AI-Driven ADMET Prediction for Herbal Compounds: Accelerating Discovery and De-risking Development
This article provides a comprehensive analysis of how artificial intelligence (AI) and machine learning (ML) are transforming the prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties for herbal...
AI-Driven ADMET Prediction for Herbal Compounds: Accelerating Discovery and De-risking Development
Abstract
This article provides a comprehensive analysis of how artificial intelligence (AI) and machine learning (ML) are transforming the prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties for herbal compounds. Targeting researchers and drug development professionals, it explores the foundational challenges of herbal ADMET, detailing the application of modern computational techniques like graph neural networks and multi-task learning. It further addresses critical methodological hurdles such as data scarcity and model interpretability, offers practical troubleshooting strategies, and evaluates model validation through competitive benchmarks and real-world case studies. The review synthesizes how these AI-guided approaches are creating a new paradigm for the efficient, evidence-based translation of traditional herbal knowledge into modern therapeutics, while also considering future regulatory and ethical directions.
The Herbal ADMET Challenge: Why AI is Essential for Modernizing Traditional Medicine Research
Herbal medicinal products present a formidable challenge for modern pharmacological research and drug development due to their intrinsic multi-component nature and variable composition [1]. Unlike single-entity pharmaceutical drugs, herbal products contain complex mixtures of bioactive phytochemicals, each with its own pharmacokinetic (PK) and pharmacodynamic (PD) profile. This complexity is compounded by variability between batches of the same herb, arising from factors such as plant origin, harvesting conditions, and processing methods [1]. Consequently, predicting their absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles—a cornerstone of drug development—becomes exceptionally difficult using traditional experimental approaches alone.
Artificial Intelligence (AI) has emerged as a transformative force in this domain. AI and machine learning (ML) algorithms are capable of managing and integrating large, diverse datasets—including cheminformatic data, pharmacological pathways, genomic information, and real-world clinical evidence [1] [2]. This computational power allows researchers to analyze the complex, multi-parameter space of herbal compounds, predict potential herb-drug interactions (HDIs), and optimize formulations, thereby bridging the gap between traditional phytotherapy and precision medicine [3] [4]. These tools are scalable and can screen large libraries, prioritizing candidates for costly experimental validation and reducing the time and resources spent on non-viable compounds [1] [5].
Computational Prediction of ADMET Properties
The initial evaluation of any therapeutic compound involves profiling its ADMET characteristics. For novel herbal compounds, in silico prediction is a critical first step to prioritize candidates for further study.
Core Predictive Models and Data Integration
AI-driven ADMET prediction utilizes various models that integrate chemical, biological, and phenotypic data. Key approaches include:
- Similarity-based methods: Infer ADMET properties by evaluating structural or target-based similarity to compounds with known profiles [1].
- Machine Learning/Deep Learning models: Integrate diverse data sources (e.g., molecular descriptors, bioassay results) to predict complex endpoints like metabolic stability, permeability, and toxicity [1] [2]. Deep learning architectures, such as graph neural networks (GNNs), are particularly adept at handling the complex molecular structures of natural products [6].
- Knowledge Graphs: Represent relationships between herbs, their constituent compounds, protein targets, and diseases as a network. This enables the prediction of multi-target mechanisms and synergistic effects within herbal formulations [7].
Table 1: Key Computational Tools for Herbal Compound ADMET Prediction
| Tool Type | Specific Model/Approach | Primary Application in Herbal Research | Key Advantage |
|---|---|---|---|
| Quantitative Structure-Activity Relationship (QSAR) | Random Forest, Support Vector Machines | Predicting toxicity, metabolic lability, and plasma protein binding from molecular structure [2]. | Interpretability, works well with smaller datasets. |
| Deep Learning for Molecules | Graph Neural Networks (GNNs), Transformers | Predicting complex ADMET endpoints for novel, structurally unique phytochemicals [6]. | Captures intricate structural patterns without manual feature engineering. |
| Network Pharmacology | Herb-Ingredient-Target-Pathway Networks | Uncovering the synergistic "multi-component, multi-target" mechanisms of herbal formulas [6] [4]. | Provides systems-level biological context, not just single-target predictions. |
| Knowledge Graph | Neo4j-based graphs with custom scoring systems [7] | Identifying synergistic herbal combinations and predicting their phenotypic effects (e.g., anti-inflammatory). | Integrates disparate data types (chemical, genomic, clinical) into a unified, queryable framework. |
Case Study: ADMET Prediction for Chamuangone
A 2025 study on chamuangone (CHM), a bioactive compound from Garcinia cowa, exemplifies the integrated computational-experimental protocol [8]. Computational predictions revealed a complex ADMET profile:
- Drug-likeness: CHM violated traditional rules (Lipinski, Pfizer) but showed high natural product-likeness and structural novelty [8].
- Absorption & Distribution: Conflicting predictions on permeability (high Caco-2, low PAMPA), high plasma protein binding (~96%), and low potential for blood-brain barrier penetration [8].
- Metabolism & Toxicity: High probability of being a P-glycoprotein inhibitor, low probability of being a substrate. Alert for potential reactive substructures (ALARM NMR) [8].
Table 2: Summary of Predicted ADMET Properties for Chamuangone (CHM) [8]
| ADMET Property Category | Specific Parameter | Predicted Value/Outcome | Interpretation & Implication |
|---|---|---|---|
| Physicochemical | LogP / LogD7.4 | Not in optimal range | May challenge solubility and formulation. |
| Drug-likeness | QED (Quantitative Estimate) | < 0.34 | Low drug-likeness per desirability concept; high complexity. |
| Synthetic Accessibility | Conflicting (Easy per SAScore, Hard per GASA) | Uncertainty in feasible synthesis. | |
| Absorption | Pgp Substrate Score | 0.0 | Very low probability of being effluxed by Pgp. |
| Pgp Inhibitor Score | 0.927 | High probability of inhibiting Pgp, risking drug-drug interactions. | |
| Human Intestinal Absorption | High | Likely excellent oral absorption. | |
| Distribution | Plasma Protein Binding (PPB) | 95.877% | High binding may reduce free, active drug concentration. |
| Blood-Brain Barrier Penetration | Score = 0.004 | Effectively does not cross BBB; limits CNS side effects. | |
| Toxicity | ALARM NMR Rule | Alert triggered | Contains substructure potentially reactive with thiols. |
| Chelator Rule | 2 Alerts | Contains two substructures that may chelate metal ions. |
Experimental Protocols for AI-Guided Validation
Predictions from AI models require rigorous experimental validation. The following protocols detail standardized methodologies for this critical phase.
Protocol 1: In Silico Molecular Docking and Dynamics for Mechanism Proposal
Objective: To validate AI-predicted targets and propose a mechanism of action for a phytochemical (e.g., Chamuangone's anti-inflammatory effect [8]).
- Target Preparation: Retrieve 3D crystal structures of key inflammatory targets (e.g., TNF-α, TLR4, iNOS, COX-2, p65 subunit of NF-κB) from the Protein Data Bank (PDB). Remove water molecules and co-crystallized ligands. Add polar hydrogens and assign Kollman/CHARMm charges.
- Ligand Preparation: Generate 3D conformers of the phytochemical (e.g., Chamuangone) from its SMILES notation. Optimize geometry using molecular mechanics (MMFF94) and assign Gasteiger charges.
- Molecular Docking: Perform flexible-ligand docking into the active/allosteric site of each prepared target using software like AutoDock Vina or Glide. Set an exhaustiveness value ≥ 50. Run docking in triplicate.
- Analysis: Rank poses by binding affinity (kcal/mol). Select the top pose for each target based on score and geometric fit. Analyze key binding interactions (hydrogen bonds, hydrophobic contacts, pi-stacking).
- Molecular Dynamics (MD) Simulation (Optional, for refinement): Solvate the best docked complex in a water box. Neutralize with ions. Run a minimization and equilibration protocol (NVT and NPT ensembles). Conduct a production MD run (e.g., 100 ns) using AMBER or GROMACS. Analyze root-mean-square deviation (RMSD), binding free energy (via MM/PBSA), and interaction persistence.
Protocol 2: In Vitro Validation of Anti-inflammatory Activity in Macrophages
Objective: To experimentally confirm the anti-inflammatory activity predicted by docking studies [8].
- Cell Culture: Maintain RAW264.7 murine macrophages in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C, 5% CO₂.
- Cell Viability Assay (Pre-requisite): Seed cells in a 96-well plate. Treat with a logarithmic concentration range of the phytochemical (e.g., 1-100 µM CHM) for 24 hours. Assess viability using the MTT assay. Calculate the IC₅₀ and select non-toxic concentrations for subsequent assays.
- Inflammation Induction and Treatment: Seed cells and pre-treat with selected non-toxic concentrations of the phytochemical for 2 hours. Induce inflammation by adding lipopolysaccharide (LPS) (e.g., 1 µg/mL) to all wells except the vehicle control. Incubate for an additional 18-24 hours.
- Measurement of Inflammatory Markers:
- Nitric Oxide (NO): Collect culture supernatant. Mix with Griess reagent. Measure absorbance at 540 nm and quantify nitrite concentration against a sodium nitrite standard curve.
- Pro-inflammatory Cytokines (TNF-α, IL-6): Use the collected supernatant in commercially available ELISA kits according to the manufacturer's protocol.
- Protein Expression (iNOS, COX-2): Lyse cells to extract protein. Perform Western Blotting using specific primary antibodies against iNOS and COX-2, with β-actin as a loading control.
- Data Analysis: Express data as mean ± SEM. Use one-way ANOVA followed by a post-hoc test (e.g., Dunnett's) to compare treatment groups to the LPS-only control. A p-value < 0.05 indicates statistical significance.
Protocol 3: Clinical Validation of an AI-Identified Herbal Combination
Objective: To evaluate the clinical efficacy of a herbal drug combination identified via a knowledge graph scoring system [7].
- Trial Design: Implement a randomized, parallel-group, open-label design. Recruit eligible patients diagnosed with the target condition (e.g., Plasma Cell Mastitis).
- Intervention: Prepare the herbal combination (e.g., Taraxacum, Fructus forsythiae, Honeysuckle, etc. [7]) as standardized granules following Good Manufacturing Practice (GMP).
- Experimental Group: Oral administration of herbal granule decoction (e.g., 20g/bag, twice daily).
- Control Group: Standard of care (e.g., methylprednisolone tablets, 20 mg/day).
- Outcome Measures: Collect blood serum samples at baseline and post-treatment (e.g., 2 months).
- Primary Endpoints: Reduction in specific inflammatory cytokines (e.g., IL-6, TNF-α) measured via ELISA.
- Secondary Endpoints: Clinical symptom score, recurrence rate, and overall health status.
- Statistical Analysis: Perform intention-to-treat analysis. Compare within-group and between-group changes using paired and independent t-tests (or non-parametric equivalents). A p-value < 0.05 is considered statistically significant.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Materials for Herbal Compound ADMET Research
| Reagent/Material | Function in Research | Key Application Example |
|---|---|---|
| Lipopolysaccharide (LPS) | A potent inflammatory stimulant used to induce a consistent pro-inflammatory state in immune cells in vitro. | Activating RAW264.7 macrophages to study the anti-inflammatory effects of compounds like Chamuangone [8]. |
| MTT Reagent | A colorimetric indicator of cell metabolic activity. Its reduction to formazan is used to quantify cell viability and cytotoxicity. | Determining the non-toxic concentration range of a herbal extract before functional assays [8]. |
| Griess Reagent | A chemical assay system for the detection and quantification of nitrite, a stable breakdown product of nitric oxide (NO). | Measuring NO production as a key readout of macrophage-mediated inflammation [8]. |
| ELISA Kits (TNF-α, IL-6, etc.) | Highly specific immunoassays for quantifying protein concentrations in complex biological fluids (e.g., cell supernatant, serum). | Quantifying levels of specific pro-inflammatory cytokines in in vitro models or patient serum samples [8] [7]. |
| Caco-2 Cell Line | A human colon adenocarcinoma cell line that spontaneously differentiates to form monolayers with properties of intestinal enterocytes. | Assessing the intestinal permeability and absorption potential of herbal compounds in vitro [8]. |
| Standardized Herbal Extract Granules | Clinically-grade, quality-controlled preparations of single herbs or formulas with consistent phytochemical profiles. | Used as the investigational product in clinical trials to ensure reproducibility and reliability of findings [7]. |
Application in Advanced Drug Delivery & Personalization
The integration of AI extends beyond prediction into the design of optimized formulations, particularly nanocarriers, to address poor bioavailability—a common limitation of herbal compounds [3].
AI-Driven Nanocarrier Design Workflow: Machine learning models, including Gaussian process regression and neural networks, are trained on datasets containing parameters of nanocarrier composition (lipid type, polymer ratio), process conditions, and resulting outputs (particle size, encapsulation efficiency, drug release profile). Once trained, the model can inverse-design nanocarrier formulations that meet target criteria for a given phytochemical (e.g., high loading for curcumin, sustained release for quercetin) [3]. This approach personalizes delivery systems by incorporating patient-specific data, bridging phytomedicine and precision nanotechnology [3].
The research paradigm for complex herbal compounds is fundamentally shifting from a purely empirical, trial-and-error approach to a predictive, AI-guided discipline. By integrating multi-scale data—from molecular structures to clinical outcomes—into sophisticated computational models, researchers can now deconvolute the complexity of multi-component formulations, rationally predict their behavior, and design more effective and safer herbal-based therapies. The future of ethnopharmacology and phytopharmaceutical development lies in this continuous, iterative loop of in silico prediction, targeted experimental validation, and clinical translation, all accelerated by the power of artificial intelligence.
Critical Gaps in Traditional ADMET Data for Herbal Medicines
The systematic evaluation of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) is a cornerstone of modern drug development. For herbal medicines, however, this evaluation is fraught with unique and significant challenges. The global reliance on plant-based therapies is substantial, with approximately 88% of the world's population using traditional and complementary medicine for primary healthcare needs [9]. Despite this widespread use, the pharmacological and toxicological profiles of herbal compounds are often poorly characterized, creating critical data gaps that hinder safety assessment, regulatory oversight, and the integration of these remedies into evidence-based medicine [9] [10]. These gaps stem from inherent complexities such as multi-component mixtures, herb-drug interactions, and variability in preparation, which are not adequately addressed by traditional, single-compound ADMET testing paradigms [11] [6].
The thesis of this work posits that artificial intelligence (AI), particularly machine learning (ML) and generative AI (GenAI), provides a transformative framework to bridge these gaps. By leveraging in silico prediction, knowledge graph construction, and multi-omics data integration, AI can reconstruct plausible ADMET profiles for complex botanicals, prioritize experimental validation, and ultimately accelerate the development of safer, more effective herbal-derived therapeutics [11] [12] [13]. This document outlines the specific quantitative deficiencies in traditional data, provides actionable experimental and computational protocols to address them, and details the essential toolkit for implementing an AI-guided research strategy.
Quantitative Analysis of Traditional ADMET Data Gaps
The limitations of traditional ADMET data for herbal medicines can be categorized and quantified across several key dimensions. The following tables summarize these critical gaps, drawing from recent pharmacovigilance reports, computational validation studies, and analyses of regulatory submissions.
Table 1: Discrepancies Between Computational Predictions and Experimental Data for Herbal Compounds
| ADMET Property | Traditional Experimental Challenge | Computational Prediction Highlight | Example Compound & Discrepancy | Implication for Herbal Medicine |
|---|---|---|---|---|
| Intestinal Absorption | Variable results across different in vitro models (Caco-2, PAMPA) [10]. | AI models predict permeability but require high-quality data for training [14] [12]. | Chamuangone: Caco-2 model suggests excellent permeability, while PAMPA suggests poor permeability [8]. | Reliable oral bioavailability prediction for complex mixtures remains difficult. |
| Metabolism (CYP450) | Complex interplay of multiple compounds inhibiting or inducing enzymes [10]. | Tools can predict sites of metabolism and major metabolites for single compounds [14]. | Polyherbal formulations may cause unpredicted herb-drug interactions via enzyme modulation [11]. | High risk of unanticipated pharmacokinetic interactions with conventional drugs. |
| Toxicity (e.g., DILI) | Chronic and idiosyncratic toxicity hard to capture in short-term assays [10]. | ML models predict endpoints like drug-induced liver injury (DILI) from chemical structure [14]. | Metabolites of herbal compounds (e.g., aristolochic acid) can be more toxic than the parent compound [12]. | Post-market pharmacovigilance is critical, as pre-market toxicity screening is often insufficient [9]. |
| Distribution (BBB Penetration) | Limited models for predicting brain exposure of natural products [10]. | Predictors estimate blood-brain barrier penetration based on physicochemical properties [14]. | Chamuangone: Predicted to not cross the BBB, potentially avoiding CNS side effects [8]. | Enables targeted design of neuroactive or neuro-safe herbal therapeutics. |
Table 2: Documented Safety Gaps and Data Deficiencies from Pharmacovigilance
| Data Gap Category | Quantitative Measure / Evidence | Source / Context | Root Cause | AI-Guided Solution Potential |
|---|---|---|---|---|
| Under-Reporting of ADRs | Only 0.6% of all reports in WHO's VigiBase (1968-2019) involved herbal ingredients as "suspected" drugs [9]. | Global pharmacovigilance database analysis. | Lack of awareness, attribution difficulty, and weak regulatory mandates for herbal products [9]. | NLP mining of electronic health records and social media for signals of herbal ADRs [11]. |
| Herb-Drug Interaction (HDI) Risk | Patients combining Chinese herbal medicine with Western drugs had "significantly higher" likelihood of adverse events [9]. | Clinical study in Singapore TCM clinics. | Polypharmacy and lack of HDI screening in clinical practice. | Knowledge graphs linking herbal compounds, drug targets, and metabolic pathways to predict HDIs [11] [6]. |
| Variable Product Quality | Analysis of dossiers in some regions reveals "significant gaps in safety data" required for market authorization [9]. | Regulatory submission review in LMICs. | Inconsistent sourcing, processing, and lack of standardization. | AI-powered chemical fingerprinting (e.g., from HPLC/MS) to authenticate products and batch consistency [6]. |
| Lack of Chronic Toxicity Data | Prolonged TCM use may lead to chronic toxicity "not detected through short-term safety assessments" [10]. | Review of TCM ADMET research challenges. | The cost and duration of long-term animal studies. | In vitro organoid models coupled with AI-based trend analysis for long-term exposure effects [10] [15]. |
Table 3: Validation Rates of AI Predictions for Natural Product ADMET
| AI Model / Platform | Reported Performance | Key Advantage for Herbal Medicines | Study / Validation Context | Reference |
|---|---|---|---|---|
| MSformer-ADMET | Outperformed conventional models across 22 ADMET tasks from TDC [12]. | Uses fragment-based representations, better for complex natural product scaffolds. | Systematic benchmarking on curated ADMET datasets. | [12] |
| ADMET Predictor | Predicts >175 properties, with models ranked #1 in independent comparisons [14]. | Integrates predictions with high-throughput PBPK simulation for dose estimation. | Used in industry for small molecules; applicable to defined herbal compounds. | [14] |
| Generative AI (LLMs) | Can digitize and decode polyherbal formulations from traditional texts [11]. | Extracts latent ADMET-related knowledge from unstructured ethnopharmacological data. | Case studies across Ayurvedic, TCM, and other traditional systems. | [11] |
| Network Pharmacology Models | Propose synergistic effects via herb-ingredient-target-pathway graphs [6]. | Moves beyond single-compartment prediction to model systemic effects of mixtures. | Applied to predict anti-cancer, anti-inflammatory actions of herbals. | [6] |
Detailed Experimental and Computational Protocols
To address the gaps identified above, researchers must adopt standardized, multi-modal protocols. The following sections detail essential workflows.
Protocol 1: Systematic Pre-Analysis for In Silico Herbal Research (SAPPHIRE Guideline) This protocol is adapted from the SAPPHIRE guideline, which provides an eight-step checklist for a robust computational study on medicinal plants [16].
- Plant Authentication & Documentation: Collect and document the botanical specimen with a certified taxonomist. Record the plant's scientific name, author citation, plant part used, geographical location, and date of collection. Deposit a voucher specimen in a recognized herbarium.
- Extraction & Fractionation: Perform sequential extraction using solvents of increasing polarity (e.g., hexane, dichloromethane, ethyl acetate, methanol, water). Document the precise extraction method, solvent-to-material ratio, temperature, time, and yield for each fraction [16].
- Phytochemical Profiling: Analyze the active fraction using High-Performance Liquid Chromatography coupled with Mass Spectrometry (HPLC-MS) or Gas Chromatography-MS (GC-MS). Use tandem mass spectrometry (MS/MS) to obtain fragmentation patterns for compound identification [16].
- Compound Identification & Dereplication: Compare MS/MS spectra and retention indices with published databases (e.g., GNPS, METLIN). For novel or ambiguous compounds, proceed with isolation via preparative HPLC and structural elucidation using Nuclear Magnetic Resonance (NMR) spectroscopy [16] [8].
- Data Curation for AI Modeling: Compile the identified compounds into a structured database. Include for each: canonical SMILES string, molecular weight, formula, and the original plant source. This clean dataset is the essential input for all subsequent in silico ADMET predictions [12].
- In Silico ADMET Screening: Input the SMILES strings into a tiered prediction pipeline:
- Tier 1 (Drug-likeness): Apply rules like Lipinski's Rule of Five and calculate quantitative estimates (QED) [14] [8].
- Tier 2 (Property Prediction): Use software (e.g., ADMET Predictor) or advanced ML models (e.g., MSformer-ADMET) to predict key properties: solubility (logS), intestinal permeability (Caco-2), metabolic stability (CYP450), and potential toxicity alerts (e.g., Ames, DILI) [14] [12] [8].
- Tier 3 (Interaction & Systems Analysis): Employ network pharmacology tools to map compounds onto protein-target networks and disease pathways to hypothesize synergy and mechanisms of action [6].
- Priority Ranking: Rank compounds based on a composite score balancing predicted favorable ADMET properties and strong target engagement or bioactivity potential.
- Reporting: Adhere to the SAPPHIRE checklist to ensure all pre-analytical steps are fully reported, enabling reproducibility and validation of the computational findings [16].
Protocol 2: In Vitro Validation of AI-Predicted ADMET Properties This protocol outlines the experimental follow-up for compounds prioritized by in silico screening. A. Intestinal Absorption Assessment
- Model Selection: Use the Caco-2 human colorectal adenocarcinoma cell line cultured on permeable Transwell inserts for 21 days until full differentiation and tight junction formation. Confirm monolayer integrity by measuring Transepithelial Electrical Resistance (TEER) > 300 Ω·cm² [10].
- Transport Study: Add the test compound to the apical compartment. Sample from the basolateral compartment at scheduled time points (e.g., 30, 60, 90, 120 min).
- Analysis: Quantify compound concentration in samples using HPLC-UV or LC-MS/MS. Calculate the apparent permeability coefficient (Papp). Compare the directionality (A-to-B vs. B-to-A) to assess the involvement of active efflux transporters like P-glycoprotein [10].
B. Metabolic Stability & Metabolite Identification
- Incubation: Incubate the test compound (e.g., 1 µM) with human liver microsomes (HLM) or hepatocytes in a suitable buffer at 37°C. Initiate the reaction with NADPH cofactor. Use a positive control (e.g., verapamil) and a negative control (no NADPH).
- Quenching & Sample Prep: At time points (e.g., 0, 5, 15, 30, 60 min), quench the reaction with an equal volume of ice-cold acetonitrile. Centrifuge to remove precipitated protein.
- Analysis: Analyze the supernatant using LC-MS/MS. Monitor the depletion of the parent compound over time to calculate intrinsic clearance. Use high-resolution MS (HRMS) in full-scan mode with data-dependent MS/MS on samples from early time points to identify major metabolites [10].
C. Cytotoxicity & Mechanistic Toxicity Screening
- Cell Viability Assay: Treat relevant cell lines (e.g., HepG2 for liver toxicity, HEK293 for renal) with a range of compound concentrations for 24-72 hours. Assess viability using standard assays like MTT or CellTiter-Glo.
- High-Content Screening (HCS): For compounds predicted to have DILI risk, employ HCS. Stain treated HepG2 cells with fluorescent dyes for markers like mitochondrial membrane potential, reactive oxygen species (ROS), and nuclear morphology. Image and analyze to derive mechanistic toxicity profiles [14].
- Organ-on-a-Chip Validation: For high-priority compounds with predicted organ-specific toxicity, use advanced microphysiological systems (e.g., liver-chip). These systems provide dynamic flow and multi-cellular architecture, offering a more physiologically relevant toxicity readout than static cultures [10] [15].
Diagram 1: AI-Guided Framework to Bridge Herbal ADMET Data Gaps
Implementing the above protocols requires a combination of wet-lab and dry-lab tools. The following table details key resources.
Table 4: Research Reagent Solutions for Herbal ADMET Research
| Tool / Resource Category | Specific Item / Platform | Function & Application in Herbal ADMET | Key Benefit / Consideration |
|---|---|---|---|
| In Vitro ADMET Models | Caco-2 cell line (HTB-37) [10] | Gold-standard model for predicting intestinal permeability and absorption of herbal compounds. | Correlates with human oral absorption; requires long (21-day) culture for differentiation. |
| In Vitro ADMET Models | Pooled Human Liver Microsomes (HLM) or Cryopreserved Hepatocytes [10] | Essential for studying Phase I/II metabolism, metabolic stability, and metabolite identification. | Source-to-source variability exists; use pooled donors for consistency. |
| In Vitro ADMET Models | MDCK-MDR1 cell line [10] | Engineered to overexpress P-glycoprotein (P-gp). Used to assess if herbal compounds are substrates or inhibitors of this key efflux transporter. | Shorter culture time than Caco-2; specifically probes transporter-mediated interactions. |
| AI/Software Platforms | ADMET Predictor (Simulations Plus) [14] | Commercial software predicting >175 ADMET endpoints. Useful for generating initial property profiles for defined herbal compounds. | Includes "ADMET Risk" scores; integrates with PBPK modeling. Requires clear chemical structures as input [14]. |
| AI/Software Platforms | MSformer-ADMET (Open Source) [12] | Advanced, fragment-based deep learning model for ADMET property prediction. Particularly suited for complex natural product scaffolds. | Outperforms conventional models; offers better interpretability via fragment attention maps [12]. |
| AI/Software Platforms | GNPS (Global Natural Products Social Molecular Networking) | Open-access platform for community-wide organization and analysis of MS/MS spectra. Critical for compound dereplication in complex herbal extracts. | Accelerates identification of known compounds and discovery of analogues within herbal mixtures. |
| Chemical Standards & Databases | Phytochemical Reference Standards (e.g., from ChromaDex, Sigma) | Pure compounds for use as analytical standards, assay controls, and for generating training data for AI models. | Essential for quantitative analysis and method validation. |
| Chemical Standards & Databases | Traditional Medicine Global Library (TMGL) / TCM Databases | Curated knowledge bases linking herbs, compounds, targets, and indications. Serve as foundational data for building network pharmacology models and knowledge graphs [11]. | Enables systems-level analysis of herbal medicine action and interaction. |
Diagram 2: Integrated AI & Experimental Workflow for Herbal ADMET
The critical gaps in traditional ADMET data for herbal medicines—spanning mixture complexity, chronic toxicity, herb-drug interactions, and systemic under-reporting—pose a significant challenge to global public health and drug discovery. However, as detailed in these application notes, a new paradigm is emerging. The integration of robust, standardized pre-analytical protocols (like SAPPHIRE) [16] with advanced AI and ML tools (such as fragment-based transformers and network pharmacology) [11] [6] [12] creates a powerful, iterative framework for knowledge generation.
The future of the field lies in the continued convergence of high-fidelity experimental data from next-generation in vitro models (e.g., organ-on-a-chip) [10] [15] and AI-driven in silico exploration. This synergy will enable a shift from reactive risk assessment to proactive, safety-by-design for herbal medicines. By adopting the detailed protocols and toolkit presented here, researchers can systematically deconvolute the complexity of botanicals, validate AI predictions with mechanistic experiments, and ultimately contribute to building a predictive, evidence-based foundation for the safe and effective use of traditional medicines in the 21st century.
The persistent 90% failure rate of drug candidates in clinical development represents one of the most significant challenges in pharmaceutical science, translating to tremendous financial losses exceeding $1-2 billion per approved drug and wasted scientific resources [17]. Analysis of clinical trial data reveals that approximately 40-50% of failures stem from inadequate clinical efficacy, while 30% result from unmanageable toxicity—both fundamentally connected to poor ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiles [17]. For natural products and herbal compounds, these challenges are magnified by structural complexity, multi-component nature, and scarce pharmacokinetic data [18]. This document provides application notes and experimental protocols for implementing AI-guided ADMET prediction within a herbal compounds research framework, aiming to address these high-stakes failures through early, accurate pharmacokinetic profiling.
Table 1: Primary Causes of Clinical Drug Development Failure
| Failure Cause | Percentage of Failures | Key ADMET Components Involved |
|---|---|---|
| Lack of Clinical Efficacy | 40-50% | Absorption, Distribution, Metabolism |
| Unmanageable Toxicity | 30% | Toxicity, Metabolism, Distribution |
| Poor Drug-like Properties | 10-15% | Absorption, Solubility, Permeability |
| Commercial/Strategic Issues | ~10% | Not applicable |
The ADMET Failure Landscape in Drug Development
Quantitative Analysis of Clinical Trial Attrition
Despite rigorous optimization in preclinical stages, nine out of ten drug candidates fail after entering clinical studies [17]. This attrition occurs primarily during Phase I, II, and III trials, with ADMET-related issues contributing to approximately 60-80% of these failures when considering both efficacy and toxicity shortcomings [17] [19]. The transition from preclinical to clinical stages represents the most costly point of failure, with investments often exceeding hundreds of millions of dollars before a compound reaches human trials.
The pharmaceutical industry has responded by implementing earlier ADMET screening, significantly reducing failures due to poor drug-like properties from 30-40% in the 1990s to 10-15% today [17]. This improvement demonstrates that strategic early intervention in ADMET assessment can substantially impact development success rates. However, natural products present unique challenges as they frequently violate conventional drug-likeness rules (such as Lipinski's Rule of Five) while maintaining therapeutic potential [20].
The Tissue Exposure Gap in Current Optimization Paradigms
Current drug optimization overwhelmingly emphasizes potency and specificity through structure-activity relationship (SAR) studies while overlooking tissue exposure and selectivity [17]. This imbalance may mislead candidate selection and impact the clinical balance of dose, efficacy, and toxicity. The Structure–Tissue Exposure/Selectivity–Activity Relationship (STAR) framework addresses this gap by classifying drug candidates based on both potency/specificity and tissue exposure/selectivity [17].
Table 2: STAR Classification Framework for Drug Candidates
| Class | Specificity/Potency | Tissue Exposure/Selectivity | Clinical Dose Implication | Success Probability |
|---|---|---|---|---|
| I | High | High | Low dose needed | High |
| II | High | Low | High dose with high toxicity | Requires cautious evaluation |
| III | Adequate | High | Low dose with manageable toxicity | Often overlooked, moderate |
| IV | Low | Low | Inadequate efficacy/safety | Should be terminated early |
Class III compounds—those with adequate specificity but high tissue exposure—represent particularly promising yet frequently overlooked candidates for natural products, which may exhibit moderate target affinity but favorable distribution profiles [17].
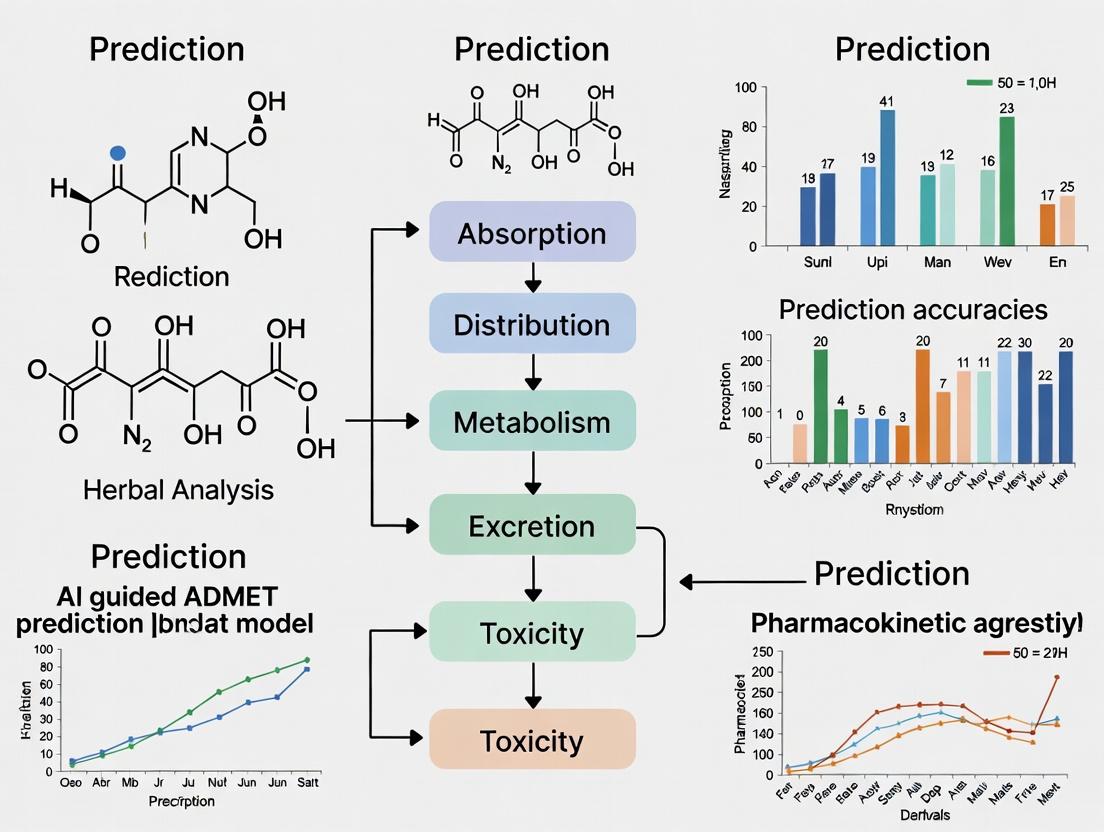
AI-Guided ADMET Prediction: Framework and Workflows
Conceptual Framework for Herbal Compound ADMET Prediction
The complex, multi-constituent nature of herbal medicines necessitates an integrated computational-experimental framework. Unlike single-entity pharmaceuticals, herbal products contain mixtures of bioactive compounds with potentially synergistic or antagonistic effects on ADMET properties [18]. AI-guided approaches must account for this complexity through multi-scale modeling that integrates chemical structure, biological activity, and pharmacokinetic parameters.
AI-Guided ADMET Prediction Workflow for Herbal Compounds
Transformer-Based Models for Multi-Task ADMET Prediction
Recent advancements in transformer architectures enable simultaneous prediction of multiple ADMET properties directly from molecular representations, bypassing manual feature engineering [19]. These models generate molecular embeddings from SMILES (Simplified Molecular Input Line Entry System) sequences using self-attention mechanisms, capturing intricate molecular features without predefined descriptors.
The transformer model processes chemical compounds through 12 encoder layers, tokenizing SMILES strings into discrete elements (typically individual atoms) that are embedded into continuous numerical space [19]. Rotary Positional Embedding (RoPE) captures spatial relationships between atoms, while linear attention mechanisms improve computational efficiency for large molecular sequences. The resulting embeddings are passed through feed-forward networks to predict diverse ADMET properties including solubility, permeability, metabolic stability, and toxicity endpoints.
Key Implementation Protocol:
- Data Preparation: Curate dataset from PharmaBench (52,482 entries across 11 ADMET properties) or similar resources [21]
- SMILES Standardization: Apply RDKit to normalize molecular representations
- Model Architecture: Implement 12-layer transformer encoder with 768-dimensional embeddings
- Multi-Task Heads: Configure separate prediction heads for each ADMET property
- Training Regimen: Pre-train on 1.8 billion molecules from ZINC and PubChem, fine-tune on ADMET-specific datasets [19]
- Validation: Apply scaffold splitting to ensure generalization to novel chemical structures
Large Language Models for Experimental Data Curation
The variability and inconsistency of experimental ADMET data present significant challenges for model training. LLM-based multi-agent systems address this by extracting and standardizing experimental conditions from unstructured assay descriptions [21]. The PharmaBench development employed a three-agent system: Keyword Extraction Agent (KEA) identifies key experimental conditions, Example Forming Agent (EFA) generates structured examples, and Data Mining Agent (DMA) extracts conditions from full datasets.
Application Protocol for LLM-Assisted Data Curation:
- Assay Description Collection: Aggregate bioassay descriptions from ChEMBL, PubChem, and literature
- Few-Shot Prompt Engineering: Develop prompts with clear instructions and examples for each ADMET assay type
- Multi-Agent Implementation: Deploy KEA, EFA, and DMA agents using GPT-4 or domain-specific LLMs
- Human Validation: Implement manual review of extracted conditions for critical assays
- Data Integration: Merge results across sources, resolving conflicts through predefined rules
- Benchmark Creation: Compile standardized datasets with consistent experimental conditions
Experimental Protocols for ADMET Validation
Integrated Computational-Experimental Workflow
Validation of AI-predicted ADMET properties requires a tiered experimental approach, particularly crucial for herbal compounds with limited existing pharmacokinetic data [8]. The following workflow integrates computational screening with progressively complex experimental validation.
Three-Tier ADMET Validation Protocol for Herbal Compounds
Protocol: In Silico ADMET Profiling of Phytochemical Libraries
Objective: Rapid screening of phytochemical libraries for favorable ADMET properties using computational tools.
Materials:
- Phytochemical library (SMILES or structure files)
- SwissADME web tool or local installation
- pkCMS software for pharmacokinetic prediction
- Toxicity prediction tools (ProTox, ADMETlab)
Procedure:
- Structure Preparation: Convert all compounds to standardized SMILES format. For ionizable compounds, generate relevant protomeric states at physiological pH (7.4).
- Physicochemical Property Calculation: Use SwissADME to compute:
- Lipophilicity (Log P/Log D)
- Water solubility (Log S)
- Molecular weight and polar surface area
- Hydrogen bond donors/acceptors
- Rotatable bonds
- Pharmacokinetic Prediction: Apply pkCMS to estimate:
- Human intestinal absorption
- Blood-brain barrier penetration
- Plasma protein binding
- Volume of distribution
- Clearance mechanisms
- Toxicity Screening: Evaluate:
- hERG channel inhibition potential
- Hepatotoxicity
- Mutagenicity and carcinogenicity
- Phospholipidosis potential
- Drug-likeness Evaluation: Assess against multiple criteria:
- Lipinski's Rule of Five
- Veber's criteria
- Ghose filter
- Natural product-likeness score
- Priority Ranking: Score compounds based on composite ADMET profile, prioritizing those with balanced properties.
Validation Reference: In a study of 308 Dracaena phytochemicals, this protocol identified 12 compounds with favorable ADMET profiles, representing 3.9% of the library [22]. Key findings included 50.3% with high gastrointestinal absorption and 89% without hepatotoxicity alerts.
Protocol: Experimental Validation of Predicted Absorption Properties
Objective: Experimental verification of computational absorption predictions for prioritized herbal compounds.
Materials:
- Caco-2 cell line (ATCC HTB-37)
- DMEM culture medium with 10% FBS
- Transwell inserts (3.0 μm pore size, 12 mm diameter)
- LC-MS/MS system for compound quantification
- Test compounds dissolved in DMSO (final concentration <0.5%)
Procedure:
- Cell Culture: Maintain Caco-2 cells in DMEM with 10% FBS at 37°C, 5% CO₂. Passage at 80-90% confluence.
- Monolayer Preparation: Seed cells on Transwell inserts at 1×10⁵ cells/cm². Culture for 21-28 days until transepithelial electrical resistance (TEER) exceeds 300 Ω·cm².
- Transport Studies:
- Apical-to-Basolateral (A-B): Add compound (10 μM) to apical chamber, sample basolateral side at 30, 60, 90, 120 minutes
- Basolateral-to-Apical (B-A): Add compound to basolateral chamber, sample apical side at same intervals
- Include positive controls (high permeability: propranolol; low permeability: atenolol)
- Sample Analysis: Quantify compound concentrations using LC-MS/MS with appropriate calibration curves.
- Data Calculation:
- Apparent permeability: P_app = (dQ/dt) / (A × C₀)
- Efflux ratio: ER = Papp(B-A) / Papp(A-B)
- Recovery: %R = (Total amount recovered) / (Initial amount) × 100
Interpretation: Compounds with P_app(A-B) > 10×10⁻⁶ cm/s exhibit high permeability, while ER > 2 suggests active efflux. Compare experimental results with computational predictions to validate and refine AI models.
Protocol: Metabolic Stability Assessment Using Liver Microsomes
Objective: Evaluate metabolic stability of herbal compounds in human liver microsomes.
Materials:
- Human liver microsomes (pooled, 20 mg/mL protein)
- NADPH regeneration system (Solution A: NADP+, glucose-6-phosphate; Solution B: glucose-6-phosphate dehydrogenase)
- Potassium phosphate buffer (100 mM, pH 7.4)
- Test compounds (1 mM stock in DMSO)
- LC-MS/MS for quantification
Procedure:
- Incubation Preparation: In duplicate, mix:
- 0.1 mg/mL liver microsomes
- 1 μM test compound
- NADPH regeneration system (1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase)
- Potassium phosphate buffer to final volume of 200 μL
- Time Course Experiment: Incubate at 37°C. Remove 25 μL aliquots at 0, 5, 15, 30, and 60 minutes. Quench with 50 μL ice-cold acetonitrile containing internal standard.
- Control Incubations: Include minus-NADPH controls to assess non-NADPH-dependent degradation.
- Sample Processing: Centrifuge at 14,000×g for 10 minutes. Analyze supernatant by LC-MS/MS.
- Data Analysis:
- Calculate percentage remaining at each time point relative to t=0
- Determine in vitro half-life: t₁/₂ = 0.693 / k, where k is elimination rate constant
- Calculate intrinsic clearance: CL_int = (0.693 / t₁/₂) × (Incubation volume / Microsomal protein)
Interpretation: Compounds with t₁/₂ > 30 minutes demonstrate acceptable metabolic stability. Compare with computational predictions of CYP450 metabolism to identify specific metabolic soft spots for structural optimization.
Protocol: Toxicity Screening for Herbal Compounds
Objective: Assess potential toxicity endpoints for herbal compounds prioritized by AI prediction.
Materials:
- HepG2 cells (ATCC HB-8065) for hepatotoxicity assessment
- hERG-transfected HEK293 cells for cardiotoxicity screening
- Ames test strains (TA98, TA100, TA1535, TA1537) for mutagenicity
- Cell culture media and reagents
- Test compounds with appropriate vehicle controls
Procedure: Hepatotoxicity Assessment:
- Culture HepG2 cells in EMEM with 10% FBS. Seed in 96-well plates at 10,000 cells/well.
- After 24 hours, treat with test compounds at 8 concentrations (typically 0.1-100 μM) in triplicate.
- Incubate for 48 hours, then assess viability using MTT assay.
- Calculate IC₅₀ values for cytotoxicity.
hERG Inhibition Screening:
- Culture hERG-HEK293 cells in DMEM with 10% FBS and selection antibiotics.
- Perform patch-clamp electrophysiology or use fluorescence-based assays (e.g., FluxOR Thallium kit).
- Test compounds at multiple concentrations to generate inhibition curves.
- Calculate IC₅₀ for hERG channel inhibition.
Mutagenicity (Ames Test):
- Prepare test compound in DMSO or appropriate solvent.
- Mix with overnight bacterial culture and top agar, with and without S9 metabolic activation.
- Pour onto minimal glucose agar plates, incubate at 37°C for 48-72 hours.
- Count revertant colonies; positive mutagenicity indicated by ≥2-fold increase over vehicle control with dose response.
Data Integration: Combine toxicity endpoints with computational predictions to build comprehensive toxicity profiles. Compounds with clean toxicity profiles across these assays progress to in vivo studies.
Case Studies and Practical Applications
Case Study: Chamuangone ADMET Profiling and Anti-inflammatory Validation
A comprehensive study of chamuangone (a phloroglucinol from Garcinia cowa) demonstrates the integrated AI-experimental approach [8]. Computational prediction using SwissADME suggested favorable intestinal absorption (high HIA score) and minimal blood-brain barrier penetration (BBB score = 0.004), reducing CNS side effect potential. However, predictions indicated high plasma protein binding (95.877%) and potential P-glycoprotein inhibition (Pgp inhibitor score = 0.927).
Experimental validation in LPS-induced RAW264.7 macrophages confirmed anti-inflammatory activity with inhibition of NO production and pro-inflammatory cytokines. Molecular docking revealed strong interactions with key inflammatory pathway proteins (NF-κB, MAPK), providing mechanistic insights. This case exemplifies how computational ADMET profiling can guide experimental design and prioritize compounds for resource-intensive biological assays.
Application: Drug-Herb Interaction Prediction Using AI Models
The complex multi-constituent nature of herbal products creates significant challenges for predicting drug-herb interactions (DHIs) [1]. AI models integrating multiple data sources can predict both pharmacokinetic and pharmacodynamic interactions:
Network Pharmacology Approach:
- Construct herb-ingredient-target-pathway networks using databases like TCMSP and HIT
- Identify overlapping targets between herbal constituents and conventional drugs
- Predict synergistic or antagonistic effects based on network topology
Machine Learning Framework:
- Train models on known DHI data from resources like DDI-Corpus and TWOSIDES
- Incorporate chemical, biological, and phenotypic features
- Apply graph neural networks to capture complex relationship patterns
Implementation Protocol:
- Data Collection: Aggregate known DHIs from literature and adverse event reports
- Feature Generation: Compute molecular descriptors for herbal constituents and drugs
- Model Training: Implement random forest or deep learning models with attention mechanisms
- Validation: Test predictions against clinical case reports and in vitro interaction studies
Table 3: AI Model Performance for ADMET Prediction
| Model Type | Application | Key Metrics | Reference |
|---|---|---|---|
| Transformer | Multi-task ADMET | AUC: 0.82-0.91 across properties | [19] |
| Graph Neural Network | Toxicity prediction | Accuracy: 87.5% for hepatotoxicity | [23] |
| Random Forest | Bioavailability | Q²: 0.78 for human oral absorption | [23] |
| Large Language Model | Data curation | F1-score: 0.89 for condition extraction | [21] |
Table 4: Research Reagent Solutions for ADMET Studies
| Resource Category | Specific Tools/Reagents | Function in ADMET Research | Key Providers |
|---|---|---|---|
| Computational Platforms | SwissADME, pkCMS, ADMETlab | In silico prediction of pharmacokinetic properties | Swiss Institute of Bioinformatics, Simulations Plus |
| AI/ML Frameworks | DeepChem, ChemBERTa, DGL-LifeSci | Machine learning model development for ADMET prediction | Harvard, Stanford, Amazon |
| Experimental Assays | Caco-2 cells, liver microsomes, hERG assays | Experimental validation of absorption, metabolism, and toxicity | ATCC, Corning, Thermo Fisher |
| Reference Datasets | PharmaBench, ChEMBL, Tox21 | Curated data for model training and benchmarking | MindRank AI, EMBL-EBI, NIH |
| Analytical Instruments | LC-MS/MS systems, plate readers, patch clamp | Quantification and mechanistic studies of ADMET properties | Waters, Agilent, Molecular Devices |
Future Directions and Implementation Guidelines
Advancing AI Models for Herbal Compound ADMET
Future development should focus on several critical areas:
- Multi-Constituent Modeling: Develop AI models that handle herbal mixtures rather than isolated compounds
- Dynamic ADMET Prediction: Incorporate temporal aspects of pharmacokinetics using time-series models
- Personalized ADMET: Integrate pharmacogenomic data to predict population variability
- Explainable AI: Implement interpretable models that provide mechanistic insights alongside predictions
Implementation Strategy for Research Laboratories
Phase 1: Foundation (Months 1-3)
- Establish computational infrastructure for AI-based prediction
- Curate herbal compound libraries with standardized structure representations
- Train team on essential ADMET concepts and AI tools
Phase 2: Integration (Months 4-9)
- Implement tiered screening protocol (computational → in vitro → in vivo)
- Validate predictions for representative compounds from each herbal class
- Refine models based on experimental feedback
Phase 3: Optimization (Months 10-18)
- Develop laboratory-specific models tuned to particular herbal medicine classes
- Establish high-throughput screening capabilities for prioritized compounds
- Create decision frameworks for compound progression based on ADMET profiles
Quality Control and Regulatory Considerations
As AI-guided ADMET prediction moves toward regulatory acceptance, several quality standards must be implemented:
- Model Validation: External validation using completely independent datasets
- Uncertainty Quantification: Implementation of confidence intervals for all predictions
- Applicability Domain: Clear definition of chemical space where models are reliable
- Documentation: Comprehensive records of training data, model parameters, and validation results
- Continuous Monitoring: Regular performance assessment and model updating as new data emerges
The integration of AI-guided ADMET prediction into herbal compound research represents a transformative approach to addressing the high failure rates in drug development. By implementing the protocols and frameworks described in this document, researchers can substantially de-risk the development pipeline, prioritize resources on compounds with favorable pharmacokinetic profiles, and ultimately increase the success rate of translating herbal medicines into evidence-based therapeutics.
The discovery and development of therapeutics from herbal compounds are undergoing a fundamental paradigm shift. The traditional approach, heavily reliant on labor-intensive trial-and-error screening of natural extracts, is being rapidly augmented and, in many cases, superseded by predictive, data-driven methodologies powered by Artificial Intelligence (AI) and machine learning (ML) [6]. This transformation is particularly critical in the domain of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction, where late-stage failures due to poor pharmacokinetic or safety profiles have historically been a major bottleneck [20].
AI offers a compelling solution to the unique challenges of herbal research. Natural products exhibit immense structural diversity and complexity, often defying conventional drug-likeness rules like Lipinski's Rule of Five [20]. Furthermore, they are frequently studied as complex mixtures, making it difficult to identify the active constituents and their synergistic effects [6] [24]. AI and in silico tools can analyze these complex datasets, predict bioactivity, infer mechanisms of action, and prioritize candidates for experimental validation, thereby accelerating the entire discovery pipeline [6] [25]. This document outlines the core AI methodologies, computational workflows, and experimental validation protocols that form the foundation of this new, predictive paradigm in herbal compound research.
Core AI Methodologies for Herbal Compound Analysis
The AI-guided pipeline employs a suite of computational methods, each addressing specific questions in the discovery process, from initial screening to mechanistic understanding.
- Bioactivity and Target Prediction: Supervised ML models are trained to predict the protein targets or biological activities of novel herbal compounds. As demonstrated in a key study, Random Forest classifiers achieved an average AUC of 0.9 in predicting shared protein targets by learning from multiple chemical similarity fingerprints (e.g., Morgan, MACCS) and physicochemical descriptors of known drug-target pairs [26]. This approach successfully identified 5-methoxysalicylic acid as a novel Cox-1 inhibitor [26].
- ADMET Profiling: In silico tools are essential for early pharmacokinetic and safety screening. Platforms like ADMETLab 3.0 provide predictions for crucial parameters such as human intestinal absorption, blood-brain barrier penetration, CYP450 enzyme inhibition, and acute toxicity [27]. For natural compounds, which may be available only in minute quantities, these computational methods offer a rapid and cost-effective alternative to preliminary experimental testing [20].
- Network Pharmacology and Synergy Prediction: To decipher the "multi-component, multi-target" action of herbal formulations, network-based approaches are used. Databases like HerbComb integrate herb-ingredient-target-pathway data to propose synergistic combinations and underlying mechanisms [24]. These models treat biological systems as interconnected networks, predicting how multiple compounds in a formulation collectively perturb disease networks.
- Advanced Molecular Modeling:
- Molecular Docking: This technique predicts the preferred orientation (binding pose) and affinity of a small molecule within a protein's target binding site. It is routinely used to understand and visualize potential interactions, such as the binding of curcumin analogs PGV-5 and HGV-5 to P-glycoprotein (P-gp) [27].
- Molecular Dynamics (MD) Simulations: MD goes beyond static docking by simulating the physical movements of atoms and molecules over time. This provides insights into the stability of protein-ligand complexes, conformational changes, and binding free energies, offering a more realistic assessment of interaction dynamics [20] [27].
Table 1: Key AI/ML Methodologies and Their Applications in Herbal Research
| Methodology | Primary Function | Typical Application in Herbal Research | Key Tools/Models |
|---|---|---|---|
| Random Forest / Ensemble Learning [26] | Classification & Regression | Predicting protein targets, bioactivity, and ADMET properties. | scikit-learn, R RandomForest package |
| Graph Convolutional Networks (GCN) [28] | Graph-based Classification | Classifying herbal properties (e.g., Cold/Hot, Meridian) from molecular graphs. | PyTorch Geometric, Deep Graph Library |
| Network Pharmacology [6] [24] | Systems-level Analysis | Mapping herb-ingredient-target-disease pathways, predicting synergistic combinations. | Cytoscape, HerbComb database [24] |
| Molecular Docking [20] [27] | Binding Pose Prediction | Visualizing and scoring the interaction of herbal compounds with protein targets (e.g., P-gp). | AutoDock Vina, MOE, Glide |
| Molecular Dynamics Simulations [20] [27] | Dynamic Interaction Analysis | Assessing stability of compound-target complexes and calculating binding free energies. | GROMACS, AMBER, NAMD |
Integrated AI-Guided Workflow for Herbal Drug Discovery
The predictive modeling process follows a structured, iterative workflow that integrates the methodologies above to prioritize and validate lead candidates.
AI-Guided Herbal Drug Discovery Pipeline
- Data Curation and Representation: The pipeline begins with assembling a high-quality dataset of herbal compounds, represented as molecular fingerprints (e.g., ECFP, MACCS) or graph structures [26] [28].
- AI Predictive Modeling: Multiple predictive models run in parallel.
- Bioactivity/Target Prediction: ML models screen the library for potential activity against a disease-relevant target [26].
- ADMET Profiling: Computational tools filter compounds based on predicted pharmacokinetic and safety profiles [20] [27].
- Network Analysis: For multi-herb formulations, network proximity models assess potential synergistic effects [24].
- Candidate Prioritization: Results from all modules are integrated to generate a shortlist of prioritized lead candidates with a high probability of desired bioactivity and favorable ADMET properties.
- Experimental Validation: Top-ranked candidates proceed to in vitro and/or in vivo testing to confirm predicted activity (see Section 5 for protocols).
- Mechanistic Elucidation (For Confirmed Hits): Experimentally active compounds undergo molecular docking and dynamics simulations to hypothesize binding modes and interactions with their targets [27].
- Iterative Model Refinement: Data from experimental validation is fed back into the AI models to retrain and improve their predictive accuracy for future cycles [6].
Case Studies in AI-Guided Discovery and Validation
Case Study 1: Predicting and Validating a Novel COX-1 Inhibitor from Dietary Compounds A study demonstrated a complete AI-to-lab workflow. An ML model combining multiple chemical fingerprints was trained on known drug-target pairs. When applied to ~11,000 natural compounds, it predicted 5-methoxysalicylic acid (found in tea and herbs) as a potential COX-1 inhibitor. Critically, in vitro enzymatic assays confirmed this prediction, while a structurally similar compound (4-isopropylbenzoic acid) not prioritized by the model showed no activity. This validated the model's ability to capture complex structure-activity relationships beyond simple similarity [26].
Case Study 2: ADMET-Driven Optimization of Curcumin Analogs for Multidrug-Resistant Cancer To address curcumin's poor bioavailability, researchers used integrated ADMET-toxicity profiling to evaluate analogs. In silico screening with ADMETLab 3.0 identified analogs PGV-5 and HGV-5 as promising P-glycoprotein (P-gp) inhibitors with potentially better profiles. Subsequent in vivo acute toxicity testing and histopathological analysis classified their safety. Molecular docking and dynamics simulations then confirmed their stable binding to P-gp, with HGV-5 showing superior binding free energy. This end-to-end approach identified a safer, more effective candidate for overcoming multidrug resistance [27].
Table 2: Summary of AI Model Performance in Featured Case Studies
| Study Focus | AI/ML Model Used | Key Performance Metrics | Experimental Validation Outcome |
|---|---|---|---|
| Bioactivity Prediction [26] | Random Forest Classifier | Avg. AUC: 0.90, MCC: 0.35, F1-Score: 0.33 | Confirmed novel COX-1 inhibition by 5-methoxysalicylic acid. |
| Medicinal Property Classification [28] | Graph Convolutional Network (GCN) | Accuracy: 0.836, F1-Score: 0.845 | Model classified "Cold/Hot" nature of herbs based on compound structures. |
| Meridian Prediction [29] | Machine Learning (Multiple) | Top Prediction Accuracy: 0.83 | Associated molecular fingerprints & ADME properties with TCM Meridian classes. |
Detailed Experimental Protocols for Validation
Following AI-based prioritization, experimental validation is essential. Below are detailed protocols for key validation assays.
Protocol 1:In VitroEnzymatic Inhibition Assay (e.g., COX-1)
This protocol validates AI-predicted target engagement, as performed for 5-methoxysalicylic acid [26].
5.1.1 Reagents and Materials
- Purified recombinant human COX-1 enzyme.
- Test compound (e.g., 5-methoxysalicylic acid) and control inhibitor (e.g., Aspirin).
- Arachidonic acid (substrate).
- Reaction buffer (e.g., 100 mM Tris-HCl, pH 8.0).
- Colorimetric or fluorimetric prostaglandin detection kit.
- 96-well microplate reader.
5.1.2 Procedure
- Enzyme Reaction: In a 96-well plate, mix COX-1 enzyme with a series of concentrations of the test compound (prepared in DMSO, final DMSO <1%) or vehicle control. Pre-incubate for 10 minutes at 25°C.
- Initiation: Start the reaction by adding arachidonic acid to a final concentration within the KM range.
- Incubation: Incubate the reaction mixture at 37°C for a predetermined time (e.g., 5-10 minutes).
- Termination & Detection: Stop the reaction with a stopping reagent (e.g., HCl). Add detection kit components to quantify the prostaglandin product according to the manufacturer's instructions.
- Data Analysis: Measure absorbance/fluorescence. Plot reaction velocity vs. compound concentration to determine the IC₅₀ value (concentration causing 50% inhibition) using non-linear regression software (e.g., GraphPad Prism).
Molecular Docking and Dynamics Workflow
Protocol 2: Molecular Docking and Dynamics Simulation
This protocol details the computational validation of binding interactions for a prioritized compound (e.g., HGV-5 binding to P-gp) [27].
5.2.1 System Preparation
- Protein: Obtain the 3D structure of the target protein (e.g., P-gp, PDB ID: 7A6C) from the RCSB Protein Data Bank. Prepare the protein by removing water molecules, adding hydrogen atoms, and assigning protonation states at physiological pH using software like MOE or UCSF Chimera.
- Ligand: Obtain or draw the 3D structure of the herbal compound (e.g., HGV-5). Perform geometry optimization and energy minimization using molecular mechanics (MMFF94 force field) or semi-empirical quantum mechanics (PM6 method).
5.2.2 Molecular Docking Procedure
- Define Binding Site: Identify the binding site coordinates, either from the co-crystallized ligand in the PDB file or from literature.
- Docking Execution: Perform docking simulations using software like AutoDock Vina or MOE. Set the search space (grid box) to encompass the binding site. Use default or optimized docking parameters.
- Pose Analysis: Cluster the resulting ligand poses and select the top-scoring pose(s) based on docking score (affinity in kcal/mol). Analyze key hydrogen bonds, hydrophobic interactions, and π-π stacking with amino acid residues.
5.2.3 Molecular Dynamics Simulation (Post-Docking Validation)
- System Setup: Solvate the protein-ligand complex from the best docking pose in a water box (e.g., TIP3P water model). Add ions to neutralize the system's charge.
- Energy Minimization & Equilibration: Minimize the system's energy to remove steric clashes. Then, equilibrate first with restrained protein backbone (NVT ensemble) and then without restraints (NPT ensemble) to stabilize temperature (310 K) and pressure (1 bar).
- Production Run: Run an unrestrained MD simulation for a significant timescale (e.g., 100-200 nanoseconds). Use software like GROMACS or AMBER with appropriate force fields (e.g., AMBER ff14SB for protein, GAFF2 for ligand).
- Trajectory Analysis: Analyze the stability via Root Mean Square Deviation (RMSD) of the protein backbone and ligand. Calculate the binding free energy using methods like MM-PBSA or MM-GBSA on trajectory frames to quantitatively assess interaction strength.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for AI-Guided Herbal Research Validation
| Item | Function/Description | Example Use Case/Protocol |
|---|---|---|
| Recombinant Human Enzymes (e.g., COX-1, CYP450s) | High-purity, consistent enzymatic source for in vitro inhibition or metabolism assays. | Validating predicted target engagement (Protocol 5.1.1) [26]. |
| Standardized Herbal Compound Libraries | Pre-purified, characterized natural compounds for screening and biological testing. | Providing high-quality inputs for both AI training and experimental validation [6] [26]. |
| ADMET Prediction Software (e.g., ADMETLab 3.0, SwissADME) | Integrated platforms for computational prediction of pharmacokinetic and toxicity endpoints. | Early-stage filtering of herbal compound libraries [30] [27]. |
| Molecular Docking & Simulation Software (e.g., MOE, GROMACS) | Tools for predicting ligand-protein binding and simulating dynamic interactions. | Elucidating binding mode and stability of confirmed active compounds (Protocol 5.2) [20] [27]. |
| Network Analysis & Visualization Tools (e.g., Cytoscape) | Software for constructing and analyzing herb-ingredient-target-disease networks. | Exploring multi-target synergy and mechanisms of action for herbal formulations [24] [1]. |
Building the Predictive Engine: Key AI Techniques and Workflows for Herbal ADMET Modeling
The integration of artificial intelligence (AI) into pharmacology has fundamentally transformed the landscape of drug discovery, introducing unprecedented efficiencies in molecular modeling and predictive analytics [13]. This evolution is particularly consequential for the research and development of therapeutics derived from herbal compounds, which present unique challenges due to their complex, multi-constituent nature and variable composition [6]. A critical barrier in this field is the high attrition rate of drug candidates, with more than 75% of compounds failing in clinical trials, often due to unfavorable absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles [31] [32]. The traditional experimental assessment of these properties for herbal mixtures is resource-intensive, costly, and complicated by batch variability [6] [1].
This article posits that a strategic deployment of core machine learning (ML) architectures—spanning from classical ensembles like Random Forests to advanced Graph Neural Networks (GNNs)—within a unified AI-guided framework is essential to overcome these hurdles. By enabling the early, accurate, and interpretable prediction of ADMET properties for herbal compounds, these technologies can de-risk development pipelines, prioritize sustainable candidates for experimental validation, and illuminate the mechanistic underpinnings of drug-herb interactions (DHIs). Framed within a broader thesis on AI-guided ADMET prediction, this discussion explores the specific architectures, experimental protocols, and practical toolkits that are revolutionizing herbal pharmacognosy and accelerating the translation of traditional remedies into safe, effective modern medicines [13] [6] [1].
Core Architectures and Their Quantitative Performance in ADMET Prediction
The predictive modeling of ADMET properties leverages a spectrum of ML architectures, each with distinct strengths in handling molecular data's complexity, volume, and relational structure. The selection of an architecture is guided by the specific prediction task, data availability, and the need for interpretability.
Table 1: Comparison of Core ML Architectures for Key ADMET Prediction Tasks
| Architecture | Typical Molecular Representation | Key Strengths | Common ADMET Applications | Reported Performance (Example Metric) |
|---|---|---|---|---|
| Random Forest (RF) | Molecular fingerprints (e.g., ECFP, MACCS), 2D descriptors [33] [34] | High interpretability via feature importance, robust to noise, handles non-linear relationships. | Early-stage toxicity screening (e.g., general toxicity, mutagenicity), CYP inhibition classification [32] [34]. | AUC: 0.80-0.95 on various hERG benchmarks [33]. |
| eXtreme Gradient Boosting (XGBoost) | Molecular fingerprints, curated 2D/3D descriptors [33] [35] | Superior handling of imbalanced datasets, high predictive accuracy, efficient execution. | High-precision cardiotoxicity (hERG) prediction, regression tasks for IC50 values [33] [35]. | Sensitivity: 0.83, Specificity: 0.90 for hERG [33]. |
| Graph Neural Network (GNN) | Molecular graph (atoms as nodes, bonds as edges) [31] [32] | Learns directly from molecular structure, captures topological and functional group information. | Multitask ADME prediction, metabolite formation, binding affinity for complex targets [31] [32]. | State-of-the-art on 7/10 ADME parameters vs. baselines [31]. |
| Transformer / Graph Transformer | SMILES string or Molecular graph with attention [35] | Models long-range dependencies in structure, excels in generative and multi-task settings. | De novo molecular generation with optimized properties, multi-parameter prediction [35]. | Successfully generated hERG-optimized analogs of known drugs [35]. |
| Multitask GNN | Shared molecular graph embedding across tasks [31] [36] | Shares information across related tasks, mitigates data scarcity for individual endpoints. | Simultaneous prediction of 10+ ADME parameters (e.g., solubility, permeability, clearance) [31]. | Outperformed single-task models on low-data parameters like fubrain [31]. |
For herbal compound research, GNNs and Multitask GNNs are particularly powerful. They naturally model the molecular structure of individual phytochemicals and, through network pharmacology approaches, can represent the complex herb-ingredient-target-pathway relationships characteristic of polyherbal formulations [6]. This allows for the prediction of both direct compound properties and emergent synergistic or antagonistic effects.
Table 2: Key ADMET Endpoints and Relevant AI Architectures for Herbal Compound Research
| ADMET Endpoint | Significance for Herbal Compounds | Preferred AI Architecture(s) | Public Benchmark Dataset (Example) |
|---|---|---|---|
| hERG Channel Inhibition | Predicts cardiotoxicity risk (QT prolongation); a major cause of drug attrition [33] [35]. | XGBoost, GNN, Transformer [33] [35] | hERG Central (>300,000 records) [34] |
| CYP450 Enzyme Inhibition | Predicts metabolism-based drug-herb interactions (e.g., St. John's Wort) [1] [32]. | GNN, GAT, Random Forest [32] | CYP450-specific data from ChEMBL [32] |
| Passive Permeability (e.g., Caco-2, Papp) | Indicates intestinal absorption potential for oral bioavailability [31] [37]. | Multitask GNN, GNN [31] [36] | Collected datasets (e.g., ~5,581 for Caco-2) [31] |
| Hepatic Clearance (CLint) | Predicts metabolic stability and exposure half-life [31]. | Multitask GNN [31] | Collected datasets (e.g., ~5,256 compounds) [31] |
| Plasma Protein Binding (fup) | Affects distribution, free concentration, and efficacy [31]. | Multitask GNN [31] | Collected datasets (e.g., ~3,472 for human fup) [31] |
Detailed Experimental Protocols
Protocol A: Building a Multitask Graph Neural Network for Multi-Parameter ADME Prediction
This protocol details the construction of a GNN model capable of predicting multiple ADME parameters simultaneously, leveraging shared learning to compensate for limited data on specific endpoints [31] [36].
1. Data Curation and Preparation:
- Source: Gather experimental ADME data paired with canonical SMILES strings. Public sources include ChEMBL and proprietary repositories like DruMAP [31].
- Standardization: Standardize all molecular structures using toolkits like RDKit (neutralize charges, remove salts, generate canonical tautomers) [33].
- Dataset Assembly: Compile a unified dataset for M tasks (e.g., 10 ADME parameters). For compound i, create a data pair:
(Graph G_i, Label Vector y_i), wherey_i ∈ R^Mcontains experimental values for available tasks; missing values are allowed [31].
2. Molecular Graph Representation:
- Represent each molecule as a graph
G = (V, E, X).
3. Model Architecture and Training (GNNMT+FT Strategy):
- Stage 1 - Multitask Pre-training:
- Use a GNN (e.g., Message Passing Neural Network) as a shared graph encoder
f_θ(G)to generate a molecular embeddingh_i[31]. - Attach separate task-specific prediction heads
g_θ_m(h_i)for each of the M ADME parameters. - Train the model by minimizing a multitask loss function (e.g., Smooth L1 loss) that aggregates weighted losses only over available labels for each compound [31].
- Use a GNN (e.g., Message Passing Neural Network) as a shared graph encoder
- Stage 2 - Per-Task Fine-tuning:
- Use the pre-trained shared encoder
f_θ(G)as a fixed-feature extractor or with lightly tuned weights. - Fine-tune individual task-specific heads
g_θ_mon the data for each specific ADME parameter to achieve task-optimal performance [31].
- Use the pre-trained shared encoder
4. Explainability Analysis (Integrated Gradients):
- Apply the Integrated Gradients (IG) method to the trained model.
- For a predicted ADME value, IG attributes importance scores to each input atom feature by integrating the model's gradients along a path from a baseline input to the actual input [31].
- Visualize the atomic contributions on the molecular structure to identify substructures positively or negatively associated with the predicted property, validating insights against medicinal chemistry knowledge [31] [37].
Protocol B: Predictive Modeling of hERG Toxicity with Ensemble XGBoost and Applicability Domain Mapping
This protocol outlines a robust pipeline for building a high-fidelity classifier for hERG channel inhibition, integrating advanced handling of class imbalance and applicability domain assessment [33].
1. Dataset Construction and Curation:
- Source: Use the largest public hERG dataset (e.g., from Sato et al., ~291,219 molecules) [33].
- Curation: Apply strict preprocessing: remove inorganic salts and metals, standardize tautomers and charges, deduplicate stereoisomers, and resolve conflicting activity labels [33].
- Binarization: Label compounds as "inhibitors" (IC50 ≤ 10 µM or %inhibition ≥ 50% at 10 µM) and "non-inhibitors" [33].
2. Data Splitting and Feature Calculation:
- Splitting: First, separate an external test set (e.g., 30%). From the remainder, perform a time- or scaffold-based split to create training and internal validation sets, ensuring generalization [33] [35].
- Descriptor Calculation: Compute a comprehensive set of 2D molecular descriptors and fingerprints (e.g., using RDKit, alvaDesc) including physicochemical properties, topological indices, and fingerprint bits [33].
3. Model Training with XGBoost and Ensemble Strategy:
- Feature Selection: Perform recursive feature elimination (RFE) to select the most informative descriptors, reducing noise and overfitting [33].
- Ensemble Training: To combat class imbalance, train multiple XGBoost models on balanced bootstrap samples (via undersampling majority class or SMOTE) drawn from the training data [33].
- Consensus Prediction: Aggregate predictions from the individual XGBoost models to produce a final, robust consensus prediction and probability score [33].
4. Applicability Domain Mapping with ISE:
- Employ Isometric Stratified Ensemble (ISE) mapping to define the model's reliable prediction space.
- Project the training and validation compounds into a latent space using dimensionality reduction (e.g., PCA, t-SNE).
- Stratify this space into bins. For a new compound, its prediction confidence is estimated based on the local performance (e.g., accuracy) of models within the bin where it resides [33]. This step is crucial for identifying predictions on structurally novel herbal compounds that may be outside the model's domain.
Protocol C: Network Pharmacology Analysis for Herbal Compound Synergy and Interaction Prediction
This protocol uses AI to model the polypharmacology of herbal mixtures, predicting synergistic effects and potential drug-herb interactions [6] [1].
1. Network Construction:
- Data Layer Assembly: For a given herb or formula, compile layers of data:
- Constituent Layer: List of identified phytochemicals (SMILES).
- Target Layer: Predicted and known protein targets for each constituent (from docking or target prediction models).
- Pathway Layer: Biological pathways enriched with the identified targets (from KEGG, Reactome).
- Disease Layer: Associated diseases linked to the pathways.
- Graph Database Creation: Construct a heterogeneous knowledge graph where nodes represent entities (herbs, compounds, targets, pathways, diseases) and edges represent relationships (contains, binds-to, participates-in, associates-with) [6] [1].
2. AI-Powered Relationship Inference and Prioritization:
- Link Prediction: Use Graph Neural Networks or other graph embedding techniques (e.g., TransE, Node2Vec) on the knowledge graph to infer missing links—for example, predicting novel targets for a phytochemical or identifying potential synergistic compound pairs that share common pathways [6] [1].
- Community Detection: Apply algorithms to identify tightly connected clusters (modules) within the graph. A module containing multiple compounds from an herbal mixture acting on a cohesive set of targets in a specific disease pathway provides strong evidence for a mechanistic basis of synergy [1].
3. Experimental Validation Gate:
- Prioritization: Rank predicted synergistic compound pairs or herb-target-disease linkages based on network topology scores (e.g., edge confidence, module centrality).
- Validation Design: Design in vitro experiments (e.g., combination index assays in cell models) or in silico validation (e.g., molecular dynamics simulation of predicted compound-target complexes) to test the top-ranked predictions [6].
Visualization of Core Workflows and Architectures
Diagram 1: CardioGenAI Framework for hERG Liability Reduction (100 chars)
Diagram 2: Multitask GNN Architecture for ADME Prediction (100 chars)
Diagram 3: Network Pharmacology for Herbal Compound Analysis (100 chars)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software, Datasets, and Tools for AI-Guided ADMET Research
| Tool/Reagent Name | Type | Primary Function in Research | Key Features for Herbal Research | Source/Reference |
|---|---|---|---|---|
| RDKit | Open-source Cheminformatics Library | Molecular standardization, descriptor calculation, fingerprint generation, and basic molecular operations. | Essential for preprocessing diverse and complex phytochemical structures into a consistent format for modeling [33]. | rdkit.org |
| KNIME Analytics Platform | Open-source Data Analytics Platform | Visual workflow orchestration for data blending, model training (integrating Python/R), and pipeline deployment. | Enables reproducible, customizable pipelines that integrate herb-specific data processing with ML model training [33]. | knime.com |
| ChEMBL Database | Public Bioactivity Database | A manually curated repository of bioactive molecules with drug-like properties and associated ADMET assays. | Primary source for extracting experimental ADMET data on small molecules, useful for training models applicable to phytochemicals [35] [34]. | ebi.ac.uk/chembl |
| DruMAP | Public ADME Database | Provides standardized, large-scale experimental ADME parameter data for diverse compounds. | Critical for accessing high-quality, curated ADME data to train robust multitask prediction models [31]. | nibiohn.go.jp/drumap |
| CypReact | Specialized CYP Reaction Database | Curates cytochrome P450-mediated metabolic reactions and associated data. | Invaluable for building models to predict the metabolism and potential interaction risks of herbal constituents [32]. | Literature-derived [32] |
| ADMET-AI / ChemProp | Pre-trained GNN Model | A state-of-the-art graph neural network model specifically designed and pretrained for ADMET property prediction. | Provides a powerful, readily available baseline or transfer learning starting point for predicting properties of novel herbal compounds [37]. | GitHub Repository |
| Alvascience alvaDesc | Molecular Descriptor Calculator | Computes over 5,000 molecular descriptors and fingerprints for quantitative structure-activity/property relationship (QSAR/QSPR) modeling. | Useful for generating a comprehensive numerical representation of herbal compounds for use in classical ML models like RF or XGBoost [33]. | alvascience.com |
| CardioGenAI Framework | Open-source ML Framework | Integrates generative and discriminative models for redesigning molecules to reduce hERG cardiotoxicity. | A specialized tool to virtually screen and optimize lead herbal compounds with potential cardiotoxicity risks [35]. | GitHub Repository |
The application of Artificial Intelligence (AI) to predict the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) of herbal compounds represents a frontier in modern drug discovery. Herbal products pose a unique challenge due to their multicomponent nature, variable composition, and diverse biological activities, which complicate traditional pharmacokinetic and safety assessments [1]. AI and machine learning (ML) models offer a powerful solution by analyzing complex, high-dimensional data to uncover patterns and predict interactions that are not immediately apparent through conventional methods [6] [1]. However, the predictive power, reliability, and translational potential of these models are fundamentally constrained by the quality, breadth, and relevance of the underlying data.
This article details the critical practice of data acquisition and curation, framing it within a comprehensive strategy for building robust AI models for herbal ADMET prediction. We explore the synergistic use of expansive public databases and focused proprietary datasets, such as those derived from 3D bioprinting platforms (BioPrint), which provide controlled, physiologically relevant experimental data [38] [39]. The following sections provide a comparative analysis of key data resources, detailed protocols for data processing and model training, and visual workflows that integrate these elements into a cohesive research pipeline.
Core Public Databases for ADMET and Herbal Compound Research
Public databases provide the foundational chemical and biological data required to train broad-coverage AI models. Their utility lies in volume, diversity, and accessibility.
Specialized ADMET Databases
These resources are specifically curated for pharmacokinetic and toxicological endpoint prediction.
- admetSAR3.0: A comprehensive platform hosting over 370,000 experimental ADMET data points for 104,652 unique compounds. It supports predictions for 119 endpoints, including expanded sections for environmental and cosmetic risk assessment. Its integrated optimization module (ADMETopt) suggests structural modifications to improve ADMET profiles [40].
- Therapeutics Data Commons (TDC) ADMET Leaderboard: Provides a curated benchmark suite for ADMET property prediction, facilitating the comparison of different ML models and feature representations on standardized tasks [41].
Broad-Coverage Chemical and Bioactivity Databases
These databases offer wider context, including chemical structures, bioactivities, and target information.
- ChEMBL: A large-scale, open-access database containing bioactivity data, functional screening assays, and ADMET information for drug-like molecules [40] [41].
- DrugBank: Combines detailed drug data with comprehensive drug target and pathway information, useful for understanding pharmacodynamic interactions [40].
- PubChem: A vast repository of chemical substances and their biological activities, serving as a source for solubility and other property data [41].
Table 1: Key Public Databases for AI-Guided Herbal ADMET Research
| Database | Primary Content Focus | Key Data Metrics | Relevance to Herbal ADMET |
|---|---|---|---|
| admetSAR3.0 [40] | ADMET Properties | 370,000+ data points; 119 prediction endpoints | Direct source for building and benchmarking ADMET prediction models. |
| TDC ADMET [41] | Benchmark ADMET Tasks | Curated datasets for ~20 ADMET properties | Standardized evaluation of model performance on specific pharmacokinetic tasks. |
| ChEMBL [40] [41] | Bioactivity & ADMET | Millions of activity data points | Source of complementary bioactivity and ADMET data for model training. |
| DrugBank [40] | Drug Targets & Pathways | Detailed drug-target-pathway relationships | Context for pharmacodynamic (PD) herb-drug interaction prediction [1]. |
Proprietary and Specialized Datasets: The Role of BioPrint
While public data offers breadth, proprietary and specialized experimental datasets provide depth, physiological relevance, and controlled validation. 3D bioprinting, referred to here as BioPrint, generates high-value data by creating cell-laden, three-dimensional tissue constructs that mimic in vivo microenvironments [38].
The Value of BioPrint Data
BioPrint data is crucial for:
- Mechanistic Validation: Moving beyond computational predictions to observe compound effects in a structured tissue context (e.g., toxicity to hepatic spheroids, cardiotoxicity in myocardium-on-chip models) [38].
- Capturing Complexity: Assessing how herbal mixtures affect cell viability, proliferation, and differentiation within a spatially organized, multi-cellular system, which is more informative than monolayer cultures [39].
- Providing Specialized Data: Generating high-quality data on specific endpoints (e.g., metabolic activity of printed islets, vascular network formation) that may be scarce in public sources [38].
Table 2: Exemplary BioPrint Applications Generating Relevant Pharmacological Data [38] [39]
| Bioprinted Tissue | Bioink/Cell Composition | Key Experimental Outcome | Relevance for ADMET |
|---|---|---|---|
| Endothelialized Myocardium-on-a-Chip | GelMA; HUVECs, Cardiomyocytes | Tissue contracted at ~60 bpm for 7-10 days. | Model for cardiotoxicity and drug/herb effects on heart function. |
| Vascularized Bone Niche | PEG, Laponite, Hyaluronic Acid; Osteoblasts | New bone formation in vivo after 12-week implant. | Model for compound effects on bone remodeling and mineralization. |
| Sweat Gland Morphogenesis | Gelatin-Alginate; Epidermal Progenitors | Self-organized glandular tissue formation guided by pore architecture. | Model for dermal absorption and localized toxicity screening. |
| Liver Microtissue | Alginate/Gelatin; Hepatocytes | Sustained metabolic activity (e.g., CYP450). | Prime model for metabolism (M) and hepatotoxicity (T) studies. |
Integrated Data Curation and Model Development Protocols
Protocol 1: Data Acquisition and Cleaning for Ligand-Based Models
This protocol ensures data quality before model training [41].
- Compound Standardization: Use a standardized tool (e.g., from Atkinson et al.) to canonicalize all SMILES strings. Define organic elements (H, C, N, O, F, P, S, Cl, Br, I, B, Si) and remove inorganic salts and organometallics [41].
- Parent Compound Extraction: For salt forms, programmatically extract the neutral parent organic compound to ensure consistency in representation [41].
- Tautomer Standardization: Adjust tautomers to a consistent representation to avoid treating the same compound as different entities [41].
- Deduplication & Conflict Resolution: Identify duplicates based on canonical SMILES. For regression tasks, keep the first entry if values are within 20% of the inter-quartile range; for binary tasks, keep entries only if labels are perfectly consistent. Otherwise, remove the entire conflicting group [41].
- Visual Inspection: For smaller datasets, use a tool like DataWarrior to perform final visual checks for anomalies [41].
Protocol 2: Feature Engineering and Model Training Protocol
This protocol outlines steps for creating robust predictive models [41].
- Feature Representation: Calculate multiple molecular representations for each compound:
- Classical Descriptors: RDKit descriptors (e.g., molecular weight, logP).
- Fingerprints: Morgan fingerprints (ECFP).
- Pre-trained Embeddings: Use embeddings from deep neural networks (e.g., ChemBERTa).
- Iterative Feature Selection: Systematically train baseline models (e.g., Random Forest) using individual and concatenated feature sets. Use statistical hypothesis testing (e.g., paired t-test on cross-validation scores) to identify the optimal feature combination for a given dataset [41].
- Model Selection & Hyperparameter Tuning: Evaluate a suite of algorithms (SVM, Random Forest, Gradient Boosting like LightGBM, and Message Passing Neural Networks). Perform dataset-specific hyperparameter tuning for the most promising model architecture [41].
- Rigorous Evaluation:
- Employ scaffold splitting for train/test/validation splits to assess generalization to novel chemotypes.
- Use nested cross-validation with statistical testing for reliable model comparison.
- Conduct external validation on a hold-out dataset from a different source (e.g., proprietary BioPrint data) to test real-world applicability [41].
Protocol 3: Experimental Validation Using 3D Bioprinting (BioPrint)
This protocol describes generating proprietary validation data [38] [39].
- Scaffold Design & Bioink Preparation:
- Design a 3D scaffold model (e.g., porous lattice) using CAD software.
- Prepare a cell-laden bioink: Suspend target cells (e.g., hepatocytes, cardiomyocytes) at a defined density in a hydrogel blend (e.g., GelMA, alginate-gelatin).
- Bioprinting Process:
- Use a pneumatic extrusion-based bioprinter.
- Print the construct layer-by-layer into a supportive bath or onto a temperature-controlled stage.
- Crosslink the structure (e.g., using UV light for GelMA, calcium chloride for alginate).
- Compound Treatment & Assaying:
- Culture the bioprinted tissue under physiological conditions for maturation (e.g., 7-14 days).
- Treat with the herbal compound or extract at physiologically relevant concentrations.
- Assess ADMET-relevant endpoints: Cell Viability (Live/Dead assay), Metabolic Activity (AlamarBlue, ATP assay), Functional Markers (qPCR for CYP450s, immunofluorescence for albumin/cardiac troponin), and Morphological Changes (histology, confocal microscopy).
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Reagents and Tools for Data Acquisition and Curation Workflows
| Item | Category | Function/Benefit |
|---|---|---|
| RDKit [41] | Software Library | Open-source cheminformatics toolkit for calculating molecular descriptors, fingerprints, and handling SMILES operations. |
| Standardization Tool [41] | Data Curation Software | Ensures consistent molecular representation by canonicalizing SMILES, removing salts, and standardizing tautomers. |
| DataWarrior [41] | Data Visualization | Free tool for interactive visualization and final cleanliness check of chemical datasets. |
| GelMA (Gelatin Methacryloyl) [38] [39] | Bioink Material | Photocrosslinkable hydrogel providing a biocompatible, tunable ECM-mimetic environment for bioprinting tissues. |
| Sodium Alginate [38] [39] | Bioink Material | Ionic-crosslinkable biopolymer used for its good printability and gentle gelling conditions, often blended with other materials. |
| admetSAR3.0 Web Interface [40] | Prediction Server | Provides easy access to a wide array of pre-built ADMET prediction models for initial compound profiling. |
Integrated Workflow Visualizations
Diagram 1: Integrated Workflow for AI Model Development and Validation. This diagram shows the logical flow from diverse data sources through curation and AI model training, culminating in experimental validation using BioPrint, which in turn feeds new data back into the cycle.
Diagram 2: Data Curation Pipeline. A sequential view of the critical steps required to transform raw, heterogeneous data into a clean, consistent dataset suitable for AI/ML modeling.
Diagram 3: BioPrint Experimental Protocol for Validation. This flowchart outlines the key stages in generating proprietary experimental data, from tissue construct design to compound treatment and data generation.
The integration of Artificial Intelligence (AI) into pharmacology has initiated a paradigm shift in drug discovery, particularly in the challenging field of herbal chemistry [13]. Herbal medicines exert therapeutic effects through multi-component, multi-target (MCMT) synergistic mechanisms, presenting a complex landscape for scientific analysis and drug development [42]. Unlike single-compound pharmaceuticals, the bioactive constituents within a single herb—such as flavonoids, alkaloids, and terpenoids—interact with diverse biological targets, systematically modulating complex disease networks [43]. This very complexity makes the early prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties both critically important and exceptionally difficult. Late-stage failures due to poor pharmacokinetics or toxicity remain a primary cause of attrition in drug development [12].
AI-guided ADMET prediction offers a transformative solution. By leveraging machine learning (ML) and deep learning (DL) models, researchers can now decode intricate structure-activity relationships from molecular data, enabling the in silico screening of herbal compounds for favorable safety and pharmacokinetic profiles early in the discovery pipeline [44]. The foundation of all these predictive models is an effective molecular representation—the translation of a chemical structure into a computer-readable format that a model can process [45]. The evolution from traditional descriptors and fingerprints to AI-learned embeddings is enhancing our ability to capture the nuanced features of phytochemicals, thereby accelerating the development of safer, more effective therapeutics derived from natural products [13] [45].
Classical and AI-Driven Molecular Representation Methods
The choice of molecular representation is foundational to computational analysis. Methods have evolved from manual, rule-based techniques to sophisticated, data-driven models capable of uncovering latent structural patterns.
Traditional Molecular Descriptors and Fingerprints
Traditional methods rely on expert-defined rules to extract explicit features. Molecular descriptors are numerical quantifications of a compound's physicochemical properties (e.g., molecular weight, logP, topological indices) [45]. Molecular fingerprints, such as Extended-Connectivity Fingerprints (ECFPs), are bit-string representations that encode the presence or absence of specific molecular substructures [45]. They are computationally efficient and highly interpretable, making them staples for tasks like similarity searching and quantitative structure-activity relationship (QSAR) modeling [46].
Modern Learned Embeddings
AI-driven methods utilize deep learning architectures to learn continuous, high-dimensional feature embeddings directly from data. These models capture complex, non-linear relationships that are often missed by manual features.
- Graph Neural Networks (GNNs): Model a molecule as a graph, with atoms as nodes and bonds as edges. GNNs use message-passing mechanisms to aggregate information from local atomic environments, effectively learning representations that embody both structure and chemistry [45].
- Language Model-Based Approaches: Treat simplified molecular-input line-entry system (SMILES) strings or other string-based notations as a chemical "language." Transformer architectures, like those used in natural language processing, can be trained to understand syntax and semantics of these strings, generating context-aware molecular embeddings [45] [12].
- Hybrid and Specialized Models: Emerging frameworks like MSformer-ADMET adopt a fragment-based approach. Instead of atoms or characters, they use chemically meaningful molecular fragments as basic units, which are then processed by a Transformer to build a representation that excels at predicting pharmacokinetic and toxicity endpoints [12].
Table 1: Comparison of Key Molecular Representation Methods for Herbal Chemistry
| Representation Type | Key Examples | Core Principle | Advantages | Limitations | Typical Application in Herbal Research |
|---|---|---|---|---|---|
| Physicochemical Descriptors | AlvaDesc, RDKit Descriptors | Calculates numerical properties (e.g., MW, LogP, H-bond donors) [45]. | Direct physicochemical insight; highly interpretable. | May miss complex structural patterns; feature engineering required. | Initial filtering for drug-likeness (e.g., Lipinski's Rule of Five). |
| Substructural Fingerprints | ECFP, MACCS Keys | Encodes presence/absence of predefined substructures as a bit vector [45]. | Excellent for similarity search; computationally fast. | Limited to predefined substructures; can be high-dimensional. | Clustering herbal compounds; similarity-based virtual screening. |
| Graph-Based Learning | Graph Neural Networks (GNNs), AttentiveFP | Learns embeddings by propagating information across the molecular graph [45]. | Captures topology and local chemistry inherently. | Can be computationally intensive; requires careful architecture design. | Predicting herb-target interactions and multi-target activity [42]. |
| Language Model-Based | SMILES-BERT, ChemBERTa | Learns from SMILES strings using Transformer architectures [45]. | Captures sequential "syntax" of chemistry; strong transfer learning potential. | SMILES can be ambiguous for complex stereochemistry. | Pre-training on large chemical corpora for downstream ADMET tasks. |
| Fragment-Based Learning | MSformer-ADMET [12] | Represents molecules as a collection of learned chemical fragment tokens. | Chemically intuitive; excels at modeling metabolic and toxicological outcomes. | Dependent on quality and comprehensiveness of fragment library. | High-accuracy prediction of ADMET properties for natural products. |
Evolution of Molecular Representation Methods
Application Notes: Molecular Representation for AI-Guided ADMET Prediction
Accurate ADMET prediction is paramount for de-risking herbal compound development. Different molecular representations contribute uniquely to this goal.
Descriptor-Augmented Embeddings for Enhanced Performance: While learned embeddings capture deep structural patterns, augmenting them with classical descriptors can provide complementary information. A study on Mol2Vec embeddings demonstrated that combining them with 2D molecular descriptors significantly boosted performance across 16 ADMET benchmarks, achieving top results in 10 tasks [47]. This hybrid approach leverages both the data-driven power of AI and the well-established interpretability of manual descriptors.
Fragment-Based Representations for Mechanistic Insight: Models like MSformer-ADMET use a pretrained library of molecular fragments as a vocabulary [12]. This method is particularly adept for ADMET prediction because properties like metabolism and toxicity are often governed by specific structural alerts (e.g., nitroaromatics, reactive esters). The fragment-based attention mechanism can identify these sub-structural motifs, providing a degree of post-hoc interpretability by highlighting which parts of a molecule contribute most to a predicted adverse outcome [12].
Multi-Task Learning for Holistic Profiling: Herbal compounds interact with multiple biological targets and pathways. Advanced frameworks employ multi-task learning to predict several ADMET endpoints simultaneously. This leverages shared information across related tasks (e.g., hepatic metabolism and cytotoxicity), improving generalization and efficiency compared to training separate models for each property [13] [12].
Table 2: Performance of AI-Driven Models on ADMET Prediction Tasks
| Model Name | Core Representation | Key Architectural Feature | Reported Performance (Example) | Advantage for Herbal Compounds |
|---|---|---|---|---|
| MSformer-ADMET [12] | Fragment-based Tokens | Transformer with fragment vocabulary & multi-head MLP | Outperformed SMILES and graph-based baselines on 22 TDC ADMET tasks. | Fragment attention maps offer interpretability for toxicophores. |
| Enhanced Mol2Vec [47] | Learned Embeddings + Classical Descriptors | MLP on concatenated feature vectors | Top-1 results in 10/16 ADMET benchmarks on TDC. | Hybrid approach balances predictive power with computational efficiency. |
| iCAM-Net [42] | Molecular Fingerprints + Protein Embeddings | Dual-channel hypergraph with cross-attention | AUROC > 0.977 on herb-disease association prediction. | Explicitly models multi-component, multi-target (MCMT) herb action. |
| FP-ADMET/MapLight [45] | Multiple Fingerprints & Descriptors | Feature maps processed by Convolutional Neural Networks (CNNs) | Robust prediction frameworks for wide ADMET property ranges. | Integrates diverse molecular features for comprehensive profiling. |
Experimental Protocols
Protocol 1: From Plant Material to Compound Identification via UPLC-MS/MS
This protocol outlines the initial steps for creating a dataset of herbal compounds, which serves as the essential input for all computational representations.
- Sample Preparation: Collect and authenticate plant material (e.g., stems, roots). Air-dry and exhaustively extract using a suitable solvent like 70% ethanol. Concentrate the extract using rotary evaporation [48].
- UPLC-MS/MS Analysis:
- Instrumentation: Use a UPLC system coupled with a high-resolution mass spectrometer (e.g., Q Exactive Orbitrap) [48].
- Chromatography: Employ a reversed-phase C18 column. A typical gradient uses mobile phase A (water with 0.1% formic acid) and B (acetonitrile with 0.1% formic acid), ramping from 1% to 99% B over 15-20 minutes [48].
- Mass Spectrometry: Acquire data in positive or negative ionization mode with data-independent acquisition (DIA). Set a scan range of m/z 100-1500 [48].
- Metabolite Annotation: Process raw data with software (e.g., Compound Discoverer, MZmine). Tentatively identify compounds by matching accurate mass and MS/MS fragmentation patterns against databases (e.g., GNPS, MassBank) and literature [48].
Protocol 2: Generating Representations for Computational Screening
This protocol describes how to convert identified compounds into formats ready for AI/ML modeling.
- Structure Standardization: Convert identified compounds into standardized SMILES strings using toolkits like RDKit or OpenBabel.
- Generate Multiple Representations:
- Descriptors: Calculate a suite of ~200 1D/2D descriptors (e.g., molecular weight, topological polar surface area, rotatable bonds) using RDKit or the alvaDesc software.
- Fingerprints: Generate ECFP4 fingerprints (radius=2, 2048 bits) using the RDKit cheminformatics library.
- Learned Embeddings:
- For graph-based embeddings, use the standardized SMILES to create molecular graph objects (nodes=atoms, edges=bonds) with node features (atom type, degree) and edge features (bond type).
- For pre-trained model embeddings, input the SMILES string into a model like ChemBERTa or the encoder from MSformer-ADMET to extract a feature vector [45] [12].
- Dataset Curation: Assemble a curated dataset linking each compound (with its multiple representations) to experimentally measured biological or ADMET endpoints.
Protocol 3: Validation via Molecular Docking and Dynamics
In silico validation of computational predictions is crucial.
- Target Preparation: Retrieve the 3D structure of a target protein (e.g., acetylcholinesterase for Alzheimer's research) from the Protein Data Bank (PDB). Remove water molecules and co-crystallized ligands. Add hydrogen atoms and assign protonation states using software like UCSF Chimera or Schrodinger's Protein Preparation Wizard.
- Ligand Preparation: Generate 3D structures of the herbal compounds. Minimize their energy and assign correct tautomeric and ionization states at physiological pH (e.g., using LigPrep or the Open Babel toolkit).
- Molecular Docking: Perform docking simulations to predict binding poses and affinities. Software like AutoDock Vina or Glide is commonly used. Key steps include defining the binding site grid and running the docking calculation. Validate the protocol by re-docking a known native ligand [48].
- Molecular Dynamics (MD) Simulation: For top-ranked docked complexes, run MD simulations (e.g., using GROMACS or AMBER) to assess binding stability. Solvate the system in a water box, add ions, and minimize energy. Run a production simulation for 50-100 nanoseconds. Analyze root-mean-square deviation (RMSD) and fluctuations (RMSF) to confirm complex stability [48].
AI-Guided ADMET Prediction Workflow for Herbal Compounds
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools and Resources for Molecular Representation & ADMET Modeling
| Category | Item/Software | Function | Key Features/Notes |
|---|---|---|---|
| Chemical Profiling | UPLC-MS/MS System (e.g., Waters, Thermo Q Exactive) | High-resolution separation and identification of compounds in herbal extracts [48]. | Enables untargeted metabolomics and generation of initial compound lists. |
| Cheminformatics | RDKit (Open-Source Toolkit) | Core platform for cheminformatics: SMILES parsing, descriptor calculation, fingerprint generation [45]. | Python-based; essential for converting structures to computational representations. |
| Descriptor Calculation | alvaDesc | Calculates a comprehensive suite (>5,000) molecular descriptors and fingerprints [45]. | Useful for building QSAR models and augmenting learned embeddings. |
| Graph Representation | Deep Graph Library (DGL) or PyTorch Geometric | Libraries for building and training Graph Neural Network (GNN) models on molecular graphs [45]. | Simplify the implementation of complex GNN architectures. |
| Pre-trained Models | ChemBERTa, Mol2Vec, MSformer-ADMET | Provide transferable, context-aware molecular embeddings without task-specific training [12] [47]. | Can be fine-tuned on smaller herbal datasets for specific ADMET tasks. |
| Docking & Simulation | AutoDock Vina, GROMACS | Validate predicted activities via binding pose prediction (docking) and stability assessment (MD) [48]. | In silico confirmation of AI predictions before wet-lab testing. |
| Benchmark Datasets | Therapeutics Data Commons (TDC) | Curated datasets for ADMET property prediction to train and benchmark models [12] [47]. | Provides standardized tasks for fair model comparison. |
| Toxicity Assessment (NAM) | HepaRG Cells, 3D Liver Spheroids | New Approach Methodologies (NAMs) for human-relevant in vitro toxicity testing [49]. | Used for generating experimental toxicity data and validating in silico predictions. |
The convergence of advanced molecular representation methods with AI forms a powerful engine for modernizing herbal medicine research. The path forward involves a synergistic integration of these approaches: using classical fingerprints for rapid similarity-based screening, leveraging learned embeddings for high-accuracy ADMET prediction, and employing fragment-based or graph-based models for mechanistic interpretation. Frameworks like iCAM-Net, which explicitly model the MCMT paradigm [42], and MSformer-ADMET, which provides interpretable toxicity predictions [12], exemplify this next generation of tools. By embedding these representation strategies into a cohesive workflow—from plant metabolomics and computational screening to in silico and in vitro validation—researchers can systematically decode the therapeutic potential of herbal compounds. This integrated approach accelerates the identification of promising, safe lead compounds, effectively bridging traditional herbal knowledge with contemporary, data-driven drug discovery.
The integration of herbal medicines with conventional pharmacotherapy presents a significant challenge in drug development and clinical practice, primarily due to the risk of pharmacokinetic herb-drug interactions (HDIs). A major mechanism underlying these interactions is the modulation of Cytochrome P450 (CYP450) enzymes, which are responsible for metabolizing over 75% of clinically used drugs [50]. Concurrently, predicting Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties—particularly permeability and toxicity—is essential for candidate selection. Traditional experimental methods for these endpoints, while foundational, are often resource-intensive, low-throughput, and struggle with the chemical complexity of herbal extracts [51] [52].
This creates a critical need for innovative, efficient, and predictive frameworks. Artificial Intelligence (AI) and machine learning (ML) have emerged as transformative tools, capable of analyzing large-scale biological and chemical data to uncover complex patterns [1]. This document provides detailed application notes and experimental protocols for generating and applying AI-guided predictive models for key ADMET endpoints relevant to herbal compound research. The protocols cover in vitro assay generation for data acquisition, phytochemical characterization, and the development and application of state-of-the-art graph-based AI models, forming a cohesive pipeline for safety and efficacy assessment within a modern drug discovery thesis.
Experimental Protocols for Data Generation
Protocol: In Vitro CYP450 Enzyme Inhibition and Induction Assays
Objective: To generate high-quality experimental data on the effects of herbal extracts or pure phytochemicals on the activity and expression of key CYP450 isoforms.
A. Inhibition Assay Using Human Liver Microsomes (HLMs) [51]
- Reagent Preparation: Prepare a master reaction mix containing pooled HLMs (0.2 mg/mL protein concentration) in 100 mM potassium phosphate buffer (pH 7.4). Pre-incubate with an NADPH-regenerating system (1.3 mM NADP⁺, 3.3 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase, 3.3 mM MgCl₂).
- Test Compound Addition: Add the herbal extract or phytochemical at a range of concentrations (typically 0.1-100 µM for pure compounds; µg/mL for extracts) to the pre-incubation mix. Include positive control inhibitors (e.g., ketoconazole for CYP3A4) and vehicle controls.
- Reaction Initiation & Quenching: Initiate the reaction by adding a CYP isoform-specific probe substrate (see Table 1 for examples). Incubate at 37°C for a predetermined time (e.g., 10-30 minutes). Quench the reaction with an equal volume of ice-cold acetonitrile containing an internal standard.
- Analysis: Centrifuge the quenched samples (14,000 x g, 10 min) and analyze the supernatant using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) to quantify the formation of the specific metabolite from the probe substrate.
- Data Analysis: Calculate percentage inhibition relative to vehicle control. Determine the half-maximal inhibitory concentration (IC₅₀) using non-linear regression analysis (e.g., log(inhibitor) vs. response model).
B. Induction Assay Using Hepatocyte-Derived Cell Lines [51]
- Cell Culture and Treatment: Culture human hepatoma cells (e.g., HepaRG, HepG2) in appropriate medium. Seed cells in collagen-coated plates and allow to attach for 24 hours.
- Compound Exposure: Treat cells with the test herbal extract/phytochemical at non-cytotoxic concentrations for 48-72 hours. Include a positive control inducer (e.g., rifampicin for CYP3A4 via PXR activation).
- mRNA/Protein Harvest: Lyse cells to isolate total RNA (for qPCR) or total protein (for Western blot).
- Expression Analysis:
- qPCR: Perform reverse transcription and quantitative PCR using primers specific for target CYP isoforms (CYP3A4, CYP1A2, etc.). Normalize cycle threshold (Ct) values to housekeeping genes (e.g., GAPDH) and calculate fold-change relative to vehicle-treated cells using the 2^(-ΔΔCt) method.
- Western Blot: Separate proteins by SDS-PAGE, transfer to a membrane, and probe with antibodies against specific CYP enzymes. Quantify band intensity and normalize to a loading control (e.g., β-actin).
- Functional Activity Confirmations: For significant inducers, confirm increased functional activity using the HLM inhibition assay protocol above, with microsomes isolated from treated cells.
Objective: To comprehensively identify and characterize the chemical constituents within a complex herbal extract, providing the essential input data for computational modeling.
- Sample Extraction: Prepare a dried, powdered herbal sample. Perform extraction with a clinically relevant solvent (e.g., water for teas, hydro-alcoholic solutions for tinctures) using a defined solid-to-solvent ratio, temperature, and time [53].
- Lyophilization: Filter the extract and lyophilize to obtain a dry powder. Store at -20°C in a desiccator until analysis.
- UHPLC-QTOF-MS Analysis:
- Chromatography: Reconstitute the lyophilized extract and inject onto a UHPLC system equipped with a C18 reverse-phase column (e.g., 150 mm x 2.1 mm, 1.7 µm). Use a gradient elution with mobile phases A (0.1% formic acid in water) and B (acetonitrile). Optimize gradient and flow rate (e.g., 0.3-0.35 mL/min) for compound separation [53].
- Mass Spectrometry: Couple the UPLC to a Quadrupole Time-of-Flight (QTOF) mass spectrometer. Use electrospray ionization (ESI) in both positive and negative modes. Set the acquisition range to m/z 100-1500. Use a lock-mass compound for real-time mass accuracy correction.
- Data Processing: Process raw data using dedicated software (e.g., MassLynx). Perform peak picking, alignment, and deconvolution. Identify compounds by matching accurate mass, isotopic pattern, and when possible, MS/MS fragmentation spectra against commercial spectral libraries (e.g., METLIN, MassBank) or in-house databases of known phytochemicals.
Objective: To screen identified phytochemicals for potential binding and inhibition of a specific CYP450 isoform prior to in vitro testing.
- Protein Preparation: Retrieve the 3D crystal structure of the target CYP enzyme (e.g., CYP2B6, PDB ID: 3QU8) from the Protein Data Bank. Remove water molecules and co-crystallized ligands. Add polar hydrogen atoms and assign Kollman charges using molecular visualization software (e.g., Discovery Studio, AutoDock Tools).
- Ligand Preparation: Generate 3D structures of phytochemicals identified from UHPLC-MS. Optimize geometry using energy minimization (e.g., MMFF94 force field). Assign Gasteiger charges.
- Docking Simulation: Define the active site of the CYP enzyme, typically around the heme iron. Perform molecular docking using an algorithm such as CDOCKER or AutoDock Vina. Set parameters to generate multiple poses per ligand.
- Validation: Validate the docking protocol by re-docking the native co-crystallized ligand and calculating the Root Mean Square Deviation (RMSD) between the docked and original pose. An RMSD < 2.0 Å is acceptable.
- Analysis & Scoring: Analyze the top-ranked poses for each phytochemical. Key interactions to evaluate include: coordination with the heme iron, π-π stacking with key phenylalanine residues, and hydrogen bonds within the active site. Use consensus scoring from multiple functions (e.g., -CDOCKER energy, LibDock score) to prioritize compounds for experimental testing [53].
Table 1: Key CYP450 Isoforms, Probe Substrates, and Experimental Findings for Selected Herbs
| CYP Isoform | Primary Probe Substrate | Example Herb/Extract | Reported Effect (In vitro) | Key AI-Ready Endpoint |
|---|---|---|---|---|
| CYP3A4 | Midazolam, Testosterone | Dan Shen (Salvia miltiorrhiza) aqueous extract [51] | Minimal to no inhibition in 61% of assays [51] | IC₅₀, Classification (Inhibitor/Non-inhibitor) |
| CYP3A4 | Midazolam, Testosterone | Gan Cao (Glycyrrhiza uralensis) extract [51] | Tendency for inhibition [51] | IC₅₀, Time-Dependent Inhibition (TDI) flag |
| CYP2C9 | Diclofenac, Tolbutamide | Huang Qi (Astragalus) aqueous extract [51] | Tendency for induction [51] | Fold-change in mRNA/activity |
| CYP2D6 | Dextromethorphan | Black Cohosh (Cimicifuga racemosa) ethanol extract [52] | Inhibition reported (IC₅₀: 1.8-100 µM for constituents) [52] | IC₅₀, Ki (inhibition constant) |
| CYP1A2 | Phenacetin, Caffeine | Kava (Piper methysticum) extract [52] | Significant inhibition (clinically relevant) [52] | IC₅₀, Classification |
| CYP2B6 | Bupropion, Efavirenz | Artemisia afra phytochemicals (e.g., Acacetin) [53] | Strong in silico binding predicted [53] | Docking Score (kcal/mol), Binding Pose |
AI Model Development and Application Protocols
Protocol: Building a Graph Neural Network (GNN) for CYP450 Substrate/Inhibitor Prediction
Objective: To create a predictive model that classifies whether a novel phytochemical is a substrate or inhibitor of a major CYP450 isoform.
- Data Curation: Compile a dataset from public sources (e.g., PubChem, ChEMBL) and in-house assays. For each compound, include: a) SMILES string, b) Binary labels for each CYP isoform (e.g.,
CYP3A4_substrate: 1/0,CYP3A4_inhibitor: 1/0). Ensure a balanced dataset to avoid bias [50]. - Molecular Graph Representation: Convert each SMILES string into a molecular graph. Nodes represent atoms, featurized with properties (atomic number, degree, hybridization, formal charge). Edges represent bonds, featurized with type (single, double, aromatic) and conjugation [50] [54].
- Model Architecture: Implement a Graph Attention Network (GAT) or Message Passing Neural Network (MPNN).
- The GNN layers will aggregate information from neighboring atoms to learn a context-aware representation for each atom and the whole molecule.
- Use a global pooling layer (e.g., global mean pooling) to generate a fixed-size molecular embedding from the atom-level features.
- Pass the molecular embedding through a final multi-layer perceptron (MLP) with a sigmoid output for binary classification.
- Multi-Task Training: Train the model in a multi-task setup, where a single GNN backbone shares learned features, and separate MLP heads predict outcomes for each CYP isoform (e.g., CYP3A4 inhibition, CYP2D6 substrate). This improves generalization [50].
- Model Evaluation: Split data into training, validation, and test sets (e.g., 70/15/15). Evaluate using metrics: Area Under the Receiver Operating Characteristic Curve (AUROC), precision, recall, and F1-score. Use the validation set for hyperparameter tuning.
Protocol: Implementing an End-to-End Metabolism Prediction with DeepMetab Framework
Objective: To predict the complete CYP450-mediated metabolic fate of a phytochemical, including the site of metabolism (SOM) and the structure of resulting metabolites [54].
- Framework Setup: Utilize the open-source DeepMetab framework or implement its core architecture [54]. This integrates three tasks: a) Substrate profiling, b) SOM identification, c) Metabolite generation.
- Input and Feature Infusion: For a input molecule (SMILES), the framework uses a GNN backbone enhanced with multi-scale features:
- Quantum-Informed Descriptors: Calculate partial charges, HOMO/LUMO energies (via semi-empirical methods like PM6/AM1).
- Topological Descriptors: Include molecular weight, logP, topological polar surface area.
- Dual-Labeling: The graph is labeled with atom-level (e.g., reactivity) and bond-level (e.g., bond order) information [54].
- Task-Specific Execution:
- The model first classifies the molecule as a substrate for specific CYP isoforms.
- For predicted substrates, it identifies the top-2 most likely SOM atoms.
- Finally, it applies a knowledge base of expert-derived reaction rules (e.g., aliphatic hydroxylation, O-dealkylation) to the SOM to generate plausible metabolite structures.
- Validation: Test the model on a hold-out set of known herbal compounds or recent drugs. Assess SOM prediction accuracy (Top-1, Top-2) and the validity/accuracy of generated metabolites compared to literature or experimental data [54].
Protocol: Predicting Blood-Brain Barrier (BBB) Permeability with Ensemble AI Models
Objective: To classify the BBB permeability potential of phytochemicals to assess CNS activity or neurotoxicity risk.
- Data Preparation: Use the Blood-Brain Barrier Database (B3DB), containing over 7,800 compounds with binary BBB+ (permeable)/BBB- (impermeable) labels [55].
- Feature Engineering: Generate two parallel feature sets for each compound:
- Descriptor-Based: Calculate key physicochemical descriptors: Molecular Weight (<450 Da favorable), calculated LogP (optimal range ~1-3), Topological Polar Surface Area (<90 Ų favorable), number of hydrogen bond donors/acceptors.
- Fingerprint-Based: Generate a 2048-bit Morgan Fingerprint (radius=2) from the SMILES string to capture substructural patterns [55].
- Model Training and Ensembling:
- Model A (Random Forest/XGBoost): Train a Random Forest or XGBoost classifier on the descriptor-based features. Optimize using grid search and 5-fold cross-validation.
- Model B (Transformer-Based): Use a pre-trained molecular transformer model (e.g., MegaMolBART) to convert SMILES into a learned molecular embedding. Train an XGBoost classifier on these embeddings [55].
- Ensemble Prediction: For a new phytochemical, obtain predictions from both Model A and Model B. Use a soft voting ensemble (averaging predicted probabilities) to generate a final, more robust permeability score.
- Similarity Search Integration: Implement a FAISS (Facebook AI Similarity Search) index on the B3DB Morgan fingerprints. For a query phytochemical, retrieve the k-nearest neighbors and use their known permeability labels as supporting evidence for the model's prediction [55].
Table 2: Performance Benchmarks of AI Models for Key ADMET Endpoints
| Prediction Endpoint | Model Type | Key Dataset | Reported Performance | Primary Use Case in Herbal Research |
|---|---|---|---|---|
| BBB Permeability | Random Forest on Morgan Fingerprints [55] | B3DB (7,807 compounds) [55] | ~91% Accuracy, ROC-AUC ~0.93 [55] | Prioritizing neuroactive phytochemicals or assessing neurotoxicity risk. |
| BBB Permeability | MegaMolBART + XGBoost [55] | B3DB [55] | ~88% Accuracy, ROC-AUC ~0.90 [55] | Alternative deep-learning approach capturing complex SMILES semantics. |
| CYP450 Metabolism (SOM) | DeepMetab GNN Framework [54] | Curated dataset (>3,800 substrates) [54] | 100% Top-2 Accuracy on 18 FDA-approved drugs [54] | Predicting metabolic soft spots and potential toxic metabolite formation from herbs. |
| CYP450 Inhibition | Graph Attention Network (GAT) [50] | PubChem BioAssay, ChEMBL | Varies by isoform (AUROC >0.85 common) [50] | High-throughput virtual screening of herbal compound libraries for interaction risk. |
| Molecular Docking (CYP2B6) | LibDock/Ludi 3 [53] | Phytochemicals from Artemisia afra [53] | Effective discrimination of active inhibitors (Validation: ROC analysis) [53] | Structural rationale for inhibition; prioritization for in vitro testing (e.g., Acacetin). |
The Scientist's Toolkit: Essential Research Reagents and Software
Table 3: Key Research Reagent Solutions and Materials
| Item/Category | Specification/Example | Function in Protocol |
|---|---|---|
| Human Liver Microsomes (HLMs) | Pooled, gender-mixed, 20 mg/mL protein concentration. | Source of human CYP450 enzymes for in vitro inhibition and kinetic assays [51]. |
| NADPH Regenerating System | Solution A: NADP⁺, Glucose-6-Phosphate. Solution B: Glucose-6-Phosphate Dehydrogenase in citrate buffer. | Provides a constant supply of NADPH, the essential cofactor for CYP450 enzymatic activity [51]. |
| CYP-Isozyme Specific Probe Substrates | See Table 1 (e.g., Midazolam for CYP3A4, Bupropion for CYP2B6). | Selective substrates metabolized to a unique, detectable metabolite to measure activity of a specific CYP isoform. |
| Hepatocyte Cell Line | Differentiated HepaRG cells or Primary Human Hepatocytes (PHHs). | Cellular model with intact nuclear receptor (PXR, CAR) pathways for studying enzyme induction [51]. |
| UHPLC-QTOF-MS System | e.g., Waters Acquity I-Class with Xevo G2-XS QTOF. | High-resolution separation and accurate mass identification of complex phytochemical mixtures [53]. |
| Molecular Docking Software | Discovery Studio BIOVIA, AutoDock Vina, Schrödinger Suite. | Predicts the 3D binding orientation and affinity of a phytochemical within a CYP enzyme's active site [53]. |
| Cheminformatics Library | RDKit (Open Source). | Python library for converting SMILES to graphs/descriptors, calculating molecular properties, and fingerprint generation [55]. |
| Deep Learning Framework | PyTorch Geometric (PyG) or Deep Graph Library (DGL). | Specialized libraries for efficiently building and training Graph Neural Network (GNN) models on molecular graph data [50] [54]. |
| BBB Permeability Database | Blood-Brain Barrier Database (B3DB). | Curated benchmark dataset for training and validating BBB permeability prediction models [55]. |
Integrated Workflow Visualizations
AI-Guided ADMET Prediction Workflow for Herbal Compounds
DeepMetab: GNN Framework for End-to-End Metabolism Prediction
Ensemble AI Pipeline for Blood-Brain Barrier Permeability Prediction
This protocol details the integrative methodology of Artificial Intelligence-driven Network Pharmacology (AI-NP), positioned within a research thesis focused on AI-guided ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) prediction for herbal compounds. Herbal medicines, characterized by their multi-component, multi-target, and multi-pathway nature, present a significant challenge for traditional single-target drug discovery and safety evaluation [56] [57]. Network Pharmacology (NP) provides a systems-level framework to map these complex interactions, constructing "compound-target-pathway-disease" networks [58]. However, conventional NP faces limitations in handling high-dimensional data, dynamic interactions, and predictive accuracy [56].
The integration of AI—encompassing machine learning (ML), deep learning (DL), and graph neural networks (GNNs)—revolutionizes this paradigm. AI enhances NP by enabling advanced pattern recognition, predictive modeling of system perturbations, and high-fidelity prediction of pharmacokinetic and toxicological profiles [56] [59]. This synergy creates a powerful tool for de-risking herbal drug development, moving from descriptive network maps to predictive, quantitative models that can forecast efficacy and safety outcomes at a systems level. This document provides the application notes and experimental protocols to implement this integrative approach, with a consistent view towards validating systems-level predictions through focused ADMET profiling.
Core Methodologies and Comparative Analysis
Foundational Concepts: From Traditional NP to AI-NP
Traditional NP relies on collecting data from public databases to construct static interaction networks, followed by topological analysis and enrichment studies to hypothesize mechanisms [60]. While valuable, this approach struggles with data heterogeneity, an inability to model temporal dynamics, and limited predictive power for novel interactions or clinical outcomes [56].
AI-NP represents a paradigm shift, employing algorithms to learn from complex, multi-scale data. Key AI technologies include:
- Machine Learning (ML): Algorithms like Random Forest (RF) and Support Vector Machines (SVM) are used for classification (e.g., target prediction) and regression (e.g., activity scoring) [57].
- Deep Learning (DL): Convolutional Neural Networks (CNNs) process structural data, while Recurrent Neural Networks (RNNs) handle sequential data. They excel in feature extraction from raw, high-dimensional inputs [57].
- Graph Neural Networks (GNNs): Specially designed to operate on graph-structured data, making them ideal for direct learning from biological networks (e.g., protein-protein interaction networks) and molecular graphs [56] [59].
- Generative Models: Techniques like Generative Adversarial Networks (GANs) can design novel molecular structures with optimized properties, useful for lead optimization [61].
The following table summarizes the critical evolution from conventional NP to AI-NP:
Table 1: Comparative Analysis of Conventional Network Pharmacology vs. AI-Driven Network Pharmacology [56]
| Comparison Dimension | Conventional Network Pharmacology | AI-Driven Network Pharmacology (AI-NP) | Impact on Herbal Compound ADMET Research |
|---|---|---|---|
| Data Acquisition & Integration | Relies on manual curation from fragmented public databases; limited multi-omics integration. | Automated integration of multimodal data (genomics, metabolomics, clinical records); dynamic updating. | Enables construction of comprehensive "herb-ADMET gene" networks, linking compounds to metabolizing enzymes and transporters. |
| Algorithmic Core & Predictions | Based on statistical correlation and topological metrics (e.g., centrality); descriptive in nature. | Uses ML/DL/GNN to identify non-linear, high-dimensional patterns; enables predictive simulation. | Moves from identifying potential ADMET targets to quantitatively predicting PK parameters (e.g., bioavailability, half-life). |
| Model Interpretability | High interpretability; networks are manually analyzed. | Often a "black box"; requires Explainable AI (XAI) tools (e.g., SHAP, LIME) for insight. | Critical for understanding why a compound is predicted to be hepatotoxic, ensuring findings are biologically plausible. |
| Computational Scalability | Low efficiency; manual processes limit scale. | High-throughput, parallelizable computing suitable for large chemical libraries. | Allows for the virtual screening of thousands of herbal constituents for favorable ADMET profiles prior to in vitro testing. |
| Clinical & Translational Utility | Focuses on mechanistic hypothesis generation for pre-clinical validation. | Integrates real-world data (RWD) and electronic health records (EHR) for outcome prediction. | Facilitates the prediction of herb-drug interactions and patient subgroup-specific ADMET risks. |
Successful AI-NP research requires a curated set of data resources and software tools. The table below lists key components of the research toolkit.
Table 2: The Scientist's Toolkit for AI-NP Research on Herbal Compounds
| Tool Category | Specific Tool/Resource | Function in AI-NP Workflow | Relevance to ADMET |
|---|---|---|---|
| Herbal & Compound Databases | TCMSP [58], HerbComb [24] | Provides curated data on herbal constituents, targets, and indications. | Source of chemical structures for ADMET prediction. HerbComb includes ADMET properties for combinational analysis [24]. |
| General Biological Databases | DrugBank [58], STRING [58], ChEMBL [59] | Supplies drug-target info, protein interactions, and bioactivity data. | DrugBank includes PK data; ChEMBL provides bioactivity data for model training. |
| Network Visualization & Analysis | Cytoscape [58] [60] | Visualizes and performs basic topological analysis on biological networks. | Used to visualize ADMET-related networks (e.g., compound-CYP450 enzyme interactions). |
| AI/ML Modeling Platforms | Python (scikit-learn, PyTorch, TensorFlow), DeepChem | Provides libraries for building and training ML, DL, and GNN models. | Core environment for developing custom ADMET prediction models. |
| Molecular Docking & Simulation | AutoDock Vina [58], Schrodinger Suite | Performs structure-based virtual screening and binding affinity estimation. | Validates predicted interactions between herbal compounds and ADMET-related proteins (e.g., metabolic enzymes). |
| ADMET Prediction Software | Discovery Studio TOpkAT [62], pkCSM, ADMETLab | Offers specialized modules for predicting pharmacokinetic and toxicity endpoints. | Used for generating labels for model training or as a benchmark for newly developed AI models. |
AI-NP Integration and Systems Prediction Workflow
Detailed Experimental Protocols
Protocol 1: Constructing an AI-Enhanced Herbal Compound-Target Network
Objective: To move beyond static network maps by constructing a predictive, data-integrated network that links herbal constituents to potential protein targets, prioritized by AI-driven likelihood scores.
Materials:
- Software: Python environment with libraries (pandas, NetworkX, PyTorch Geometric), Cytoscape [58].
- Data: Compound structures from TCMSP/HerbComb [24]; known drug-target pairs from DrugBank [58]; protein-protein interaction (PPI) data from STRING [58].
Procedure:
- Data Curation: For a chosen herbal formula (e.g., Maxing Shigan Decoction [58]), extract all documented chemical constituents and their SMILES notations from TCMSP. In parallel, compile a list of disease-relevant targets from GeneCards and OMIM.
- Feature Representation: Convert each compound into a numerical feature vector using molecular descriptors (e.g., Mordred) or a learned molecular fingerprint via a pre-trained neural network. Represent each protein target by its amino acid sequence or a pre-trained protein language model embedding.
- Model Training for Target Prediction: Train a supervised ML model (e.g., a Gradient Boosting model or a GNN) using known compound-target pairs from databases like ChEMBL [59] and DrugBank as positive examples. Generate negative examples through random pairing or using confirmed non-interacting pairs. The model learns to associate compound and target features with the binary outcome (interaction/non-interaction).
- Network Construction & Prioritization: Apply the trained model to predict interaction probabilities for all herb constituent-disease target pairs. Construct a bipartite network in Cytoscape where edges connect compounds to targets, weighted by the predicted probability score. Filter edges based on a defined confidence threshold (e.g., probability > 0.85).
- Contextual Enrichment: Embed the compound-target layer into a larger biological context by fetching PPI data for the predicted targets from STRING. Merge networks to create a compound-target-PPI multiplex network.
- Topological & AI-Joint Analysis: Perform traditional topological analysis (degree, betweenness centrality) on the network. Use the AI-generated probability scores as an additional filter to prioritize nodes (targets) that are both topologically central and have high-confidence predicted interactions with multiple herbal compounds.
Protocol 2: AI-Driven Predictive Modeling of ADMET Properties
Objective: To train and validate ensemble AI models for the accurate prediction of key ADMET parameters, directly supporting the safety and viability assessment of herbal constituents.
Materials:
- Software: Python with scikit-learn, XGBoost, DeepChem; discovery studio for some ADMET descriptors [62].
- Data: Curated datasets of compounds with experimentally measured ADMET properties. Primary source: ChEMBL [59]. Supplement with data from specialized studies [62].
Procedure:
- Dataset Preparation: From ChEMBL, extract compounds with reliably measured endpoints (e.g., human intestinal absorption, CYP450 inhibition, hepatotoxicity, plasma protein binding). Ensure a clean, balanced dataset. Split into training (~70%), validation (~15%), and hold-out test sets (~15%).
- Feature Engineering: Calculate an extensive set of molecular descriptors (constitutional, topological, electronic) and fingerprints (ECFP4). For advanced modeling, use graph representations where atoms are nodes and bonds are edges.
- Model Building & Ensemble Construction: Train multiple base learner models:
- Descriptor-based Models: Train Random Forest, XGBoost, and Support Vector Regressors/Classifiers on the molecular descriptor set.
- Graph-based Model: Train a Graph Neural Network (GNN) directly on the molecular graph data [59].
- Hybrid Model: Experiment with architectures that combine learned representations from both descriptors and graphs.
- Ensemble Stacking: Implement a stacking ensemble method as evidenced by high-performing models [59]. Use the predictions from the base learners (RF, XGBoost, GNN) as new feature inputs to a meta-learner (often a simpler linear model). This allows the model to capture diverse patterns from different algorithms.
- Hyperparameter Optimization & Validation: Use Bayesian optimization or grid search to tune hyperparameters for all models. Evaluate performance on the validation set using metrics appropriate for the task: R² and Mean Absolute Error (MAE) for regression; AUC-ROC, precision, recall for classification [59] [61].
- Final Evaluation & Interpretation: Apply the final stacked ensemble model to the hold-out test set. Report key performance metrics. Use Explainable AI (XAI) tools like SHAP to interpret the model, identifying which molecular features (e.g., presence of certain functional groups, logP) most strongly influence the ADMET prediction.
Table 3: Performance Benchmark of AI Models for ADMET-Related Predictions (Representative Data) [59]
| Prediction Task (Example) | Best Performing Model | Key Performance Metric | Result | Implication |
|---|---|---|---|---|
| Pharmacokinetic Parameter Regression | Stacking Ensemble (RF, XGB, GNN) | Coefficient of Determination (R²) | 0.92 [59] | Model explains 92% of variance in PK data; highly predictive. |
| Pharmacokinetic Parameter Regression | Stacking Ensemble | Mean Absolute Error (MAE) | 0.062 [59] | Low average error in predicted vs. actual values. |
| Target Interaction Prediction | Graph Neural Network (GNN) | R² (vs. traditional models) | 0.90 [59] | Superior capture of structural relationships for interaction prediction. |
Protocol 3: AI-Enhanced Quantitative Systems Pharmacology (QSP) for Herbal Medicine
Objective: To integrate AI-NP findings into a mechanistic, mathematical QSP framework for simulating the holistic, dynamic effects of herbal interventions at the tissue or organism level.
Materials:
- Software: QSP platform (e.g., MATLAB SimBiology, Julia SciML), AI/ML libraries, model reduction tools.
- Data: Outputs from Protocol 1 (validated target list, interaction strengths), systems biology models (SBML files), pharmacokinetic parameters from Protocol 2.
Procedure:
- Mechanistic Model Foundation: Develop or select an existing QSP model core relevant to the disease (e.g., a model of tumor-immune dynamics for cancer, or a liver metabolism model). This core consists of ordinary differential equations (ODEs) describing key biological processes.
- AI-Informed Parameterization & Reduction:
- Use AI to inform model parameters. For example, the binding affinity (Ki) of a herbal compound to a target predicted in Protocol 1 can be estimated via a dedicated AI model or molecular docking, then used to parameterize the drug-target interaction term in the QSP model.
- Employ AI for model reduction. Train an artificial neural network (ANN) as a surrogate (or "emulator") of the full QSP model. This surrogate approximates the input-output relationships (e.g., dose → biomarker response) with drastically reduced computational cost, enabling large-scale simulations [63].
- Virtual Patient Population Generation: To account for population variability, use generative AI models like Generative Adversarial Networks (GANs) or variational autoencoders (VAEs). Train these on distributions of key physiological and genomic parameters (e.g., enzyme expression levels, organ volumes) to generate realistic, heterogeneous virtual patient cohorts [63].
- Simulation and Prediction: Run the AI-parameterized QSP model (or its surrogate) on the virtual patient population. Simulate different dosing regimens of the herbal intervention. Outputs include time-course predictions of biomarker levels, disease progression, and the emergence of efficacy or toxicity.
- Validation and Hypothesis Testing: Compare simulation outputs with available in vitro or in vivo data for validation. Use the calibrated model to generate testable hypotheses, such as identifying patient subpopulations most likely to respond or predicting potential herb-drug interactions by simulating the co-administration of the herbal compound with a standard-of-care drug.
AI-Enhanced QSP Protocol for Systems-Level Prediction
Validation and Application Framework
Iterative Experimental Validation: Predictions from AI-NP and AI-QSP models must be rigorously validated through an iterative cycle:
- In vitro: Test prioritized compounds for binding (SPR/BLI [60]), enzyme inhibition, and cellular efficacy/toxicity in relevant cell lines.
- In vivo: Validate PK predictions and overall therapeutic efficacy/toxicity in animal models that reflect key aspects of the human QSP model.
- Clinical Data: Where possible, use real-world evidence or clinical trial data to refine and validate prediction models for patient outcomes [56].
Application in Thesis Research: Within an AI-guided ADMET thesis, these protocols provide a structured pipeline:
- Use Protocol 1 to identify which constituents in a herbal formula are most likely to interact with ADMET-relevant proteins (CYPs, transporters).
- Use Protocol 2 to quantitatively predict the ADMET profile of these constituents and the whole formula's synergy.
- Use Protocol 3 to simulate how these predicted ADMET properties, combined with pharmacodynamic effects, translate to whole-body exposure, response, and potential toxicity in a heterogeneous population, generating specific, testable hypotheses for experimental validation.
This integrative approach bridges the gap between the holistic nature of herbal medicine and the demands of modern, predictive, and precision drug development.
Overcoming Real-World Hurdles: Data Scarcity, Model Interpretability, and Translational Gaps
Strategies for Small and Imbalanced Herbal Compound Datasets
Within the broader thesis on AI-guided ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction for herbal compounds, a fundamental and pervasive challenge is the nature of the data itself. Herbal medicines (HMs) represent complex mixtures of phytochemicals whose quality and composition are influenced by numerous factors such as growing conditions, harvest timing, and post-harvest processing [64]. This inherent chemical complexity, coupled with the traditional focus on a limited number of marker compounds like flavonoids for quality control, results in small, heterogeneous, and often severely imbalanced datasets [64]. These datasets are poorly suited for conventional machine learning (ML) models, which typically require large, balanced, and homogenous data to achieve robust and generalizable predictions.
The imperative to overcome this data bottleneck is clear. AI and ML have revolutionized drug discovery, compressing early-stage timelines and enabling the design of novel therapeutics [65]. Platforms like Exscientia have demonstrated AI-driven design cycles that are significantly faster and require fewer synthesized compounds than industry norms [65]. For herbal compounds, which are a cornerstone of traditional medicine and a rich source for novel pharmacophores, applying these powerful AI tools is essential. However, their successful application hinges on developing specialized strategies to train accurate, reliable models on the suboptimal datasets that characterize the field. This document provides detailed application notes and protocols to address this critical issue, framing solutions within the context of building predictive ADMET models for herbal compound research.
The first step in developing a mitigation strategy is a thorough understanding of the data landscape. The imbalance and scarcity in herbal compound datasets are multidimensional.
Source-Driven Scarcity and Bias: High-quality, experimentally validated ADMET data for pure herbal compounds or standardized extracts is limited. Public repositories may contain data for well-studied phytochemicals (e.g., quercetin, berberine), but this creates a long-tail distribution. A few common compounds have abundant data points, while the vast majority of herbal constituents have sparse or no data [23]. Furthermore, data is often collected under non-standardized experimental conditions, introducing noise and bias.
Outcome-Based Imbalance: This is particularly acute in toxicity prediction. For any given endpoint (e.g., hERG channel blockade, hepatotoxicity), the number of confirmed toxic compounds is vastly outnumbered by those deemed safe, leading to a severe class imbalance [23] [66]. A model trained on such data can achieve high accuracy by simply predicting "safe" for all inputs, failing to identify the critical toxicants.
Chemical Space Fragmentation: Herbal compounds occupy distinct regions of chemical space compared to synthetic drug libraries. They often possess unique scaffolds, higher stereochemical complexity, and different physicochemical property profiles. Models trained primarily on synthetic molecules may fail to generalize to these structurally divergent herbal compounds, a problem known as domain shift [15].
The table below summarizes the core data challenges and their specific impacts on model development for herbal compound ADMET prediction.
Table 1: Core Data Challenges in Herbal Compound ADMET Modeling
| Challenge Dimension | Description | Impact on ML Model Development |
|---|---|---|
| Dataset Size | Limited number of total data points for unique herbal compounds or mixtures [64]. | Increased risk of overfitting; poor model generalization and high variance in performance. |
| Class Imbalance | Severe skew in labeled outcomes (e.g., 98% non-toxic vs. 2% toxic) [23]. | Model bias towards the majority class; low sensitivity/recall for the critical minority class (e.g., toxicity). |
| Data Heterogeneity | Data aggregated from disparate sources with varying experimental protocols and quality [64]. | Introduces noise and confounding patterns, reducing model accuracy and reliability. |
| Feature Representation | Difficulty in capturing synergistic effects of multi-component herbal mixtures using single-molecule descriptors [64]. | Models may fail to predict the bioactivity or ADMET profile of the whole mixture accurately. |
| Domain Shift | Herbal compounds occupy a different region of chemical space than typical synthetic drug libraries [15]. | Models pre-trained on synthetic molecules show degraded performance when applied to herbal compounds. |
Methodological Strategies for Imbalanced and Sparse Data
To build effective models despite these challenges, a multi-faceted strategy combining data-, algorithm-, and validation-level techniques is required.
Data-Centric Strategies
These approaches focus on manipulating the training dataset to create a more balanced and informative foundation for learning.
Advanced Data Augmentation: For herbal compounds, augmentation must be chemically meaningful.
- Protocol: Employ SMILES-based transformation (e.g., randomized SMILES enumeration) and scaffold-aware analogue generation. Using tools like RDKit, generate valid, similar structures by adding/substituting small functional groups (e.g., -OH, -OCH3) common in phytochemistry or by ring cleavage/formation around the core scaffold. This artificially expands the dataset while staying within plausible chemical space [15].
- Application Note: Always validate augmented structures with basic chemical rule checkers and, if possible, cross-reference against virtual libraries of natural product derivatives to maintain realism.
Strategic Oversampling & Undersampling:
- Protocol: Implement Synthetic Minority Over-sampling Technique (SMOTE) or its derivatives (e.g., Borderline-SMOTE) for the minority class (e.g., toxic compounds). Instead of duplicating samples, SMOTE creates synthetic examples by interpolating between existing minority class instances in feature space. For the majority class, apply cluster-based undersampling, which reduces data by selecting representative prototypes from clusters, preserving information while balancing class ratios [23].
- Application Note: Apply sampling only to the training set. The validation and test sets must remain untouched with their original distribution to provide a realistic assessment of model performance on real-world, imbalanced data.
Knowledge-Driven Data Fusion:
- Protocol: Integrate disparate, small datasets by incorporating auxiliary knowledge graphs. Construct a graph where nodes represent herbs, compounds, targets, pathways, and ADMET endpoints. Edges represent relationships (contains, targets, associates_with, causes). Use this graph to infer latent connections and enrich the feature representation of compounds with sparse direct data [67] [68].
- Application Note: Platforms like BenevolentAI utilize such knowledge graphs for target identification [65]. For herbal ADMET, this can link a poorly characterized compound to well-studied analogues via shared targets or biosynthetic pathways, thereby transferring information.
Algorithm-Centric Strategies
These involve selecting or modifying ML algorithms to make them inherently more robust to imbalance.
Cost-Sensitive Learning:
- Protocol: During model training, assign a higher misclassification cost (penalty) to errors involving the minority class. For example, in a toxicity classifier, the cost of falsely labeling a toxic compound as safe (false negative) should be set 5-10 times higher than the opposite error. This can be implemented via class weight parameters in algorithms like Random Forest (e.g.,
class_weight='balanced') or Support Vector Machines (SVM) [23] [66]. - Application Note: The optimal cost ratio is a hyperparameter. Use the validation set and metrics like Geometric Mean (G-Mean) to tune it, rather than simple accuracy.
- Protocol: During model training, assign a higher misclassification cost (penalty) to errors involving the minority class. For example, in a toxicity classifier, the cost of falsely labeling a toxic compound as safe (false negative) should be set 5-10 times higher than the opposite error. This can be implemented via class weight parameters in algorithms like Random Forest (e.g.,
Ensemble Methods:
- Protocol: Use ensemble techniques like Balanced Random Forest or EasyEnsemble. Balanced Random Forest undersamples the majority class in each bootstrap sample to create balanced data for each tree. EasyEnsemble creates multiple balanced subsets by undersampling the majority class and trains a classifier on each, combining their outputs [23].
- Application Note: Ensembles are particularly effective for small, imbalanced data as they reduce variance. They align with the "Centaur Chemist" paradigm, where multiple algorithmic perspectives are combined for robust decision-making [65].
Transfer & Few-Shot Learning:
- Protocol: Leverage pre-trained models on large, related chemical datasets (e.g., ChEMBL, ZINC). Use a model pre-trained to predict ADMET properties for synthetic molecules. Then, fine-tune only the final layers of this model using your small, curated dataset of herbal compounds. This allows the model to start with a strong general understanding of chemistry and adapt specifically to the herbal domain [15].
- Application Note: This is highly effective for addressing domain shift. The pre-training task should be as relevant as possible (e.g., general toxicity prediction).
The following workflow diagram integrates these data-centric and algorithm-centric strategies into a coherent pipeline for model development.
Validation & Evaluation Protocols
Using standard accuracy on imbalanced data is misleading. Rigorous, tailored evaluation is paramount.
Protocol for Metric Selection:
- Primary Metrics: Use Geometric Mean (G-Mean) and Matthews Correlation Coefficient (MCC). G-Mean = √(Sensitivity × Specificity). It provides a single metric that is high only when both class-wise performances are good. MCC is a balanced measure applicable even when classes are of very different sizes.
- Supporting Metrics: Always report the Confusion Matrix and calculate Precision, Recall (Sensitivity), and Specificity for each class independently.
- Avoided Metric: Do not rely on overall Accuracy as a primary performance indicator.
Protocol for Validation Strategy:
- Employ Stratified k-Fold Cross-Validation. Ensure each fold preserves the original dataset's class distribution. For very small datasets, use Leave-One-Out Cross-Validation (LOOCV) or repeated stratified k-fold to maximize training data.
- Hold-out Test Set: Always retain a completely held-out, chronologically separated (if possible), or rigorously curated test set that reflects the real-world imbalance. This is the final arbiter of model performance.
Protocol for Model Interpretation:
- Utilize SHAP (SHapley Additive exPlanations) or LIME (Local Interpretable Model-agnostic Explanations) to explain individual predictions. For herbal mixtures, this can help identify which constituent(s) are driving a predicted ADMET outcome [68].
- Application Note: Interpretability is crucial for building trust in AI-guided herbal research and for generating chemically actionable insights for lead optimization [13].
Implementation Protocol: Building a Herbal Compound Solubility Predictor
This protocol provides a step-by-step guide for a common ADMET endpoint: predicting aqueous solubility.
Aim: To build a robust classification model (soluble vs. insoluble) for novel flavonoid derivatives using a small, imbalanced dataset.
Materials & Data:
- Dataset: 350 flavonoid compounds with experimental solubility data (300 insoluble, 50 soluble), sourced from literature and the Therapeutics Data Commons (TDC) [23] [69].
- Software: Python with RDKit (descriptor calculation), imbalanced-learn (sampling), scikit-learn (ML algorithms), XGBoost, ADMET-AI web tool or API for benchmark comparison [69].
Step-by-Step Procedure:
Data Preparation:
- Standardize all compound structures using RDKit (neutralize charges, remove salts, generate canonical SMILES).
- Calculate a suite of 2D molecular descriptors (200+ using RDKit) and molecular fingerprints (ECFP4).
- Perform basic cleaning: remove duplicates, handle missing values (impute or remove).
Feature Engineering & Selection:
- Apply variance threshold to remove low-variance descriptors.
- Use Recursive Feature Elimination (RFE) with a Random Forest estimator to select the top 50 most predictive features for solubility. This reduces noise and overfitting [23].
Data Resampling (Training Set Only):
- Perform a stratified split: 70% train (245 cmpds), 30% test (105 cmpds). The test set remains imbalanced.
- On the training set, apply SMOTE to the minority "soluble" class and Cluster Centroids undersampling to the majority "insoluble" class to achieve a 1:1 ratio.
Model Training with Transfer Learning:
- Option A (From Scratch): Train a Cost-Sensitive XGBoost model on the resampled training data. Set
scale_pos_weightparameter to the inverse of the original class ratio. - Option B (Transfer Learning): Use a pre-trained graph neural network from the ADMET-AI platform (trained on 41 ADMET datasets) [69]. Fine-tune the last two layers using your balanced flavonoid training data.
- Option A (From Scratch): Train a Cost-Sensitive XGBoost model on the resampled training data. Set
Hyperparameter Optimization:
- Use Bayesian Optimization or Grid Search with 5-fold stratified cross-validation on the resampled training set.
- Optimize for G-Mean, not accuracy.
Evaluation:
- Predict on the original, untouched, imbalanced test set.
- Generate the confusion matrix and report: G-Mean, MCC, Recall for the "soluble" class, and Specificity.
- Benchmark against a) a model trained without resampling, and b) predictions from the base ADMET-AI model without fine-tuning.
Deployment & Iteration:
- Package the best model into a simple web interface or Python API for predicting solubility of newly designed flavonoid analogues.
- As new experimental data is generated, retrain the model periodically in a Design-Build-Test-Learn (DBTL) cycle, as exemplified by AI platforms like Exscientia's automated workflow [65].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational & Experimental Tools for Herbal ADMET Research
| Tool/Resource Name | Type | Primary Function in Herbal ADMET Research | Key Consideration |
|---|---|---|---|
| RDKit | Software Library | Calculates molecular descriptors and fingerprints; performs chemical transformations for data augmentation [23]. | Open-source. Essential for standardizing compound representation and generating features. |
| Therapeutics Data Commons (TDC) | Data Repository | Provides curated, publicly available ADMET datasets for model training and benchmarking [23] [69]. | Useful for finding auxiliary data for pre-training or transfer learning. |
| ADMET-AI | Web Platform / Model | Provides state-of-the-art graph neural network predictions for 41 ADMET endpoints; offers a benchmark for model performance [69]. | Can be used as a baseline predictor or as a source of pre-trained models for fine-tuning. |
| imbalanced-learn | Python Library | Implements advanced resampling techniques (SMOTE, cluster-based undersampling) to handle class imbalance [23]. | Critical for preparing training data; should only be applied to the training set. |
| SHAP/LIME | Interpretation Library | Explains individual model predictions, identifying which chemical features contribute to an ADMET outcome [68]. | Vital for moving from "black box" predictions to chemically interpretable insights. |
| UHPLC-QTOF-MS | Analytical Instrument | Provides high-resolution chemical fingerprinting and metabolomics data for herbal extracts, enabling holistic quality control [64]. | Generates the complex, multi-constituent data that models must ultimately interpret. |
| Caco-2/ PAMPA Assay Kits | In Vitro Assay | Provides experimental measurement of intestinal permeability (absorption) for validation of computational predictions [23]. | Essential for generating high-quality ground-truth data to feed and validate ML models. |
Effectively leveraging AI for herbal compound ADMET prediction necessitates a deliberate shift from standard ML workflows to strategies specifically engineered for data scarcity and imbalance. By integrating data augmentation with chemical intelligence, algorithmic techniques like cost-sensitive and ensemble learning, and rigorous, imbalance-aware validation, researchers can build models with practical utility. The integration of these models into a closed-loop DBTL cycle—where predictions guide the design of new experiments, and experimental results refine the model—represents the future of rational, AI-guided herbal medicine research [65]. As the field progresses, the creation of large, standardized, and openly accessible herbal compound ADMET databases will be the single most impactful development, allowing these sophisticated strategies to reach their full potential in accelerating the discovery and development of safe and effective plant-derived therapeutics.
The integration of Explainable Artificial Intelligence (XAI) into the prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties represents a transformative advancement in the field of herbal compound research [23]. Herbal medicines, characterized by their complex multi-component nature, present unique challenges for traditional drug development pipelines, where a lack of pharmacokinetic data and unclear mechanisms often hinder progress [1]. The application of opaque "black-box" machine learning (ML) models, while powerful, fails to provide the mechanistic insights and scientific rationale necessary for researchers to trust and act upon computational predictions [70]. This opacity is a significant barrier in a field that requires understanding not just if a compound is active, but why.
XAI directly addresses this critical gap by making the decision-making process of AI models transparent, interpretable, and actionable [71]. For scientists working with herbal compounds, XAI techniques can illuminate which specific phytochemical substructures contribute to a predicted ADMET outcome—such as poor intestinal absorption, high hepatic metabolism, or potential toxicity [70]. This translucency is essential for guiding the rational optimization of herbal formulations, prioritizing compounds for costly experimental validation, and building confidence in AI-driven pipelines. Furthermore, as regulatory bodies emphasize the need for understanding AI-based tools in healthcare, XAI provides the necessary documentation and evidence to support computational findings [72]. This document outlines key protocols and applications of XAI, framing them within a research thesis focused on building trustworthy, AI-guided ADMET prediction systems for herbal medicine discovery.
Core XAI Methodologies and Their Application to ADMET
The selection of an XAI technique depends on the model type and the specific interpretability question. The methodologies can be broadly categorized into model-agnostic and model-specific approaches.
Model-Agnostic Techniques: These methods can be applied to any ML model after it has been trained, treating the model as a "black box."
- SHapley Additive exPlanations (SHAP): Based on cooperative game theory, SHAP assigns each input feature (e.g., a molecular descriptor) an importance value for a particular prediction [72]. It quantifies how much each feature contributes to moving the model's output from a baseline expectation. In ADMET, SHAP can reveal that a specific range of logP (lipophilicity) values or the presence of a reactive ester group is the primary driver behind a predicted high clearance or toxicity alert [70].
- Local Interpretable Model-agnostic Explanations (LIME): LIME approximates a complex model locally around a single prediction with a simple, interpretable model (like linear regression) [70]. It answers: "For this specific herbal compound, which features were most important for this prediction?" This is invaluable for explaining outlier predictions or understanding the profile of a single lead candidate.
Model-Specific Techniques: These are built into certain model architectures.
- Attention Mechanisms: Used in advanced deep learning models like Graph Neural Networks (GNNs), attention mechanisms allow the model to focus on specific parts of the input data [73]. In a molecular graph, the attention weights can highlight which atoms or bonds the model "attended to" when making a prediction, directly linking chemical structure to ADMET property.
- Gradient-Based Methods: For neural networks, techniques like Grad-CAM use gradients flowing back into the final layers to produce a heatmap of important input regions [70]. While common in image analysis, analogous approaches for chemistry can highlight critical structural motifs.
The following workflow diagram illustrates how these XAI techniques integrate into a standard ADMET prediction pipeline for herbal compounds.
Diagram 1: XAI-Integrated ADMET Prediction Workflow for Herbal Compounds (Max Width: 760px)
Experimental Protocols for XAI-Guided ADMET Research
Protocol: Benchmarking ML Models and Feature Representations for Herbal ADMET Prediction
Objective: To systematically evaluate the performance and interpretability of different machine learning algorithms and molecular feature representations in predicting a specific ADMET endpoint relevant to herbal compounds (e.g., human liver microsomal clearance) [41].
Materials: Software: Python with libraries (scikit-learn, RDKit, DeepChem, SHAP). Data: Curated dataset from public sources like the Therapeutics Data Commons (TDC) or ChEMBL, ensuring inclusion of known phytochemicals [41].
Procedure:
- Data Curation and Cleaning: Standardize SMILES strings of compounds. Remove inorganic salts and metals. Resolve tautomers and deduplicate entries, keeping consistent measurements [41].
- Feature Generation: Compute diverse molecular representations for each compound:
- Classical Descriptors: 200+ RDKit descriptors (e.g., molecular weight, logP, topological polar surface area).
- Fingerprints: Morgan fingerprints (radius=2, nBits=2048).
- Learned Representations: Pre-trained molecular embeddings (e.g., from Mol2Vec or a GNN) [41].
- Model Training & Hyperparameter Optimization:
- Split data using scaffold splitting to ensure generalizability to novel chemotypes [41].
- Train multiple models: Random Forest (RF), Gradient Boosting (XGBoost/LightGBM), Support Vector Machine (SVM), and a Graph Neural Network (GNN).
- Perform hyperparameter tuning for each model using Bayesian optimization within a 5-fold cross-validation framework.
- Model Evaluation & XAI Application:
- Evaluate models on a held-out test set using metrics: R² (regression) or AUC-ROC (classification), MAE, RMSE.
- Apply SHAP (TreeExplainer for RF/GB, KernelExplainer for SVM) to the best-performing model. Calculate global feature importance and generate local explanations for key herbal compound predictions.
- Analysis: Correlate top SHAP features with known medicinal chemistry principles. Identify if the model is using chemically meaningful signals (e.g., high logP leading to high clearance) or potential data artifacts.
Protocol: XAI-Driven Mechanistic Elucidation of Drug-Herb Interactions (DHI)
Objective: To use XAI to uncover and visualize the potential mechanisms (Pharmacokinetic/PK or Pharmacodynamic/PD) by which a specific herbal extract or constituent may interact with a conventional drug [1].
Materials: Software: KNIME or Python with network analysis tools (Cytoscape), molecular docking software (AutoDock Vina), ADMET prediction platforms (e.g., pkCSM). Data: Constituent list of the herbal extract, target protein structures (e.g., CYP3A4, P-gp), known drug interaction networks.
Procedure:
- Constituent Profiling & ADMET Prediction: Compile a comprehensive list of major bioactive constituents in the herbal extract. Use in silico models to predict their key ADMET properties, focusing on interaction-prone profiles: CYP450 enzyme inhibition/induction, P-glycoprotein substrate/inhibition, and plasma protein binding [74].
- Multi-Model Interaction Prediction: Employ different AI models:
- A similarity-based model to infer interactions based on structural similarity to known interactors [1].
- A network-based model integrating drug-target and protein-protein interaction networks to predict indirect effects [1].
- A knowledge-graph model linking herbs, compounds, genes, enzymes, and pathways.
- XAI Integration & Mechanistic Hypothesis Generation:
- Apply LIME or attention mechanisms to the network/knowledge-graph model to explain why a DHI was predicted.
- The explanation should highlight the most influential path in the network (e.g., "Herbal Constituent A → inhibits CYP2C9 → reduces metabolism of Drug X").
- Use SHAP on the ADMET predictors to identify which molecular features of the constituent contribute to the inhibition prediction.
- In Silico & In Vitro Validation: Prioritize the top mechanistic hypothesis. Perform molecular docking of the key constituent against the implicated target (e.g., CYP enzyme) to assess binding affinity and pose [74]. Design a focused in vitro assay (e.g., CYP inhibition assay) to experimentally confirm the predicted interaction.
Performance Data and Model Comparisons
Table 1: Benchmark Performance of ML Models on Selected ADMET Tasks (Regression - R² Score) [23] [73] [41]
| ADMET Endpoint | Dataset Size | Random Forest | Gradient Boosting | Support Vector Machine | Graph Neural Network | Key Molecular Features (via SHAP) |
|---|---|---|---|---|---|---|
| Human Liver Microsomal Clearance | ~1,200 compounds | 0.68 | 0.71 | 0.62 | 0.75 | logP, #Rotatable Bonds, H-bond acceptors, CYP3A4 substrate probability |
| Caco-2 Permeability (logPapp) | ~900 compounds | 0.72 | 0.74 | 0.65 | 0.77 | Polar Surface Area (PSA), Molecular Weight, Number of H-bond donors |
| Plasma Protein Binding (%) | ~1,500 compounds | 0.81 | 0.79 | 0.70 | 0.80 | logD, #Aromatic rings, Acidic pKa |
| hERG Inhibition Risk (Binary) | ~10,000 compounds | 0.85 (AUC) | 0.87 (AUC) | 0.82 (AUC) | 0.86 (AUC) | Basic pKa, logP, Presence of a aromatic amine |
Table 2: Bibliometric Analysis of XAI in Drug Research (Top Contributing Countries, 2002-2024) [72]
| Country | Total Publications (TP) | Total Citations (TC) | TC/TP (Avg. Citation/Paper) | Notable Research Focus |
|---|---|---|---|---|
| China | 212 | 2,949 | 13.91 | Broad applications, including TCM compound analysis [72]. |
| United States | 145 | 2,920 | 20.14 | Foundational algorithms and translational applications. |
| Germany | 48 | 1,491 | 31.06 | Early pioneer (since 2002), multi-target compounds [72]. |
| Switzerland | 19 | 645 | 33.95 | Molecular property prediction and drug safety [72]. |
| Thailand | 19 | 508 | 26.74 | Applications in biologics and herbal medicine research [72] [74]. |
Visualizing Mechanisms: Drug-Herb Interaction Pathway
A critical application of XAI is deconstructing the complex mechanisms of Drug-Herb Interactions (DHIs). The following diagram illustrates a PK-based DHI pathway elucidated through an XAI-informed analysis, showing how explanations can be traced from a model's prediction back to specific herbal constituents and their biological targets [1].
Diagram 2: XAI-Educated PK Mechanism of a Drug-Herb Interaction (Max Width: 760px)
Table 3: Key Software, Databases, and Experimental Resources for XAI-ADMET Research on Herbal Compounds
| Category | Resource Name | Primary Function in Research | Key Utility for Herbal Studies |
|---|---|---|---|
| Public Databases | Therapeutics Data Commons (TDC) | Curated benchmark datasets and leaderboards for ADMET prediction tasks [41]. | Provides standardized datasets to train and benchmark models applicable to phytochemical space. |
| ChEMBL | Large-scale bioactivity database for drug-like molecules [41]. | Source of experimental ADMET data for known natural products and analogs. | |
| Cheminformatics Software | RDKit | Open-source toolkit for cheminformatics and descriptor calculation [41]. | Calculates thousands of molecular descriptors and fingerprints for herbal constituents. |
| Molinspiration / DataWarrior | Platforms for calculating physicochemical properties and bioactivity scores [41] [74]. | Rapid profiling of drug-likeness and lead-likeness of herbal compounds. | |
| ML/XAI Frameworks | scikit-learn | Python library for classic ML algorithms (RF, SVM, etc.) [41]. | Core framework for building baseline predictive models. |
| SHAP & LIME Libraries | Python libraries for model-agnostic explainability [72] [70]. | Generates global and local explanations for any ADMET model's predictions. | |
| DeepChem / PyTorch Geometric | Libraries for deep learning on molecular graphs [73] [41]. | Enables building of GNNs that learn directly from molecular structure. | |
| In Silico Prediction Suites | SwissADME / pkCSM | Free web tools for predicting key ADMET and physicochemical properties [74]. | Provides quick, accessible first-pass ADMET profiling for herbal compound lists. |
| Experimental Assay Kits | P450-Glo CYP450 Assay | Luminescent in vitro assay kit for CYP450 enzyme inhibition/induction [1]. | Critical for validating XAI-predicted PK interactions (e.g., herbal inhibition of CYP3A4). |
| MTS/PrestoBlue Cell Viability Assay | Colorimetric/fluorimetric assay for cytotoxicity screening [74]. | Tests predicted herbal compound or extract toxicity in cell models (e.g., hepatocytes). | |
| Caco-2 Cell Line | Human colon carcinoma cell line model for intestinal permeability studies [23]. | Gold-standard in vitro model to experimentally verify predicted absorption properties. |
Abstract The integration of Artificial Intelligence (AI) for predicting the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties of herbal compounds presents a transformative opportunity for drug discovery. However, the inherent chemical complexity, batch variability, and sparse experimental data associated with natural products pose significant challenges to model reliability. This article establishes that the rigorous definition of a model's Applicability Domain (AD) is the critical determinant of trustworthiness in this context. We provide detailed application notes and protocols for defining, evaluating, and documenting the AD within AI-guided herbal compound research. This includes standardized methodologies for chemical data curation, the implementation of distance- and probability-based AD methods, and a tiered experimental validation strategy. By framing these protocols within a comprehensive trust assessment framework, we equip researchers with the tools to discern when a predictive model can be confidently applied to novel herbal derivatives and when its predictions require stringent experimental verification.
The pursuit of herbal compounds as leads for modern therapeutics is revitalized by AI, which can navigate their vast and intricate chemical space to predict pharmacological and safety profiles [6] [75]. A primary application is the in silico prediction of ADMET properties, a historically costly attrition point in drug development [23]. Machine learning (ML) models, including graph neural networks and ensemble methods, have demonstrated superior performance over traditional quantitative structure-activity relationship (QSAR) models for many ADMET endpoints [23] [41].
Despite this promise, the direct application of models trained primarily on synthetic chemical libraries to herbal compounds is fraught with risk. Herbal chemical space is characterized by unique scaffolds, stereochemical complexity, and the prevalence of mixtures—factors often underrepresented in public ADMET datasets [6] [76]. Consequently, predictions for novel natural products frequently constitute extrapolation beyond a model's trained experience, leading to potential failures and lost resources [77] [78].
The Applicability Domain is the established concept to mitigate this risk. It is defined as the "theoretical space defined by relevant structural features, physicochemical descriptor values, or the range of prediction end points, in which the chemical of interest... is compliant with the model’s specifications" [78]. According to OECD validation principles, defining the AD is a mandatory prerequisite for the regulatory acceptance of any (Q)SAR model [77]. Within AI-guided herbal research, the AD is not a limitation but a essential confidence metric. It provides a systematic, quantifiable answer to the core question of trust: when a model's prediction is an informed interpolation within its learned domain, and when it is a speculative extrapolation that must be flagged for cautious interpretation and prioritization for experimental testing.
Foundational Concepts and Data Requirements
The Chemical Space of Herbal Compounds and ADMET Data Landscape
Herbal compounds (natural products) occupy a region of chemical space distinct from synthetic drug-like molecules, often exhibiting greater structural complexity, molecular rigidity, and a higher prevalence of oxygen atoms [75]. This uniqueness is a source of therapeutic potential but also of domain shift for ML models. The AD must therefore account for this shift by explicitly mapping the coverage of natural product features.
Public and proprietary ADMET datasets form the backbone of model training. Key resources include the Therapeutics Data Commons (TDC) ADMET benchmark group, datasets from PubChem, and specialized collections like those from Biogen [41]. For herbal informatics, natural product-specific databases (e.g., COCONUT, NPASS) are crucial, though they often lack extensive ADMET annotations [76]. The quality and relevance of the training data directly dictate the scope and robustness of the derived AD.
Table 1: Key Public ADMET Datasets for Model Development and Benchmarking
| Dataset Name/Source | Primary ADMET Endpoints Covered | Notable Characteristics | Relevance to Herbal Compounds |
|---|---|---|---|
| TDC ADMET Benchmark Group [41] | Solubility, Permeability (Caco-2, Pgp-inh), Microsomal Clearance, Toxicity (hERG, Ames) | Curated, scaffold-split benchmarks for ML. | General drug-like space; baseline for domain gap analysis. |
| Biogen In-house ADME Dataset [41] | Kinetic solubility, Metabolic stability, Permeability | High-quality, experimentally consistent data on ~3000 purchasable compounds. | Useful for hybrid models; assesses extrapolation to new scaffolds. |
| NIH Solubility Dataset (PubChem) [41] | Kinetic solubility | Large public dataset. | Requires careful cleaning for salt forms. |
| Natural Product Databases (e.g., COCONUT, NPASS) [76] | Structural information, limited bioactivity | Extensive collections of unique natural product scaffolds. | Essential for characterizing domain coverage and identifying underrepresented regions. |
Defining the Applicability Domain: Core Methodologies
AD methods can be categorized by their underlying algorithm. The choice of method depends on the model type, descriptor set, and desired strictness.
Table 2: Core Methodologies for Defining the Applicability Domain (AD)
| Method Category | Description | Key Algorithms/Measures | Advantages | Limitations |
|---|---|---|---|---|
| Range-Based | Defines AD based on min/max values of model descriptors. | Bounding Box, PCA Bounding Box [77]. | Simple, fast to compute. | Cannot identify empty regions within bounds; overly conservative. |
| Geometric | Defines the convex hull containing the training set. | Convex Hull [77]. | Clear geometric interpretation. | Computationally intensive in high dimensions; ignores internal density. |
| Distance-Based | Calculates distance of query compound to training set centroid or neighbors. | Leverage (Mahalanobis distance), Euclidean, City Block [77]. | Handles correlated descriptors (Mahalanobis); intuitive. | Threshold definition is critical and often arbitrary. |
| Probability-Density Based | Estimates the probability density of the training set; queries in low-density regions are outside AD. | Probability density functions, Parzen windows [77]. | Reflects the actual distribution of training data. | Computationally demanding; requires sufficient data for reliable density estimation. |
| Structural Fragment-Based | Flags queries containing sub-structures not present in the training set. | Fingerprint sub-structure keys [78]. | Highly interpretable; flags "true" structural novelties. | May be too restrictive if model generalizes well beyond specific fragments. |
For herbal compounds, a consensus approach is recommended. A query should be considered within the AD only if it passes a combination of criteria: e.g., its Mahalanobis distance is below a defined threshold and it contains no critical unobserved substructures and it falls within a region of sufficient training data density.
Experimental Protocols for AD Assessment and Validation
Protocol 1: Data Curation and Feature Engineering for Herbal Compounds
Objective: To create a standardized, reproducible pipeline for cleaning chemical data and generating representative molecular features for ADMET model training and AD definition.
Materials & Software: RDKit or OpenBabel cheminformatics toolkits; Standardizer tools (e.g., [41]); Dataset-specific SMILES lists.
Procedure:
- Data Cleaning:
- Standardize Representations: Input SMILES are canonicalized. Tautomers are normalized to a consistent representation [41].
- Handle Salts and Mixtures: Remove inorganic counterions. For organometallic or complex mixtures common in herbal extracts, flag the entry for potential exclusion or specialized treatment [41].
- Deduplicate: Remove exact duplicates. For entries with the same structure but conflicting property measurements, apply a consistency rule (e.g., remove the entire group if values differ by more than 20% of the inter-quartile range for continuous variables) [41].
- Feature Engineering:
- Calculate Descriptors: Generate a comprehensive set of 1D/2D molecular descriptors (e.g., using RDKit) capturing physicochemical properties (LogP, molecular weight, H-bond donors/acceptors, topological surface area).
- Generate Fingerprints: Compute structural fingerprints such as Morgan fingerprints (ECFP4), which are crucial for distance-based AD methods like Tanimoto similarity [79].
- Feature Selection: Apply filter methods (e.g., correlation-based) or embedded methods to reduce dimensionality and retain features most relevant to the ADMET endpoint, improving model performance and AD clarity [23].
Deliverable: A curated dataset in a standardized format (e.g., CSV) with associated molecular descriptor and fingerprint matrices.
Protocol 2: Implementing a Tiered Applicability Domain Assessment
Objective: To systematically evaluate whether a novel herbal compound falls within the AD of a pre-trained ADMET model.
Pre-requisite: A trained ML model (e.g., Random Forest, Graph Neural Network) and its defined training set chemical space.
Procedure:
- Tier 1: Structural Alert Check.
- Generate the Morgan fingerprint (radius=2) for the query compound.
- Compute the maximum Tanimoto similarity between the query fingerprint and all fingerprints in the training set.
- Decision Point: If maximum similarity < 0.4 (conservative) or 0.6 (moderate) [79], flag the compound as "High-Risk Extrapolation" and proceed to Tier 3. If similarity is above threshold, proceed to Tier 2.
- Tier 2: Descriptor Space Distance Check.
- Project the query and training set into the space of the most relevant molecular descriptors (e.g., using PCA).
- Calculate the leverage (Mahalanobis distance) of the query compound relative to the training set centroid.
- Decision Point: If the leverage value is greater than the critical value (typically 3 times the mean leverage of the training set) [77], flag as "Outside Geometric AD". Proceed to Tier 3.
- Tier 3: Experimental Validation Gating.
- Compounds flagged in Tiers 1 or 2 are automatically prioritized for targeted experimental validation.
- Design a minimal experimental assay (e.g., kinetic solubility, microsomal stability) to test the model's prediction.
- Use the discrepancy between prediction and experimental result to iteratively refine the AD thresholds.
Deliverable: An AD assessment report classifying the query as "Within AD (High Confidence)", "Borderline (Moderate Confidence)", or "Outside AD (Experimental Verification Required)".
Protocol 3: External Validation and "Registered Model" Framework
Objective: To conduct an unbiased, conclusive evaluation of model and AD performance on a fully independent dataset of herbal compounds.
Rationale: Internal cross-validation can yield optimistic performance estimates due to data leakage or overfitting [80]. External validation is the gold standard for establishing generalizability.
Materials: An independent set of herbal compounds with experimentally measured ADMET properties, not used in any model discovery step.
Procedure (Adaptive Registered Model Framework) [80]:
- Model Discovery & Preregistration:
- Finalize the model architecture, feature processing pipeline, and AD definition logic using the discovery dataset.
- Publicly preregister or archive the finalized model weights, all hyperparameters, and the complete feature engineering code before any exposure to the external validation set.
- Adaptive External Validation:
- Acquire external validation data prospectively or use a strictly held-out set.
- Apply the registered, frozen model to predict the properties of the external compounds.
- Use the tiered AD protocol to classify each external compound.
- Analyze performance metrics (e.g., RMSE, AUC-ROC) separately for compounds predicted to be inside vs. outside the AD. A trustworthy model will show significantly better performance for the "Inside AD" group.
Deliverable: A validation report quantifying model predictive performance stratified by AD membership, providing empirical evidence for the utility of the defined AD.
Visualization of Workflows and Decision Logic
Tiered AD Assessment Workflow for Herbal Compounds
Logic Tree for Assessing Model Trustworthiness
Table 3: Essential Toolkit for ADMET Model Development and AD Assessment
| Category | Tool/Reagent | Specific Example/Product | Function in AD/ADMET Research |
|---|---|---|---|
| Computational Cheminformatics | Molecular Descriptor & Fingerprint Calculator | RDKit, OpenBabel, PaDEL-Descriptor | Generates numerical representations (descriptors, ECFP fingerprints) of compounds for model training and distance calculations in AD methods. |
| Computational Modeling | Machine Learning Framework | Scikit-learn, DeepChem, XGBoost, PyTorch | Provides algorithms (Random Forest, Neural Networks) to build predictive ADMET models and enables custom implementation of AD logic. |
| Data Curation | Chemical Standardization Tool | Standardizer (e.g., from Atkinson et al. [41]), MolVS | Cleans and canonicalizes chemical structure data (SMILES) to ensure consistency before model training and AD definition. |
| AD Calculation | Specialized AD Software | QSARINS, AMBIT, KNIME ADMET nodes | Implements standardized range, geometric, and distance-based methods (e.g., Leverage, PCA) to define and visualize the AD. |
| Experimental Validation (In Vitro) | Caco-2 Cell Line | ATCC HTB-37 | Measures intestinal permeability (absorption) to validate predictions for novel herbal compounds flagged outside AD. |
| Experimental Validation (In Vitro) | Human Liver Microsomes (HLM) | Commercially available pooled HLM (e.g., from Corning) | Assesses metabolic stability (Phase I metabolism) to verify model predictions for compounds with unfamiliar scaffolds. |
| Experimental Validation (In Vitro) | Sens-Is Assay Components | Keratinocyte cell line (e.g., HaCaT), specific cytokine ELISA kits [81] | Validates skin sensitization toxicity predictions, particularly important for topical herbal product development. |
Trust in AI-guided ADMET predictions for herbal compounds is not a binary state but a continuum informed by rigorous AD assessment. The protocols and frameworks outlined herein provide a actionable path forward. The future of reliable natural product drug discovery lies in the iterative cycle of in silico prediction, explicit AD evaluation, targeted experimental validation, and model refinement. By adopting a disciplined approach to defining and respecting the Applicability Domain, researchers can transform AI from a black-box oracle into a calibrated, trustworthy partner in navigating the complex landscape of herbal medicine.
The integration of Artificial Intelligence (AI) into the discovery and development of drugs from herbal compounds represents a paradigm shift, aiming to address the persistent high failure rates in pharmaceutical research [25]. Within this broader thesis, the core challenge is effectively bridging in-silico predictions with tangible experimental outcomes, particularly for Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties [82]. Herbal compounds, or phytochemicals, offer a vast and structurally diverse resource for new therapeutics but are accompanied by significant complexity due to multi-component mixtures, batch variability, and a frequent lack of comprehensive pharmacokinetic data [6] [83].
This document establishes a framework for a Synthesis Feedback Loop, a cyclical and iterative process where AI predictions guide experimental design, and experimental results, in turn, refine and validate the AI models. The loop is designed to accelerate the identification of promising herbal-derived lead compounds with favorable ADMET profiles and proven experimental feasibility, thereby de-risking the development pipeline [84] [85].
Foundational Data & AI Model Landscape
The efficacy of the feedback loop is contingent on the quality of foundational data and the sophistication of the AI models employed. The following sections outline the current landscape.
Phytochemical Databases for AI Training
A critical first step is sourcing high-quality, curated structural and bioactivity data. The following table summarizes key phytochemical databases essential for training robust AI/ML models for ADMET prediction [86].
Table 1: Key Phytochemical Structure and Activity Databases for AI Model Training
| Database Name | Primary Focus / Region | Key Features for AI | Access |
|---|---|---|---|
| COCONUT | Comprehensive Open Natural Products Database | Vast collection of unique natural product structures; enables diversity analysis and novel scaffold identification [86]. | Open Access, Bulk Download |
| NPACT | Natural Products Anticancer Compound Database | Curated anticancer activity data; useful for training target-specific activity models [86]. | Open Access |
| TCMID | Traditional Chinese Medicine Integrated Database | Integrates herbal formulas, ingredients, targets, and diseases; essential for network pharmacology approaches [86]. | Open Access |
| IMPPAT | Indian Medicinal Plants Phytochemistry and Therapeutics | Curated phytochemicals from Indian medicinal plants with associated therapeutic uses [86]. | Open Access |
| NuBBE DB | Nucleus of Brazilian Bioactive Compounds Database | Bioactive compounds from Brazilian biodiversity with associated experimental data [86]. | Open Access |
AI/ML Techniques for ADMET Prediction
Different AI techniques are applied across the discovery pipeline, from initial screening to lead optimization [15].
Table 2: Core AI/ML Techniques in Herbal Compound ADMET Prediction
| AI Category | Key Techniques | Application in Herbal ADMET | Typical Output |
|---|---|---|---|
| Supervised Learning | Random Forest, Support Vector Machines (SVM), Deep Neural Networks (DNN) | Building Quantitative Structure-Activity Relationship (QSAR) models to predict properties like solubility, metabolic stability, or toxicity from molecular descriptors [87] [15]. | Classification (e.g., toxic/non-toxic) or regression (e.g., predicted IC50 value) models. |
| Unsupervised Learning | Clustering (k-means), Principal Component Analysis (PCA) | Exploring chemical space of phytochemical databases, identifying inherent clusters or patterns without pre-defined labels, assessing dataset diversity [15]. | Compound clusters, dimensionality-reduced visualizations of chemical space. |
| Deep Learning (Generative) | Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs) | De novo generation of novel molecular structures inspired by phytochemical scaffolds, optimized for desired ADMET properties [15]. | Novel, synthetically plausible molecular structures (e.g., in SMILES format). |
| Graph Neural Networks (GNNs) | Message Passing Neural Networks (MPNN) | Directly learning from molecular graph structures (atoms as nodes, bonds as edges) to predict activity or properties, capturing complex structural information [6]. | Predictions based on holistic molecular representation. |
The Synthesis Feedback Loop: Core Workflow
The Synthesis Feedback Loop is an iterative process comprising four interconnected phases. The following diagram illustrates the workflow and its cyclical nature.
Diagram 1: The Synthesis Feedback Loop in AI-Guided Herbal Research (Max Width: 760px)
Phase 1: AI-Prioritization & Virtual Screening
Objective: To computationally screen vast phytochemical libraries and prioritize a shortlist of candidates with high predicted bioactivity and desirable ADMET profiles.
Protocol 1.1: Multi-Parameter Virtual Screening Workflow
- Library Curation: Compile a focused library from databases in Table 1. Standardize structures (e.g., remove salts, neutralize charges) and generate molecular descriptors/fingerprints [86].
- Model Deployment: Employ a suite of pre-trained or custom-built AI models:
- Activity Prediction: Use a QSAR or DTI (Drug-Target Interaction) prediction model for the therapeutic target of interest (e.g., PD-L1 inhibitor for immunomodulation) [15].
- ADMET Profiling: Simultaneously predict key properties using specialized models: Human Intestinal Absorption (HIA), Cytochrome P450 inhibition (e.g., CYP3A4), hERG channel blockage risk, and aqueous solubility [82] [85].
- Multi-Objective Ranking: Apply a scoring function or Pareto front analysis to rank compounds that balance predicted potency with optimal ADMET characteristics and synthetic accessibility scores [15] [84].
- Output: A prioritized list of 20-50 phytochemicals or derived scaffolds for experimental validation.
Phase 2: Experimental Feasibility Assays
Objective: To validate the AI predictions using standardized in vitro assays, generating reliable experimental ADMET data.
Protocol 2.1: Core In Vitro ADME Assay Suite
- Absorption - Caco-2 Permeability Assay:
- Principle: Measures apparent permeability (Papp) across a monolayer of human colon adenocarcinoma cells, modeling intestinal absorption [83].
- Procedure: Culture Caco-2 cells on transwell inserts for 21 days. Apply the test phytochemical (e.g., 10 µM) to the apical chamber. Sample from basolateral chamber over 2 hours. Analyze compound concentration by LC-MS/MS. Calculate Papp and assess efflux ratio if transport is studied in both directions [83].
- Metabolism - Microsomal Stability Assay:
- Principle: Evaluates metabolic clearance using human liver microsomes (HLM), a source of cytochrome P450 enzymes [83].
- Procedure: Incubate test compound (1 µM) with HLM (0.5 mg/mL) and NADPH cofactor in potassium phosphate buffer. Aliquot reactions at time points (0, 5, 15, 30, 60 min). Stop reaction with cold acetonitrile. Quantify remaining parent compound via LC-MS/MS. Calculate half-life (t1/2) and intrinsic clearance (CLint) [83].
- Toxicity Screening - hERG Inhibition Patch Clamp:
- Principle: Assess risk for cardiac arrhythmia by measuring inhibition of the hERG potassium channel current [85].
- Procedure: Use a stable cell line expressing hERG channels. Employ whole-cell patch clamp electrophysiology. Hold cells at -80 mV, step to +20 mV to activate channels, then step to -50 mV to elicit tail current. Apply test compound cumulatively and measure inhibition of tail current amplitude (IC50 determination is ideal) [85].
Table 3: Representative Experimental Validation Outcomes from a Feedback Loop Cycle
| Phytochemical (Source) | AI Prediction | Experimental Result | Outcome & Action |
|---|---|---|---|
| Curcumin (Curcuma longa) | High predicted solubility; Moderate CYP3A4 inhibition risk [82]. | Low Caco-2 Papp (poor permeability); Confirmed moderate CYP3A4 inhibition. | Prediction Partially Validated. Action: Enter optimization loop (Phase 4) to design analogs with improved permeability. |
| Piperine (Piper nigrum) | High predicted permeability; High hERG risk alert [82]. | High Caco-2 Papp confirmed; hERG IC50 < 10 µM (high risk). | ADMET Risk Confirmed. Action: Depotentiate or deprioritize due to toxicity. Data used to refine hERG model. |
| Withaferin A (Withania somnifera) | Moderate predicted metabolic stability; High predicted activity for target X. | Moderate HLM stability (t1/2 = 25 min); High potency in target assay (IC50 = 0.1 µM). | Promising Lead. Action: Progress to advanced in vivo PK studies. Data added to training set for stability models. |
Phase 3: Data Integration & Model Refinement
Objective: To use experimental results to assess AI model performance, identify biases, and iteratively improve predictive accuracy.
Protocol 3.1: Model Performance Analysis & Retraining
- Discrepancy Analysis: Systematically compare predicted vs. experimental values for all assayed compounds. Calculate performance metrics (e.g., Root Mean Square Error (RMSE) for regression, Area Under the Curve (AUC) for classification).
- Chemical Space Interrogation: Use PCA or t-SNE plots to visualize the chemical space of tested compounds. Identify regions where model predictions consistently fail (e.g., specific scaffolds or physicochemical property ranges associated with prediction error).
- Model Retraining: Augment the original training dataset with the new, high-quality experimental data from Phase 2. Retrain the AI models using cross-validation to ensure improved generalizability and reduced overfitting [85].
- Output: A new, refined version of the predictive model with an expanded applicability domain and updated documentation on its performance and limitations.
Phase 4: Synthetic & Structural Optimization
Objective: To design new, improved compounds based on experimental insights and refined AI models.
Protocol 4.1: AI-Driven Analog Design
- Define Optimization Goals: Based on Phase 2 results, set clear multi-parameter objectives (e.g., increase permeability of Curcumin analog by 5x while maintaining potency and reducing CYP inhibition).
- Generative AI Design: Use a generative model (e.g., VAE or GAN) conditioned on the desired properties. Input a promising but flawed parent compound (e.g., Curcumin) and let the AI propose structural modifications [15].
- Synthetic Accessibility Filtering: Pass generated structures through a retrosynthetic analysis algorithm (e.g., using a rule-based system or a trained neural network) to filter for synthetically feasible compounds [85].
- Output: A set of novel, designed analog structures predicted to overcome the identified ADMET shortcomings, which are then fed back into Phase 1 for the next loop iteration.
Network Pharmacology & Pathway Analysis
For herbal compounds, which often exert effects via polypharmacology, network pharmacology is a crucial component of the feedback loop [6]. This involves mapping compounds to targets and affected signaling pathways.
Protocol 4.2: Network Pharmacology Workflow
- Target Prediction: Use AI-based DTI prediction tools to identify potential protein targets for the active phytochemical(s).
- Pathway Enrichment Analysis: Input the list of predicted and known targets into bioinformatics databases (e.g., KEGG, Reactome) to identify statistically enriched biological pathways.
- Mechanistic Hypothesis Generation: Formulate a testable hypothesis about the compound's mechanism of action based on the central nodes in the enriched pathways (e.g., "Compound X modulates the JAK-STAT pathway via targets A and B") [15].
The following diagram illustrates a key signaling pathway frequently targeted by immunomodulatory phytochemicals, such as those affecting PD-L1 expression, which can be investigated within this workflow [15].
Diagram 2: JAK-STAT-IRF1-PD-L1 Pathway for Phytochemical Intervention (Max Width: 760px)
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 4: Key Research Reagents and Materials for the Feedback Loop
| Reagent/Material | Function in the Loop | Application Example |
|---|---|---|
| Human Liver Microsomes (HLM) | Source of CYP450 enzymes for in vitro metabolism studies (Phase 2) [83]. | Determining metabolic stability (t1/2, CLint) of AI-prioritized phytochemicals. |
| Caco-2 Cell Line | Differentiated intestinal epithelial cell model for assessing passive and active transport (Phase 2) [83]. | Measuring apparent permeability (Papp) to predict oral absorption potential. |
| Recombinant hERG-Expressing Cell Line | Stable cell line for reliable, reproducible assessment of cardiotoxicity risk (Phase 2) [85]. | Patch-clamp electrophysiology to determine hERG channel inhibition potency (IC50). |
| LC-MS/MS System | High-sensitivity analytical instrument for quantitation of compounds in complex biological matrices [83]. | Quantifying parent compound loss in metabolic assays or transport in permeability assays. |
| Curated Phytochemical Library | Physically available collection of pure phytochemicals for experimental screening [86]. | Providing the tangible compounds for testing after AI virtual screening (Phase 1 to 2 handoff). |
| NADPH Regenerating System | Biochemical cofactor system essential for CYP450 enzyme activity in microsomal incubations [83]. | Supporting phase I oxidative metabolism reactions in HLM stability assays. |
The convergence of artificial intelligence (AI) and herbal medicine research represents a paradigm shift in the discovery and development of plant-based therapeutics. This integration directly addresses critical challenges in modern pharmacology, including the high cost and prolonged timelines of drug development, where less than 10% of new entities reach the market, with oncology success rates even lower [25]. For herbal research, which deals with chemically complex mixtures and vast, often unstructured traditional knowledge, AI offers transformative capabilities in predictive modeling, virtual screening, and multi-parameter optimization [88] [15].
Framed within a broader thesis on AI-guided ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction, this article posits that open-source AI tools are democratizing the field. They enable researchers to systematically evaluate the pharmacokinetic and safety profiles of herbal compounds early in the discovery pipeline. This is crucial, as illustrated by clinical cases where adulterated herbal preparations caused lead toxicity and adrenal insufficiency, underscoring the non-negotiable need for rigorous safety prediction [89]. By leveraging open-source platforms, researchers can accelerate the translation of traditional herbal knowledge—such as that from the Fertile Crescent or Ayurveda—into evidence-based, safe, and effective leads for pressing global health challenges, from cancer to cognitive decline [90] [91] [25].
AI Application Notes in Herbal Research Workflow
AI technologies are being integrated across the entire herbal research value chain, from initial plant identification to final lead optimization. The following table summarizes key AI applications and their impact on specific research phases.
Table 1: Key AI Applications Across the Herbal Research Pipeline
| Research Phase | Specific AI Application | Function & Benefit | Relevant Open-Source Tool/Approach |
|---|---|---|---|
| Plant Identification & Data Aggregation | Computer Vision for species ID [88]; NLP for literature mining [91] [88] | Automates species classification from images; extracts structured data on traditional uses from texts. | Deep learning models (e.g., CNN architectures in TensorFlow/PyTorch); NLP libraries (spaCy, NLTK). |
| Phytochemical Profiling | Metabolomics data analysis [88]; Spectral pattern recognition (MS, NMR) | Identifies and quantifies bioactive compounds in complex plant extracts. | Tools for chemoinformatics (RDKit); ML libraries (scikit-learn) for pattern analysis. |
| Bioactivity Prediction & Virtual Screening | QSAR modeling [13]; Deep learning for binding affinity prediction [15] | Predicts biological activity (e.g., anticancer, antimicrobial) against specific targets, prioritizing compounds for lab testing. | DeepChem; KNIME with cheminformatics extensions. |
| ADMET Prediction | Predictive modeling of pharmacokinetics and toxicity [13] [15] | Forecasts human absorption, metabolism, potential toxicity, and drug-likeness, filtering out problematic leads early. | ADMET prediction platforms (e.g., Deep-PK, DeepTox concepts); QSAR toolkits. |
| De Novo Design & Optimization | Generative AI (VAEs, GANs) [13] [15]; Multi-parameter optimization | Designs novel, synthetically accessible molecules with desired bioactivity and ADMET profiles inspired by herbal scaffolds. | PyTorch/TensorFlow for building generative models; Reinforcement learning frameworks. |
2.1. Plant Identification and Ethnobotanical Data Mining Computer vision algorithms, particularly Convolutional Neural Networks (CNNs), can be trained on curated image datasets (leaves, flowers, roots) to achieve high-accuracy identification of medicinal plant species, aiding field research and combating adulteration [88]. Concurrently, Natural Language Processing (NLP) techniques like named entity recognition can mine historical texts, clinical case reports, and modern literature to build structured databases linking plants, their traditional uses, and reported phytochemicals [91] [88]. For instance, an AI-aided scoping review of the Fertile Crescent's medicinal plants efficiently categorized research focus areas, demonstrating how AI can map a fragmented knowledge landscape [91].
2.2. Predictive Bioactivity and Multi-Target Screening The polypharmacological nature of herbal extracts—where multiple compounds act on multiple targets—is a key challenge. AI models excel here. Quantitative Structure-Activity Relationship (QSAR) models and more advanced graph neural networks can predict the interaction of phytochemicals with protein targets. For example, compounds can be screened in silico against immunomodulatory targets like PD-L1 or IDO1, which are critical in cancer immunotherapy [15]. This allows researchers to hypothesize and validate the mechanistic basis for traditional uses, such as identifying potential cognitive enhancers from a library of natural extracts [90].
2.3. AI-Guided ADMET Prediction for Herbal Compounds This is the core application for de-risking herbal drug development. Open-source AI models predict critical ADMET endpoints from molecular structure. Key predictive tasks include:
- Absorption & Permeability: Predicting Caco-2 cell permeability or human intestinal absorption.
- Metabolism: Forecasting sites of metabolism and interaction with cytochrome P450 enzymes.
- Toxicity: Identifying structural alerts for hepatotoxicity, cardiotoxicity, or genotoxicity [13].
These predictions are vital for prioritizing compounds. For instance, while a herbal compound may show potent activity in vitro, an AI model might flag a high risk of hepatotoxicity or poor oral bioavailability, guiding chemists to modify the structure or deprioritize it before costly laboratory experiments [13] [15].
Table 2: Key ADMET Endpoints for Herbal Compound Prioritization
| ADMET Property | Prediction Goal | Importance for Herbal Leads |
|---|---|---|
| Lipinski's Rule of Five | Drug-likeness filter. | Assesses oral bioavailability potential of isolated pure compounds. |
| Caco-2 Permeability | Estimates intestinal absorption. | Critical for orally administered herbal formulas. |
| Cytochrome P450 Inhibition | Predicts drug-metabolizing enzyme interactions. | Flags potential herb-drug interactions, a major safety concern. |
| hERG Channel Inhibition | Predicts cardiotoxicity risk. | Identifies compounds with potential for fatal arrhythmias. |
| Hepatotoxicity | Predicts liver injury risk. | Screens for a common toxicity issue in drug development. |
| AMES Test | Predicts mutagenic potential. | Assesses genotoxicity safety. |
Detailed Experimental Protocols
3.1. Protocol: AI-Assisted Virtual Screening of Herbal Compound Libraries for a Novel Target This protocol outlines a computational workflow to identify potential hit compounds from a herbal phytochemical library.
Objective: To screen an in silico library of phytochemicals against a defined protein target (e.g., IDO1 for immunomodulation [15]) using open-source docking and AI-based scoring. Materials/Software:
- Target Protein: PDB file of the target protein (e.g., from RCSB PDB).
- Compound Library: SDF file of phytochemicals (e.g., from databases like CMAUP or TCMSP).
- Software: RDKit (cheminformatics), AutoDock Vina or GNINA (docking), PyTorch/TensorFlow (for pre-trained scoring models).
- Computing Environment: Linux-based system or high-performance computing cluster.
Procedure:
- Library Preparation:
- Use RDKit to load the SDF library. Standardize molecules (neutralize charges, add hydrogens).
- Generate low-energy 3D conformers for each compound.
- Filter library based on basic drug-likeness rules (e.g., Lipinski's Rule of Five).
Target Preparation:
- Prepare the protein PDB file: remove water molecules, add polar hydrogens, define Gasteiger charges.
- Define the docking search space (grid box) centered on the target's known active site.
Molecular Docking:
- Execute docking simulations using AutoDock Vina for all library compounds.
- Output and rank compounds by docking score (estimated binding affinity in kcal/mol).
AI-Powered Re-scoring & Filtering:
- Apply a pre-trained graph neural network-based scoring model (e.g., implemented in DeepChem) to re-score the top 1000 docking hits. This model, trained on known binding data, often outperforms classical scoring functions [13].
- Filter the re-ranked list using open-source ADMET predictors (e.g., for hERG inhibition, hepatotoxicity).
- Output: A final prioritized list of 50-100 phytochemical hits with predicted strong binding and favorable safety profiles for in vitro validation.
3.2. Protocol: Building a Predictive ADMET Model for Herbal Compounds Objective: To train a machine learning model to predict a specific ADMET property (e.g., aqueous solubility) using a public dataset. Materials/Software:
- Dataset: Curated public dataset (e.g., from ChEMBL or ADMET benchmark datasets).
- Software: Python with scikit-learn, DeepChem, and RDKit libraries.
- Descriptors: Molecular fingerprints (e.g., Morgan fingerprints) or graph-based representations.
Procedure:
- Data Curation:
- Collect and merge data from reliable sources. Ensure consistent measurement units and endpoint values.
- Clean data: remove duplicates, handle missing values, correct obvious errors.
- Use RDKit to compute molecular fingerprints (e.g., 2048-bit Morgan fingerprint) for each compound as feature vectors (X). The measured ADMET endpoint (e.g., logS) is the label (y).
Model Training & Validation:
- Split data into training (70%), validation (15%), and test (15%) sets.
- Train multiple algorithms (e.g., Random Forest, Gradient Boosting, Graph Convolutional Network) on the training set.
- Optimize hyperparameters using the validation set via grid or random search.
- Evaluate model performance on the held-out test set using metrics like Mean Absolute Error (MAE) or Area Under the ROC Curve (AUC-ROC) for classification tasks.
Model Application & Interpretation:
- Save the best-performing model as a reusable file (e.g., using joblib).
- Use the model to predict the ADMET property for novel herbal compounds.
- Employ model interpretation tools (e.g., SHAP analysis) to identify which molecular substructures contribute positively or negatively to the prediction, guiding medicinal chemistry optimization [13].
Visualizing Workflows and Relationships
AI-Driven Herbal Discovery Workflow
AI-ADMET Prediction Integration
The Scientist's Toolkit: Essential Research Reagents & Materials
This table lists critical reagents, materials, and software resources for implementing the AI-driven protocols described, emphasizing open-source and widely accessible components.
Table 3: Essential Research Reagent Solutions for AI-Guided Herbal Research
| Item Name | Category | Function in Research | Example/Note |
|---|---|---|---|
| Herbal Phytochemical Library (Digital) | Digital Resource | Provides structured, machine-readable molecular data for virtual screening. | Libraries from CMAUP, TCMSP, NPASS. Format: SDF or SMILES. |
| Curated ADMET Datasets | Digital Resource | Serves as labeled training data for building or benchmarking predictive AI models. | Datasets from ChEMBL, Tox21, ADMETlab. |
| RDKit | Open-Source Software | Core cheminformatics toolkit for molecule manipulation, descriptor calculation, and fingerprint generation. | Python library. Essential for preprocessing steps before AI modeling. |
| DeepChem | Open-Source Software | Deep learning library specifically designed for chemoinformatics and drug discovery tasks. | Provides out-of-the-box models for toxicity prediction and molecular property analysis. |
| AutoDock Vina / GNINA | Open-Source Software | Performs molecular docking to predict how herbal compounds bind to protein targets. | GNINA incorporates CNN-based scoring for improved accuracy [13]. |
| Standardized Plant Extracts & Pure Phytochemicals | Physical Reagent | Provides material for in vitro and in vivo validation of AI predictions. | Critical for moving from in silico hits to experimental confirmation. Commercial suppliers or in-house isolation. |
| In Vitro ADMET Assay Kits | Physical Reagent | Validates AI predictions of absorption, metabolism, and toxicity in the laboratory. | Examples: Caco-2 permeability assay kits, CYP450 inhibition kits, hERG binding assays. |
| High-Performance Computing (HPC) Resources | Infrastructure | Provides the computational power needed for training large AI models and screening massive libraries. | Cloud platforms (Google Colab Pro, AWS) or institutional HPC clusters. |
Benchmarks, Blinded Trials, and Real-World Impact: Validating AI Models for Herbal ADMET
The integration of herbal medicine into modern therapeutics necessitates a foundational shift from traditional use to evidence-based validation. A central challenge in this endeavor is the efficient and accurate prediction of the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiles of complex herbal compounds [92]. Poor ADMET properties are a leading cause of failure in drug development, making early and reliable prediction critical for prioritizing promising herbal leads [23].
This document provides detailed application notes and protocols for constructing rigorous validation frameworks within the context of a broader thesis on AI-guided ADMET prediction for herbal compounds. Machine learning (ML) models offer powerful tools for this task, but their reliability is contingent upon stringent validation to avoid over-optimistic performance estimates and ensure generalizability to new, unseen chemical entities [93]. We focus on three pillars of robust validation: Cross-Validation for robust internal performance estimation, External Test Sets for assessing real-world generalizability, and comprehensive Performance Metrics for nuanced model evaluation [41]. These frameworks are designed to provide researchers with methodologies to build credible, reproducible, and clinically translatable predictive models for herbal drug discovery.
Core Validation Framework Components
Cross-Validation: Protocols for Robust Internal Validation
Cross-validation (CV) is a foundational technique for estimating model performance when a single, dedicated external test set is not available or must be preserved. It mitigates the bias and variance associated with a single random train-test split [94].
Protocol 2.1.1: Stratified K-Fold Cross-Validation for Herbal ADMET Classification
- Objective: To obtain a reliable, bias-reduced estimate of model performance for binary or multi-class ADMET endpoints (e.g., hepatotoxicity, CYP450 inhibition) while preserving class distribution.
- Materials: Pre-processed dataset of herbal compound molecular representations (e.g., fingerprints, descriptors) and associated ADMET labels.
- Procedure:
- Data Preparation: Ensure the dataset is cleaned (deduplicated, standardized) and featurized [41].
- Fold Generation: Initialize
StratifiedKFoldfromscikit-learnwithn_splits=5or10. This algorithm partitions the data into k folds, ensuring each fold maintains the same proportion of class labels as the original dataset [94]. - Iterative Training & Validation: For each unique fold
i:- Designate fold
ias the validation set. - Designate the remaining k-1 folds as the training set.
- Train the ML model (e.g., Random Forest, XGBoost) on the training set.
- Predict on the validation set and compute the chosen metric(s) (e.g., AUC-ROC, Balanced Accuracy).
- Designate fold
- Performance Aggregation: Store the metric from each iteration. The final performance estimate is the mean ± standard deviation across all k folds. The standard deviation indicates the model's sensitivity to specific data splits [93].
Protocol 2.1.2: Nested Cross-Validation for Hyperparameter Tuning and Model Selection
- Objective: To perform model selection and hyperparameter optimization without data leakage, providing an unbiased estimate of the performance of the best-found model pipeline.
- Procedure:
- Define Outer Loop: Split data into K outer folds (e.g., 5). Each fold will serve once as the holdout test set.
- Define Inner Loop: For each outer training set, perform a second, independent CV (e.g., 3-fold) to tune hyperparameters.
- Optimize: For each hyperparameter candidate, evaluate its average performance across the inner CV folds. Select the best parameters.
- Retrain & Evaluate: Retrain a model on the entire outer training set using the best parameters. Evaluate it on the held-out outer test set.
- Final Estimate: The average performance across all K outer test folds is the unbiased estimate of the tuned model's performance [93].
Comparative Analysis of Cross-Validation Methods Table 1: Suitability of cross-validation methods for herbal ADMET modeling.
| Method | Key Principle | Advantages | Disadvantages | Recommended Use Case |
|---|---|---|---|---|
| K-Fold [94] | Randomly split data into K equal folds. | Reduces variance from a single split; uses all data for validation. | Can create imbalanced folds for skewed datasets. | Preliminary regression tasks with balanced data. |
| Stratified K-Fold [94] | K-Fold preserving class distribution in each fold. | Essential for imbalanced classification tasks. | Only applicable to classification problems. | Binary ADMET classification (e.g., toxicity). |
| Leave-One-Out (LOOCV) [94] | Each sample is a validation fold; model trained on all others. | Low bias; uses maximum data for training. | High computational cost; high variance in estimate. | Very small datasets (<100 samples). |
| Nested CV [93] | Separate loops for parameter tuning (inner) and error estimation (outer). | Prevents data leakage; most unbiased performance estimate. | Very high computational cost. | Final model evaluation & reporting. |
External Test Sets: The Gold Standard for Generalizability
An external test set is data that is completely withheld from the model development and tuning process, often sourced from a different study, laboratory, or time period. It is the ultimate test of a model's utility for prospective prediction [41].
Protocol 2.2.1: Construction and Use of an External Test Set
- Objective: To evaluate the model's ability to generalize to novel herbal compounds outside its training distribution.
- Protocol:
- Source Identification: Procure an external dataset for the same ADMET endpoint from an independent source (e.g., a different public database, literature, or in-house assay) [41]. For herbal compounds, this may involve separate phytochemical studies.
- Rigorous Withholding: This external set must never be used for feature selection, parameter tuning, or any aspect of model training. It should be locked away until the final model is fully specified [93].
- Preprocessing Consistency: Apply the exact same preprocessing steps (e.g., featurization, normalization, cleaning rules) to the external data as were applied to the training data.
- Blinded Evaluation: Use the finalized, frozen model to predict the external test set. Compare predictions against the ground truth using comprehensive metrics. A significant drop in performance from CV to external testing indicates overfitting and limited generalizability [41].
Performance Metrics: Beyond Simple Accuracy
Selecting appropriate metrics is critical for accurate model assessment, especially for imbalanced datasets common in ADMET prediction (e.g., where toxic compounds are rare) [23].
Protocol 2.3.1: Metric Selection and Interpretation
- For Classification Tasks (e.g., Toxic/Non-Toxic):
- Primary Metric: Use the Area Under the Receiver Operating Characteristic Curve (AUC-ROC). It evaluates the model's ability to rank positive and negative instances across all classification thresholds and is robust to class imbalance.
- Supporting Metrics: Report Precision, Recall (Sensitivity), Specificity, and F1-Score at a defined probability threshold (e.g., 0.5). A confusion matrix should be generated to visualize these metrics.
- For Regression Tasks (e.g., Solubility, Clearance Value):
- Primary Metrics: Report Mean Absolute Error (MAE) for interpretability and Root Mean Squared Error (RMSE) for penalizing larger errors.
- Supporting Metric: Report the Coefficient of Determination (R²) to indicate the proportion of variance explained by the model.
Comparative Analysis of Model Performance Metrics Table 2: Key performance metrics for evaluating herbal ADMET prediction models.
| Task Type | Metric | Formula / Principle | Interpretation | When to Use | ||
|---|---|---|---|---|---|---|
| Classification | AUC-ROC | Area under the True Positive Rate vs. False Positive Rate curve. | 1.0 = perfect classifier; 0.5 = random guess. Robust to imbalance. | Primary metric for imbalanced ADMET classification. | ||
| Classification | F1-Score | Harmonic mean of Precision and Recall: 2*(Precision*Recall)/(Precision+Recall) |
Balances false positives and false negatives. Best for skewed classes. | When both Precision and Recall are important. | ||
| Classification | Balanced Accuracy | (Sensitivity + Specificity) / 2 |
Accuracy adjusted for class imbalance. | Better than standard accuracy for imbalanced data. | ||
| Regression | Root Mean Squared Error (RMSE) | sqrt(mean((y_true - y_pred)^2)) |
Punishes large errors more severely. In target variable units. | Primary metric for penalizing large prediction errors. | ||
| Regression | Mean Absolute Error (MAE) | `mean( | ytrue - ypred | )` | Average magnitude of error. Easier to interpret. | Primary metric for interpretability of average error. |
| Regression | R-squared (R²) | 1 - (SS_res / SS_tot) |
Proportion of variance explained. 1.0 = perfect fit. | To understand how well the model captures data variance. |
Integrated Validation Workflow for Herbal ADMET Prediction
This section synthesizes the core components into a complete, sequential experimental protocol for a thesis research project.
Protocol 3.1: Comprehensive Validation of an AI Model for Herbal Compound Hepatotoxicity Prediction
- Aim: To develop and rigorously validate a binary classifier predicting hepatotoxicity of herbal phytochemicals.
- Phase 1: Data Curation & Partitioning
- Data Collection: Aggregate herbal compound structures and hepatotoxicity labels from public sources (e.g., TDC, PubChem) and proprietary thesis research [41] [23].
- Data Cleaning: Standardize SMILES, remove duplicates and inorganic salts, and resolve inconsistent measurements as per protocol in [41].
- Featurization: Compute molecular descriptors (e.g., RDKit) and fingerprints (e.g., Morgan) for all compounds.
- Strategic Splitting:
- External Test Set: Immediately withhold 15-20% of the data, ensuring it is from a distinct chemical scaffold or data source. Lock this away.
- Development Set: Use the remaining 80-85% for all model development.
- Phase 2: Model Development with Nested CV
- On the Development Set, run a Nested Cross-Validation (Protocol 2.1.2) to:
- Test multiple algorithms (e.g., Random Forest vs. SVM vs. GNN).
- Tune their hyperparameters.
- Select the best model based on the mean outer fold AUC-ROC.
- On the Development Set, run a Nested Cross-Validation (Protocol 2.1.2) to:
- Phase 3: Final Model Training & Evaluation
- Train the final model on the entire Development Set using the optimal algorithm and hyperparameters identified in Phase 2.
- Blinded External Evaluation: Apply the final model to the locked External Test Set. Record AUC-ROC, F1-Score, Precision, and Recall.
- Internal-External Validation (if possible): As a final stress test, train the model on all internal data and evaluate it on a completely independent dataset from another lab or publication [41].
- Deliverables:
- A trained, ready-to-use model.
- A validation report detailing: CV performance (mean ± std), final external test performance, and a clear analysis of the performance gap (if any).
- A SHAP or LIME analysis for model interpretability, highlighting chemical features driving toxicity predictions.
Visualization of the Validation Framework
Diagram 1: Comprehensive validation workflow for AI-guided herbal ADMET prediction.
Table 3: Essential software, databases, and resources for building herbal ADMET prediction models.
| Category | Item / Software | Primary Function | Application in Herbal ADMET Research |
|---|---|---|---|
| Cheminformatics & Featurization | RDKit (Open-source) | Calculation of molecular descriptors and fingerprints [41]. | Generating numerical representations (features) from herbal compound structures (SMILES). |
| Machine Learning Frameworks | scikit-learn (Python) | Provides implementations of classic ML algorithms (RF, SVM) and validation tools (CV splitters) [94]. | Building and validating baseline predictive models. |
| Deep Learning Frameworks | PyTorch / TensorFlow | Flexible frameworks for building deep neural networks (DNNs) and graph neural networks (GNNs). | Implementing advanced architectures for learning directly from molecular graphs. |
| Specialized ADMET Modeling | Chemprop (DGL) | A message-passing neural network (MPNN) specifically designed for molecular property prediction [41]. | State-of-the-art prediction of ADMET properties from molecular structures. |
| Data Sources & Benchmarks | Therapeutics Data Commons (TDC) | Curated benchmarks and datasets for ADMET prediction tasks [41]. | Accessing standardized datasets for training and benchmarking models. |
| Data Sources & Benchmarks | PubChem | Public repository of chemical structures, bioactivities, and assays [41]. | Sourcing experimental ADMET data for herbal and synthetic compounds. |
| Validation & Statistics | SciPy / StatsModels | Libraries for statistical testing and analysis. | Performing hypothesis tests (e.g., paired t-test) to statistically compare model performances from CV [41]. |
| Visualization & Reporting | Matplotlib / Seaborn | Python libraries for creating static, publication-quality plots and charts [95]. | Generating performance plots (ROC curves, scatter plots), confusion matrices, and result figures. |
The ASAP-Polaris-OpenADMET Blind Challenge represents a paradigm shift in evaluating computational methods for drug discovery [96]. Organized by the NIH-funded ASAP Discovery Consortium, the Polaris benchmarking platform, and the ARPA-H-funded OpenADMET project, this community-wide initiative provided a rare opportunity to test machine learning (ML) and physics-based models against real, undisclosed preclinical data from a pan-coronavirus antiviral program [97] [98]. For researchers focused on AI-guided ADMET prediction for herbal compounds—a field often hampered by small, inconsistent datasets—the insights from this rigorous benchmark are invaluable [6]. The challenge's structure around potency, ADMET, and ligand posing directly mirrors the core triage steps in natural product lead optimization, where understanding bioavailability and safety is as critical as confirming activity [99] [100]. This analysis translates the challenge's key findings into actionable protocols and perspectives for advancing the prediction of herbal compound pharmacokinetics and toxicity.
Quantitative Benchmarking Results and Performance Analysis
The challenge attracted 66 international teams, whose submissions created a clear landscape of the state-of-the-art [96] [98]. The performance data underscores both the promise and the persistent gaps in predictive modeling.
Table 1: Summary of Key Performance Metrics from the Blind Challenge
| Sub-Challenge | Primary Evaluation Metric | Top-Performing Result / Key Benchmark | Implication for Herbal Compound Research |
|---|---|---|---|
| Biochemical Potency (pIC50) | Mean Absolute Error (MAE) | Best models achieved MAE of ~0.5 log units [98]. Simple local models were highly competitive [101]. | Potency prediction for novel natural product scaffolds may not require complex AI; robust local models can be effective. |
| ADMET Endpoints | MAE on log-transformed data [97] | Winning model used external ADMET data. Error was 23-41% lower than models without it [101]. | Data quality and relevance are paramount. Integrating high-quality external ADMET data is crucial for herbal libraries. |
| Ligand Pose Prediction | % of poses with RMSD < 2Å [97] | Best methods achieved >80% success rate [98]. Performance varied by target and chemotype. | Predicting how complex herbal metabolites bind to targets or off-targets (e.g., hERG) remains a significant challenge. |
A deeper analysis of ADMET model performance reveals critical dependencies on data strategy and chemical space.
Table 2: Impact of Modeling Strategy on ADMET Prediction Performance
| Modeling Strategy | Description | Relative Performance (vs. Winning Model) | Key Insight for Herbal Informatics |
|---|---|---|---|
| Global Model + External ADMET Data | Model trained on challenge data plus additional, curated ADMET datasets. | Baseline (Best Performance) [101] | Demonstrates the value of augmenting limited program-specific data with high-quality, task-specific external data. |
| Large Non-Task-Specific Pretrained Model | Model pre-trained on massive chemical datasets (e.g., MolE, MolGPS) without ADMET labels. | 37% higher error [101] | General chemical representation learning, without domain-specific fine-tuning, offers limited direct benefit for ADMET tasks. |
| Local Model (Descriptors/Fingerprints) | Traditional ML (e.g., Random Forest) using only the provided challenge training data. | 53-60% higher error [101] | Highlights the limitation of small, localized datasets common in natural product projects. |
Crucially, performance was highly variable across different ADMET endpoints and chemical series [101]. For example, predicting MDR1-MDCKII permeability was easier on the challenge test set due to its specific chemical series composition, while kinetic solubility was harder because most data clustered at the assay's upper limit [101]. This program-dependence of model performance is a critical caveat: a method that excels on one herbal chemical series (e.g., flavonoids) may not generalize well to another (e.g., terpenoids) [6].
Experimental Protocols from the Challenge
The reliability of the benchmark stems from the high-quality, standardized experimental data generated by the ASAP consortium. These protocols serve as a gold standard for generating data to train predictive models for herbal compounds.
Protocol for Biochemical Potency Assay (pIC50 Determination)
- Objective: To measure the half-maximal inhibitory concentration (IC50) of compounds against SARS-CoV-2 and MERS-CoV Main Protease (Mpro) [100].
- Materials:
- Procedure:
- Serially dilute test compounds in DMSO and then in assay buffer.
- In a black 384-well plate, mix enzyme with compound solution and incubate.
- Initiate the reaction by adding the fluorogenic substrate.
- Measure fluorescence increase (excitation ~360 nm, emission ~460 nm) continuously for 30-60 minutes.
- Calculate reaction velocity for each well.
- Fit dose-response curves to determine IC50 values, which are then converted to pIC50 (-log10 IC50) for modeling [100].
Protocol for Key ADMET Endpoints
The challenge evaluated multiple ADMET properties; the following are particularly relevant for herbal compound profiling [97] [101].
Human Liver Microsomal (HLM) Stability
- Objective: Measure metabolic clearance in vitro.
- Method: Incubate test compound with pooled human liver microsomes and NADPH cofactor. Quantify parent compound loss over time via LC-MS/MS to determine intrinsic clearance [101].
Kinetic Solubility (PBS, pH 7.4)
- Objective: Determine equilibrium solubility in physiologically relevant buffer.
- Method: Disperse solid compound in phosphate-buffered saline, shake for a defined period (e.g., 24 hours), filter, and quantify concentration in the filtrate via UV or CLD (chemiluminescent nitrogen detection) [101].
MDR1-MDCKII Apparent Permeability (Papp)
- Objective: Assess cell membrane permeability and P-glycoprotein efflux liability.
- Method: Grow MDCKII cells overexpressing human MDR1 on transwell filters. Apply compound to the donor chamber (apical-to-basolateral for Papp A-B, and vice-versa for Papp B-A). Measure compound appearance in the receiver chamber by LC-MS/MS to calculate apparent permeability and efflux ratio [101].
Protocol for X-ray Crystallography (Ligand Pose Determination)
- Objective: Determine the three-dimensional atomic structure of Mpro in complex with an inhibitor [97].
- Procedure:
- Protein Purification & Crystallization: Purify recombinant Mpro and grow crystals using vapor diffusion methods.
- Ligand Soaking/Co-crystallization: Introduce the small molecule inhibitor into the crystal by soaking or co-crystallization.
- Data Collection: Flash-freeze the crystal and collect X-ray diffraction data at a synchrotron source (e.g., Diamond Light Source) [97].
- Structure Solution: Use molecular replacement to solve the phase problem, followed by iterative model building and refinement to produce the final electron density map and atomic coordinates, which serve as the ground truth for ligand pose [96].
Visualizing Workflows and Insights
Diagram 1: ASAP-Polaris-OpenADMET Challenge Workflow & Herbal Research Integration
Diagram 2: Modeling Strategy Performance Comparison
Diagram 3: Translating Challenge Insights to Herbal Compound Research
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for Emulating Challenge-Quality Experiments
| Item / Reagent | Function in the Challenge Context | Relevance to Herbal Compound ADMET Research |
|---|---|---|
| Recombinant Viral Proteases (e.g., SARS-CoV-2 Mpro) | Target protein for biochemical potency assays [100]. | Can be substituted with recombinant human ADME-relevant enzymes (CYPs, UGTs) or toxicity targets (e.g., hERG channel protein) for herbal metabolite screening. |
| Fluorogenic Peptide Substrates | Enable real-time, high-throughput kinetic measurement of protease inhibition [97]. | Representative of robust, quantitative assay reagents needed to generate high-quality data for model training. |
| Pooled Human Liver Microsomes (HLM) | In vitro system for Phase I metabolic stability assessment [101]. | Critical reagent for predicting herbal compound metabolism and potential drug-drug interactions. |
| MDR1-MDCKII Cell Line | Cell monolayer model for assessing permeability and P-gp efflux liability [101]. | Standard system for evaluating intestinal absorption and blood-brain barrier penetration of herbal metabolites. |
| Crystallography-Grade Protein & Crystallization Kits | Enabling determination of 3D ligand-protein structures for pose validation [97] [96]. | For structural biology efforts on herbal compounds binding to proteins involved in ADMET (e.g., CYP3A4, hERG). |
| CDD Vault Public / Polaris Hub | Platforms for collaborative, secure data management and public dataset access [102] [103]. | Essential for curating, sharing, and finding high-quality herbal compound bioactivity and ADMET data to build better models. |
Discussion: Implications for AI-Guided Herbal Compound Research
The blind challenge conclusively demonstrates that data quality and strategic curation are more impactful than algorithmic complexity for ADMET prediction [101] [99]. This is a pivotal lesson for herbal informatics, where data is often the primary bottleneck [6]. The superior performance of models augmented with external ADMET data argues for a concerted effort to create and standardize high-throughput ADMET profiles for key herbal metabolite scaffolds. Furthermore, the observed program-dependence of model performance mandates a focus on defining applicability domains for any model applied to novel herbal chemistries [101] [99].
Future research should adopt the challenge's "blind" prospective evaluation paradigm, using temporal splits of herbal compound data to simulate real-world discovery [100]. The OpenADMET project's ongoing mission to generate open datasets and models for the "avoidome"—targets to be avoided for safety—is directly aligned with the needs of herbal medicine research to predict and mitigate off-target toxicity [99] [102]. By embracing the collaborative, open-science principles and rigorous benchmarking standards exemplified by the ASAP-Polaris-OpenADMET challenge, the field of AI-guided herbal compound research can accelerate the transformation of traditional remedies into safe, effective, and well-characterized modern therapeutics.
The integration of artificial intelligence (AI) into natural product (NP) discovery represents a paradigm shift, moving the field from manual, trial-and-error processes to data-driven, predictive pipelines [104]. This transformation is critically important within the broader thesis on AI-guided ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) prediction for herbal compounds. Herbal medicines, with their complex mixtures and multi-target pharmacology, present unique challenges for standard pharmacokinetic and safety evaluation [6]. AI not only accelerates the identification of bioactive NPs but also provides a powerful framework for early ADMET profiling, thereby de-risking the development pipeline and bridging the gap between traditional herbal medicine and modern drug development standards [105] [74].
AI-driven NP discovery employs a suite of machine learning (ML) and deep learning (DL) techniques to navigate the vast, complex chemical space of natural compounds [105]. Key methodologies include:
- Predictive Modeling & Virtual Screening: ML models trained on NP databases can predict biological activity, enabling the ultra-large virtual screening of millions of compounds to prioritize candidates for experimental testing [104] [84].
- Generative AI for Molecular Design: Generative models, including Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Transformer-based architectures, can design novel "NP-inspired" scaffolds that retain desirable bioactivity while improving synthetic feasibility and drug-like properties [104] [106].
- Knowledge Graph & Multimodal Fusion: Integrating disparate data types—chemical structures, genomic data from biosynthetic gene clusters (BGCs), spectral information (NMR, MS), and biological assay results—into connected knowledge graphs enables sophisticated target fishing, mechanism inference, and repurposing predictions [104] [6].
- ADMET & Property Prediction: Supervised learning models are trained on curated datasets to predict critical pharmacokinetic and safety endpoints, allowing for the early prioritization of compounds with a higher probability of clinical success [84] [74].
The following diagram illustrates the logical workflow of an integrated AI-driven NP discovery campaign, highlighting the critical role of ADMET prediction within the broader thesis context.
AI-Driven Natural Product Discovery Workflow
Case Studies of Successful AI-Driven Campaigns
Case Study: AI-ADMET-Guided Discovery of Acetylcholinesterase Inhibitors from a Thai Herbal Formulation
This study exemplifies the thesis focus on AI-guided ADMET prediction for herbal compounds [74].
- Objective: To identify neuroprotective components from the multi-herb formulation Suk-Saiyasna and evaluate their potential as acetylcholinesterase (AChE) inhibitors for Alzheimer's disease.
- AI/Computational Methodology:
- Molecular Docking: 167 ligands (cannabinoids, flavonoids, terpenoids, alkaloids) from the formulation were docked into the human AChE crystal structure (PDB: 4EY7).
- In Silico ADMET Prediction: Key pharmacokinetic and toxicity properties (e.g., GI absorption, BBB permeability, CYP inhibition, mutagenicity) were predicted using the pkCSM and ProTox-II platforms.
- Key Findings & Validation:
- The Suk-Saiyasna extract showed significant in vitro AChE inhibitory activity (IC~50~ = 1.25 ± 0.35 mg/mL) and protected SH-SY5Y cells from amyloid-β42-induced cytotoxicity.
- Docking identified Δ9-THC, mesuaferrone B, piperine, β-sitosterol, and chlorogenic acid as top binders, with binding energies superior to standard drugs (galantamine, rivastigmine).
- ADMET predictions were crucial for prioritization: Δ9-THC and piperine showed favorable profiles, including high BBB permeability and no predicted neurotoxicity for Δ9-THC.
Table 1: Key Quantitative Results from Suk-Saiyasna Study [74]
| Assay/Parameter | Result | Implication |
|---|---|---|
| DPPH Radical Scavenging (IC~50~) | 27.40 ± 1.15 µg/mL | Confirms direct antioxidant activity of the extract. |
| In Vitro AChE Inhibition (IC~50~) | 1.25 ± 0.35 mg/mL | Validates the primary therapeutic mechanism of action. |
| Cell Viability (Aβ42-induced stress) | Significant protection at 1 µg/mL | Demonstrates neuroprotective effect at a low concentration. |
| Top Docking Score (Δ9-THC) | -10.4 kcal/mol | Stronger predicted binding affinity than reference drugs. |
| Predicted BBB Permeability (Δ9-THC) | High (LogBB > 0.3) | AI-ADMET prediction suggests compound can reach brain target. |
Case Study: Autonomous AI Platform for Enzyme Engineering in NP Biosynthesis
While focused on protein engineering, this study provides a transferable protocol for closed-loop, AI-driven optimization relevant to engineering biosynthetic pathways for NP production [107].
- Objective: To develop a generalized autonomous platform for rapid enzyme engineering using a Design-Build-Test-Learn (DBTL) cycle.
- AI/Experimental Methodology:
- Design: Variant libraries were designed using a protein Large Language Model (ESM-2) and an epistasis model (EVmutation).
- Build & Test: An automated biofoundry (iBioFAB) performed HiFi-assembly mutagenesis, protein expression, and high-throughput enzymatic assays.
- Learn: Assay data trained a low-N machine learning model to predict fitness and guide the design of the next iterative cycle.
- Key Outcome: The platform successfully engineered a halide methyltransferase (AtHMT) for a 16-fold improvement in ethyltransferase activity within 4 weeks and fewer than 500 variants tested, showcasing the dramatic acceleration possible through AI-automation integration.
Table 2: Performance of AI Models in NP & Small-Molecule Discovery [104] [84] [106]
| AI Application Area | Model/Platform Type | Reported Performance/Outcome | Validation Stage |
|---|---|---|---|
| Virtual Screening | Deep Learning QSAR / Neural Network Scoring | >75% hit validation rate in some campaigns; outperforms classical docking [106]. | In vitro validation |
| Generative Design | Conditional VAE (CVAE) | Generated 3,040 molecules; identified 15 dual-active CDK2/PPARγ inhibitors; 30-fold selectivity gain [106]. | Preclinical (IND-enabling) |
| Generative Design | Reinforcement Learning (ReLeaSE) | Generated 50,000 JAK2 inhibitor scaffolds; 12 with IC~50~ ≤ 1 µM; 85% had improved CYP450 profiles [106]. | In vivo (xenograft) |
| Property Optimization | AI-ADMET Prediction Models | Enables early filtering, reducing late-stage attrition due to PK/toxicity issues [84] [105]. | In silico, guides experimental design |
Application Notes & Experimental Protocols
Protocol: Integrated In Silico & In Vitro Workflow for Herbal Compound Screening
This protocol is adapted from the Suk-Saiyasna case study and is tailored for research on herbal compounds [74].
A. In Silico Screening & ADMET Profiling
- Compound Library Preparation:
- Curate a 2D/3D structural library of known constituents from the herbal source using literature mining and databases (e.g., PubChem, NPASS).
- Prepare ligands: Optimize geometries and assign charges using tools like Open Babel or RDKit.
- Molecular Docking:
- Target Preparation: Retrieve the 3D protein structure (e.g., from PDB). Remove water, add hydrogens, and define the binding site grid.
- Docking Execution: Perform docking simulations using AutoDock Vina, Glide, or similar. Use standard protocols with appropriate exhaustiveness.
- Analysis: Rank compounds by binding affinity (kcal/mol). Visually inspect top poses for key interactions (H-bonds, π-stacking, hydrophobic contacts).
- In Silico ADMET Prediction:
- Use platforms such as pkCSM, SwissADME, or ProTox-II.
- Input SMILES strings of top-ranked docked compounds. Predict and record key parameters: Water solubility, Caco-2 permeability, BBB penetration, CYP450 inhibition, hepatotoxicity, Ames mutagenicity.
- Filter compounds based on a balanced profile of good binding energy and favorable predicted ADMET properties.
B. In Vitro Experimental Validation
- Bioassay-Guided Fractionation:
- Prepare crude extract of the herbal material. Use the primary target assay (e.g., AChE inhibition) to guide the fractionation of active extracts via column chromatography.
- Enzyme Inhibition Assay (e.g., AChE):
- Follow Ellman's method. In brief, mix test compound/extract with AChE enzyme, DTNB, and acetylthiocholine iodide in buffer.
- Incubate and measure the increase in absorbance at 412 nm from the reaction product.
- Calculate % inhibition and IC~50~ values using non-linear regression.
- Cytotoxicity & Neuroprotection Assay:
- Culture relevant cell lines (e.g., SH-SY5Y neuroblastoma). Pre-treat cells with test compounds, then induce stress (e.g., with amyloid-β42).
- Measure cell viability using MTT or resazurin assays after 24-48 hours.
Protocol: Design-Build-Test-Learn (DBTL) Cycle for AI-Driven Optimization
This protocol is derived from the autonomous enzyme engineering platform and can be adapted for optimizing NP-producing pathways [107].
The following diagram visualizes the iterative DBTL cycle, which is central to modern AI-driven discovery platforms.
AI-Optimization DBTL Cycle
- Design Phase:
- Define the objective (e.g., improve enzyme activity, increase NP yield).
- Use generative or predictive ML models (e.g., protein LLMs for enzymes, VAEs for small molecules) to propose a focused, diverse library of variants/conditions.
- Build Phase:
- Automate the construction process. For enzymes: use robotic liquid handlers for site-directed mutagenesis PCR and transformation. For NP pathways: automate plasmid assembly for biosynthetic gene cluster refactoring.
- Test Phase:
- Implement automated, miniaturized, high-throughput assays (e.g., microplate-based enzymatic assays, LC-MS for product quantification) to generate fitness data for each variant.
- Learn Phase:
- Aggregate the experimental data. Train or fine-tune the initial AI model with the new results to improve its predictive power.
- Use the updated model to design the next, more informed library, closing the loop.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for AI-Driven NP Discovery Campaigns
| Category | Item/Resource | Function & Application in NP Research | Example/Note |
|---|---|---|---|
| Software & Databases | NP-Specific Databases (e.g., NPASS, COCONUT, LOTUS) | Provide curated chemical structures and associated bioactivity data for model training and dereplication [104]. | Critical for building NP-aware AI models. |
| Docking Software (AutoDock Vina, Glide, MOE) | Predict binding pose and affinity of NP constituents against protein targets [74]. | First step in virtual screening workflows. | |
| ADMET Prediction Platforms (pkCSM, SwissADME, ProTox-II) | Provide early in silico estimates of pharmacokinetics and toxicity for prioritization [74]. | Core to the thesis focus on ADMET prediction. | |
| Generative AI Platforms (Chemistry42, REINVENT) | De novo design of novel molecules with specified properties, inspired by NP scaffolds [104] [106]. | Used for lead generation and optimization. | |
| Laboratory Materials | Automated Liquid Handling Systems | Enable high-throughput preparation of assays, fractionation plates, and PCR reactions for DBTL cycles [107]. | Essential for scaling experimental validation. |
| High-Content Screening Assay Kits | Provide standardized, robust biochemical (e.g., AChE inhibition) or cellular assays for testing NP bioactivity [74]. | Key for the "Test" phase of DBTL. | |
| LC-MS/MS Systems | Essential for dereplication (identifying known compounds), quantifying NP yields, and analyzing complex mixtures [104]. | Bridges analytical chemistry and bioinformatics. | |
| AI/Computational Infrastructure | GPU Clusters | Accelerate the training of deep learning models (e.g., GNNs, Transformers) on large chemical datasets [105]. | Required for complex generative or predictive tasks. |
| Cloud-Based ML Services (AWS SageMaker, Google Vertex AI) | Provide scalable environments for building, training, and deploying custom AI models without local hardware constraints. | Facilitates collaboration and reproducibility. |
The integration of Artificial Intelligence (AI) into drug discovery represents a paradigm shift, particularly for the complex domain of herbal compound research. Herbal medicines, characterized by their multi-component, multi-target nature and inherent chemical variability, present unique challenges for predicting Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) using traditional methods [6] [1]. This article frames the comparative performance of AI models, traditional Quantitative Structure-Activity Relationship (QSAR), and experimental methods within the context of a broader thesis on AI-guided ADMET prediction. The thesis posits that AI and machine learning (ML) are not merely incremental improvements but are essential for deconvoluting the synergistic pharmacology and polypharmacology of herbal extracts, enabling the transition from phenotypic observations to mechanistically grounded, personalized therapeutics [6] [4]. By bridging traditional computational chemistry with contemporary AI, researchers can establish innovative workflows to accelerate the discovery and safety profiling of natural product-derived therapeutics [108] [109].
Performance Comparison: Quantitative Metrics and Qualitative Insights
The performance of computational and experimental methods can be evaluated across dimensions of speed, cost, accuracy, and applicability to herbal compounds. The following table summarizes a comparative analysis.
Table 1: Comparative Performance of ADMET Prediction Methodologies for Herbal Compound Research
| Method Category | Key Techniques/Examples | Typical Time Scale | Relative Cost | Key Strengths | Primary Limitations for Herbal Research |
|---|---|---|---|---|---|
| Traditional QSAR & Molecular Modeling | MLR, PLS, Molecular Docking (e.g., AutoDock), Pharmacophore Mapping [108] [110]. | Days to weeks (per model/screen). | Low to Moderate (computational resources). | High interpretability; strong theoretical foundation; excellent for lead optimization of single compounds [110]. | Struggles with multi-component mixtures; requires curated, congeneric datasets; cannot handle "chemistry-agnostic" data like omics [6]. |
| Contemporary AI/ML Models | Graph Neural Networks (GNNs), Transformer Models, Deep Generative Models (VAEs, GANs), Ensemble Methods [110] [15]. | Hours to days (after model training). | Moderate (high initial compute for training). | Can model complex, non-linear relationships; integrates multi-modal data (e.g., structures, omics); suitable for de novo design and polypharmacology prediction [6] [15]. | "Black-box" nature requires XAI; dependent on large, high-quality datasets; risk of bias and domain shift with heterogeneous herbal data [6] [1]. |
| Experimental In Vitro Methods | Caco-2 assays (absorption), microsomal stability assays (metabolism), hERG patch clamp (toxicity), CYP450 inhibition assays [1]. | Weeks to months (per assay series). | High (reagents, lab equipment, personnel). | Considered gold standard; provides direct biological measurement; essential for regulatory validation. | Low-throughput for complex mixtures; cannot screen virtual libraries; difficult to deduce mechanism from phenotype alone [1]. |
| Experimental In Vivo Methods | Pharmacokinetic studies in animal models, toxicology profiling [108]. | Months to years. | Very High (animal husbandry, ethical oversight). | Provides holistic, systemic ADMET insight; required for preclinical drug development. | Extreme cost and time; ethical concerns; interspecies translational limitations; impractical for early-stage screening of numerous herbal constituents [108]. |
| Hybrid AI-Experimental Workflows | AI-prioritized candidates validated in targeted in vitro assays; network pharmacology guided by multi-omics data [6] [4]. | Variable (accelerated by AI triage). | Moderate to High (integrated cost). | Maximizes resource efficiency; generates iterative, data-rich feedback loops; enables validation of AI predictions. | Requires interdisciplinary expertise; integration of data streams from different sources can be complex [6]. |
A precise comparison of molecular target prediction methods highlights the performance variance within AI tools themselves. A 2025 benchmark study of seven prediction methods (including MolTarPred, RF-QSAR, and TargetNet) on a dataset of FDA-approved drugs found that the ligand-centric method MolTarPred demonstrated superior performance in identifying correct targets [111]. The study also revealed that using high-confidence interaction filters and optimized molecular fingerprints (e.g., Morgan fingerprints) significantly enhances prediction reliability, though it may reduce recall—a critical consideration for drug repurposing campaigns in herbal research [111].
Application Notes & Detailed Protocols
This section provides detailed methodologies for implementing key computational and experimental workflows relevant to AI-guided herbal ADMET research.
Protocol 1: Building a Hybrid QSAR-AI Model for Herbal Constituent Activity Prediction Objective: To create a predictive model for a specific biological activity (e.g., CYP3A4 inhibition) using a library of isolated herbal constituents.
- Data Curation & Descriptor Calculation:
- Source bioactivity data (e.g., IC50 for CYP3A4 inhibition) from public databases like ChEMBL or specialized natural product libraries [111].
- Standardize chemical structures (e.g., using RDKit). Generate a comprehensive set of molecular descriptors (1D-3D) using software like Dragon or PaDEL [110]. For AI-ready input, also generate learned representations such as molecular fingerprints (ECFP) or graph embeddings [110].
- Feature Selection & Data Splitting:
- Apply dimensionality reduction (e.g., Principal Component Analysis) or feature importance algorithms (e.g., from Random Forest) to select the most relevant descriptors [110].
- Split the dataset into training (70-80%), validation (10-15%), and hold-out test sets (10-15%). Use scaffold splitting to ensure structural diversity between sets, which is critical for assessing generalizability to novel herbal scaffolds [6].
- Model Training & Hybridization:
- Traditional QSAR Arm: Train a classical model (e.g., Partial Least Squares regression) using the selected descriptors.
- AI/ML Arm: Train a machine learning model (e.g., Gradient Boosting Machine or a Graph Neural Network) using the same data.
- Hybridization: Use a stacking ensemble method. The predictions from the QSAR and AI models become meta-features input to a final "blender" model (e.g., a linear regression) that produces the final activity prediction [110].
- Validation & Interpretation:
- Validate models rigorously using the hold-out test set and external validation sets if available. Report standard metrics: R², Q², RMSE [110].
- Employ Explainable AI (XAI) techniques like SHAP (SHapley Additive exPlanations) on the AI model to interpret which structural features contribute most to the predicted activity, bridging the gap between traditional QSAR interpretability and AI power [110].
Protocol 2: AI-Driven Network Pharmacology for Herbal Formulation ADMET Profiling Objective: To predict potential herb-drug interactions (HDIs) and systemic ADMET effects of a multi-herb formulation.
- Constituent-Target Mapping:
- Network Expansion & Pathway Analysis:
- Interaction & ADMET Risk Prediction:
- Pharmacokinetic (PK) Risk: Identify predicted targets that are key ADMET proteins (e.g., CYP450s, P-glycoprotein). Use specialized AI models trained on drug interaction data to predict the likelihood and direction (inhibition/induction) of modulation by herbal constituents [1].
- Pharmacodynamic (PD) Risk: Analyze if the modulated pathways intersect with the pathways of co-administered conventional drugs. Predict synergistic or antagonistic effects that could alter drug efficacy or cause toxicity [1].
- Experimental Triaging & Validation:
Protocol 3: Experimental Validation of AI-Predicted Herb-Drug Interactions Objective: To validate a predicted pharmacokinetic herb-drug interaction in vitro.
- AI Prediction: The workflow from Protocol 2 identifies a high-risk prediction: "Kaempferol (from Ginkgo biloba) is a predicted inhibitor of CYP3A4."
- In Vitro CYP Inhibition Assay:
- Materials: Human liver microsomes (HLMs), NADPH regeneration system, fluorogenic CYP3A4 substrate (e.g., 7-benzyloxy-4-trifluoromethylcoumarin, BFC), positive control inhibitor (e.g., Ketoconazole), test compound (Kaempferol) [1].
- Procedure: Incubate HLMs with a range of Kaempferol concentrations, the substrate, and NADPH. Use a fluorescence plate reader to measure metabolite formation over time.
- Analysis: Calculate the remaining enzyme activity (%) compared to a vehicle control. Determine the half-maximal inhibitory concentration (IC50) via non-linear regression.
- Data Integration & Model Refinement:
- The experimentally derived IC50 value serves as a high-quality data point to retrain or fine-tune the initial AI prediction model, closing the iterative loop between in silico prediction and in vitro validation [6].
Visualizing Workflows and Pathways
Diagram 1: AI-Guided ADMET Workflow for Herbal Compounds
Diagram 2: Key Signaling Pathways in Herbal ADMET: CYP450 & P-gp
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Research Reagent Solutions for Herbal ADMET Research
| Tool/Reagent Category | Specific Examples | Primary Function in Herbal ADMET Research |
|---|---|---|
| Chemical & Bioactivity Databases | ChEMBL [111], TCMSP [4], HIT (Herbal Ingredients' Targets), DrugBank [108]. | Provide curated structural and bioactivity data for herbal constituents and drugs to train and validate AI/QSAR models. |
| Cheminformatics & Modeling Software | RDKit [110], Schrödinger Suite [108], AutoDock Vina [108], PyTorch/TensorFlow for DL [15]. | Generate molecular descriptors, perform molecular docking, and build/train custom AI models for activity and property prediction. |
| AI Target Prediction Services | MolTarPred (stand-alone) [111], SuperPred (web server) [111], PPB2 (Polypharmacology Browser) [111]. | Perform ligand-centric target "fishing" to identify potential protein targets for novel herbal constituents, enabling network pharmacology. |
| In Vitro ADMET Assay Kits | P450-Glo CYP450 Inhibition Assays, Caco-2 permeability assay kits, MDR1-MDCK II cells for P-gp transport studies [1]. | Provide standardized, reproducible systems for experimental validation of AI-predicted interactions related to metabolism, absorption, and efflux. |
| Biological Reagents | Human liver microsomes (HLMs), recombinant human CYP450 enzymes, transfected cells overexpressing specific transporters (e.g., OATP1B1, P-gp) [1]. | Essential for conducting mechanistically clear in vitro studies to confirm and quantify interactions with key ADMET proteins. |
| Multi-Omics Data Resources | GEO (Gene Expression Omnibus), CPTAC (Proteomic Data), HMDB (Metabolomics) [6] [15]. | Provide systems biology data to inform network pharmacology models and connect herbal constituent targets to broader disease or toxicity pathways. |
The integration of Artificial Intelligence (AI) into traditional medicine, particularly for the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) prediction of herbal compounds, presents a transformative opportunity for modernizing and standardizing ancient practices. This convergence offers the potential to accelerate natural product discovery, enhance the precision of herbal formulations, and provide mechanistic insights into complex drug-herb interactions [6] [1]. However, this integration occurs within a complex and often fragmented landscape of regulatory frameworks, ethical challenges, and technical limitations [112]. The development of robust, culturally sensitive standards is not merely a technical prerequisite but a fundamental requirement to ensure safety, efficacy, equity, and trust. This article provides detailed application notes and protocols to guide researchers and drug development professionals in navigating this evolving domain, ensuring that AI applications in traditional medicine are both scientifically rigorous and ethically sound.
Analysis of Current Regulatory Frameworks & Governance Challenges
The regulatory environment for AI in traditional medicine is nascent and characterized by a patchwork of international guidelines and national regulations. Effective governance must address the dual complexities of AI as a novel technology and traditional medicine as a diverse, holistic practice.
International and National Regulatory Postures
Globally, regulatory bodies are taking initial steps to define pathways for AI in health. The U.S. Food and Drug Administration (FDA) has proposed a predetermined change control plan framework for AI/ML-based Software as a Medical Device (SaMD), allowing for iterative model updates under a reviewed plan [113]. The European Union’s AI Act introduces a risk-based classification system, where AI tools for health are typically deemed "high-risk," mandating rigorous conformity assessments, data governance, and post-market monitoring [114]. The World Health Organization (WHO) emphasizes a holistic governance strategy that integrates ethical principles, data privacy, and the need to preserve the integrity of traditional knowledge systems [112].
A significant governance gap exists in assigning legal accountability for AI-driven decisions in traditional medicine practice. Clear frameworks are needed to determine liability among developers, practitioners, and healthcare institutions in cases of error or adverse outcomes [112].
Table 1: Comparative Analysis of Regulatory Frameworks for AI in Traditional Medicine
| Regulatory Body/Initiative | Core Approach | Key Requirements/Principles | Relevance to AI for Herbal ADMET |
|---|---|---|---|
| U.S. FDA (AI/ML SaMD Action Plan) [113] | Premarket review with lifecycle oversight (Predetermined Change Control Plans). | Safety, effectiveness, transparency, real-world performance monitoring. | Applicable to AI tools intended for clinical diagnostic or treatment decisions based on ADMET predictions. |
| EU AI Act [114] | Risk-based classification; "high-risk" AI systems require conformity assessment. | Data quality, technical documentation, record-keeping, human oversight, cybersecurity. | Directly governs AI systems used for safety screening of herbal compounds within the EU. |
| WHO Global Strategy [112] | Guidance and policy development for member states, focusing on integration and ethics. | Safety, efficacy, quality, access, rational use, respect for intellectual property and traditional knowledge. | Provides the overarching ethical and policy context for developing and deploying AI tools globally. |
| International Coalition (CISA, NSA, FBI) [115] | Cybersecurity best practices for AI data and systems. | Securing training data pipelines, protecting model integrity, ensuring resilient operation. | Critical for protecting proprietary herbal compound libraries and sensitive patient data used in model training. |
Diagram 1: Decision Pathway for Regulatory Strategy (78 characters)
Standardization of Data and Terminology
A foundational challenge is the scarcity of standardized, high-quality data. Traditional medicine encompasses diverse systems with inconsistent terminologies and limited structured electronic records [112]. For AI models, this leads to issues of data imbalance, domain shift, and poor generalizability [6]. Initiatives like India’s Traditional Knowledge Digital Library (TKDL) and efforts to create "Minimal Information for AI on Natural Product Metadata" are crucial steps toward creating interoperable, FAIR (Findable, Accessible, Interoperable, Reusable) data resources [6] [112].
Core Ethical Principles and Implementation Protocols
Ethical integration requires moving beyond abstract principles to implementable protocols that address bias, equity, transparency, and respect for traditional knowledge.
Ethical Framework and Operational Risks
Key ethical risks include algorithmic bias from unrepresentative training data, cultural erosion from decontextualized digitalization, and biopiracy where AI is used to exploit traditional knowledge without fair benefit-sharing [112]. Furthermore, the "black-box" nature of complex AI models like deep neural networks conflicts with the need for mechanistic understanding in pharmacology and regulatory review [6].
Table 2: Key Ethical Risks & Mitigation Protocols for AI in Herbal ADMET Research
| Ethical Risk | Potential Impact | Recommended Mitigation Protocol |
|---|---|---|
| Algorithmic Bias & Inequity | Models perform poorly for underrepresented ethnic groups or herbal traditions, exacerbating health disparities. | Implement "bias audits" during development using diverse compound/outcome datasets. Apply fairness constraints in model training. |
| Exploitation of Traditional Knowledge | Uncompensated commercial use of indigenous knowledge digitized and analyzed by AI. | Adhere to Nagoya Protocol principles. Implement provenance-aware data systems that track origin and access terms [6] [112]. |
| Lack of Transparency/Explainability | Inability to understand model predictions undermines scientific trust and clinical adoption. | Integrate Explainable AI (XAI) techniques (e.g., SHAP, LIME) into workflows. Generate mechanistic hypotheses (e.g., likely target pathways) for validation [6]. |
| Data Privacy & Security | Breach of sensitive patient genomic or health data used in personalized Ayurgenomics or pharmacovigilance models [112]. | Employ privacy-by-design approaches: federated learning, differential privacy, and strict access controls aligned with GDPR/HIPAA [116] [114]. |
| Erosion of Humanistic Practice | AI tools may displace the holistic, empathetic patient-practitioner relationship central to traditional medicine. | Design AI as a decision-support tool, not a replacement. Protocols must mandate human-in-the-loop review and preserve time for patient interaction [112]. |
Diagram 2: From Ethical Principles to Operational Protocols (80 characters)
Protocol for Ethical Data Sourcing and Governance
Objective: To establish a compliant and ethical pipeline for acquiring and managing data for AI model development in traditional medicine research.
- Provenance Documentation: For any traditional knowledge or herbal formula data, create immutable records detailing the geographical origin, cultural source, custodians, and access conditions. Link this metadata to all derivative digital data [6].
- Prior Informed Consent (PIC): When collecting clinical or patient data (e.g., for pharmacogenomics or outcome studies), implement a dynamic consent process that clearly explains AI-specific uses, including potential for model training and secondary research [112].
- Data Security: Implement a Zero Trust Architecture (ZTA) for data infrastructure. Encrypt data at rest and in transit. Employ data loss prevention (DLP) tools and strict access controls following the principle of least privilege [115] [116].
- Bias Mitigation: At the data curation stage, statistically analyze datasets for representation across relevant demographics and herbal traditions. Use techniques like synthetic minority oversampling or informed undersampling to address imbalances before model training [6].
Technical Protocols for AI-Guided ADMET Prediction
Robust, validated experimental protocols are essential to ensure the scientific credibility of AI predictions for herbal compounds, which are often complex mixtures with limited prior data.
Protocol for Benchmarking & Validating Computational ADMET Tools
Objective: To evaluate and select the most reliable computational tools for predicting key ADMET properties of herbal compounds, as part of a New Approach Methodology (NAM) pipeline. Background: Studies have benchmarked software using curated external validation datasets, finding that models for physicochemical properties (average R² = 0.717) often outperform those for toxicokinetic properties (average R² = 0.639) [117]. Herbal compounds' novelty often places them outside standard models' applicability domains (AD), necessitating rigorous checking [6] [117]. Materials:
- Compound Library: Standardized SMILES representations of herbal constituents.
- Software Tools: A selection of benchmarked tools (e.g., ADMETlab 2.0, which predicts 88 endpoints) [118].
- Validation Datasets: Curated, experimental datasets for key endpoints (e.g., LogP, Caco-2 permeability, CYP inhibition) from literature [117].
Procedure:
- Data Curation & Standardization:
- Convert all compounds to canonical isomeric SMILES.
- Standardize structures (e.g., neutralize salts, remove duplicates) using toolkits like RDKit [117].
- For validation data, identify and remove "inter-outliers" (compounds with inconsistent values across datasets) using standardized deviation thresholds [117].
- Applicability Domain (AD) Assessment:
- For each tool and property, determine if the target herbal compound falls within the model's AD (e.g., based on chemical descriptor ranges of the training set). Flag all out-of-AD predictions as unreliable [117].
- Prediction & Performance Validation:
- Run predictions for the curated validation set compounds.
- Calculate standard performance metrics: R² (regression) or Balanced Accuracy (classification), Sensitivity, Specificity.
- Compare performance against published benchmark values for industrial chemicals/drugs to gauge potential performance drop-off for natural products [117].
- Uncertainty Quantification:
- Utilize tools that provide prediction confidence intervals or implement ensemble methods to generate uncertainty estimates. Prioritize compounds where multiple models/approaches yield concordant predictions.
Table 3: Summary of Key ADMET Endpoints & Validation Performance Benchmarks [117]
| Property Category | Example Endpoints | Typical Benchmark Performance (External Validation) | Critical Consideration for Herbal Compounds |
|---|---|---|---|
| Physicochemical (PC) | LogP, Water Solubility, pKa | R² Average: ~0.717 | Foundation for bioavailability; predictions generally reliable but check for glycosides & complex polyphenols. |
| Pharmacokinetic/Toxicokinetic (TK) | Caco-2 Permeability, BBB Penetration, P-gp Substrate | BA* Avg: ~0.78; R² Avg: ~0.639 | Critical for interaction prediction (e.g., P-gp). Performance more variable; essential to use AD. |
| Metabolism | CYP450 Inhibition (e.g., 3A4, 2D6) | Classification Accuracy Varies | Central to drug-herb interaction risk. Seek models trained on diverse chemical space, including natural products. |
| Toxicity | hERG inhibition, Ames mutagenicity | Classification Accuracy Varies | High-stakes endpoint. Use as a sensitive initial filter; always require experimental follow-up. |
BA: Balanced Accuracy
Protocol for Predicting Drug-Herb Interactions (DHIs) Using AI
Objective: To use AI models to predict potential pharmacokinetic (PK) and pharmacodynamic (PD) interactions between a conventional drug and an herbal compound or formulation. Background: DHIs are complex due to multi-constituent herbs and multi-mechanism actions (e.g., St. John's Wort induces CYP3A4 and P-gp) [1]. AI methods like network pharmacology and graph neural networks can integrate chemical, target, and pathway data to infer interactions [6] [1]. Materials:
- AI Platform: Access to a network pharmacology or DHI prediction tool (e.g., integrating knowledge graphs).
- Databases: Chemical structure databases, protein-target databases (e.g., UniProt), pathway databases (e.g., KEGG). Procedure:
- Data Integration:
- For the herbal product, create a comprehensive list of known or predicted bioactive constituents (from Protocol 3.1 or literature).
- For each constituent and the co-administered drug, gather or predict: molecular targets, involvement in metabolic pathways (CYPs, UGTs), and transporter substrates/inhibition profiles.
- Network Construction:
- Build a herb-ingredient-target-pathway network. Connect herbs to their ingredients, ingredients to protein targets/enzymes/transporters, and these to biological pathways [6].
- Overlay the drug's interaction network onto this graph.
- Interaction Inference:
- Use graph algorithms or ML classifiers to identify shared nodes (e.g., common CYP enzyme) or adjacent nodes (e.g., interacting proteins in a pathway) that indicate potential interaction points.
- Prioritize interactions based on network topology metrics (e.g., centrality) and biological plausibility.
- Mechanistic Hypothesis Generation:
- Output should not be a binary prediction but a set of testable mechanistic hypotheses (e.g., "Constituent X may inhibit CYP2C9, potentially increasing plasma levels of Drug Y"). This aligns with the need for explainability and guides validation experiments [1].
Diagram 3: AI-Driven ADMET Prediction & DHI Screening Workflow (86 characters)
The Scientist's Toolkit: Essential Research Reagent Solutions
This table details key resources for implementing the aforementioned protocols.
Table 4: Research Reagent Solutions for AI-Guided Herbal ADMET Research
| Tool/Resource Category | Example/Product | Primary Function in Research | Key Consideration |
|---|---|---|---|
| ADMET Prediction Platforms | ADMETlab 2.0 [118], SwissADME, pkCSM | Provides a comprehensive suite of web-based models for predicting key physicochemical, pharmacokinetic, and toxicity endpoints. | Evaluate based on benchmark performance [117], transparency of models, and applicability domain description. |
| Cheminformatics Toolkits | RDKit (Open-Source), KNIME, ChemAxon | Enables critical data preparation: SMILES standardization, molecular descriptor calculation, fingerprint generation, and dataset curation. | Essential for preprocessing herbal compound libraries before feeding into AI models and for curating validation datasets [117]. |
| Network Analysis & Visualization | Cytoscape, Gephi, NetworkX (Python) | Constructs and analyzes herb-ingredient-target-pathway networks for mechanistic DHI prediction and hypothesis generation [6] [1]. | Look for plugins that integrate biological databases (KEGG, Reactome) to automate network building. |
| Explainable AI (XAI) Libraries | SHAP (SHapley Additive exPlanations), LIME, Captum | Interprets "black-box" ML model predictions by quantifying feature importance, helping to translate AI output into biologically intelligible insights [6]. | Crucial for building trust and meeting regulatory demands for transparency. |
| Data Security & Governance | Zero Trust Network Access (ZTNA) solutions, Data Encryption tools, Federated Learning frameworks (e.g., PySyft). | Protects sensitive intellectual property (herbal libraries) and patient data throughout the AI lifecycle, enabling secure collaborative research [115] [116] [114]. | Must be designed into the research infrastructure from the start, not added as an afterthought. |
Cybersecurity & Lifecycle Management Protocols
AI models are dynamic assets requiring continuous oversight. Cybersecurity is integral to scientific integrity and patient safety, not just IT compliance.
Protocol for Securing the AI/ML Lifecycle
Objective: To implement cybersecurity best practices across the development, deployment, and maintenance of AI models for herbal medicine research. Procedure:
- Development & Training Phase:
- Secure Training Pipelines: Use encrypted data storage and integrity checks (e.g., hashing) to prevent data poisoning [115] [114]. Maintain immutable data lineage records.
- Adversarial Testing (Red Teaming): Before deployment, systematically probe the model with crafted inputs designed to cause prediction errors or reveal sensitive training data [116] [114].
- Deployment & Operational Phase:
- Runtime Monitoring: Deploy anomaly detection systems to monitor model performance and input data for significant drift or suspicious patterns indicative of an attack [114].
- Model Access Controls: Implement strict authentication and authorization for APIs serving model predictions. Use API rate limiting to prevent model extraction attacks [114].
- Maintenance & Update Phase:
- Model Versioning & Provenance: Maintain an immutable registry tracking every model version, its training data hash, hyperparameters, and performance metrics [114]. This is vital for auditability and reproducibility.
- Compliance Automation: Use automated tools to continuously check model operations against relevant regulatory and security policy requirements (e.g., GDPR, EU AI Act) [116].
The establishment of standards for AI in traditional medicine is an interdisciplinary imperative. It requires the fusion of advanced computational techniques with deep pharmacological knowledge, all within a framework built on rigorous ethics, adaptable regulation, and resilient cybersecurity. The protocols outlined here provide a concrete starting point for researchers to build credible, reproducible, and responsible AI applications for herbal ADMET prediction. The future trajectory must involve collaborative international efforts, such as those spearheaded by WHO, to harmonize data standards, validate methodologies across diverse medical traditions, and create governance models that protect both innovation and the invaluable heritage of traditional knowledge systems [112]. By proactively addressing these regulatory and ethical considerations, the scientific community can ensure that AI fulfills its potential as a force for advancing global health through the intelligent integration of traditional and modern medicine.
Conclusion
The integration of AI into herbal ADMET prediction represents a transformative convergence of computational power and traditional pharmacopeia. By systematically addressing foundational data gaps, applying sophisticated ML methodologies, implementing robust troubleshooting for real-world challenges, and adhering to rigorous validation standards, researchers can de-risk and accelerate the development of herbal-based therapeutics. Successful case studies and competitive benchmarks demonstrate that AI models can achieve laboratory-grade precision for key properties[citation:8], offering a powerful tool for prioritizing compounds and predicting complex interactions[citation:4]. The future trajectory points toward more holistic, ethically grounded frameworks that incorporate multi-omics data, digital twins[citation:1], and patient-specific factors for personalized medicine, all while respecting data sovereignty and traditional knowledge[citation:7]. For the field to mature, continued collaboration between computational scientists, ethnopharmacologists, chemists, and regulators is essential to build trustworthy, transparent, and impactful AI systems that unlock the full potential of herbal medicine for global health.
References
- 1. Artificial Intelligence Models and Tools for the Assessment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11944892/]
- 2. Opportunities and challenges in application of artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9838466/]
- 3. Artificial intelligence in predicting personalized nanocarrier ... [https://www.sciencedirect.com/science/article/pii/S2949866X25000504]
- 4. Integrating artificial intelligence into the modernization of ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1181183/full]
- 5. Fixing drug discovery's most persistent problem with AI [https://www.drugtargetreview.com/article/168391/fixing-drug-discoverys-most-persistent-problem-with-ai/]
- 6. Artificial intelligence for natural product drug discovery and ... [https://www.sciencedirect.com/science/article/pii/S2666521225001206]
- 7. An herbal drug combination identified by knowledge graph ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10063228/]
- 8. Prediction of ADMET profile and anti-inflammatory potential ... [https://www.nature.com/articles/s41598-025-86809-y]
- 9. The safety of herbal medicines in low- and middle-income ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551009/]
- 10. In vitro technology and ADMET research in traditional ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1605330/full]
- 11. Reimagining Ethnopharmacology with Generative AI [https://www.sciencedirect.com/science/article/pii/S104366182500427X]
- 12. Advancing ADMET prediction through multiscale fragment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12478026/]
- 13. Revolutionizing pharmacology: AI-powered approaches in ... [https://www.sciencedirect.com/science/article/pii/S259009862500020X]
- 14. ADMET Predictor® Modules [https://www.simulations-plus.com/software/admetpredictor/]
- 15. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 16. SAPPHIRE: Gupta and Jachak guideline for a Pre-Step ... [https://www.sciencedirect.com/science/article/pii/S2667031325000922]
- 17. Why 90% of clinical drug development fails and how to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9293739/]
- 18. ADME properties of herbal medicines in humans [https://pubmed.ncbi.nlm.nih.gov/21385154/]
- 19. Enhancing drug screening and pharmacokinetic predictions [https://www.sciencedirect.com/science/article/abs/pii/S1385894725012288]
- 20. In Silico ADME Methods Used in the Evaluation of Natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389637/]
- 21. PharmaBench: Enhancing ADMET benchmarks with large ... [https://www.nature.com/articles/s41597-024-03793-0]
- 22. In-silico pharmacokinetics ADME/Tox analysis of ... [https://www.sciencedirect.com/science/article/pii/S2468227625002650]
- 23. Leveraging machine learning models in evaluating ADMET ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205928/]
- 24. HerbComb: an integrated database for the discovery of ... [https://www.sciencedirect.com/science/article/pii/S2001037025004696]
- 25. Exploring Artificial Intelligence's Potential to Enhance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12582507/]
- 26. Bioactivity assessment of natural compounds using machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9071136/]
- 27. Integrated molecular and ADME-toxicity profiling identifies ... [https://www.nature.com/articles/s41598-025-02858-3]
- 28. Classification of cold and hot medicinal properties ... [https://www.sciencedirect.com/science/article/pii/S2589377725000084]
- 29. Predicting Meridian in Chinese traditional medicine using ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007249]
- 30. Herbal concoction Unveiled: A computational analysis of ... [https://www.sciencedirect.com/science/article/pii/S2666027X23000166]
- 31. Improving ADME Prediction with Multitask Graph Neural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606635/]
- 32. Advancing ADMET for major CYP450 isoforms... prediction [https://biomedical-engineering-online.biomedcentral.com/articles/10.1186/s12938-025-01412-6]
- 33. hERG toxicity prediction in early drug discovery using ... [https://www.nature.com/articles/s41598-025-99766-3]
- 34. Frontiers | Recent advances in AI -based toxicity prediction for drug... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1632046/full]
- 35. CardioGenAI: a machine learning-based framework for re ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11881490/]
- 36. Improving ADME Prediction with Multitask Graph Neural ... [https://chemrxiv.org/engage/chemrxiv/article-details/68d60081f2aff16770a09ee9]
- 37. Predicting ADMET On Rowan - Rowan Newsletter - Substack [https://rowansci.substack.com/p/predicting-admet-on-rowan]
- 38. An Introduction to 3D Bioprinting: Possibilities, Challenges ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6266989/]
- 39. 3D bioprinting matrices with controlled pore structure and ... [https://www.nature.com/articles/srep34410]
- 40. admetSAR3.0: a comprehensive platform for exploration ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11223829/]
- 41. Benchmarking ML in ADMET predictions: the practical impact ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01041-0]
- 42. iCAM-Net: Interpretable Herb-Disease Association ... [https://www.sciencedirect.com/science/article/abs/pii/S0944711325011286]
- 43. Current Status on Phytochemicals Classification, Structure ... [https://phcogrev.com/article/2025/19/38/105530phrev20252344]
- 44. Progress of AI-Driven Drug–Target Interaction Prediction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563295/]
- 45. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 46. From Structure to Function: Biological Activity Prediction ... [https://www.jsr.org/hs/index.php/path/article/view/8615]
- 47. Improving ADMET prediction with descriptor augmentation of ... [https://sciety-labs.elifesciences.org/articles/by?article_doi=10.1101/2025.07.14.664363]
- 48. Machine learning tools for the characterization of bioactive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11973551/]
- 49. Machine Learning for Toxicity Prediction Using Chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12093382/]
- 50. Advancing ADMET prediction for major CYP450 isoforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12288271/]
- 51. Cardioprotective Chinese herbs and antiretroviral drug ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1627058/full]
- 52. Cytochrome P450 enzyme mediated herbal drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4464477/]
- 53. Phytochemical profiling and in silico prediction of ... [https://link.springer.com/article/10.1007/s42452-025-07685-9]
- 54. DeepMetab: a comprehensive and mechanistically informed ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04631a]
- 55. AI in drug discovery for CNS: blood-brain barrier ... [https://deepnote.com/explore/ai-in-drug-discovery-for-cns-blood-brain-barrier-permeability-prediction]
- 56. AI driven network pharmacology: Multi-scale mechanisms of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12663848/]
- 57. Decoding Herbal Medicine: AI-Powered Omics and ... [https://www.sciencedirect.com/science/article/abs/pii/S0944711325010906]
- 58. Leveraging network pharmacology for drug discovery [https://www.sciencedirect.com/science/article/pii/S259009862500017X]
- 59. Enhancing drug discovery with AI: Predictive modeling of ... [https://www.sciencedirect.com/science/article/pii/S2949866X24001187]
- 60. Network Pharmacology: A New Approach for Chinese Herbal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3671675/]
- 61. Artificial Intelligence (AI) Applications in Drug Discovery and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11510778/]
- 62. ADMET profile and virtual screening of plant and microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7561576/]
- 63. 将AI与定量系统药理学相结合,革新药物发现 [https://hub.baai.ac.cn/view/48376]
- 64. Flavonoids as Markers in Herbal Medicine Quality Control [https://www.mdpi.com/2297-8739/12/11/289]
- 65. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 66. Leveraging machine learning models in evaluating ADMET properties for drug discovery and development - PubMed [https://pubmed.ncbi.nlm.nih.gov/40585410/]
- 67. Leveraging artificial intelligence in antibody-drug conjugate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12638810/]
- 68. Artificial Intelligence Meets Drug Discovery: A Systematic ... [https://www.preprints.org/manuscript/202503.0912]
- 69. ADMET-AI [https://admet.ai.greenstonebio.com/]
- 70. Explainable Artificial Intelligence: A Perspective on Drug ... [https://www.mdpi.com/1999-4923/17/9/1119]
- 71. Explainable AI's Role in Revolutionizing Drug Discovery [https://www.grgonline.com/post/cracking-the-code-explainable-ai-s-role-in-revolutionizing-drug-discovery]
- 72. Explainable Artificial Intelligence in the Field of Drug Research [https://pmc.ncbi.nlm.nih.gov/articles/PMC12129466/]
- 73. Machine learning approaches for next-generation ADMET ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625002004]
- 74. Utilizing ADMET Analysis and Molecular Docking to ... [https://www.mdpi.com/1422-0067/26/7/3189]
- 75. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 76. Challenges in natural product-based drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10613855/]
- 77. Comparison of Different Approaches to Define the ... [https://www.mdpi.com/1420-3049/17/5/4791]
- 78. Chemical Space Covered by Applicability Domains of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10882972/]
- 79. Applicability domains are common in QSAR but irrelevant ... [https://variational.ai/blog/applicability-domains-are-common-in-qsar-but-irrelevant-for-conventional-ml-tasks/]
- 80. External validation of machine learning models—registered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12077397/]
- 81. Applicability Domain of the Sens-Is In Vitro Assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12115193/]
- 82. Herbal concoction Unveiled: A computational analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10440360/]
- 83. The In Vitro Pharmacokinetics of Medicinal Plants: A Review [https://www.mdpi.com/1424-8247/18/4/551]
- 84. The Future of Drug Discovery: Why AI Is More Than Just a ... [https://www.gmp-journal.com/current-articles/details/the-future-of-drug-discovery-why-ai-is-more-than-just-a-buzzword.html]
- 85. AI in Drug Discovery 2025: Real-World Impact & ... [https://dr7.ai/blog/model/ai-in-drug-discovery-2025-real-world-impact-regulatory-truth/]
- 86. Systematic review on phytochemicals structure and activity ... [https://www.sciencedirect.com/science/article/pii/S2667031324001180]
- 87. Artificial intelligence in drug discovery and development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7577280/]
- 88. Advancing Medicinal Plant Research with Artificial ... [https://www.nrfhh.com/Advancing-Medicinal-Plant-Research-with-Artificial-Intelligence-Key-Applications,206025,0,2.html]
- 89. 'Ayurveda', hyperemesis and anaemia in pregnancy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12440907/]
- 90. Effects of natural extracts in cognitive function of healthy ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1573034/full]
- 91. An artificial intelligence-aided scoping review of medicinal ... [https://pubmed.ncbi.nlm.nih.gov/40529497/]
- 92. Herbal Medicine: Scientific Validation and Future Prospects [https://www.sciencepublishinggroup.com/article/10.11648/j.ijpc.20251103.12]
- 93. Practical Considerations and Applied Examples of Cross ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11041453/]
- 94. An intuitive guide to understanding Cross-validation [https://medium.com/learning-data/an-intuitive-guide-to-understanding-cross-validation-abd01bf5e879]
- 95. Guide to Data Mastery | Atlassian Chart [https://www.atlassian.com/data/charts]
- 96. A Computational Community Blind Challenge on Pan ... [https://chemrxiv.org/engage/chemrxiv/article-details/68ac00d1728bf9025e22fe45]
- 97. Antiviral Competition: Advancing Open Science with ASAP ... [https://polarishub.io/blog/antiviral-competition]
- 98. Pan-Coronavirus Drug Discovery Gets a Boost from ... [https://globalbiodefense.com/2025/07/28/pan-coronavirus-ai-drug-discovery-blind-challenge-results/]
- 99. Time For a New Adventure - Practical Cheminformatics [https://patwalters.github.io/Time-For-a-New-Adventure/]
- 100. antiviral-potency-2025 [https://polarishub.io/competitions/asap-discovery/antiviral-potency-2025]
- 101. Lessons from the Polaris ADMET competition [https://www.inductive.bio/blog/lessons-from-the-polaris-admet-competition]
- 102. Blind Challenges [https://openadmet.org/community/events/blind-challenges/]
- 103. Polaris - The benchmarking platform for drug discovery [https://polarishub.io/]
- 104. AI in Natural Product Drug Discovery: Now & Next [https://ai.creative-biolabs.com/blog/ai-in-natural-product-drug-discovery-now-next/]
- 105. Artificial Intelligence in Natural Product Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC11874025/]
- 106. Applications of Artificial Intelligence in Biotech Drug Discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12308071/]
- 107. A generalized platform for artificial intelligence-powered ... [https://www.nature.com/articles/s41467-025-61209-y]
- 108. opportunities for innovation in drug discovery - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12641420/]
- 109. opportunities for innovation in drug discovery [https://pubs.rsc.org/en/content/articlelanding/2025/md/d5md00700c]
- 110. AI-Integrated QSAR Modeling for Enhanced Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525248/]
- 111. A precise comparison of molecular target prediction methods [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00199d]
- 112. Artificial intelligence in traditional medicine: policy and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12578530/]
- 113. Artificial Intelligence in Software as a Medical Device [https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-software-medical-device]
- 114. AI Security Best Practices: 12 Essential Ways to Protect ML [https://www.sentinelone.com/cybersecurity-101/data-and-ai/ai-security-best-practices/]
- 115. New Best Practices Guide for Securing AI Data Released [https://www.cisa.gov/news-events/alerts/2025/05/22/new-best-practices-guide-securing-ai-data-released]
- 116. 10 Cybersecurity Best Practices in the Age of AI (2025) [https://www.sisainfosec.com/blogs/10-cybersecurity-best-practices-in-the-age-of-ai-2025/]
- 117. Comprehensive benchmarking of computational tools for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11674477/]
- 118. ADMETlab 2.0 [https://admetmesh.scbdd.com/]