applications-and-advancements-405121]
- 11. Advances in high-throughput drug screening based on ... [https://www.sciencedirect.com/science/article/pii/S2090123225006897]
- 12. Navigating structure-based drug discovery with emerging ... [https://www.nature.com/articles/s44386-025-00031-4]
- 13. MIBiG 3.0: a community-driven effort to annotate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9825592/]
- 14. Dereplication of microbial metabolites through database ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6168521/]
- 15. HypoRiPPAtlas as an Atlas of hypothetical... | Nature Communications [https://www.nature.com/articles/s41467-023-39905-4?error=cookies_not_supported&code=b3474464-c06f-4449-8dc8-224dcf0bcd78]
- 16. 7.0: new and improved predictions for detection, regulation... antiSMASH [https://pmc.ncbi.nlm.nih.gov/articles/PMC10320115/]
- 17. Genome mining reveals the distribution of biosynthetic gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278603/]
- 18. The Deep Mining Era: Genomic, Metabolomic, and Integrative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299652/]
- 19. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 20. Comment on “Using genomic data and machine learning to ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013673]
- 21. Genome mining reveals novel biosynthetic gene clusters in ... [https://www.nature.com/articles/s41598-023-47121-9]
- 22. Dereplication, Sequencing and Identification of Peptidic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5590107/]
- 23. Dereplication of microbial metabolites through database ... [https://www.nature.com/articles/s41467-018-06082-8]
- 24. Genome Mining and Comparative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8303946/]
- 25. The Integration of Genome Mining, Comparative ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.600116/full]
- 26. Accelerating discovery of bioactive ligands with ... [https://www.nature.com/articles/s41467-025-56349-0]
- 27. Dereplication of peptidic natural products through database ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5409158/]
- 28. Molecular networking: An efficient tool for discovering and ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708525000822]
- 29. An integrated multi-omic approach for discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560952/]
- 30. EasyGeSe – a resource for benchmarking genomic prediction ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-12129-0]
- 31. Performance Comparison of Genomic Best Linear ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11816165/]
- 32. Natural Products Dereplication: Databases and Analytical ... [https://pubmed.ncbi.nlm.nih.gov/39101983/]
- 33. Dereplication - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/dereplication]
- 34. skDER & CiDDER: two scalable approaches for microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10690176/]
- 35. Comparing Genomic Prediction Models by Means of Cross ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8639521/]
- 36. Genomic prediction and cross-validation [https://bio-protocol.org/exchange/minidetail?id=715319&type=30]
- 37. Recovery of genomes from metagenomes via a ... [https://www.nature.com/articles/s41564-018-0171-1]
- 38. Natural product discovery in soil actinomycetes: unlocking ... [https://www.sciencedirect.com/science/article/pii/S1369527424000638]
- 39. Targeted genome mining with GATOR-GC maps ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11888242/]
- 40. Advanced Methods for Natural Products Discovery: Bioactivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10222211/]
- 41. Mining Actinomycetes' Metabolomes and Genomes for Anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12663769/]
- 42. Genome mining and metabolite profiling illuminate the ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-03801-2]
- 43. Biological activity of secondary metabolites ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1550516/full]
- 44. Genomes of 211 Actinomycete Strains from Diverse ... [https://www.nature.com/articles/s41597-025-05567-8]
- 45. Navigating and expanding the roadmap of natural product ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9749553/]
- 46. Quality assessment of peptide tandem mass spectra - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2423436/]
- 47. The most complete view of the human genome yet sets ... [https://www.jax.org/news-and-insights/2025/july/the-most-complete-view-of-the-human-genome-yet-sets-new-standard-for-use-in-precision-medicine]
- 48. Complex genetic variation in nearly complete human ... [https://www.nature.com/articles/s41586-025-09140-6]
- 49. Beyond clinical genomics: addressing critical gaps in One ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12066572/]
- 50. Benchmarking metagenomic binning tools on real datasets ... [https://www.nature.com/articles/s41467-025-57957-6]
- 51. Correlative metabologenomics of 110 fungi reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10313767/]
- 52. New AI-Powered Metagenomics Toolkit Enhances Large- ... [https://www.bluetools-project.eu/new-ai-powered-metagenomics-toolkit-enhances-large-scale-microbialanalysis/]
- 53. Metagenomics-Toolkit: the flexible and efficient cloud ... [https://pubmed.ncbi.nlm.nih.gov/40677915/]
- 54. Benchmarking Metagenomic Pipelines for the Detection of ... [https://www.sciencedirect.com/science/article/pii/S0362028X25001358]
- 55. Computational Tools and Resources for Long-read ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12631790/]
- 56. Deep Dive into Metagenomics Kits [https://www.marketreportanalytics.com/reports/metagenomics-kits-312062]
- 57. Targeted genome mining for natural product discovery [https://www.sciencedirect.com/science/article/abs/pii/S0076687925001624]
- 58. RFBGCpred: A random forest based tool for prediction of ... [https://www.sciencedirect.com/science/article/abs/pii/S0304416525001047]
- 59. Dereplication of secondary metabolites from Sophora ... [https://www.nature.com/articles/s41598-025-94958-3]
- 60. Metagenomics-Toolkit: the flexible and efficient cloud-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12267984/]
- 61. Automated genome mining predicts structural diversity and ... [https://elifesciences.org/reviewed-preprints/109154/reviews]
- 62. Genome Analysis of Alternaria alstroemeriae L6 ... [https://www.mdpi.com/2309-608X/11/10/710]
- 63. Untargeted metabolomics screening reveals unique ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1038299/full]
- 64. Genome mining reveals the distribution of biosynthetic ... [https://pubmed.ncbi.nlm.nih.gov/40691532/]
- 65. Comparative genome features and secondary metabolite ... [https://www.nature.com/articles/s41598-023-36039-x]
- 66. Artificial intelligence for natural product drug discovery and ... [https://www.sciencedirect.com/science/article/pii/S2666521225001206]
- 67. lightweight nucleotide sequence search and dereplication [https://arxiv.org/html/2511.19769v1]
- 68. Genome mining based on transcriptional regulatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12161575/]
- 69. Genomic structure predicts metabolite dynamics in ... [https://www.sciencedirect.com/science/article/pii/S0092867421015427]
- 70. MINE: a new way to design genetics experiments for discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12001805/]
- 71. Heterologous expression, purification and function of the ... [https://pubmed.ncbi.nlm.nih.gov/29202831/]
- 72. Recent Advances in Genome Editing and Bioinformatics [https://pmc.ncbi.nlm.nih.gov/articles/PMC11989416/]