Natural Product-Based Drug Design and Scaffold Hopping: From Foundational Principles to AI-Driven Discovery
This article provides a comprehensive overview of natural product-based drug design, with a specific focus on the strategy of scaffold hopping.
Natural Product-Based Drug Design and Scaffold Hopping: From Foundational Principles to AI-Driven Discovery
Abstract
This article provides a comprehensive overview of natural product-based drug design, with a specific focus on the strategy of scaffold hopping. It explores the foundational role of natural products as biologically prevalidated starting points for drug discovery, detailing the principles of scaffold hopping as defined by its key objective: retaining biological activity while altering the core molecular structure. The content covers a spectrum of methodological approaches, from traditional bioisosteric replacements and pharmacophore-based searches to modern, AI-driven generative models. It further addresses common challenges in the field, such as balancing structural novelty with maintained activity and navigating intellectual property, and presents validation frameworks through case studies and comparative analyses of different techniques. Aimed at researchers, scientists, and drug development professionals, this review synthesizes historical context, current state-of-the-art technologies, and future directions, offering a practical guide for leveraging natural product-inspired design to discover novel therapeutic candidates with improved properties.
The Unparalleled Role of Natural Products as a Foundation for Drug Discovery
Why Natural Products? Historical Success and Inherent Bioactivity
Natural products (NPs) are chemical compounds derived from natural sources such as plants, microorganisms, marine organisms, and fungi. These molecules have served as a major source of chemically novel, bioactive therapeutics throughout human history and continue to play a pivotal role in modern drug discovery [1] [2]. Their structural diversity and evolutionary refinement make them indispensable for tackling complex medical challenges, particularly for cancer and infectious diseases [3] [4].
The historical use of natural products dates back to ancient civilizations, with the earliest records depicted on clay tablets in cuneiform from Mesopotamia (2600 B.C.) documenting oils from Cupressus sempervirens (Cypress) and Commiphora species (myrrh) for treating coughs, colds, and inflammation [1]. The continued relevance of these natural compounds in modern medicine underscores their inherent bioactivity and therapeutic value, validated by both traditional use and contemporary scientific research [1] [5].
Historical Success of Natural Products
Traditional Medicine and Early Discoveries
Traditional medicinal practices have formed the foundation of most early medicines, with subsequent clinical, pharmacological, and chemical studies validating their efficacy [1]. Ancient records including the Ebers Papyrus (2900 B.C.), Chinese Materia Medica (1100 B.C.), and the works of Greek physician Dioscorides (100 A.D.) documented hundreds of plant-based drugs that established the foundation for modern pharmacotherapy [1].
Probably the most famous example is the development of acetylsalicyclic acid (aspirin) derived from the natural product salicin isolated from the bark of the willow tree Salix alba L. [1]. Similarly, investigation of Papaver somniferum L. (opium poppy) resulted in the isolation of several alkaloids including morphine, first reported in 1803, which remains a commercially important analgesic drug [1].
Natural Products in Modern Drug Discovery
Natural products and their structural analogues have historically made a major contribution to pharmacotherapy, especially for cancer and infectious diseases [4]. Despite a decline in their pursuit by the pharmaceutical industry from the 1990s onwards, recent technological developments have revitalized interest in natural product-based drug discovery [3] [4].
Table 1: Historically Significant Natural Product-Derived Drugs
| Natural Product | Source Organism | Therapeutic Application | Discovery Timeline |
|---|---|---|---|
| Salicin | Willow tree (Salix alba L.) | Anti-inflammatory (precursor to aspirin) | Ancient use, isolated 1828 |
| Morphine | Opium poppy (Papaver somniferum) | Analgesic | Isolated 1803 |
| Artemisinin | Sweet wormwood (Artemisia annua) | Antimalarial | Discovered 1972 |
| Paclitaxel | Pacific yew tree (Taxus brevifolia) | Anticancer | Discovered 1971 |
| Teixobactin | Bacterium (Eleftheria terrae) | Antibiotic | Discovered 2015 |
Natural products continue to provide unique structural diversity in comparison to standard combinatorial chemistry, which presents opportunities for discovering novel low molecular weight lead compounds [1]. With less than 10% of the world's biodiversity evaluated for potential biological activity, numerous useful natural lead compounds await discovery [1].
Inherent Bioactivity of Natural Products
Evolutionary Advantages
Natural products distinguish themselves from synthetic libraries through their elevated molecular complexity, including higher proportions of sp3-hybridized carbon atoms, increased oxygenation, and decreased halogen and nitrogen content [2]. This chemical richness is coupled with rigid molecular frameworks and lower lipophilicity, traits that facilitate favorable interactions with biological targets, particularly those elusive to synthetic small molecules [2].
What sets NPs apart most profoundly is their evolutionary purpose [6]. These molecules function as defense chemicals, signaling agents, and ecological mediators, fine-tuned for optimal interactions with living systems through millions of years of evolutionary refinement [6] [2]. This natural selection has endowed NPs with mechanisms of action that exploit biological vulnerabilities, particularly in pathogens and cancer cells [2].
Diverse Biological Functions
Natural products possess several innate functions, including the ability to allosterically alter the catalytic activity of enzymes, promote or disrupt macromolecular interactions, act as chemical messengers between cells, participate in inter-kingdom signaling, and serve as toxins for defense [6]. They can even carry out some protein-like functions of their own [6].
The biochemical diversity of natural products presents both a challenge and source of inspiration for biologists and chemists across the globe [6]. This diversity enables them to target specific pathways implicated in disease processes, offering tailored therapeutic strategies [5]. Moreover, the synergy observed within natural extracts—where multiple bioactive compounds act collaboratively—enhances their overall efficacy and broadens their therapeutic potential [5].
Table 2: Innate Functions of Natural Products in Producing Organisms
| Function | Mechanism | Example Natural Products |
|---|---|---|
| Defense | Deter herbivory through bitter or toxic compounds | Pyrrolizidine alkaloids, glucosinolates |
| Signaling | Act as chemical messengers between cells | Flavonoids, strigolactones |
| Pollination | Attract pollinators through chromo-pigments | Carotenoids, anthocyanins |
| Symbiosis | Facilitate ecological associations | Nod factors in rhizobia-legume symbiosis |
| Environmental Adaptation | Protect against biotic and abiotic stresses | Osmoprotectants, phytoalexins |
Current Research Methodologies
Advanced Screening and Analytical Techniques
The field of natural product drug discovery is experiencing a paradigm shift due to advanced technologies that increase speed, accuracy, and sustainability [2]. Traditional discovery workflows are being enhanced by high-throughput screening, artificial intelligence, machine learning, and omics technologies, which collectively streamline compound identification and development [2].
Advanced analytical techniques including ultrasonic-assisted extraction, supercritical fluid extraction, and various chromatographic methods have revolutionized the isolation and purification of natural bioactive compounds [5]. Characterization techniques such as mass spectrometry, nuclear magnetic resonance spectroscopy, and high-performance liquid chromatography provide detailed insights into chemical composition and structural elucidation [5].
Natural Product Drug Discovery Workflow. The diagram outlines the standard pipeline for discovering bioactive natural products, from initial extraction to final compound optimization. Key steps include bioassay-guided fractionation and target identification to ensure therapeutic relevance. UAE: Ultrasound-Assisted Extraction; SFE: Supercritical Fluid Extraction; MS: Mass Spectrometry; NMR: Nuclear Magnetic Resonance; HPLC: High-Performance Liquid Chromatography.
Genomics and Bioinformatics Approaches
The integration of genome mining and biosynthetic engineering has revolutionized natural product discovery, offering solutions to longstanding challenges in the field [2]. Advances in understanding NP biosynthetic pathways, coupled with sophisticated genomic analysis tools, have paved the way for systematic exploration of microbial genomes [2].
Tools such as CRISPR-Cas systems, artificial intelligence, and bioinformatics platforms are accelerating hit discovery, de-replication, and biosynthetic pathway engineering, overcoming long-standing barriers to NP research [2]. Genome mining tools like DeepBGC and AntiSMASH enable rapid prediction and characterization of biosynthetic gene clusters, facilitating the discovery of novel compounds [2].
Experimental Protocols
Protocol 1: Bioactivity-Guided Fractionation of Plant Extracts
Principle: This protocol outlines a standardized approach for the extraction, fractionation, and identification of bioactive compounds from plant material using bioactivity-guided fractionation to isolate natural products with therapeutic potential [7] [5].
Materials:
- Plant material (dried and powdered)
- Extraction solvents (methanol, ethanol, ethyl acetate, hexane)
- Chromatography media (silica gel, Sephadex LH-20, C18 reverse-phase resin)
- Cell lines for bioassays (e.g., cancer cells, microbial strains)
- Analytical instruments (HPLC, MS, NMR)
Procedure:
- Extraction: Macerate 500g of dried plant material in 2L of methanol for 24 hours at room temperature with occasional stirring. Filter through Whatman No. 1 filter paper and concentrate under reduced pressure at 40°C to obtain crude extract.
- Bioactivity Screening: Test crude extract for desired biological activity (e.g., cytotoxicity, antimicrobial activity) using appropriate assays. Proceed with fractionation only if significant activity (IC50 < 100 μg/mL) is observed.
- Solvent Partitioning: Suspend crude extract in 90% aqueous methanol and partition successively with hexane, ethyl acetate, and n-butanol (3 × 200 mL each). Concentrate each fraction under reduced pressure.
- Bioassay-Guided Fractionation: Test all fractions for bioactivity and select the most active for further separation. For the active ethyl acetate fraction (2g):
- Subject to vacuum liquid chromatography on silica gel (200-400 mesh) with step gradient elution from hexane to methanol.
- Combine similar fractions based on TLC profiling to yield 8-10 primary fractions.
- Test each primary fraction for bioactivity.
- Further Purification: For active primary fractions:
- Purify by Sephadex LH-20 column chromatography using methanol as eluent.
- Further separate by semi-preparative HPLC (C18 column, gradient elution with acetonitrile-water).
- Monitor purity by analytical HPLC and confirm structure by NMR and MS.
Troubleshooting:
- If fractionation yields inactive compounds, consider synergistic effects and test combinations of fractions.
- For complex mixtures, employ hyphen techniques like LC-MS-NMR for dereplication [4].
Protocol 2: Genome Mining for Natural Product Discovery
Principle: This protocol utilizes bioinformatics tools to identify biosynthetic gene clusters in microbial genomes, followed by heterologous expression to discover novel natural products [4] [2].
Materials:
- Bacterial/fungal strains or environmental DNA samples
- Bioinformatics software (AntiSMASH, DeepBGC)
- PCR reagents and cloning vectors
- Heterologous expression host (e.g., Streptomyces coelicolor)
- Fermentation equipment
Procedure:
- Genome Sequencing and Analysis:
- Sequence genome of target organism using Illumina or PacBio platforms.
- Annotate genome using RAST or Prokka.
- Biosynthetic Gene Cluster Identification:
- Submit annotated genome to AntiSMASH for identification of biosynthetic gene clusters (BGCs).
- Analyze results for novel or silent BGCs with potential novel chemistry.
- Cluster Activation:
- Design primers to amplify entire BGC or key regulatory genes.
- Clone BGC into appropriate expression vector (e.g., BAC, cosmic).
- Introduce construct into heterologous host for expression.
- Metabolite Analysis:
- Culture recombinant strains under various conditions (media, temperature, aeration).
- Extract metabolites with organic solvents and analyze by LC-HRMS.
- Compare metabolic profiles with wild-type strains to identify novel compounds.
- Structure Elucidation:
- Ispute novel compounds using preparative HPLC.
- Determine structures using NMR (1D and 2D) and high-resolution MS.
- Test compounds for biological activity in relevant assays.
Troubleshooting:
- For silent clusters, consider promoter engineering or co-cultivation to activate expression.
- If heterologous expression fails, optimize codon usage or refactor cluster design [2].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Natural Product Research
| Reagent/Resource | Function | Application Examples |
|---|---|---|
| AntiSMASH | Identifies biosynthetic gene clusters in genomic data | Genome mining for novel natural products [2] |
| GNPS (Global Natural Products Social Molecular Networking) | Facilitates mass spectrometry data sharing and annotation | Dereplication and compound identification [4] |
| Sephadex LH-20 | Size exclusion chromatography medium for natural product separation | Fractionation of crude extracts by molecular size [5] |
| CRISPR-Cas Systems | Genome editing for pathway engineering | Activation of silent biosynthetic gene clusters [2] |
| HPLC-MS Systems | High-performance liquid chromatography coupled with mass spectrometry | Compound separation, quantification, and identification [5] |
| Dihydroevocarpine | Dihydroevocarpine, CAS:15266-35-0, MF:C23H35NO, MW:341.5 g/mol | Chemical Reagent |
| Nisamycin | Nisamycin, CAS:150829-93-9, MF:C24H27NO6, MW:425.5 g/mol | Chemical Reagent |
- Induced Pluripotent Stem Cells (iPSCs): Used in phenotypic screening platforms for evaluating natural product bioactivity and toxicity in human-relevant systems [2].
- C18 Reverse-Phase Resin: Essential for final purification steps of medium to non-polar natural products through preparative HPLC [5].
Application in Scaffold Hopping Research
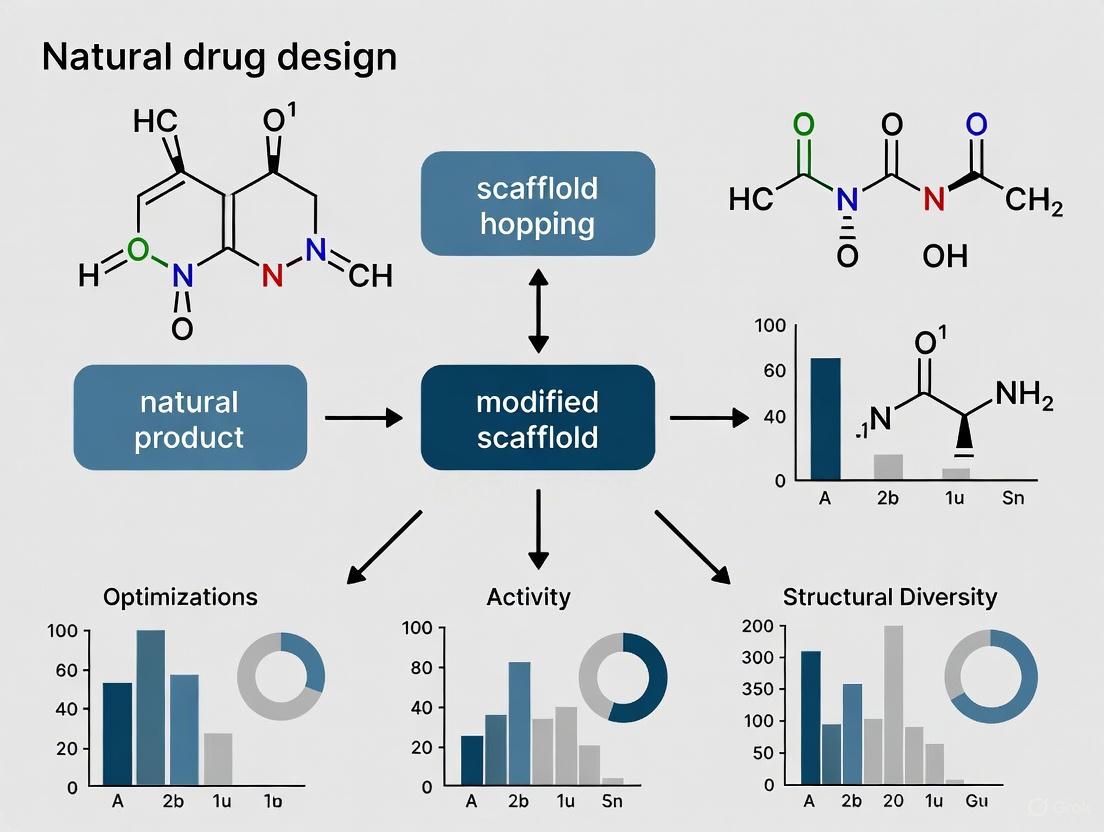
Natural products serve as excellent starting points for scaffold hopping approaches in drug discovery [8]. Scaffold hopping involves modifications to the core structure of an existing bioactive molecule to create new patentable molecules with potentially improved properties [8].
The complex molecular architectures of natural products, honed by evolutionary selection for bioactivity, provide privileged scaffolds that can be optimized through various hopping strategies [8]. These include heterocycle replacement, ring opening or closure, and peptidomimetics to enhance drug-like properties while maintaining biological activity [8].
Scaffold Hopping Strategy for Natural Products. This diagram illustrates how natural product scaffolds can be modified through various hopping approaches to generate diverse analog libraries for lead optimization. Multiple structural modification strategies are employed to enhance drug-like properties while maintaining core bioactivity.
Table 4: Successful Natural Product-Derived Drugs Developed Through Scaffold Hopping
| Original Natural Product | Scaffold Hopping Approach | Resulting Drug/Candidate | Therapeutic Application |
|---|---|---|---|
| Roxadustat | Heterocycle replacement | Novel HIF-PHD inhibitors | Renal anemia treatment [8] |
| GLPG1837 | Ring closure and expansion | SBD-100 | Cystic fibrosis (enhanced potency) [8] |
| Imidazo[1,2-a]pyrazine TTK inhibitors | Iterative heterocycle replacement | CFI-402257 | Anticancer agent (improved exposure) [8] |
| Sorafenib | Ring opening and amide bond modification | Quinazoline-2-carboxamide analogs | Enhanced VEGFR2 inhibition [8] |
Natural products remain a cornerstone of pharmaceutical innovation due to their unparalleled structural diversity, evolutionary optimization, and proven therapeutic potential [3] [2]. Their historical success in treating various diseases, combined with inherent bioactivity honed through millions of years of evolution, positions them as invaluable resources for addressing current and future health challenges [6] [4].
The integration of modern technologies—including genomics, artificial intelligence, and advanced analytical techniques—with traditional knowledge is revitalizing natural product research [4] [2]. These developments are overcoming historical limitations and creating new opportunities to harness nature's chemical ingenuity for drug discovery, particularly through strategies such as scaffold hopping that optimize natural scaffolds for clinical application [8]. As global health challenges continue to evolve, natural products will undoubtedly remain at the forefront of therapeutic development.
Scaffold hopping is a fundamental strategy in modern medicinal chemistry and drug discovery. It is defined as the process of modifying the central molecular core, or scaffold, of a known bioactive compound to generate a novel chemotype while preserving or improving its biological activity [9] [10]. The core objective is to identify isofunctional molecular structures with significantly different molecular backbones [11].
This approach is pivotal for overcoming limitations associated with existing lead compounds, such as poor pharmacokinetics, toxicity, or intellectual property restrictions [12] [10]. By generating structurally novel compounds that retain the desired biological function, scaffold hopping enables researchers to create "me-better" or "fast-follower" drugs, expand the intellectual property space, and explore uncharted regions of chemical space for bioactive molecules [12] [13]. Its application is particularly valuable in natural product-based drug design, where complex structures often require optimization for drug-like properties [12] [14].
A Classification System for Scaffold Hops
The structural changes in scaffold hopping can be systematically categorized based on the degree and nature of the modification to the parent molecule. The following table outlines a widely used classification system.
Table 1: A Classification Framework for Scaffold Hopping
| Degree of Hop | Core Modification | Key Characteristics | Exemplar Case |
|---|---|---|---|
| 1° (Heterocyclic Replacement) | Replacement, addition, or swap of heteroatoms within a ring system [9] [10]. | Retains the core spatial pharmacophore arrangement; tunes physicochemical properties [10]. | Sildenafil to Vardenafil: Swap of carbon and nitrogen atoms in a fused ring system [9] [10]. |
| 2° (Ring Opening or Closure) | Breaking a ring to create an acyclic chain or forming a new ring to rigidify a structure [9]. | Can significantly alter molecular flexibility and entropic penalties for binding [9]. | Morphine to Tramadol: Ring opening of a rigid, fused system to a more flexible molecule [9]. |
| 3° (Peptidomimetics) | Replacement of a peptide backbone with non-peptide moieties [9]. | Mimics the topology of a peptide while improving metabolic stability and oral bioavailability [9]. | Conversion of a therapeutic peptide (AMP1) to a small, non-peptide synthetic mimetic [12]. |
| 4° (Topology-Based Hopping) | Identification of cores with similar shape and pharmacophore features but distinct atomic connectivity [9]. | Leads to the highest degree of structural novelty; often relies on 3D shape and electrostatic similarity [9] [11]. | Use of the FTrees method to find distant chemical relatives based on "fuzzy pharmacophores" [11]. |
Computational Protocols for Scaffold Hopping
Several computational methodologies have been developed to facilitate scaffold hopping. The protocols below detail two primary approaches: one leveraging deep learning and another based on virtual screening with pharmacophore constraints.
â—ˆ Protocol 1: Deep Learning-Based Scaffold Hopping with DeepHop
DeepHop formulates scaffold hopping as a supervised molecule-to-molecule translation task, conditioned on a specific protein target [13].
- Objective: To generate a "hopped" molecule (Y) from a reference molecule (X) for a target (Z), such that Y has improved bioactivity, high 3D similarity, but low 2D similarity to X [13].
- Data Preparation and Model Training:
- Data Curation: From a bioactivity database (e.g., ChEMBL), curate pairs of molecules ((X; Y)\|Z) where, for a given target Z, molecule Y has significantly improved bioactivity (e.g., pChEMBL value ≥ 1) over X, with a 2D scaffold Tanimoto similarity ≤ 0.6 and a 3D shape similarity ≥ 0.6 [13].
- Model Architecture: Employ a multimodal Transformer neural network. The model integrates:
- Molecular 2D graph information.
- Molecular 3D conformer information processed by a spatial graph neural network.
- Protein sequence information processed by a Transformer encoder [13].
- Training: Train the model to translate the reference molecule X to the improved molecule Y using the constructed paired dataset [13].
- Application and Fine-Tuning:
- Input: A reference molecule (X) and its target protein (Z).
- Generation: The trained DeepHop model generates novel candidate molecules (Y).
- Validation: A deep QSAR model (e.g., Multi-Task Deep Neural Network) is used for rapid virtual profiling of the generated molecules to predict bioactivity [13].
- Generalization: The model can be fine-tuned on a small set of active compounds for a new target protein not included in the original training [13].
The workflow for this deep learning approach is standardized as follows:
â—ˆ Protocol 2: Virtual Screening with Pharmacophore Constraints
This protocol uses structure-based virtual screening, enhanced with pharmacophore constraints, to identify scaffold hops from commercial or internal compound libraries [11].
- Objective: To identify novel chemical entities that bind to the same target as a known ligand, utilizing a defined protein binding pocket.
- Methodology:
- Target Structure Preparation:
- Obtain a 3D structure of the target protein (e.g., from X-ray crystallography, NMR, or the PDB).
- Prepare the structure by adding hydrogen atoms, assigning correct protonation states, and optimizing side-chain conformations.
- Pharmacophore Definition:
- Based on the binding mode of a known ligand or analysis of the binding site, define key pharmacophore features. These may include: Hydrogen Bond Donor, Hydrogen Bond Acceptor, Positive Ionizable, Hydrophobic Region, Aromatic Ring [11].
- Virtual Screening Workflow:
- Docking: Perform molecular docking of a compound library (e.g., ZINC, PubChem) into the target's binding site using software like SeeSAR [11].
- Pharmacophore Constraint: Apply the predefined pharmacophore features as a filter. Only consider docking poses that match these critical interactions for further analysis [11].
- Scoring and Ranking: Rank the filtered compounds based on their predicted binding affinity (docking score) and the quality of the pharmacophore fit.
- Post-Screening Analysis:
- Visually inspect the top-ranked compounds to verify the binding mode and interactions.
- Analyze the scaffold of the hits to confirm novelty compared to the original lead.
- Target Structure Preparation:
The following diagram illustrates the logical decision process within the virtual screening workflow:
Experimental Validation of Scaffold Hops
Computational predictions require rigorous experimental validation to confirm successful scaffold hopping. The following table outlines key biophysical and cellular assays used for this purpose.
Table 2: Key Assays for Experimental Validation of Scaffold Hops
| Assay Type | Measured Parameter | Protocol Summary | Application in Validation |
|---|---|---|---|
| Intact Mass Spectrometry | Direct detection of ligand-bound protein complex and binding stoichiometry [15]. | Protein-ligand complexes are buffer-exchanged into volatile ammonium acetate and analyzed by native mass spectrometry [15]. | Confirms stabilization of a protein-protein interaction (PPI) by a molecular glue, demonstrating cooperative binding [15]. |
| Time-Resolved FRET (TR-FRET) | Change in fluorescence resonance energy transfer between labeled binding partners [15]. | A fluorescent donor and acceptor are attached to the two interacting proteins. Ligand-induced proximity increases FRET efficiency, measured over time [15]. | Quantifies the potency (EC50) of a scaffold hop in stabilizing or inhibiting a PPI in a purified system [15]. |
| Surface Plasmon Resonance (SPR) | Binding kinetics (association rate kon, dissociation rate koff) and affinity (K_D) [15]. | One binding partner is immobilized on a sensor chip. Analyte containing the other partner flows over it, and binding-induced refractive index changes are monitored in real-time [15]. | Determines if the scaffold hop maintains or improves binding affinity and residence time compared to the original ligand. |
| NanoBRET | Bioluminescence resonance energy transfer in live cells [15]. | Proteins of interest are tagged with NanoLuc luciferase (donor) and HaloTag (acceptor). Ligand-induced interaction is measured via BRET signal in live cells [15]. | Validates target engagement and functional efficacy (e.g., PPI stabilization) of scaffold hops in a physiologically relevant, cellular context [15]. |
The Scientist's Toolkit: Essential Research Reagents and Software
Successful execution of scaffold hopping campaigns relies on a suite of computational tools and chemical resources.
Table 3: Essential Research Reagents and Software for Scaffold Hopping
| Tool / Resource | Type | Primary Function in Scaffold Hopping |
|---|---|---|
| Cresset Blaze / Spark [12] | Software | Blaze: Virtual screening of vendor libraries for whole-molecule replacement. Spark: Fragment replacement to generate ideas for synthesis [12]. |
| AnchorQuery [15] | Software / Virtual Library | Pharmacophore-based screening of a >31 million compound library of readily synthesizable (via MCR chemistry) scaffolds [15]. |
| SeeSAR & ReCore [11] | Software | SeeSAR: Interactive structure-based design and docking. ReCore: Topological replacement of molecular fragments based on 3D vector geometry [11]. |
| FTrees / infiniSee [11] | Software | FTrees: Similarity searching based on "Feature Trees" (fuzzy pharmacophores) to find distant chemical relatives in large chemical spaces [11]. |
| Scaffold Hunter [16] | Software | A visual analytics framework for analyzing chemical compound data, featuring scaffold tree visualization, clustering, and dataset comparison [16]. |
| ChEMBL Database [13] [17] | Database | A manually curated database of bioactive molecules with drug-like properties, used for training models and extracting bioactivity data [13] [17]. |
| ZINC Database [11] | Database | A freely available database of commercially available compounds for virtual screening [11]. |
| Groebke-Blackburn-Bienaymé (GBB) Reaction [15] | Chemical Reaction | A multi-component reaction used to rapidly synthesize drug-like imidazo[1,2-a]pyridine scaffolds identified through computational design [15]. |
| Trovafloxacin mesylate | Trovafloxacin mesylate, MF:C21H19F3N4O6S, MW:512.5 g/mol | Chemical Reagent |
| Lethedoside A | Lethedoside A | Lethedoside A is a natural flavone for cancer research. This product is for Research Use Only (RUO), not for human or veterinary use. |
In the pursuit of novel therapeutics, the strategy of scaffold hopping has become a cornerstone of modern medicinal chemistry, particularly within natural product-based drug design. This approach, defined as the modification of a compound's central core structure to generate a novel chemotype while retaining or improving biological activity, serves as a powerful method to overcome limitations of original leads [18] [9]. The ultimate goal is to discover structurally novel compounds that maintain efficacy against a biological target while achieving superior pharmacological properties [9].
The conceptual foundation of scaffold hopping, introduced in 1999, emphasizes two key components: different core structures and similar biological activities relative to the parent compound [18] [9]. This strategy appears to challenge the traditional similarity-property principle but is instead enabled by a more sophisticated understanding of molecular recognition. Ligands that fit the same protein pocket often share essential three-dimensional features—such as shape and electrostatio potential surface—even if their underlying two-dimensional architectures belong to different chemotypes [18].
This application note systematically classifies scaffold hopping approaches, provides detailed experimental protocols, and frames these methodologies within the context of advancing natural product-based drug discovery.
Established Classification of Scaffold Hopping Approaches
Scaffold hopping strategies are broadly categorized into four distinct classes based on the degree and nature of structural modification applied to the parent molecule [18] [19] [9]. These classes represent a spectrum of structural change, from minor atomic substitutions to complete topological reorganization.
Table 1: Fundamental Classification of Scaffold Hopping Approaches
| Hop Class | Degree of Structural Novelty | Core Methodology | Primary Application Context |
|---|---|---|---|
| 1° Hop: Heterocycle Replacement | Low | Swapping or replacing atoms (e.g., C, N, O, S) within a ring system. | Lead optimization, patent circumvention, improving physicochemical properties like solubility [18] [9]. |
| 2° Hop: Ring Opening or Closure | Low to Medium | Breaking bonds to open fused rings or forming new bonds to create ring systems and control molecular flexibility [18] [9]. | Modifying pharmacokinetic profiles, enhancing potency by reducing entropy loss upon binding [18]. |
| 3° Hop: Peptidomimetics | Medium | Replacing peptide backbones with non-peptide moieties to mimic the spatial arrangement of key pharmacophoric groups [18] [9]. | Developing drug-like molecules from bioactive but metabolically unstable peptides [18]. |
| 4° Hop: Topology-Based Hopping | High | Identifying or designing cores with different connectivity but similar shape and pharmacophore alignment in 3D space [18] [19] [9]. | Discovering truly novel chemotypes, high-risk lead hopping for challenging targets [18]. |
The following diagram illustrates the logical relationships and decision-making pathways connecting these four classes of scaffold hops:
Experimental Protocols & Case Studies
Protocol 1: Topology-Based Scaffold Hopping for Molecular Glues
Molecular glues, which stabilize protein-protein interactions (PPIs), represent a challenging and promising frontier. A 2025 study detailed a scaffold-hopping approach to develop molecular glues for the 14-3-3σ/ERα complex, starting from a known covalent molecular glue [20].
Objective: To design a novel, non-covalent molecular glue scaffold with improved drug-like properties using a computational topology-based hopping approach followed by synthesis via multicomponent reactions (MCRs) [20].
Table 2: Key Research Reagents & Solutions for Molecular Glue Development
| Reagent/Solution | Function/Description | Application in Protocol |
|---|---|---|
| AnchorQuery Software | Pharmacophore-based screening tool for a 31M+ compound library synthesizable via one-step MCRs [20]. | Virtual screening to identify novel scaffolds based on anchor and pharmacophore points from a known ligand. |
| GBB-3CR Reaction Components | Groebke-Blackburn-Bienaymé multicomponent reaction using aldehydes, 2-aminopyridines, and isocyanides [20]. | Rapid synthesis of the proposed imidazo[1,2-a]pyridine scaffold, enabling generation of diverse analogs. |
| TR-FRET Assay Kit | Time-Resolved Förster Resonance Energy Transfer assay for measuring PPI stabilization in a biochemical setting. | Biophysical validation of molecular glue efficacy in stabilizing the 14-3-3σ/ERα interaction. |
| NanoBRET Assay System | Bioluminescence Resonance Energy Transfer assay configured for live-cell PPI analysis. | Cellular confirmation of target engagement and PPI stabilization under physiological conditions. |
Methodology:
- Template Definition: Use a high-resolution crystal structure of the lead compound (e.g., compound 127, PDB: 8ALW) bound to the target PPI interface [20].
- Anchor Identification: Define a deeply buried, critical structural motif (e.g., a p-chloro-phenyl ring) as the "anchor." This anchor is kept constant during the virtual screen [20].
- Pharmacophore Query: From the original ligand's binding pose, define a set of three additional key pharmacophore points (e.g., hydrogen bond donors/acceptors, hydrophobic regions). This 3-point pharmacophore, combined with the anchor, is used to query the MCR virtual library in AnchorQuery [20].
- Hit Selection & Synthesis: Rank proposed scaffolds by RMSD fit to the original pharmacophore. Synthesize top-ranking scaffolds, prioritizing those like the imidazo[1,2-a]pyridines accessible via the GBB-3CR, allowing for rapid derivatization [20].
- Biophysical & Cellular Validation: Profile synthesized analogs using orthogonal assays:
- Intact Mass Spectrometry: Confirm binding and stoichiometry.
- Surface Plasmon Resonance (SPR): Quantify binding affinity and kinetics.
- TR-FRET: Measure PPI stabilization in a biochemical context.
- NanoBRET: Confirm functional PPI stabilization in live cells [20].
The workflow for this protocol is summarized in the following diagram:
Protocol 2: Enzyme-Enabled Scaffold Hopping in Terpenoid Synthesis
A groundbreaking 2024 study demonstrated a hybrid enzymatic-chemical strategy for scaffold hopping in complex natural product synthesis, moving beyond purely computational designs [14].
Objective: To efficiently generate diverse terpenoid natural product scaffolds from a single, commercially available terpenoid precursor via enzymatic oxidation followed by selective chemical rearrangement [14].
Methodology:
- Selection of Platform Scaffold: Choose a synthetically accessible and versatile natural product scaffold as the starting point (e.g., sclareolide, a commercially available sesquiterpene lactone) [14].
- Enzymatic Functionalization: Employ engineered cytochrome P450 enzymes to perform site-selective oxidation (e.g., at the C3 position of sclareolide) that is challenging to achieve with traditional synthetic chemistry. This creates a key oxygenated intermediate [14].
- Scaffold Diversification: Use the introduced oxygen functionality as a chemical handle to direct subsequent rearrangement reactions (e.g., Wagner-Meerwein shifts, cyclizations). By varying the reaction conditions, this single intermediate can be diverted down multiple synthetic pathways [14].
- Target Synthesis: Apply this strategy to synthesize distinct terpenoid natural products with unique carbon frameworks from the common intermediate. The study successfully synthesized merosterolic acid B, cochlioquinone B, (+)-daucene, and dolasta-1(15),8-diene from oxidized sclareolide [14].
This protocol challenges traditional retrosynthetic analysis by establishing a shared, enzyme-generated intermediate for multiple target scaffolds, significantly improving synthetic efficiency.
The Evolving Toolbox: AI-Driven Molecular Representation
Modern scaffold hopping is increasingly powered by artificial intelligence (AI) and advanced molecular representation methods, which move beyond traditional fingerprint-based approaches [19].
- Language Model-Based Representations: Models like transformers treat molecular strings (e.g., SMILES) as a chemical language, learning contextual relationships between atoms and functional groups to generate novel, valid structures [19].
- Graph-Based Representations: Graph Neural Networks (GNNs) natively represent molecules as graphs (atoms as nodes, bonds as edges), enabling them to learn from both local atomic environments and global topological structure, which is crucial for recognizing scaffold-level similarities [19] [21].
- 3D Interaction-Driven Models: For target-informed hopping, models like DeepFrag and FRAME leverage 3D structural data of protein-ligand complexes. DeepFrag frames the problem as a classification task, predicting optimal fragments to fill a binding pocket, while FRAME uses equivariant neural networks to explicitly model protein-ligand interactions and select optimal connection points [21].
These AI-driven strategies can be categorized based on their approach to structural modification, as shown in the table below.
Table 3: AI-Driven Models for Molecular Modification in Scaffold Hopping
| Model Name | Core Architecture | Modification Strategy | Key Application Note |
|---|---|---|---|
| DeepFrag [21] | 3D Deep Convolutional Neural Network (DCNN) | Fragment Splicing | Treats fragment replacement as a classification task based on the protein-ligand complex structure. |
| FREED/FREED++ [21] | Graph CNN + Reinforcement Learning (RL) | Fragment Splicing | Uses RL to efficiently explore chemical space and generate molecules with high docking scores. |
| FRAME [21] | SE(3)-Equivariant Neural Network | Fragment Splicing | Explicitly models 3D protein-ligand interactions (H-bonds, π-π) for dynamic fragment selection. |
| MolEdit3D [21] | 3D Graph Neural Network | Fragment Editing & Splicing | A 3D graph editing model allowing for precise atomic and fragment-level modifications. |
| TACOGFN [21] | GFlowNet + Graph Transformer | Fragment Splicing | Incorporates target pocket information into a generative flow network for guided fragment addition. |
The systematic classification of scaffold hops into heterocycle replacements, ring operations, peptidomimetics, and topology-based changes provides a robust conceptual framework for medicinal chemists. This is particularly valuable in natural product research, where the goal is often to translate complex, bioactive scaffolds into viable drug leads. The integration of computational protocols—ranging from pharmacophore-based MCR screening to FEP-guided design—with innovative experimental strategies, such as enzyme-enabled diversification, is redefining the scaffold hopping landscape. As AI-driven molecular representation and generation models continue to mature, they promise to further accelerate the discovery of novel chemotypes, enhancing our ability to explore chemical space and address unmet medical needs through natural product-inspired design.
Application Notes
The strategic modification of natural product (NP) scaffolds is a cornerstone of modern drug discovery, enabling the optimization of bioactive compounds for enhanced efficacy and druggability. This approach leverages the inherent, evolutionarily refined biological activities of NPs while overcoming limitations such as poor bioavailability, low potency, or high toxicity. The following application notes detail a transformative protocol for the diversification of terpenoid scaffolds, a class of NPs renowned for their structural complexity and broad bioactivity.
A pioneering application of enzyme-enabled scaffold hopping is demonstrated in the work of Renata and colleagues, who developed a versatile chemoenzymatic strategy to generate diverse terpenoid frameworks from a common precursor [14]. This method challenges the conventional retrosynthetic paradigm of designing a custom synthesis for each distinct molecular target. Instead of viewing an enzymatic modification as a final step, the team treated a biocatalytically installed functional group as a handle for subsequent abiotic skeletal rearrangements [22] [23].
Core Application Workflow: The process begins with sclareolide, a commercially available sesquiterpene lactone with a drimane-type skeleton [14]. The key enabling step involves the highly selective oxidation of a single carbon atom (C-3) on this scaffold using engineered cytochrome P450 enzymes (CYP450s) [14]. This biocatalytic transformation, difficult to achieve with traditional chemical methods, produces an alcohol intermediate. This intermediate is not an end-product but a versatile platform for "scaffold hopping" – a process that intentionally alters the core connectivity of the molecule [23]. Through carefully designed chemical reactions (e.g., ring-opening, rearrangement, and cyclization sequences), this single oxidized intermediate can be diverted down multiple synthetic pathways to produce terpenoids with vastly different carbon skeletons [14] [22].
Outcomes and Significance: Using this strategy, the research team successfully synthesized four distinct terpenoid natural products from sclareolide: merosterolic acid B, cochlioquinone B, (+)-daucene, and dolasta-1(15),8-diene [14] [22]. This demonstrates the remarkable structural divergence achievable from a single starting point. The implications for drug discovery are profound, as this platform technology significantly enhances synthetic efficiency. It saves time and cost by providing a shared entry point with branching pathways, thereby accelerating the exploration of structure-activity relationships (SAR) around complex terpenoid scaffolds for medicinal chemistry programs [14].
Table 1: Terpenoid Natural Products Synthesized via Enzyme-Enabled Scaffold Hopping
| Natural Product | Class | Key Structural Features Achieved | Potential/Reported Bioactivity |
|---|---|---|---|
| Merosterolic Acid B | Meroterpenoid | Complex ring system integrated with non-terpenoid structural units | Not specified in search results |
| Cochlioquinone B | Sesquiterpene-quinone | Fused quinonoid moiety installed via oxidation and rearrangement [23] | Environmentally relevant terpenoid [23] |
| (+)-Daucene | Sesquiterpene hydrocarbon | Altered ring junctions from the original drimane core [23] | Serves as a critical biosynthetic intermediate [23] |
| Dolasta-1(15),8-diene | Diterpene hydrocarbon | Unique double bond placements and ring fusion patterns [23] | Not specified in search results |
Protocols
Protocol 1: Enzyme-Enabled Scaffold Hopping for Terpenoid Diversification
This protocol outlines the detailed methodology for the oxidative diversification of sclareolide and subsequent abiotic rearrangements to access distinct terpenoid skeletons, as pioneered by Renata's group [14] [22] [23].
I. Materials and Equipment
Research Reagent Solutions
| Reagent/Material | Function/Explanation |
|---|---|
| Sclareolide | The starting material; a commercially available sesquiterpene lactone providing a defined drimane-type scaffold. |
| Engineered Cytochrome P450 Enzymes (CYP450) | Biocatalysts engineered for high regio- and stereoselective oxidation of inert C-H bonds, installing a handle (alcohol) for further functionalization. |
| Cofactor System (e.g., NADPH) | Required for the enzymatic activity of CYP450s to drive the oxidation reaction. |
| Appropriate Buffer | To maintain optimal pH and ionic strength for enzymatic stability and activity. |
| Abiotic Reagents | Chemical reagents (e.g., acids, bases, catalysts) for skeletal rearrangements post-oxidation, enabling scaffold hopping. |
Essential Equipment: Bioreactor or controlled environment shaker for enzymatic reactions, standard organic synthesis glassware, analytical instruments (HPLC, LC-MS, NMR) for reaction monitoring and purification.
II. Experimental Procedure
Step 1: Enzymatic C-H Hydroxylation of Sclareolide
- Reaction Setup: Prepare a reaction mixture containing sclareolide (e.g., 1.0 mmol) in a suitable aqueous-organic buffer system. Add the engineered CYP450 enzyme and a cofactor regeneration system (e.g., NADP+, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase) to sustain the catalytic cycle.
- Incubation: Incubate the reaction mixture with gentle agitation at a temperature and pH optimized for the specific engineered enzyme (e.g., 30°C, pH 7.4) for a predetermined time (e.g., 12-24 hours) to achieve high conversion.
- Monitoring and Work-up: Monitor the reaction progress by LC-MS or TLC. Upon completion, extract the product using an organic solvent (e.g., ethyl acetate). Purify the crude extract via flash chromatography to isolate the C-3 hydroxylated sclareolide intermediate. Confirm the structure and regiochemistry by ( ^1 \text{H} ) and ( ^13\text{C} ) NMR spectroscopy [14].
Step 2: Abiotic Skeletal Rearrangement (Scaffold Hopping) This step is target-dependent. The following are generalized pathways based on the synthesized products.
- Pathway to Merosterolic Acid B/Cochlioquinone B:
- Subject the alcohol intermediate to an oxidative ring-opening reaction.
- Employ tailored conditions (e.g., specific oxidizing agents, controlled temperatures) to guide the formation of a new reactive species.
- Initiate cyclization and rearrangement sequences to form the distinct carbon frameworks characteristic of merosterolic acid and cochlioquinone B, the latter involving installation of a quinone moiety [23].
- Pathway to (+)-Daucene/Dolasta-diene:
- Activate the alcohol group (e.g., via mesylation or tosylation) to form a better leaving group.
- Treat the activated intermediate with a strong base or Lewis acid to promote a concerted ring contraction/expansion or Wagner-Meerwein-type rearrangement.
- Carefully control reaction parameters (e.g., solvent, temperature, concentration) to steer the rearrangement towards the desired hydrocarbon scaffold of (+)-daucene or dolasta-diene [23].
Step 3: Purification and Characterization
- Purify each final terpenoid product using techniques such as preparative thin-layer chromatography (PTLC) or HPLC.
- Characterize each compound fully using high-resolution mass spectrometry (HRMS) and 1D/2D NMR spectroscopy. Compare the spectral data with literature values to confirm identity and purity [14] [22].
III. Data Analysis and Interpretation
The success of the protocol is determined by the structural confirmation of all final products. The key data for comparison is summarized below.
Table 2: Quantitative Data for Synthesized Terpenoids from Sclareolide
| Natural Product | Number of Synthetic Steps from Sclareolide (Representative) | Overall Yield (Representative, Over Multiple Steps) | Key Analytical Data (e.g., Specific Rotation [α]D) |
|---|---|---|---|
| Merosterolic Acid B | Not specified in search results | Not specified in search results | Not specified in search results |
| Cochlioquinone B | Not specified in search results | Not specified in search results | Not specified in search results |
| (+)-Daucene | Not specified in search results | Not specified in search results | The "(+)" designation indicates the compound is dextrorotatory. |
| Dolasta-1(15),8-diene | Not specified in search results | Not specified in search results | Not specified in search results |
Note: The source articles announce the achievement of the synthesis but do not report detailed quantitative yield data or step counts. The primary analytical confirmation is based on NMR and MS comparison with authentic samples or literature data [14] [22].
IV. Visual Workflow
The following diagram illustrates the logical workflow of the enzyme-enabled scaffold hopping strategy.
The Scientist's Toolkit
Table 3: Essential Reagents for Enzyme-Enabled Terpenoid Diversification
| Tool/Reagent | Category | Function in Research |
|---|---|---|
| Engineered Cytochrome P450s | Biocatalyst | Key to regioselective C-H activation; provides the critical "handle" for downstream diversification that is unattainable purely chemically [14]. |
| Sclareolide | Natural Product Scaffold | Commercially available, complex starting material that serves as a versatile and privileged platform for generating structural diversity [14] [23]. |
| Cofactor Regeneration System | Biochemical Reagent | Maintains the activity of oxidative enzymes like CYP450s over prolonged reactions, improving efficiency and atom economy. |
| Molecular Generative AI Models | Computational Tool | For target-unknown scenarios, these AIDD models can propose novel structural modifications and predict bioactivity, guiding scaffold hopping efforts [21]. |
| Fragment Hotspot Maps (FHMs) | Computational Tool | Used in silico to identify optimal sites on a protein target for fragment binding, informing the design of new scaffolds for specific targets [21]. |
| Aprindine | Aprindine, CAS:33237-74-0, MF:C22H30N2, MW:322.5 g/mol | Chemical Reagent |
| Dodecamethylpentasiloxane | Dodecamethylpentasiloxane, CAS:141-63-9, MF:C12H36O4Si5, MW:384.84 g/mol | Chemical Reagent |
Methodologies for Scaffold Hopping: From Traditional Rules to AI-Driven Generation
In the field of natural product-based drug design, scaffold hopping—the identification of isofunctional molecular structures with significantly different molecular backbones—is a central strategy for discovering novel lead compounds with improved properties and intellectual potential [9]. This process enables researchers to transition from complex natural product scaffolds to synthetically accessible mimetics while preserving biological activity [24]. Traditional computational methods for scaffold hopping primarily rely on 2D molecular fingerprints and 3D pharmacophore models, each offering distinct advantages for exploring the complex chemical space of natural products [25] [26]. This application note details standardized protocols for employing these methods within natural product research programs, complete with performance benchmarks and implementation workflows.
Molecular Fingerprints for Natural Product Analysis
Molecular fingerprints are vector representations that encode molecular structures into binary, count, or categorical formats based on specific structural or chemical patterns. They are predominantly used for rapid similarity searching and quantitative structure-activity relationship (QSAR) modeling [26].
Key Fingerprint Types and Their Performance on Natural Products
Natural products often possess distinct chemical characteristics compared to synthetic drug-like molecules, including higher molecular complexity, more stereocenters, and a greater fraction of sp³-hybridized carbons [26]. These differences can significantly impact the performance of various fingerprinting algorithms. The table below summarizes the performance of major fingerprint categories on natural product datasets.
Table 1: Performance Evaluation of Molecular Fingerprints on Natural Product Datasets
| Fingerprint Category | Representative Examples | Basis of Calculation | Performance on Natural Products |
|---|---|---|---|
| Circular Fingerprints | ECFP, FCFP [26] | Atom-centered radial substructures | Good performance, but can be outperformed by other fingerprints for specific NP bioactivity prediction tasks [26]. |
| Path-Based Fingerprints | Atom Pairs (AP), Depth First Search (DFS) [26] | Linear paths through molecular graph | Performance varies; requires benchmarking for specific NP datasets [26]. |
| Pharmacophore Fingerprints | Pharmacophore Pairs (PH2), Triplets (PH3) [26] | Topological distances between pharmacophoric points | Can provide an abstract, interaction-based representation less dependent on specific scaffold [26]. |
| Substructure-Based Fingerprints | MACCS, PubChem [26] | Presence of predefined structural keys | Limited by predefined fragment dictionary, potentially hampering scaffold hopping [27]. |
| String-Based Fingerprints | MHFP, MAP4 [26] | Fragmentation of SMILES strings | MAP4 has shown competitive or superior performance to ECFP in NP bioactivity prediction [26]. |
Experimental Protocol: Fingerprint-Based Virtual Screening for Scaffold Hopping
Principle: This ligand-based protocol uses a known active natural product as a query to identify structurally different compounds with similar bioactivity by calculating molecular similarity in a 2D descriptor space [24].
Materials & Software:
- Query Compound: A known bioactive natural product (e.g., from COCONUT or CMNPD databases) [26].
- Screening Database: A database of candidate compounds (e.g., commercial screening libraries, in-house collections).
- Software: Cheminformatics toolkits (e.g., RDKit, OpenBabel) for fingerprint calculation and similarity searching.
Procedure:
- Data Preparation: Standardize all molecular structures (query and database compounds). This includes removing salts, neutralizing charges, and generating canonical SMILES [28] [26].
- Fingerprint Selection and Calculation: Based on the benchmarking data in Table 1, select one or more fingerprint types (e.g., MAP4, ECFP, PH2). Calculate fingerprints for both the query natural product and all compounds in the screening database.
- Similarity Calculation: Compute the pairwise similarity between the query fingerprint and every database compound fingerprint. The Tanimoto coefficient (Jaccard similarity) is the most common metric for this purpose [26].
- Ranking and Analysis: Rank the database compounds based on their similarity score to the query. Visually inspect the top-ranking compounds to identify those that maintain core pharmacophoric elements but possess a novel scaffold (Murcko scaffold) [28] [27].
3D Pharmacophore Models for Scaffold Hopping
A pharmacophore is defined as "the ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target" [25] [29]. Pharmacophore modeling abstracts a molecule from its specific chemical structure to a set of generalized features essential for binding, making it inherently powerful for scaffold hopping [25].
Pharmacophore Feature Types and Generation Methods
Table 2: Core Pharmacophore Features and Model Generation Approaches
| Feature Type | Geometric Representation | Interaction Type | Generation Method | Description & Utility |
|---|---|---|---|---|
| H-Bond Donor (HBD) | Vector or Sphere [25] | Hydrogen Bonding | Structure-Based [25] | Derived from protein-ligand complex crystal structure. Most reliable method. |
| H-Bond Acceptor (HBA) | Vector or Sphere [25] | Hydrogen Bonding | Ligand-Based [25] | Generated from a set of aligned active ligands. Requires known actives. |
| Positive/Negative Ionizable (PI/NI) | Sphere [25] | Ionic | Manual [25] | Built based on deep expert knowledge of target and ligands. |
| Aromatic (AR) | Plane or Sphere [25] | π-Stacking, Cation-π | - | - |
| Hydrophobic (H) | Sphere [25] | Hydrophobic Contact | - | - |
| Exclusion Volumes | Sphere [25] | Steric Clash Prevention | - | Define regions the ligand must not occupy; often from protein structure [25]. |
Experimental Protocol: Structure-Based Pharmacophore Generation and Screening
Principle: This protocol uses the 3D structure of a biological target (often with a bound natural product ligand) to derive a set of essential interaction features, which are then used as a query to screen for novel scaffolds [25].
Materials & Software:
- Protein Structure: A high-resolution 3D structure of the target (e.g., from PDB), preferably in complex with a ligand.
- Software: Molecular visualization software (e.g., PyMOL); pharmacophore modeling software (e.g., MOE, LigandScout, Schrödinger's Phase).
Procedure:
- Structure Preparation: Prepare the protein-ligand complex structure by adding hydrogen atoms, assigning correct protonation states, and optimizing hydrogen bonds.
- Interaction Analysis: Analyze the binding site to identify key interactions between the ligand (e.g., a natural product) and the protein. Note hydrogen bonds, ionic interactions, and hydrophobic regions.
- Feature Mapping: Map the identified interactions to abstract pharmacophore features (HBA, HBD, H, etc.) and place them in 3D space. Add exclusion volume spheres to represent forbidden regions of the binding site [25].
- Model Validation (Optional but Recommended): Validate the initial model by screening a small dataset of known actives and inactives to ensure it can discriminate between them.
- Virtual Screening: Use the validated pharmacophore model as a query to screen a large, conformationally-enriched database of compounds. The search will identify molecules that can spatially and electronically align with all critical features of the query.
- Post-Processing: The resulting hits, which often possess diverse scaffolds, should be further evaluated by molecular docking and visual inspection [30].
Integrated Workflow and Benchmarking
Combining 2D and 3D Methods
A synergistic approach that leverages the speed of 2D fingerprints and the scaffold-hopping power of 3D pharmacophores is often most effective. A common strategy is to use a 2D similarity pre-filter to narrow down a large database, followed by a more computationally intensive 3D pharmacophore screen on the resulting subset [30]. Furthermore, holistic 3D descriptors like WHALES (Weighted Holistic Atom Localization and Entity Shape) have been developed specifically to bridge the gap between natural products and synthetic mimetics by simultaneously encoding pharmacophore, shape, and partial charge distribution [24] [27].
Performance Benchmarking
The table below provides a comparative overview of the scaffold-hopping performance of different molecular representations, highlighting the effectiveness of 3D methods.
Table 3: Benchmarking the Scaffold-Hopping Performance of Different Molecular Representations
| Molecular Representation | Dimension | Core Principle | Reported Scaffold-Hopping Performance (SDA%)* | Key Advantage for NPs |
|---|---|---|---|---|
| ECFPs | 2D | Circular substructures | 73 ± 12 [27] | Fast, widely used, good overall performance [26]. |
| MACCS Keys | 2D | Predefined fragments | 75 ± 12 [27] | Interpretable, but limited by predefined dictionary [27]. |
| CATS2 | 2D | Topological pharmacophore pairs | - | Abstract representation aids hopping [27]. |
| WHIM | 3D | Statistical projection of 3D coordinates | - | Captures overall molecular shape [27]. |
| WHALES | 3D | Holistic integration of shape & pharmacophore | Outperformed benchmarks in 89% of 182 targets [27] | Specifically designed for NP-to-synthetic hopping; high success rate [24] [27]. |
*SDA% (Scaffold Diversity of Actives): The ratio of unique Murcko scaffolds to the number of active compounds retrieved in the top 5% of a virtual screening rank, with higher values indicating better scaffold-hopping ability [27].
The Scientist's Toolkit: Essential Research Reagents and Software
Table 4: Key Software Tools for Molecular Fingerprint and Pharmacophore Modeling
| Tool Name | Category | Primary Function | Application in Protocol |
|---|---|---|---|
| RDKit | Cheminformatics | Open-source toolkit for cheminformatics | Fingerprint calculation (ECFP, etc.), molecular standardization, and similarity searching [28] [26]. |
| FTrees/InfiniSee | Virtual Screening | Pharmacophore-based similarity search | Algorithm behind the "Scaffold Hopper Mode" to find compounds related by pharmacophore features [30]. |
| ReCore (SeeSAR) | Structure-Based Design | Structure-based core replacement | Suggests molecular motifs that replace a scaffold while maintaining binding interactions and side chains [30]. |
| FlexS | Molecular Alignment | 3D ligand alignment | Used to align candidate compounds to a reference pharmacophore for similarity assessment [30]. |
| MOE | Integrated Suite | Molecular modeling and simulation | Comprehensive environment for pharmacophore model creation (both structure- and ligand-based) and virtual screening [9]. |
| Cetefloxacin | Cetefloxacin|CAS 141725-88-4|RUO | Cetefloxacin is a synthetic fluoroquinolone antibiotic for research use only (RUO). Inhibits DNA gyrase. Not for human or veterinary use. | Bench Chemicals |
| Penethamate hydriodide | Penethamate hydriodide, CAS:808-71-9, MF:C22H32IN3O4S, MW:561.5 g/mol | Chemical Reagent | Bench Chemicals |
The Weighted Holistic Atom Localization and Entity Shape (WHALES) descriptors represent an advanced molecular representation technique designed to facilitate scaffold hopping in computer-assisted drug discovery. Unlike traditional fingerprints that focus on molecular connectivity or presence of specific fragments, WHALES descriptors simultaneously capture critical information about molecular shape and partial charge distribution in a holistic manner. Originally developed to translate structural and pharmacophore information from bioactive natural products to synthetically accessible isofunctional compounds, WHALES has demonstrated remarkable capability in identifying novel ligand chemotypes that populate uncharted regions of the chemical space while maintaining desired biological activity [31] [32].
The fundamental innovation of WHALES lies in its integration of geometric interatomic distances with atomic physicochemical properties, enabling the identification of structurally diverse compounds that share similar bioactivity profiles. This approach has proven particularly valuable in natural product-based drug design, where complex molecular architectures often serve as inspiration for developing synthetically tractable compounds with improved drug-like properties. By enabling efficient navigation through chemical space, WHALES addresses a key challenge in medicinal chemistry: the discovery of novel bioactive chemotypes through straightforward similarity searching [32].
Theoretical Foundation and Calculation Protocol
Conceptual Framework
WHALES descriptors encode molecular information through a sophisticated algorithm that transforms three-dimensional molecular structures and their electronic properties into a numerical representation. The methodology employs weighted locally-centred atom distances computed for each atom position in a three-dimensional molecular conformation, creating a comprehensive profile that captures molecular shape and charge distribution simultaneously. This holistic approach allows WHALES to identify structurally diverse compounds that share similar steric and electronic properties, making it particularly effective for scaffold hopping applications where traditional fragment-based methods often fail [32].
The theoretical foundation of WHALES rests on the calculation of an atom-centred covariance matrix for each non-hydrogen atom, which captures the distribution of atoms and their partial charges around each atomic center. This matrix incorporates both spatial atomic coordinates and their associated partial charges, creating a weighted representation of the molecular environment that forms the basis for subsequent distance calculations and descriptor generation [32].
Step-by-Step Calculation Protocol
Step 1: Molecular Preparation and Partial Charge Calculation
- Generate a 3D molecular conformation using energy minimization with the MMFF94 force field or similar reliable methods [32]
- Calculate partial atomic charges using either:
Step 2: Atom-Centred Covariance Matrix Computation
For each non-hydrogen atom (j) in the molecule, compute the weighted covariance matrix using the formula:
$${{\bf{S}}}{w(j)}=\frac{{\sum }{i=1}^{n}\,|{\delta }{i}|\cdot ({{\bf{x}}}{i}-{{\bf{x}}}{j}){({{\bf{x}}}{i}-{{\bf{x}}}{j})}^{{\rm{T}}}}{{\sum }{i=1}^{n}\,|{\delta }_{i}|}$$
where:
- (xáµ¢ - xâ±¼) represents the differences between the 3D coordinates of the j-th atomic center and those of any i-th non-hydrogen atom
- |δᵢ| is the absolute value of the partial charge of the i-th atom [32]
Step 3: Atom-Centred Mahalanobis Distance (ACM) Calculation
Compute the ACM distance matrix with the equation:
$${\bf{A}}{\bf{C}}{\bf{M}}\,(i,j)={({{\bf{x}}}{i}-{{\bf{x}}}{j})}^{{\rm{T}}}\cdot {{\bf{S}}}{w(j)}^{-1}\cdot ({{\bf{x}}}{i}-{{\bf{x}}}_{j})$$
This matrix collects all pairwise normalized interatomic distances according to the atom-centred covariance matrix, where atoms located in directions of high variance have smaller distances from the atomic center than those in low-variance regions [32].
Step 4: Atomic Parameter Calculation
From the ACM matrix (excluding diagonal elements), calculate:
- Remoteness degree: Row average of the ACM matrix
- Isolation degree: Column minimum of the ACM matrix
- Ratio values: Isolation degree divided by remoteness value Assign negative values for these parameters for negatively-charged atoms [32].
Step 5: Molecular Descriptor Vector Generation
Calculate the distribution statistics of atomic remoteness, isolation degree, and their ratios to generate 33 WHALES descriptors comprising:
- Minimum and maximum values
- Decile values (10th, 20th, ..., 90th percentiles) This fixed-length descriptor vector enables molecular similarity comparisons independent of molecular size [32].
Table 1: WHALES Descriptor Variants Based on Partial Charge Calculation Methods
| Descriptor Version | Partial Charge Method | Complexity Level | Key Applications |
|---|---|---|---|
| WHALES-DFTB+ | DFTB+ accelerated quantum mechanical simulation | High chemical detail | High-precision scaffold hopping |
| WHALES-GM | Gasteiger-Marsili connectivity-based method | Medium chemical detail | Large-scale virtual screening |
| WHALES-shape | δᵢ = 1 for all atoms (no charge information) | Basic shape-based representation | Shape-focused similarity searching |
Workflow Visualization
Application Notes: WHALES for Scaffold Hopping
Retrospective Virtual Screening Benchmark
WHALES descriptors have been rigorously evaluated for their scaffold-hopping potential through comprehensive benchmarking studies. In a systematic analysis comparing WHALES with seven state-of-the-art molecular representations across 30,000 bioactive compounds and 182 biological targets, WHALES demonstrated superior performance in identifying structurally diverse active compounds [32].
The benchmark included molecular descriptors spanning different dimensionalities and chemical information domains:
- 0D/1D descriptors: Constitutional descriptors capturing basic structural properties
- 1D fingerprints: MACCS 166 keys and Extended Connectivity Fingerprints (ECFPs)
- 2D descriptors: CATS2 pharmacophore pairs and matrix-based descriptors
- 3D descriptors: WHIM and GETAWAY capturing three-dimensional molecular features
Performance was quantified using the Scaffold Diversity of Actives (SDA%) metric, calculated as:
$$S{D}_{A} \% =\frac{ns}{na}\cdot 100$$
where ns represents the number of unique Murcko scaffolds identified in the top 5% of the ranked list, and na is the number of actives present in that same portion [32].
Table 2: Performance Comparison of Molecular Descriptors in Scaffold Hopping
| Molecular Representation | Dimensionality | Mean SDA% ± Standard Deviation | Key Strengths |
|---|---|---|---|
| WHALES-DFTB+ | 3D + Electronic | 89 ± 9 | Best overall scaffold hopping |
| WHALES-GM | 3D + Electronic | 87 ± 10 | Balance of speed and performance |
| WHALES-shape | 3D Shape | 85 ± 11 | Pure shape similarity |
| GETAWAY | 3D | 84 ± 11 | Atom-weighted 3D descriptors |
| WHIM | 3D | 82 ± 12 | Principal axes molecular properties |
| CATS2 | 2D | 80 ± 13 | Pharmacophore pairs |
| Matrix-based descriptors | 2D | 78 ± 13 | Graph theory-based |
| MACCS FP | 1D | 75 ± 12 | 166 predefined substructures |
| ECFPs | 1D | 73 ± 12 | Atom-centered radial fragments |
| Constitutional descriptors | 0D/1D | 76 ± 13 | Basic molecular properties |
The benchmark results demonstrated that all three WHALES versions outperformed state-of-the-art methods in 89% of the tested biological targets, with WHALES-DFTB+ showing the highest scaffold-hopping ability [32]. This superior performance highlights WHALES' capacity to identify structurally diverse compounds that maintain similar bioactivity, a crucial capability in natural product-inspired drug design where structural complexity often necessitates simplification while retaining activity.
Prospective Application: Discovery of Novel RXR Modulators
The scaffold-hopping capability of WHALES was validated in a prospective application targeting the retinoid X receptor (RXR). Using known RXR modulators as queries, WHALES descriptors identified four novel agonists with innovative molecular scaffolds that populated previously uncharted regions of the chemical space [32].
Notably, one identified agonist possessed a rare non-acidic chemotype that exhibited:
- High selectivity across 12 nuclear receptors
- Comparable efficacy to bexarotene in inducing ABCA1 (ATP-binding cassette transporter A1)
- Effective induction of angiopoietin-like protein 4 and apolipoprotein E expression
This successful prospective application confirmed WHALES' ability to detect novel bioactive chemotypes through straightforward similarity searching, demonstrating its practical utility in hit identification and lead optimization campaigns [32].
Recent Application: hDAT-Targeted Drug Repurposing
A 2025 study further validated WHALES' utility in drug repurposing for the human dopamine transporter (hDAT). Researchers employed WHALES descriptors to identify novel atypical inhibitors that bind to hDAT's allosteric site, using four benztropine-like atypical inhibitors as templates [33].
The workflow encompassed:
- Similarity screening of 4,921 marketed and clinically tested drugs using WHALES
- ADMET prediction for 27 identified candidates
- Induced-fit docking to estimate binding affinities
- In vitro validation of six selected compounds
This integrated approach successfully identified three compounds with significant hDAT inhibitory potency (IC₅₀ values of 0.753 μM, 0.542 μM, and 1.210 μM, respectively), demonstrating WHALES' effectiveness in prospective drug discovery applications [33].
Experimental Protocols
Protocol 1: Virtual Screening with WHALES Descriptors
Purpose: To identify novel chemotypes with similar biological activity to a query compound through WHALES-based similarity searching.
Materials and Software:
- Chemical database: Collections such as ChEMBL, ZINC, or in-house compound libraries
- WHALES calculation code: Available from github.com/ETHmodlab/scaffoldhoppingwhales [31]
- 3D conformation generator: RDKit, OpenBabel, or similar cheminformatics toolkit
- Partial charge calculator: DFTB+ for high accuracy or Gasteiger-Marsili for rapid calculation [32]
Procedure:
- Query molecule preparation:
- Select a known active compound as the query structure
- Generate an energy-minimized 3D conformation using MMFF94 or similar force field
- Calculate partial charges using DFTB+ or Gasteiger-Marsili method
Database preparation:
- Curate database molecules by removing duplicates and undesirable compounds
- Generate 3D conformations for all database compounds
- Calculate partial charges using a consistent method
WHALES descriptor calculation:
- Compute WHALES descriptors for query and all database compounds
- Standardize descriptor values using z-score normalization if necessary
Similarity searching:
- Calculate similarity between query and database compounds using Euclidean or Mahalanobis distance
- Rank database compounds by descending similarity (or ascending distance)
- Apply scaffold analysis to identify diverse chemotypes in top rankings
Result analysis:
- Extract top-ranked compounds for visual inspection
- Calculate scaffold diversity metrics (SDA%) to evaluate scaffold-hopping performance
- Select promising candidates for experimental validation
Troubleshooting:
- If results show insufficient scaffold diversity, try WHALES-shape version to emphasize structural over electronic similarity
- If computational resources are limited, use Gasteiger-Marsili partial charges instead of DFTB+
- Ensure consistent protonation states for all compounds at physiological pH
Protocol 2: Natural Product-Inspired Scaffold Hopping
Purpose: To translate structural information from complex natural products to synthetically accessible compounds using WHALES descriptors.
Materials and Software:
- Natural product database: Such as COCONUT, NPASS, or in-house collections
- Synthetic compound library: Commercially available screening compounds or synthetically feasible virtual libraries
- WHALES descriptors with DFTB+ partial charges for maximum sensitivity [32]
Procedure:
- Natural product selection:
- Identify bioactive natural product with desired biological activity
- Generate representative 3D conformation accounting for conformational flexibility
- Calculate WHALES descriptors for the natural product
Synthetic library screening:
- Curate library of synthetically accessible compounds
- Compute WHALES descriptors for all library compounds
- Perform similarity search using natural product as query
Scaffold hopping analysis:
- Identify top-ranking synthetic compounds with high WHALES similarity
- Analyze Murcko scaffolds to confirm structural novelty compared to natural product
- Evaluate synthetic accessibility of identified hits
Hit validation:
- Select diverse chemotypes for experimental testing
- Prioritize compounds with favorable drug-like properties
- Validate biological activity through in vitro assays
Validation: In the proof-of-concept study, WHALES identified seven natural-product-inspired synthetic compounds that modulated the cannabinoid receptor, featuring innovative scaffolds compared to actives annotated in ChEMBL [32].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for WHALES Applications
| Tool/Resource | Type | Function in WHALES Workflow | Availability |
|---|---|---|---|
| DFTB+ | Software | Calculates accurate partial charges for WHALES-DFTB+ | Freely available |
| Gasteiger-Marsili Method | Algorithm | Rapid partial charge calculation for large datasets | Implemented in RDKit, OpenBabel |
| MMFF94 Force Field | Parameter Set | Generates energy-minimized 3D conformations | Implemented in most cheminformatics packages |
| RDKit | Cheminformatics Library | Handles molecular I/O, conformation generation, and descriptor calculation | Open-source Python library |
| - WHALES Implementation | Code Repository | Complete code for WHALES calculation and screening | github.com/ETHmodlab/scaffoldhoppingwhales [31] |
| ChEMBL Database | Chemical Database | Source of bioactive compounds for benchmarking and queries | Publicly available |
| - Murcko Scaffold Analysis | Algorithm | Quantifies scaffold diversity in screening results | Implemented in RDKit |
| Ziconotide | Ziconotide (Prialt) | Ziconotide is a synthetic ω-conotoxin for research into chronic pain. It is a selective, non-opioid N-type calcium channel blocker. For Research Use Only. Not for human use. | Bench Chemicals |
| Chloranil | Chloranil, CAS:118-75-2, MF:C6Cl4O2, MW:245.9 g/mol | Chemical Reagent | Bench Chemicals |
WHALES descriptors represent a significant advancement in molecular representation for scaffold hopping and natural product-inspired drug design. By simultaneously capturing molecular shape and charge distribution, WHALES enables efficient identification of structurally diverse compounds with similar biological activity, addressing a critical challenge in medicinal chemistry. The robust performance of WHALES across retrospective benchmarks and prospective applications highlights its value in hit identification and lead optimization workflows. As computational drug discovery continues to evolve, holistic representations like WHALES will play an increasingly important role in bridging the gap between complex natural product architectures and synthetically accessible therapeutic compounds.
Natural products (NPs) are invaluable resources for drug discovery, characterized by their intricate scaffolds and diverse bioactivities. However, their structural complexity often leads to undesirable properties such as toxicity, metabolic instability, or poor pharmacokinetic profiles [21]. Scaffold hopping has emerged as a critical strategy to overcome these limitations by designing molecules with novel core structures (scaffolds) that retain the desired biological activity of the original natural product [19]. This approach not only helps optimize drug-like properties but also facilitates the creation of novel chemical entities with freedom-to-operate advantages [24].
The rapid evolution of artificial intelligence (AI), particularly deep learning, has revolutionized the computational approach to scaffold hopping. Traditional methods relied on searching predefined databases using molecular fingerprints or pharmacophore models, inherently limiting exploration to known chemical space [28]. AI-driven methods, especially transformer networks and graph-based models, now enable generative exploration of vast chemical spaces, facilitating the discovery of novel scaffolds absent from existing libraries [19] [34]. These approaches have positioned AI-assisted drug design as a prominent research area, significantly accelerating early screening and lead compound identification [19].
Molecular Representation: The Foundation of AI-Driven Scaffold Hopping
A prerequisite for effective AI-driven scaffold hopping is translating molecular structures into computer-readable formats, known as molecular representations [19]. These representations bridge the gap between chemical structures and their biological, chemical, or physical properties [19].
Evolution of Molecular Representation Methods
Traditional molecular representations include:
- String-based encodings: Simplified Molecular-Input Line-Entry System (SMILES) and Self-Referencing Embedded Strings (SELFIES) provide compact string-based representations of molecular structures [19] [34].
- Molecular fingerprints: Extended-connectivity fingerprints (ECFPs) encode substructural information as binary strings or numerical values [19] [28].
- Molecular descriptors: Quantify physical or chemical properties of molecules, such as molecular weight, hydrophobicity, or topological indices [19].
- Holistic representations: Methods like WHALES (Weighted Holistic Atom Localization and Entity Shape) descriptors incorporate pharmacophore and shape patterns, capturing partial charge, atom distributions, and molecular shape simultaneously [24].
AI-driven molecular representations employ deep learning techniques to learn continuous, high-dimensional feature embeddings directly from large datasets [19]. These include:
- Language model-based approaches: Treat molecular sequences (e.g., SMILES) as chemical language [19].
- Graph-based representations: Model molecules as graphs with atoms as nodes and bonds as edges [19].
- Multimodal and contrastive learning frameworks: Integrate multiple representation types for enhanced learning [19].
Table 1: Comparison of Molecular Representation Methods for Scaffold Hopping
| Representation Type | Key Examples | Advantages | Limitations |
|---|---|---|---|
| String-Based | SMILES, SELFIES | Simple, compact, human-readable [19] | Limited in capturing molecular complexity [19] |
| Fingerprint-Based | ECFP, Morgan Fingerprints [28] | Computationally efficient, effective for similarity search [19] | Predefined features, may overlook subtle structural relationships [19] |
| 3D Holistic | WHALES [24] | Encodes shape and pharmacophore information | Requires 3D conformer generation |
| Graph-Based | Graph Neural Networks [19] | Naturally represents molecular structure | Requires specialized architectures |
| Language Model-Based | Molecular Transformers [19] | Leverages NLP advances, captures sequential patterns | SMILES syntax may not reflect structural similarity |
Transformer Networks in Scaffold Hopping
Transformer networks, originally developed for natural language processing, have been successfully adapted for molecular design tasks including scaffold hopping. Their self-attention mechanism enables modeling of long-range dependencies and complex structural relationships in molecular data [35].
DeepHop: A Multimodal Transformer Framework
The DeepHop model represents a significant advancement in scaffold hopping by reformulating the task as a supervised molecule-to-molecule translation problem [28]. This multimodal architecture integrates molecular 3D conformer information through a spatial graph neural network and protein sequence information through a transformer model [28].
Architecture and Training:
- DeepHop was trained on over 50,000 scaffold-hopping pairs curated from ChEMBL20, spanning 40 kinases [28].
- Training pairs were constructed with strict similarity conditions: 2D scaffold similarity (Tanimoto score on Morgan fingerprints of Bemis-Murcko scaffolds) ≤ 0.6, and 3D similarity (shape and color similarity score) ≥ 0.6 [28].
- The model incorporates a multi-task deep neural network (MTDNN) as a virtual profiling model to predict bioactivity of generated molecules [28].
Performance Metrics:
- DeepHop generates approximately 70% of molecules with improved bioactivity, high 3D similarity, but low 2D scaffold similarity to template molecules [28].
- This success rate is 1.9 times higher than other state-of-the-art deep learning methods and rule-based virtual screening approaches [28].
- The model demonstrates generalization capability to new target proteins through fine-tuning with small sets of active compounds [28].
Figure 1: DeepHop Multimodal Transformer Architecture for Scaffold Hopping
Experimental Protocol: Implementing DeepHop for Natural Product Optimization
Data Preparation:
- Input Requirements: Provide natural product query as SMILES string with associated target protein sequence [28].
- Conformer Generation: Generate 100 conformations for each molecule using RDKit MMFF94 force field [28].
- Similarity Calculation:
Model Application:
- Fine-tuning: For new target proteins, fine-tune pre-trained DeepHop model with small set (50-100) of active compounds [28].
- Generation: Generate candidate molecules with the conditioned transformer model.
- Validation: Assess generated molecules using virtual profiling model (MTDNN) to predict bioactivity improvement [28].
Graph-Based Models in Scaffold Hopping
Graph-based models, particularly Graph Neural Networks (GNNs), provide a natural representation of molecular structure by treating atoms as nodes and bonds as edges in a graph [19]. These models have demonstrated remarkable capability in capturing both local and global molecular features essential for effective scaffold hopping [36].
3D-Aware Graph Models for Structure-Based Scaffold Hopping
Recent advancements in graph-based approaches have incorporated 3D structural information to enhance scaffold hopping performance:
FRAME utilizes SE(3)-equivariant neural networks to explicitly model protein-ligand interactions (hydrogen bonds, π-π stacking) and dynamically select optimal connection points and fragments from starting molecules [21].
D3FG employs a diffusion model with rigid functional group definitions to preserve complex fragment structures during generation, capturing spatial relationships and interactions between proteins and ligands via graph neural networks [21].
STRIFE dynamically guides starting molecule expansion by extracting fragment hotspot maps (FHMs) from protein targets, using a combination of GNN and CNN architectures [21].
Experimental Protocol: Fragment-Based Scaffold Hopping with Graph Networks
Fragment Library Preparation:
- Source Compounds: Curate fragments from synthesis-validated sources like ChEMBL (e.g., 3+ million unique scaffolds) [37].
- Fragmentation: Apply HierS algorithm to systematically decompose molecules into ring systems, side chains, and linkers [37].
- Descriptor Calculation: Generate molecular fingerprints (ECFP) and 3D shape descriptors for similarity assessment [37].
Model Implementation:
- Graph Encoding: Represent molecules as graphs with atom features (element type, hybridization, valence) and bond features (bond type, conjugation) [21].
- 3D Integration: Incorporate spatial coordinates using SE(3)-equivariant networks to maintain geometric constraints [21].
- Generation: Employ reinforcement learning or generative flow networks (GFlowNets) to sequentially add fragments while optimizing binding affinity [21].
Validation:
- Virtual Screening: Assess generated molecules using docking simulations and molecular dynamics [21].
- Similarity Assessment: Calculate Tanimoto similarity (2D) and electron shape similarity (3D) to ensure novel scaffolds maintain pharmacophores [37].
Table 2: Performance Comparison of AI-Driven Scaffold Hopping Methods
| Method | Architecture | 2D Similarity Range | 3D Similarity Range | Success Rate | Key Applications |
|---|---|---|---|---|---|
| DeepHop [28] | Multimodal Transformer | ≤0.6 (Tanimoto) | ≥0.6 (SC Score) | 70% (Improved bioactivity) | Kinase inhibitors |
| WHALES [24] | Holistic Descriptors | Variable | High shape similarity | 35% (Experimental active) | Cannabinoid receptor modulators |
| ChemBounce [37] | Fragment Replacement | Tunable threshold | Electron shape similarity | Varies by target | General synthetic mimetics |
| FREED [21] | GCN + Reinforcement Learning | Not specified | Not specified | High docking scores | Target-informed design |
Figure 2: Graph-Based Fragment Replacement Workflow for Scaffold Hopping
Table 3: Research Reagent Solutions for Scaffold Hopping Implementation
| Resource Category | Specific Tools/Libraries | Function | Application Context |
|---|---|---|---|
| Molecular Representation | RDKit, ODDT (with ElectroShape) [37] | Chemical informatics and descriptor calculation | Preprocessing, similarity assessment |
| Fragment Libraries | ChEMBL-derived scaffolds (3.2M+ unique) [37], VEHICLe database [28] | Source of replaceable molecular fragments | Fragment-based scaffold replacement |
| Deep Learning Frameworks | PyTorch, TensorFlow, PyTorch Geometric [35] | Implementation of GNNs and transformers | Model development and training |
| Benchmarking Platforms | OpenGT [35], GuacaMol, MOSES [34] | Standardized evaluation of generative models | Performance validation |
| Property Prediction | MTDNN [28], DMPNN [28], FP-ADMET [19] | Virtual profiling of generated molecules | Bioactivity and ADMET assessment |
AI-driven scaffold hopping, particularly through transformer networks and graph-based models, has fundamentally transformed natural product-based drug design. These approaches enable systematic exploration of chemical space beyond the limitations of traditional database searching, facilitating the discovery of novel scaffolds with maintained bioactivity and improved drug-like properties [19] [34].
The integration of 3D structural information through spatial graph networks and the application of multimodal learning represent significant advancements in the field [21] [28]. Models like DeepHop demonstrate that combining molecular structure with target protein information yields substantially higher success rates in generating viable scaffold hops compared to traditional methods [28].
Future developments will likely focus on improving synthetic accessibility of AI-generated molecules, enhancing model interpretability, and addressing data scarcity for novel targets [34] [38]. As these computational methods continue to evolve, they will further accelerate the transformation of natural product inspiration into viable therapeutic candidates, bridging the gap between nature's chemical diversity and modern drug discovery needs.
Tuberculosis (TB), caused by the bacterial pathogen Mycobacterium tuberculosis (Mtb), remains a leading cause of death worldwide, with the emergence of multi-drug resistant (MDR) and extensively drug-resistant (XDR) TB posing significant challenges to global eradication efforts [39] [40]. The complexity of Mtb's physiology, including its lipid-rich cell envelope and extensive metabolic adaptation capabilities, necessitates novel treatment strategies and therapeutic targets [40]. Protein kinases (PKs), key regulators of cellular processes across all life forms, have emerged as major targets in anti-TB drug discovery due to their essential roles in Mtb survival, metabolism, and pathogenesis [39] [40].
The Mtb genome encodes approximately 60 different protein kinases, including 11 two-component regulatory systems (2CRS) and 11 serine/threonine protein kinases (STPKs), along with numerous metabolic kinases essential for fundamental biosynthetic pathways [40]. This application note presents practical case studies and detailed protocols for targeting Mtb kinases, with a specific focus on scaffold hopping strategies to discover novel chemotypes with improved properties.
Case Study 1: Targeting Thymidylate Kinase (TMPKmt) in Mtb
Target Validation and Biological Significance
Thymidylate kinase of Mtb (TMPKmt) catalyzes the phosphorylation of dTMP to dTDP in the pyrimidine biosynthesis pathway, an essential step in DNA synthesis [39] [40]. TMPKmt is mechanistically and structurally unrelated to the human enzyme, making it an attractive target for developing selective antitubercular agents with minimal host toxicity [40]. Gene essentiality studies have confirmed that TMPKmt is indispensable for mycobacterial survival, further validating its therapeutic potential [39].
Scaffold Hopping Strategies for TMPKmt Inhibitors
Scaffold hopping from known natural product inhibitors of TMPKmt has yielded promising synthetic analogs with retained activity and improved synthetic accessibility. The application of holistic molecular similarity approaches, such as Weighted Holistic Atom Localization and Entity Shape (WHALES) descriptors, has enabled successful scaffold hopping by simultaneously capturing pharmacophore features, partial charge distributions, and molecular shape [24]. These descriptors facilitate the identification of isofunctional synthetic compounds that maintain key interaction patterns while reducing structural complexity compared to natural product templates [24].
Table 1: Classification of Scaffold Hopping Approaches for Kinase Inhibitor Design
| Hop Category | Structural Change | Degree of Novelty | Example Application |
|---|---|---|---|
| 1° Hop | Heteroatom replacements or swaps in backbone rings | Low | Carbon-nitrogen swaps in aromatic systems [9] |
| 2° Hop | Ring opening or closure | Medium | Morphine to tramadol transformation [9] |
| 3° Hop | Peptidomimetics | Medium-High | Replacement of peptide backbones with non-peptidic moieties [9] |
| 4° Hop | Topology-based changes | High | Field-based scaffold hopping using electrostatic similarity [12] |
Case Study 2: Host-Directed Therapy Using Kinase Inhibitors
Screening Kinase Inhibitor Libraries for Host-Directed Therapies
Host-directed therapy (HDT) represents a promising alternative strategy for combating intracellular bacteria like Mtb by targeting host cellular mechanisms that support bacterial survival [41]. A recent high-throughput screen of 827 ATP-competitive kinase inhibitors from the Published Kinase Inhibitor Sets (PKIS1 and PKIS2) identified multiple compounds effective against intracellular Mtb in human cell lines and primary macrophages [41].
The screening workflow employed flow cytometry-based infection models using HeLa and MelJuSo cell lines infected with DsRed-expressing Mtb. Two distinct populations of infected cells were observed: DsRed-dim (containing few bacteria) and DsRed-bright (containing replicating bacteria, with 142 times more viable bacteria than dim cells) [41]. Compounds reducing the DsRed-bright population without host cell cytotoxicity were prioritized for further development.
Experimental Protocol: Kinase Inhibitor Screening for Anti-Mtb Activity
Protocol 1: Primary Screening of Kinase Inhibitor Libraries
- Cell preparation: Seed HeLa or MelJuSo cells in 96-well plates at 2×10ⴠcells/well and incubate for 24h at 37°C, 5% CO₂
- Infection: Infect cells with DsRed-expressing Mtb at MOI 10:1 for 4h, then remove extracellular bacteria with gentamicin (10μg/mL) treatment
- Compound treatment: Add kinase inhibitors at 10μM concentration in duplicate, include H-89 (PKA/AKT inhibitor) as positive control and DMSO as negative control
- Incubation: Maintain cells for 48h post-infection
- Analysis: Harvest cells, fix with 4% paraformaldehyde, and analyze by flow cytometry measuring DsRed fluorescence and cell counts
- Hit selection: Calculate z-scores, select compounds with z-score < -2 for DsRed-bright population and z-score > -3 for cell count (indicating no cytotoxicity) [41]
Table 2: Promising Kinase Inhibitor Chemotypes Identified as HDT Candidates Against Mtb
| Compound Chemotype | Primary Kinase Targets | Mtb Inhibition (Rescreen Z-score) | Cytotoxicity | Therapeutic Potential |
|---|---|---|---|---|
| Morpholino-imidazo/triazolo-pyrimidinones | PIK3CB | < -2 | Non-cytotoxic | PI3K/AKT pathway modulation [41] |
| 2-Aminobenzimidazoles | ABL1 | < -2 | Non-cytotoxic | Lysosomal acidification restoration [41] |
| 2-Anilino-4-pyrrolidinopyrimidines | JAK2, AAK1 | < -2 | Non-cytotoxic | Intracellular trafficking modulation [41] |
| 4-Anilinoquinolines | MAP2K5, RIPK2, RSK4 | < -2 | Non-cytotoxic | Multiple signaling pathway disruption [41] |
Case Study 3: Target-Based Discovery of Shikimate Kinase Inhibitors
Shikimate Kinase as a Selective Anti-TB Target
Shikimate kinase (SK), the fifth enzyme in the shikimate pathway, catalyzes the ATP-dependent phosphorylation of shikimate to shikimate-3-phosphate [42]. This pathway is essential in bacteria, fungi, and plants but absent in mammals, making it an attractive target for selective antimicrobial development [42]. Mtb SK (MtSK) is encoded by the aroK gene and has been validated as essential for bacterial growth through gene knockout studies [42].
Experimental Protocol: LC-MS-Based Functional Screening for MtSK Inhibitors
Protocol 2: LC-MS Screening for Shikimate Kinase Inhibitors
- Enzyme preparation: Express and purify recombinant MtSK using E. coli expression system
- Compound library: Select 404 compounds from NIH TAACF phenotypic screens with known anti-Mtb activity (IC₉₀ <5 μg/mL against H37Rv)
- Reaction conditions: In 50μL total volume, combine 50mM Tris-HCl (pH 7.5), 10mM MgCl₂, 2.5mM ATP, 0.5mM shikimic acid, test compound (10μM), and MtSK (0.5μg)
- Incubation: Conduct reactions at 37°C for 30min, terminate by heating at 95°C for 5min
- LC-MS analysis:
- Chromatography: C18 column (150×2.1mm, 3.5μm), mobile phase 10mM ammonium acetate (pH 5.5) and acetonitrile gradient
- Mass detection: Negative ion mode, MRM transition m/z 233→97 for shikimate-3-phosphate (S3P)
- Quantification: Measure S3P formation in test samples compared to no-inhibitor controls
- Hit validation: Determine ICâ‚…â‚€ values for compounds showing >50% inhibition in primary screen [42]
This target-based approach identified 14 novel MtSK inhibitors with IC₉₀ values <5 μg/mL against Mtb H37Rv and favorable selectivity indices (SI >10), demonstrating the utility of combining phenotypic screening with target-based validation [42].
Computational Approaches for Scaffold Hopping in Kinase Inhibitor Design
WHALES Descriptors for Natural Product-Inspired Scaffold Hopping
The WHALES (Weighted Holistic Atom Localization and Entity Shape) molecular representation enables scaffold hopping from complex natural products to synthetically accessible mimetics by simultaneously encoding pharmacophore features, atomic distributions, and molecular shape [24]. The descriptor calculation involves four key steps:
- Atom-centered covariance matrix calculation: Computes weighted covariance around each atom using partial charges as weights
- Atom-centered Mahalanobis distance calculation: Determines normalized interatomic distances based on local feature distributions
- Atomic indices calculation: Derives remoteness, isolation degree, and their ratio for each atom
- WHALES descriptor generation: Applies binning procedure to obtain fixed-length representation (33 descriptors) enabling comparison of diverse molecules [24]
In prospective applications using natural cannabinoids as queries, this approach achieved 35% success rate in identifying novel synthetic modulators of human cannabinoid receptors, with identified modulators being structurally less complex than their natural product templates [24].
Field-Based Scaffold Hopping for Kinase Inhibitors
Field-based molecular similarity methods provide another powerful approach for scaffold hopping in kinase inhibitor design. These methods compare molecules based on their electrostatic, steric, and hydrophobic fields rather than structural frameworks, enabling identification of diverse chemotypes with conserved interaction potential [12]. Successful applications include:
- Peptide to small molecule hops: Transforming therapeutically interesting peptide analogs into non-peptide synthetic mimetics with maintained field characteristics
- Whole molecule replacement: Using software such as Blaze to search commercial compound collections for suitable replacements
- Fragment replacement: Employing tools like Spark to systematically replace molecular components while maintaining key interaction features [12]
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Kinase-Focused Anti-TB Drug Discovery
| Reagent/Material | Specifications | Experimental Function | Application Examples |
|---|---|---|---|
| PKIS Libraries | PKIS1 & PKIS2 (827 ATP-competitive kinase inhibitors) | Source of chemically diverse, well-characterized kinase inhibitors with known target profiles | Host-directed therapy screening against intracellular Mtb [41] |
| DsRed-Expressing Mtb | Recombinant Mtb H37Rv expressing DsRed fluorescent protein | Enables quantification of intracellular bacterial burden via flow cytometry | High-throughput screening in host cell infection models [41] |
| Recombinant Kinases | Purified Mtb kinases (TMPKmt, MtSK) expressed in E. coli | Target proteins for biochemical inhibition assays | Enzyme activity assays, inhibitor characterization [39] [42] |
| LC-MS System | HPLC coupled to mass spectrometer with C18 column | Quantification of reaction substrates and products in enzyme assays | Shikimate-3-phosphate detection in MtSK inhibition assays [42] |
| 2-Undecanone | 2-Undecanone, CAS:112-12-9, MF:C11H22O, MW:170.29 g/mol | Chemical Reagent | Bench Chemicals |
Kinase inhibitors represent promising candidates for direct antitubercular activity and host-directed therapy approaches. The case studies and protocols presented herein demonstrate practical applications of kinase-focused drug discovery, highlighting the utility of scaffold hopping strategies to generate novel chemotypes with maintained activity against Mtb targets. Integration of computational design methods with robust experimental screening protocols provides a powerful framework for advancing new therapeutic candidates against drug-resistant tuberculosis.
Overcoming Challenges in Natural Product-Based Scaffold Hopping
The pursuit of novel therapeutic agents requires researchers to navigate a fundamental tension: the need for structural innovation against the imperative to maintain biological activity. This challenge is particularly acute in natural product-based drug design, where the complex scaffolds of natural products offer rich starting points for drug discovery but often require optimization to improve drug-like properties [24]. Scaffold hopping, defined as the strategic modification of a molecule's core structure to generate novel chemotypes while preserving bioactivity, has emerged as a powerful methodology to address this challenge [9] [18].
The similarity property principle posits that structurally similar molecules tend to exhibit similar biological activities [9]. However, scaffold hopping operates at the boundaries of this principle, demonstrating that significant structural changes can retain or even enhance desired activity when key pharmacophore elements are conserved [24]. This application note provides a structured framework for researchers to balance structural novelty with biological activity through scaffold hopping approaches, with particular emphasis on natural product-inspired design.
Classification of Scaffold Hopping Approaches
Scaffold hopping strategies can be systematically categorized into distinct classes based on the degree and nature of structural modification [9] [18]. Understanding this classification helps researchers select appropriate strategies for their specific design goals. The following table summarizes the four primary scaffold hopping categories:
Table 1: Classification of Scaffold Hopping Approaches
| Hop Category | Structural Change | Novelty Level | Success Rate | Primary Application |
|---|---|---|---|---|
| 1° Heterocycle Replacements | Swapping atoms in aromatic rings or replacing carbon with heteroatoms | Low | High | Lead optimization, patent protection |
| 2° Ring Opening/Closure | Breaking or forming ring systems to control molecular flexibility | Medium | Medium | Modifying pharmacodynamic and pharmacokinetic properties |
| 3° Peptidomimetics | Replacing peptide backbones with non-peptidic moieties | Medium-High | Medium | Converting bioactive peptides into drug-like molecules |
| 4° Topology-Based Hopping | Identifying chemotypes with similar shape and pharmacophores but different atomic connectivity | High | Low | Discovering truly novel chemotypes from natural products |
The trade-off between structural novelty and success rate is evident across these categories [9]. Small-step hops (1° and some 2°) generally offer higher probabilities of maintaining bioactivity but yield more modest structural innovations. Large-step hops (4° and some 3°), while offering greater novelty, present higher risks of activity loss [18]. This relationship must be carefully considered when planning scaffold hopping campaigns.
Quantitative Framework for Assessing the Trade-off
The strategic implementation of scaffold hopping requires quantitative assessment of both structural novelty and biological activity. The following experimental workflow establishes a standardized approach for evaluating this balance:
Molecular Descriptors for Quantitative Analysis
Different molecular descriptors capture complementary aspects of structural similarity and novelty [24]:
WHALES (Weighted Holistic Atom Localization and Entity Shape) descriptors: Encode information on geometric interatomic distances, molecular shape, and atomic properties in a holistic way. These descriptors are particularly effective for scaffold hopping from natural products to synthetic mimetics [24].
ECFP (Extended-Connectivity Fingerprints): Fragment-based representations that capture local atomic environments. While intuitive and widely used, they may be less effective for detecting similarities between structurally diverse natural products and synthetic compounds [24].
Pharmacophore descriptors: Focus on the spatial arrangement of functional groups essential for biological activity.
Table 2: Quantitative Data Analysis in Natural Product Scaffold Hopping
| Analysis Type | Statistical Methods | Application Example | Key Outcome Metrics |
|---|---|---|---|
| Dose-Response Analysis | ANOVA, Regression analysis | Natural product trials in rat models for neuroinflammation and memory deficits [43] | IC50, EC50, Hill coefficient |
| Correlation Analysis | Pearson/Spearman correlation | Assessing relationship between compound concentration and inflammatory marker levels [43] | Correlation coefficient (r), p-value |
| Longitudinal Analysis | Repeated measures ANOVA, mixed models | Monitoring disease progression in chronic disease models [43] | Slope of progression, treatment effect over time |
| Survival Analysis | Kaplan-Meier curves, log-rank test | Evaluating anti-cancer properties in xenograft models [43] | Hazard ratio, median survival time |
| Multivariate Analysis | PCA, PLS-DA | Accounting for age, sex, and housing conditions in in vivo tests [43] | Variable importance, clustering patterns |
Experimental Protocols for Scaffold Hopping
Protocol 1: WHALES Descriptor Calculation for Natural Product Scaffold Hopping
Purpose: To compute holistic molecular descriptors that facilitate scaffold hopping from natural products to synthetic mimetics [24].
Materials:
- Energy-minimized 3D molecular structures (MMFF94 or similar force field)
- Partial atomic charges (Gasteiger-Marsili or quantum-chemistry derived)
- Computational chemistry software (e.g., Molecular Operating Environment)
Procedure:
- Generate 3D Conformations: Obtain energy-minimized 3D structures for all query natural products and database compounds.
- Calculate Partial Charges: Compute partial atomic charges using the chosen method.
- Compute Atom-Centered Covariance Matrix: For each non-hydrogen atom j, calculate the weighted covariance matrix using the formula:
Sw(j) = Σ[|δi| · (xi - xj)(xi - xj)T] / Σ|δi|where δi is the partial charge of atom i, and xi represents 3D coordinates [24]. - Calculate Atom-Centered Mahalanobis (ACM) Distances:
ACM(i,j) = (xi - xj)T · Sw(j)-1 · (xi - xj) - Derive Atomic Indices: Compute remoteness (row-average of ACM matrix), isolation degree (column minimum), and isolation-remoteness ratio for each atom.
- Generate WHALES Descriptors: Apply binning procedure to obtain fixed-length representation (11 values for each atomic index, total 33 descriptors).
Validation: In prospective application, this approach achieved 35% success rate in identifying novel synthetic modulators of human cannabinoid receptors using natural cannabinoids as queries [24].
Protocol 2: Biological Validation of Scaffold-Hopped Compounds
Purpose: To experimentally validate the biological activity of scaffold-hopped compounds derived from natural products.
Materials:
- Scaffold-hopped compounds and natural product reference
- Relevant cell lines or animal models
- Assay reagents specific to target pathway
- HPLC system for compound quantification (for nanocarrier delivery studies) [43]
Procedure:
- In Vitro Binding Assays:
- Conduct competitive binding assays with target receptor
- Determine IC50 values using non-linear regression
- Compare potency relative to natural product lead
Functional Activity Assays:
- Measure efficacy (Emax) and potency (EC50) in cell-based functional assays
- Assess selectivity against related targets
- Perform dose-response analysis with appropriate statistical testing (ANOVA with post-hoc tests) [43]
In Vivo Efficacy Studies:
- Administer compounds in relevant disease models (e.g., neuroinflammation, xenograft)
- Include multiple dose levels to establish dose-response relationship
- Use appropriate sample sizes and randomization procedures
- Apply longitudinal analysis for chronic studies [43]
ADMET Profiling:
- Assess metabolic stability in liver microsomes
- Determine permeability in Caco-2 or MDCK cell models
- Evaluate cytotoxicity in relevant cell lines
Case Studies in Natural Product Scaffold Hopping
Morphine to Tramadol: Ring Opening Strategy
The transformation from morphine to tramadol represents a classic example of ring opening scaffold hopping [9] [18]. Morphine's rigid 'T'-shaped structure with multiple fused rings was modified by breaking six ring bonds, resulting in tramadol's more flexible structure. Despite significantly different 2D structures, 3D superposition conserves key pharmacophore features: the positively charged tertiary amine, aromatic ring, and hydroxyl group. This scaffold hop reduced potency but improved oral bioavailability and created a safer analgesic profile with reduced addiction potential [9].
Natural Cannabinoids to Synthetic Mimetics: Topology-Based Hopping
A prospective study demonstrated successful scaffold hopping from natural cannabinoids to synthetic modulators using WHALES descriptors [24]. This holistic molecular representation captured pharmacophore and shape patterns, enabling identification of synthetic compounds with low-micromolar potency at human cannabinoid receptors CB1 and CB2. Of the selected compounds, 35% were experimentally confirmed as active, with five representing novel scaffolds not found in existing cannabinoid ligand databases [24].
Antihistamine Development: Ring Closure and Heterocycle Replacement
The evolution of antihistamines illustrates multiple scaffold hopping strategies [9] [18]:
- Ring closure transformed pheniramine into cyproheptadine, locking aromatic rings in active conformations and improving binding affinity.
- Heterocycle replacement in cyproheptadine produced pizotifen (phenyl to thiophene) and azatadine (phenyl to pyrimidine), the latter improving solubility. These modifications demonstrate how small structural changes can yield different activity profiles and medical uses while reducing molecular flexibility to potentially decrease entropy loss upon target binding [9].
Research Reagent Solutions
Table 3: Essential Research Reagents for Scaffold Hopping Studies
| Reagent/Category | Function/Application | Example Use Cases |
|---|---|---|
| WHALES Descriptors | Holistic molecular representation for scaffold hopping | Identifying synthetic mimetics of natural products [24] |
| Molecular Operating Environment (MOE) | Flexible Alignment program for 3D molecular superposition | Pharmacophore comparison between morphine and tramadol [9] |
| Liposomal Nanocarriers | Improve bioavailability of natural compounds | Delivery of antiviral natural compounds in in vivo models [43] |
| High-Throughput Screening Assays | Rapid activity assessment of compound libraries | Identifying active compounds from large collections [44] |
| qPCR Reagents | Quantify gene expression changes in disease models | Measuring inflammation-related gene expression in rat models [43] |
| MMFF94 Force Field | Energy minimization for 3D structure preparation | Conformational analysis for WHALES descriptor calculation [24] |
Successfully navigating the trade-off between structural novelty and biological activity requires a methodical approach that integrates computational design with experimental validation. The scaffold hopping classification system provides a strategic framework for selecting appropriate modification strategies based on project goals. Natural products continue to offer rich structural templates for drug discovery, with computational methods like WHALES descriptors enabling more effective translation of their complex architectures into synthetically accessible compounds with improved drug-like properties.
The experimental protocols and case studies presented herein demonstrate that strategic structural modifications, when guided by pharmacophore conservation principles and validated through rigorous biological testing, can yield novel chemotypes with maintained or improved therapeutic potential. As drug discovery faces increasing challenges, these scaffold hopping approaches will remain essential tools for expanding the structural diversity of chemical probes and therapeutic agents.
Addressing Complexity and Synthetic Accessibility of Natural Product Scaffolds
Natural products (NPs) are invaluable resources for drug discovery, characterized by their intricate scaffolds and diverse bioactivities [21]. However, their clinical application often faces challenges due to inherent complexities, including unfavorable ADMET properties, violation of Lipinski's rule of five, and low oral bioavailability [21]. Structural modification of NPs through scaffold hopping has emerged as a critical strategy to overcome these limitations while preserving biological activity [37]. This Application Note provides detailed protocols for addressing NP complexity and synthetic accessibility through computational scaffold hopping, enabling researchers to generate novel, patentable drug candidates with improved drug-like properties.
Quantitative Analysis of Natural Product Complexity
Molecular Complexity Metrics
Table 1 summarizes key molecular properties that contribute to the complexity of natural products and their implications for drug discovery.
Table 1: Molecular Complexity Metrics of Natural Products and Implications for Drug Discovery
| Molecular Property | Typical NP Profile | Synthetic Compound Profile | Impact on Drug Discovery |
|---|---|---|---|
| sp³-hybridized carbon atoms | Higher proportion | Lower proportion | Increased 3D structural complexity, improved target selectivity [2] |
| Oxygen content | Increased | Decreased | Enhanced hydrogen bonding capacity, improved solubility [2] |
| Nitrogen/halogen content | Decreased | Increased | Reduced toxicity potential [2] |
| Molecular rigidity | Increased | Variable | Improved binding affinity, pre-organized bioactive conformation [2] |
| Lipophilicity (cLogP) | Lower | Variable | Improved solubility, reduced metabolic clearance [2] |
| Molecular weight | Often >500 Da | Typically <500 Da | Potential challenges with oral bioavailability despite successful examples [21] |
| Structural complexity | High (multiple stereocenters, macrocycles) | Moderate to low | Synthetic challenges, requires specialized strategies [21] |
Performance Benchmarking of Scaffold Hopping Tools
Table 2 compares the performance of various computational tools for scaffold hopping, highlighting their advantages in generating synthetically accessible compounds.
Table 2: Performance Comparison of Scaffold Hopping Tools for Natural Product Optimization
| Tool/Platform | Methodology | Scaffold Library Size | Key Advantages for NP Optimization | Synthetic Accessibility Metrics |
|---|---|---|---|---|
| ChemBounce [37] | Fragment replacement, shape similarity | 3.2 million (ChEMBL-derived) | Open-source, high synthetic accessibility, ElectroShape similarity | Lower SAscore, higher QED values compared to commercial tools |
| FREED/FREED++ [21] | Reinforcement learning + GCN | Predefined fragments | Target-aware generation, explores diverse chemical space | Pharmacochemically acceptable molecules with high docking scores |
| D3FG [21] | Diffusion + SE(3)-equivariant GNN | Rigid functional groups | Preserves complex fragment structures, captures protein-ligand interactions | Maintains structural integrity of complex NP fragments |
| TACOGFN [21] | GFlowNet + graph transformer | 72 fragment types | Target pocket information integration, gradual fragment addition | Controlled growth ensures synthetic feasibility |
| DeepFrag [21] | 3D DCNN classification | Predefined fragments | Considers receptor pocket and full ligand context | Fragment-based approach ensures chemical authenticity |
Experimental Protocols
Protocol 1: Scaffold Hopping with ChemBounce
Materials and Requirements
- Input: Valid SMILES string of natural product (pre-processed to remove salts and validate structure)
- Software: ChemBounce (available at https://github.com/jyryu3161/chembounce [37])
- Computational Environment: Python 3.7+ or Google Colaboratory notebook [37]
- Library: Default ChEMBL-derived scaffold library (3,231,556 unique scaffolds) or custom library [37]
Step-by-Step Procedure
Input Preparation and Validation
- Convert NP structure to canonical SMILES format
- Validate SMILES syntax, atomic symbols, and valence rules
- Remove counterions, solvents, and multiple components separated by "."
- Pre-process complex stereochemistry representations
Scaffold Identification
Scaffold Replacement and Filtering
Output Analysis
- Review generated structures in output directory
- Assess synthetic accessibility scores (SAscore) and drug-likeness (QED)
- Select top candidates for further evaluation
Workflow Visualization
Protocol 2: Target-Aware Scaffold Optimization
Materials and Requirements
- Input: NP structure with known target protein (PDB structure or homology model)
- Software: Target-interaction-driven models (DeepFrag, FREED++, D3FG) [21]
- Libraries: Predefined fragment libraries specific to tool selection
- Docking Software: AutoDock Vina, Glide, or similar for validation
Step-by-Step Procedure
Protein-Ligand Complex Preparation
- Obtain 3D structure of NP bound to target (experimental or docked)
- Prepare protein structure: add hydrogens, assign charges, optimize side chains
- Define binding site coordinates based on NP binding mode
Fragment-Based Optimization
Interaction-Preserving Generation
- Maintain critical hydrogen bonds, hydrophobic contacts, and π-π stacking
- Optimize new interactions with subpockets and peripheral regions
- Preserve key pharmacophore features essential for biological activity
Multi-parameter Optimization
- Balance binding affinity with drug-like properties
- Apply synthetic accessibility constraints throughout generation
- Prioritize compounds with favorable QED and SAscore profiles
Target-Driven Optimization Visualization
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for NP Scaffold Optimization
| Tool/Reagent | Type | Function in NP Scaffold Optimization | Example Sources/Platforms |
|---|---|---|---|
| ChEMBL-derived Fragment Library | Chemical Library | 3.2 million synthesis-validated scaffolds for replacement [37] | Curated from ChEMBL database [37] |
| COCONUT | NP Database | Over 400,000 non-redundant natural products for reference and inspiration [45] | Zenodo (open access) [45] |
| ScaffoldGraph | Software Library | Implements HierS algorithm for systematic scaffold decomposition [37] | Python package [37] |
| Open Drug Discovery Toolkit (ODDT) | Software Library | Provides ElectroShape implementation for shape similarity calculations [37] | Python library [37] |
| Predefined Fragment Sets | Chemical Building Blocks | 72+ fragment types for target-aware structure generation [21] | Various commercial and open sources [21] |
| SE(3)-Equivariant Neural Networks | Algorithmic Framework | Captures 3D spatial relationships in protein-ligand complexes [21] | D3FG, FRAME, other structure-based tools [21] |
| Reinforcement Learning Frameworks | Algorithmic Framework | Explores chemical space while optimizing multiple objectives [21] | FREED, FREED++ implementations [21] |
Implementation Considerations
Handling Complex NP Scaffolds
When working with structurally complex natural products such as macrocycles, high molecular weight peptides, or compounds with multiple stereocenters, specific considerations apply:
- Performance Scaling: Processing times vary from seconds for simple compounds to 21 minutes for complex structures exceeding 4000 Da [37]
- Stereochemistry Preservation: Approximately 12% of collected NP structures lack stereochemistry information despite having stereocenters – manual validation recommended [45]
- Structural Simplification: For NPs violating multiple drug-like property rules, consider structure simplification approaches while preserving key pharmacophores [21]
Customization for Specific NP Classes
The protocols can be customized for different natural product classes:
- Peptides and Macrocyclic Compounds: Utilize shape-based similarity constraints with increased weight on 3D conformation [37]
- Polyketides and Terpenoids: Focus on scaffold hopping with rigid functional group preservation [21]
- Alkaloids and Aromatics: Prioritize Tanimoto similarity for planar structure conservation [37]
The integration of computational scaffold hopping approaches with natural product research provides a powerful framework for addressing the inherent complexity and synthetic challenges of NPs. The protocols outlined in this Application Note enable systematic exploration of chemical space while maintaining biological relevance and ensuring synthetic feasibility. By leveraging these methodologies, researchers can accelerate the transformation of complex natural products into viable drug candidates with optimized properties, bridging the gap between nature's chemical diversity and modern pharmaceutical requirements.
Optimizing Pharmacokinetics and Reducing Toxicity through Core Modifications
Natural products (NPs) and their derivatives represent a cornerstone of drug discovery, particularly in therapeutic areas such as oncology and anti-infectives, accounting for approximately 30% of FDA-approved drugs over recent decades [21] [46]. However, their inherent structural complexity often leads to suboptimal pharmacokinetic (PK) profiles and toxicity, which present significant barriers to clinical application [21] [46]. Unmodified NPs frequently exhibit unfavorable ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties, including poor oral bioavailability, low specificity, and high toxicity [21]. Consequently, strategic structural modification of the core scaffold is not merely beneficial but essential to transform these naturally occurring compounds into viable therapeutic agents.
The process of "scaffold hopping" – the purposeful alteration of a molecule's core structure – has emerged as a powerful strategy to overcome these limitations while preserving or enhancing desired pharmacodynamic effects [46] [15]. This approach allows medicinal chemists to navigate the complex chemical space of natural products, systematically improving drug-like characteristics. The ultimate goal is to achieve an optimal balance within the "P3 properties model", which integrates Pharmacodynamic (efficacy), Physicochemical, and Pharmacokinetic/ADMET profiles through deliberate molecular design [46]. This Application Note provides detailed protocols and frameworks for employing core modifications to optimize PK and reduce toxicity within the context of modern natural product-based drug design.
Core Principles: Pharmacokinetic and Toxicity Challenges
Physiological Alterations Influencing Pharmacokinetics
Critically ill patients, such as those with sepsis or cancer, undergo significant physiological changes that profoundly impact drug PK. Understanding these alterations is critical for effective drug design and dosing. The major factors are summarized below [47]:
- Volume of Distribution (Vd): Aggressive fluid resuscitation and third-spacing of fluids in critically ill patients increase the Vd for hydrophilic antimicrobials (e.g., aminoglycosides, β-lactams, glycopeptides), leading to decreased plasma concentrations with standard dosing.
- Protein Binding: Hypoalbuminemia is common and can increase the Vd and elimination of unbound, acidic, highly protein-bound drugs (e.g., ceftriaxone, ertapenem). Conversely, increased α1-acid glycoprotein can bind basic drugs and decrease free concentrations.
- Organ Dysfunction: Hepatic hypoperfusion ("shock liver") alters drug metabolism, while acute kidney injury (AKI) reduces the clearance of hydrophilic drugs. Conversely, some patients may experience Augmented Renal Clearance (ARC), leading to subtherapeutic drug levels.
- Timely Administration: Early and appropriate administration of therapeutics is critical. In sepsis, for example, each hour delay in antimicrobial administration is associated with an average 7.6% decrease in survival [47].
Common Toxicity Mechanisms in Natural Products
Natural products often contain structural motifs that, while biologically active, can cause off-target toxic effects. Key strategies to mitigate these include [46]:
- Eliminating Toxicophores: Identifying and modifying functional groups responsible for non-specific binding and toxicity. For example, the α,β-unsaturated carbonyl motif in Apratoxin A undergoes irreversible Michael addition with cellular nucleophiles, causing in vivo toxicity. Saturation of this double bond in Apratoxin S4 reduced this nonspecific binding and improved the therapeutic index [46].
- Modifying Basic Side Chains: Aliphatic basic side chains can impart off-target cytotoxicity and cardiotoxicity. Their modulation, as demonstrated with indoloquinoline alkaloids, can significantly reduce these adverse effects while maintaining target potency [46].
The following tables consolidate key quantitative relationships and structural modifications that impact pharmacokinetics and toxicity.
Table 1: Impact of Physiological Alterations on Pharmacokinetic Parameters of Selected Drug Classes [47]
| Physiological Alteration | Impacted PK Parameter | Effect on Hydrophilic Drugs (e.g., β-lactams, Aminoglycosides) | Effect on Lipophilic Drugs (e.g., Azithromycin, Tigecycline) |
|---|---|---|---|
| Fluid Overload / Third-Spacing | Volume of Distribution (Vd) | Significantly Increased | Minimal Change |
| Hypoalbuminemia | Free Drug Concentration (Unbound Vd) | Increased for acidic, highly protein-bound drugs (e.g., Ceftriaxone) | Variable |
| Augmented Renal Clearance (ARC) | Clearance (CL) | Significantly Increased | Minimal Change |
| Acute Kidney Injury (AKI) | Clearance (CL) | Significantly Decreased | Minimal Change |
| Hepatic Dysfunction | Clearance (CL) | Minimal Change (for renally cleared) | Significantly Decreased |
Table 2: Representative Structural Modifications and Their Effects on Efficacy and Toxicity [46] [48]
| Natural Product / Compound | Core Modification | Impact on Efficacy | Impact on Toxicity / PK |
|---|---|---|---|
| Apratoxin A | Saturation of α,β-unsaturated carbonyl (to Apratoxin S4) | Retained potent cytotoxic activity | Reduced in vivo toxicity; eliminated nonspecific Michael addition [46] |
| Podophyllotoxin | Introduction of N-heterocyclic rings (e.g., β-Carboline) | Converted to Topo II inhibitor; retained anticancer activity | Reduced toxicity compared to original tubulin inhibitor [46] |
| Platinum Nanodrug (DACHPt/HANP) | Nanoformulation + Intralipid pre-treatment | Improved tumor targeting via EPR effect | Reduced liver/spleen/kidney accumulation; reduced organ toxicity [48] |
| Tanshinone I | Hybridization with piperidine scaffold | Enhanced anti-inflammatory activity (NLRP3 inhibition) | Improved drug-like properties [46] |
Experimental Protocols
Protocol 4.1: AI-Guided Functional Group Modification for a Known Target
This protocol uses target-interaction-driven molecular generative models to optimize a natural product lead when the protein target is known [21] [49].
I. Research Reagent Solutions
- Target Protein: Purified recombinant protein of interest (e.g., Topoisomerase IIα).
- Ligand Structure: 3D molecular structure of the natural product lead (e.g., in SDF or MOL2 format).
- Structural Data: High-resolution crystal structure of the target protein or a reliable homology model (e.g., PDB file).
- Software Tools:
- DeepFrag: An open-source model that uses a 3D Deep Convolutional Neural Network (3D DCNN) to suggest optimal fragment replacements by treating generation as a classification task [21] [49].
- Molecular Docking Software (e.g., AutoDock Vina, Glide) for validating proposed modifications.
- CHEMBL or PubChem: Databases for accessing bioactivity data of similar compounds.
II. Step-by-Step Methodology
- Data Preparation: Prepare the protein-ligand complex file. Remove the specific fragment from the lead compound that is targeted for modification within the binding pocket.
- Model Query: Input the modified complex into DeepFrag. The model will analyze the created void and the chemical environment of the pocket to propose a ranked list of optimal fragments from its library to replace the removed one.
- In-silico Validation: Dock the top-generated molecules (the lead with suggested fragments) back into the target's binding site. Analyze key interactions, such as hydrogen bonds, hydrophobic contacts, and pi-stacking.
- Potency Prediction: Use integrated scoring functions or external QSAR models to predict the binding affinity (e.g., pIC50) of the modified molecules.
- ADMET Screening: Employ in-silico ADMET prediction tools (e.g., SwissADME, pkCSM) on the top candidates to filter out those with predicted poor pharmacokinetics or high toxicity.
- Output: A curated list of synthesized and experimentally tested candidate molecules for further validation.
Protocol 4.2: Enzyme-Enabled Scaffold Hopping for Terpenoid Diversification
This protocol describes a hybrid enzymatic-chemical approach for scaffold hopping in terpenoid natural products, moving beyond traditional total synthesis [14].
I. Research Reagent Solutions
- Starting Material: Commercially available terpenoid scaffold (e.g., Sclareolide).
- Enzymes: Engineered Cytochrome P450 enzymes (e.g., from Bacillus megaterium) expressed and purified.
- Chemical Reagents: Standard organic synthesis reagents and catalysts for downstream transformations (e.g., Grignard reagents, oxidants, reducing agents).
- Analytical Equipment: HPLC-MS, NMR (¹H, ¹³C) for structural characterization.
II. Step-by-Step Methodology
- Enzymatic Oxidation:
- Set up a reaction containing the terpenoid scaffold (e.g., Sclareolide), the engineered cytochrome P450 enzyme, and necessary cofactors (NADPH, etc.) in an appropriate buffer.
- Incubate with shaking to achieve site-selective oxidation (e.g., at the C3 position of sclareolide). Monitor reaction completion by TLC or LC-MS.
- Purify the oxidized intermediate (e.g., alcohol) using flash chromatography.
- Scaffold Diversification:
- Use the functional handle introduced by enzymatic oxidation (e.g., alcohol group) for chemical transformations. This can include oxidation to aldehydes, alkylation, ring-opening, or cyclization reactions.
- Employ different synthetic pathways from the same intermediate to access distinct molecular architectures (scaffold hops). For example, the same oxidized sclareolide intermediate was used to synthesize merosterolic acid B, cochlioquinone B, (+)-daucene, and dolasta-1(15),8-diene [14].
- Characterization: Purify all final compounds and confirm their structures and stereochemistry using comprehensive NMR and MS analysis.
Protocol 4.3: In Vivo Evaluation of Toxicity and Bioavailability for a Novel Nanoformulation
This protocol assesses the in vivo performance of a novel nanodrug, using a platinum-based anti-cancer nanodrug as an example, and a strategy to reduce its RES-mediated toxicity [48].
I. Research Reagent Solutions
- Test Article: Platinum-loaded, polymer-coated nanoparticle (e.g., DACHPt/HANP).
- Intervention: FDA-approved fat emulsion (e.g., Intralipid 20%).
- Animal Model: Sprague Dawley rats.
- Key Reagents: Assay kits for serum ALT (liver toxicity) and creatinine (kidney toxicity). Reagents for H&E staining and TUNEL assay (apoptosis detection).
- Analytical Instrument: ICP-MS for quantifying platinum biodistribution.
II. Step-by-Step Methodology
- Experimental Groups:
- Group 1: Naïve control (no treatment).
- Group 2: Intralipid control (2 g/kg, i.v.).
- Group 3: DACHPt/HANP only (i.v.).
- Group 4: Intralipid (2 g/kg, i.v.) pre-treatment 1 hour before DACHPt/HANP injection.
- Toxicity and Biodistribution Study:
- Administer treatments via tail vein injection.
- At 24 and 72 hours post-nanodrug injection, collect blood samples via retro-orbital bleeding or cardiac puncture. Separate serum.
- Euthanize animals and harvest organs (liver, spleen, kidney, tumor if applicable). Weigh organs immediately.
- Serum Biochemistry: Quantify ALT and creatinine levels using commercial kits per manufacturer's instructions.
- Histopathology: Fix organ tissues in formalin, embed in paraffin, section, and perform H&E and TUNEL staining. Analyze for necrosis and apoptosis under a light microscope.
- Biodistribution: Digest weighed portions of tissues in nitric acid. Analyze Pt content using ICP-MS to determine drug accumulation.
- Data Analysis:
- Compare organ weight/body weight ratios between groups.
- Statistically compare serum biomarker levels and tissue Pt concentrations between Group 3 and Group 4 to determine the effect of Intralipid pre-treatment.
Visualization of Workflows and Pathways
The following diagrams illustrate the core experimental and conceptual frameworks described in this note.
AI-Guided Optimization Workflow
Enzyme-Enabled Scaffold Hopping
Table 3: Key Research Reagent Solutions for Core Modification Studies
| Reagent / Resource | Function / Application | Example(s) |
|---|---|---|
| Engineered Cytochrome P450s | Enables site-selective oxidation of inert C-H bonds in complex NPs, creating handles for diversification. | Engineered P450 from B. megaterium for sclareolide C3 oxidation [14]. |
| Open-Source AI Models | Computational guidance for rational structure-based design via fragment replacement or scaffold hopping. | DeepFrag (group modification), ScaffoldGVAE (scaffold hopping) [21] [49]. |
| Intralipid 20% | An FDA-approved fat emulsion used to transiently blunt RES function, reducing nanodrug accumulation in off-target organs. | Pre-treatment to reduce liver/spleen/kidney toxicity of Pt-nanodrugs [48]. |
| Groebke-Blackburn-Bienaymé (GBB) Reaction | A multi-component reaction to rapidly generate complex, drug-like scaffolds (e.g., imidazo[1,2-a]pyridines) for scaffold hopping. | Used to develop novel molecular glues for the 14-3-3/ERα complex [15]. |
| Molecular Docking Software | Validates the predicted binding mode of modified compounds to the target protein. | AutoDock Vina, Glide, GOLD. |
| In-silico ADMET Platforms | Predicts pharmacokinetic and toxicity profiles of designed compounds prior to synthesis. | SwissADME, pkCSM, ProTox-II. |
Strategic Use of Multimodal Data and Constraints to Guide Successful Hops
Scaffold hopping, the practice of designing compounds with novel core structures that retain the biological activity of a parent molecule, is a crucial strategy in natural product-based drug design for overcoming limitations such as poor pharmacokinetics, toxicity, or intellectual property constraints [9] [18]. The core challenge lies in maintaining essential interactions with the biological target while significantly altering the molecular framework [50]. This application note details protocols for employing advanced computational models that strategically integrate multimodal data—including 2D molecular graphs, 3D structural information, and protein target data—to guide successful scaffold hops. By framing these methodologies within the context of natural product optimization, we provide researchers with a structured framework for discovering novel chemotypes with improved properties.
Key Computational Approaches and Their Performance
The following models represent the state-of-the-art in data-driven scaffold hopping, each utilizing different types of data and architectural principles.
Table 1: Performance Comparison of Deep Learning Models for Scaffold Hopping
| Model Name | Core Architecture | Primary Data Modalities | Reported Key Performance Metric | Applicable Scenario in NP Optimization |
|---|---|---|---|---|
| DeepHop [28] | Multimodal Transformer | Molecular Sequence, 2D Graph, 3D Conformer, Protein Sequence | ~70% of generated molecules had improved bioactivity, high 3D, but low 2D similarity (1.9x higher than other methods) | Target-informed hopping for NPs with known protein targets |
| DeLinker [51] | 3D-aware Graph Neural Network (GGNN) | 2D Molecular Graph, 3D Distance & Orientation | Designed 60% more molecules with high 3D similarity vs. baseline; 200% outperformance for linkers ≥5 atoms | Fragment linking in NP-derived partial structures |
| ScaffoldGVAE [17] | Graph Variational Autoencoder (GVAE) | 2D Molecular Graph (scaffold & side-chain views) | Generated validated novel inhibitors for LRRK2; superior scores on 4 scaffold-hopping specific metrics | Generating novel scaffolds while preserving NP-derived side chains |
| WHALES [32] | 3D Molecular Descriptors | 3D Molecular Conformation, Partial Atomic Charges | Outperformed 7 state-of-the-art descriptors in scaffold-hopping ability (SDA%) for 89% of 182 biological targets | Ligand-based hopping when 3D structure of NP is available |
Experimental Protocols for Key Methodologies
Protocol: Implementing a 3D-Aware Generative Model (DeLinker)
This protocol is designed for scaffold hopping and linker design in natural product-derived fragments [51].
Research Reagent Solutions:
- Software Framework: Python with PyTorch or TensorFlow.
- Cheminformatics Library: RDKit (for molecule handling, conformer generation, and descriptor calculation).
- 3D Structure Generator: RDKit's ETKDG method or OMEGA for conformer generation.
- Data Source: ChEMBL database or in-house database of natural product structures and their fragments.
Procedure:
- Input Preparation:
- Define the two molecular fragments (FragA and FragB) to be linked, derived from the natural product.
- Generate a 3D conformation for the complete target molecule or specify the desired 3D constraints (distance and relative orientation) between FragA and FragB.
- Model Configuration:
- Initialize the DeLinker model, which uses a Gated Graph Neural Network (GGNN) [51].
- Set the maximum linker length (number of atoms to be generated).
- Encoding:
- The model converts the input fragments into a graph representation where atoms are nodes and bonds are edges.
- The graph is passed through the GGNN to update node hidden states, incorporating the local chemical environment of each atom.
- The 3D structural information (distance and angle between fragments) is integrated into the model to guide the generation.
- Iterative Generation:
- The model initializes a set of "expansion nodes" with hidden states sampled from a normal distribution.
- In a breadth-first manner, the model iteratively: a. Selects a node from a queue (initially populated with the fragments' exit vectors). b. Labels the node with an atom type based on its hidden state and the 3D constraints. c. Adds edges (bonds) between the current node and other nodes in the graph until a "stop" edge is selected.
- Output and Validation:
- The process results in a fully connected molecular graph incorporating both input fragments.
- Validate the generated molecules for chemical validity (e.g., using RDKit's sanitization checks) and evaluate 3D similarity to the original natural product's binding pose if available.
Protocol: Employing a Multimodal Transformer for Target-Aware Hopping (DeepHop)
This protocol uses a supervised translation approach to generate scaffold-hopped molecules with improved bioactivity for a specific protein target [28].
Research Reagent Solutions:
- Software Framework: Python with PyTorch/TensorFlow and Transformers library.
- Molecular Representation: SMILES strings and 3D conformers.
- Protein Representation: Amino acid sequence or structural data.
- Bioactivity Data: Public databases (e.g., ChEMBL) or proprietary data for model fine-tuning.
Procedure:
- Data Curation and Preprocessing:
- From a bioactivity database (e.g., ChEMBL), extract pairs of molecules (
X,Y) where moleculeYhas significantly improved activity (e.g., pChEMBL value ≥ 1) overXagainst the same target (Z), while having low 2D scaffold similarity (Tanimoto score ≤ 0.6) but high 3D similarity (SC score ≥ 0.6) [28]. - Preprocess SMILES strings (normalize, remove salts, neutralize charges) using RDKit.
- Generate 3D conformers for each molecule.
- From a bioactivity database (e.g., ChEMBL), extract pairs of molecules (
- Model Training/Finetuning:
- The DeepHop model is built on a Transformer architecture.
- Multimodal Integration: The model encodes:
- Molecular Sequence: The SMILES string of the input molecule
X. - Molecular 3D Graph: A Spatial Graph Neural Network encodes the 3D conformer of
X. - Protein Target: The amino acid sequence of target
Zis encoded via a Protein Transformer.
- Molecular Sequence: The SMILES string of the input molecule
- The model is trained to translate the input molecule
Xinto the output moleculeYconditioned on the target and structural information.
- Inference and Generation:
- Input a natural product (or any known active)
Xand the target proteinZinto the trained model. - The model generates a hopped molecule
Ypredicted to have improved bioactivity againstZ, a novel 2D scaffold, and a similar 3D profile toX.
- Input a natural product (or any known active)
- Virtual Profiling:
- Use a pre-trained deep QSAR model (e.g., Multi-Task DNN) to rapidly predict the bioactivity of the generated molecules against the target of interest [28].
Protocol: Leveraging 3D Molecular Descriptors for Scaffold Hopping (WHALES)
This protocol is for ligand-based scaffold hopping using advanced 3D molecular descriptors, useful when the target structure is unknown but a 3D conformation of the active natural product is available [32].
Research Reagent Solutions:
- Software Environment: Python with RDKit and NumPy/SciPy.
- Conformer Generation: RDKit's MMFF94 force field implementation.
- Partial Charge Calculation: DFTB+ (for high detail) or Gasteiger-Marsili method (for speed).
Procedure:
- Query Molecule Preparation:
- Obtain a 3D conformation of the natural product query molecule. Energy minimization using the MMFF94 force field is recommended [32].
- Calculate partial atomic charges using a chosen method (e.g., Gasteiger-Marsili).
- WHALES Descriptor Calculation: a. Compute the weighted, atom-centred covariance matrix ( \mathbf{S}_{w(j)} ) for each non-hydrogen atom ( j ), incorporating atomic coordinates and partial charges [32]. b. Calculate the Atom-Centred Mahalanobis distance (ACM) matrix, which encodes normalized interatomic distances based on the local chemical environment [32]. c. From the ACM matrix, derive atomic parameters (remoteness and isolation degrees) for each atom. d. Aggregate these atomic parameters into a fixed-size molecular descriptor vector by computing the minimum, maximum, and decile values of their distributions.
- Similarity Searching:
- Calculate WHALES descriptors for each molecule in a screening database (e.g., a diverse virtual compound library or a database of synthetic fragments).
- Perform a similarity search (e.g., using Euclidean or Cosine distance) to rank database molecules by their WHALES descriptor similarity to the natural product query.
- Hit Analysis:
- Inspect the top-ranked molecules. These are predicted to share similar 3D shape and pharmacophoric features with the query but may possess structurally distinct (hopped) scaffolds.
- The scaffold-hopping ability can be quantified by the Scaffold Diversity of Actives (SDA%), which measures the number of unique scaffolds found among the top-ranking active molecules [32].
Workflow Visualization
The following diagram illustrates the integrated workflow for multimodal scaffold hopping, synthesizing the key elements from the protocols above.
Diagram 1: Integrated Multimodal Scaffold Hopping Workflow. This diagram outlines the core process where a natural product input is decomposed into multiple data modalities that inform a generative model to produce novel, validated compound designs. The process is iterative, leveraging validation results to refine subsequent design cycles.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Item/Tool Name | Category | Function in Scaffold Hopping | Example/Note |
|---|---|---|---|
| RDKit | Cheminformatics Library | Handles molecule I/O, SMILES processing, fingerprint generation, scaffold analysis, and 3D conformer generation. | Open-source; essential for preprocessing and feature extraction [28] [17]. |
| ChEMBL Database | Bioactivity Data | Provides curated bioactivity data for constructing training pairs and benchmarking. | Used in DeepHop, ScaffoldGVAE, and WHALES studies [28] [32] [17]. |
| PyTorch / TensorFlow | Deep Learning Framework | Provides the environment for building, training, and deploying complex generative models (GNNs, Transformers, VAEs). | Standard frameworks for implementing models like DeLinker and DeepHop [51] [28]. |
| ETKDG / OMEGA | 3D Conformer Generator | Generates realistic 3D molecular conformations required for 3D-aware models and descriptor calculations. | RDKit's ETKDG method; critical for DeLinker and WHALES inputs [51] [32]. |
| MMFF94 | Force Field | Used for energy minimization and geometry optimization of generated 3D structures. | Ensures generated conformers are energetically reasonable [28] [32]. |
| DFTB+ / Gasteiger-Marsili | Partial Charge Method | Calculates atomic partial charges, a key input for detailed 3D descriptors like WHALES. | DFTB+ for accuracy; Gasteiger for speed [32]. |
| ScaffoldGraph | Scaffold Analysis | Systematically decomposes molecules to extract hierarchical scaffolds for model training and analysis. | Used in ChemBounce and ScaffoldGVAE for scaffold definition [37] [17]. |
Validating Success: Assessing Scaffold Hops from Activity to Practical Impact
The drug discovery landscape is undergoing a profound transformation, moving from traditional, labor-intensive processes to modern, technology-driven approaches. Within natural product-based drug design and scaffold hopping research, understanding the quantitative performance differences between these paradigms is crucial for strategic research planning. Traditional drug discovery remains an arduous endeavor, typically requiring 10-15 years and exceeding $1-2 billion per approved therapy, with a dismally low success rate of fewer than 10% of candidates entering Phase I trials ultimately gaining approval [52]. Modern approaches, particularly those leveraging artificial intelligence (AI) and structured scaffold hopping, demonstrate potential to compress timelines to 12-18 months for early discovery phases and significantly improve efficiency [52] [53]. This application note provides a quantitative benchmark and detailed experimental protocols to guide researchers in adopting these advanced methodologies.
Quantitative Benchmarking of Discovery Approaches
Table 1: Performance Metrics of Traditional vs. Modern Drug Discovery Methods
| Performance Metric | Traditional Methods | Modern AI-Driven Methods | Scaffold Hopping Approaches |
|---|---|---|---|
| Typical Discovery Timeline | 3-6 years (to preclinical candidate) [52] | 12-18 months (to preclinical candidate) [52] [53] | Varies (builds on existing molecules) [8] |
| Clinical Success Rate | <10% (Phase I to approval) [52] | Under evaluation; early data shows improved efficiency [52] [54] | Often higher (starting from validated starting points) [8] |
| Key Supporting Technologies | High-Throughput Screening (HTS), Structure-Based Design [55] | Machine Learning (ML), Deep Learning (DL), Multi-modal Data Fusion [52] [53] [56] | Computational Design (e.g., MORPH software), Advanced Synthetic Chemistry [8] |
| Representative Case Study | Conventional HTS for lead identification | Insilico Medicine's AI-generated anti-fibrotic (18 months to PCC) [52] | Roxadustat analogs developed via heterocycle replacement [8] |
Table 2: Analysis of AI Methodologies in Modern Drug Discovery (Analysis of 173 Studies, 2015-2025) [52]
| AI Methodology | Adoption Rate | Primary Application in Discovery |
|---|---|---|
| Machine Learning (ML) | 40.9% | Target identification, compound potency/ADMET prediction [52] [56] |
| Molecular Modeling & Simulation (MMS) | 20.7% | Molecular docking, binding affinity prediction, protein-ligand interactions [52] |
| Deep Learning (DL) | 10.3% | De novo molecular design, analysis of complex data (e.g., histopathology images) [52] [56] |
| Reinforcement Learning (RL) | Not quantified | Multi-objective optimization in generative chemistry [53] |
| Natural Language Processing (NLP) | Not quantified | Mining scientific literature and patents for target identification [53] [56] |
Experimental Protocols
Protocol 1: Traditional Bioactivity-Guided Natural Product Fractionation
This protocol describes the classical approach to isolating active compounds from natural sources, which serves as a foundational starting point for many scaffold-hopping campaigns.
3.1.1 Materials and Reagents
- Crude natural extract (e.g., plant, marine organism)
- Series of organic solvents (hexane, dichloromethane, ethyl acetate, n-butanol)
- Chromatography media: Silica gel, Sephadex LH-20, C18 reverse-phase resin
- Cell-based or biochemical assay for target activity
- Analytical standards (for TLC and HPLC)
3.1.2 Step-by-Step Procedure
- Primary Fractionation: Subject the crude extract to liquid-liquid partitioning using solvents of increasing polarity (e.g., hexane, DCM, ethyl acetate, n-butanol) to obtain primary fractions.
- Bioactivity Screening: Test all primary fractions in a relevant pharmacological assay. Select the most active fraction for further separation.
- Column Chromatography: Load the active fraction onto a normal-phase (Silica gel) or size-exclusion (Sephadex LH-20) column. Elute with appropriate solvent systems and collect sequential sub-fractions.
- Thin-Layer Chromatography (TLC): Analyze all sub-fractions by TLC to profile chemical composition. Pool sub-fractions with similar TLC profiles.
- Secondary Bioassay: Screen the pooled sub-fractions for bioactivity. Take the active, chemically complex pools for further purification.
- High-Performance Liquid Chromatography (HPLC): Use analytical and preparative HPLC (typically reverse-phase C18) to isolate pure compounds from the active pool.
- Structure Elucidation: Determine the chemical structure of active pure compounds using spectroscopic techniques (NMR, MS).
- Validation: Confirm the biological activity of the isolated pure compound in a dose-dependent manner.
Protocol 2: AI-Driven Target Identification from Natural Product MoA
This modern protocol uses AI to hypothesize and prioritize molecular targets for a natural product with a known phenotypic effect but an unknown mechanism of action (MoA).
3.2.1 Materials and Software
- Multi-omics data (transcriptomics, proteomics) from compound-treated vs. control cells
- Access to an AI target discovery platform (e.g., Insilico Medicine's PandaOmics [53] or Verge Genomics' CONVERGE [53])
- Literature and patent corpus (integrated within platform)
- Knock-out/down models (e.g., CRISPR) for experimental validation
3.2.2 Step-by-Step Procedure
- Data Generation and Curation: Treat a relevant human cell line with the natural product and generate high-quality transcriptomic (RNA-seq) and/or proteomic data. Prepare a robust dataset of differential gene/protein expression.
- Platform Interrogation: Input the differential expression profile into the AI target identification platform. The platform, such as PandaOmics, uses its knowledge graph—built from millions of samples and documents—to identify upstream targets that could explain the observed gene expression changes [53].
- Target Prioritization: Apply the platform's ranking algorithms, which typically use composite scores based on gene expression reverse causal reasoning, pathway analysis, literature mining (NLP), and novelty assessment [53].
- In Silico Validation: Use the platform's integrated tools to explore the biological network surrounding the top-predicted targets, assessing their connectivity to the disease pathway of interest.
- Hypothesis Generation: The output is a ranked list of potential protein targets with associated confidence scores and mechanistic hypotheses.
- Experimental Validation: Select the top 3-5 predicted targets for experimental validation using techniques like CRISPR knock-down followed by a phenotypic assay to confirm sensitization/resistance to the natural product.
Protocol 3: Scaffold Hopping from a Natural Product Lead
This protocol outlines a systematic approach, enhanced by computational chemistry, to generate novel chemical entities based on the core structure of an active natural product.
3.3.1 Materials and Software
- 3D structure of the natural product lead compound
- Computational software for scaffold hopping (e.g., MORPH) [8] and molecular docking
- Information on the known or predicted pharmacophore
- Synthetic chemistry equipment and reagents
3.3.2 Step-by-Step Procedure
- Pharmacophore Definition: Analyze the structure-activity relationship (SAR) of the natural product lead to define the essential chemical features (pharmacophore) required for activity.
- Scaffold Disconnection: Identify the central core (scaffold) and the peripheral substituents of the lead molecule.
- In Silico Scaffold Replacement: Use computational tools (e.g., MORPH software) to perform systematic in silico modifications of the central core. Common strategies include [8]:
- Heterocycle Replacement (1°): Swapping a core heterocycle (e.g., pyridine) for a bioisostere (e.g., pyrimidine).
- Ring Closure (2°): Introducing a new ring bridge within the existing scaffold.
- Ring Fusion (3°): Adding a new ring system adjacent to the original core.
- Virtual Library Generation & Screening: Generate a virtual library of scaffold-hopped analogs. Screen this library in silico using molecular docking to the target protein (if structure is known) and predictive models for ADMET properties.
- Synthesis: Prioritize the top-ranking virtual analogs based on predicted activity, synthetic accessibility, and novelty. Synthesize these compounds.
- Biological Evaluation: Test the synthesized scaffold-hopped analogs in biological assays to evaluate potency, selectivity, and improved pharmaceutical properties (P3: Pharmacodynamics, Physicochemical, Pharmacokinetic) [8].
Workflow Visualization
Scaffold Hopping from a Natural Product Lead
AI-Driven Target Deconvolution for Natural Products
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Platforms for Modern Discovery
| Tool / Reagent | Function / Application | Key Characteristic / Benefit |
|---|---|---|
| PandaOmics (Insilico Medicine) [53] | AI-powered target discovery; identifies and prioritizes novel therapeutic targets from multi-omics data. | Leverages knowledge graphs from 1.9 trillion data points and NLP on 40M+ documents for holistic target assessment. |
| Chemistry42 (Insilico Medicine) [53] | Generative chemistry AI platform for de novo molecular design and lead optimization. | Uses generative adversarial networks (GANs) and reinforcement learning to balance potency, selectivity, and ADMET. |
| Recursion OS Platform [53] | Maps trillions of biological relationships using proprietary data and AI models (e.g., Phenom-2, MolPhenix). | Integrates automated wet-lab biology with AI computational models for phenotypic drug discovery. |
| Organ-on-a-Chip Models [57] | Microphysiological systems for human-relevant ADMET and efficacy testing (a key New Approach Methodology - NAM). | Provides more predictive human toxicity and PK data than animal models, reducing late-stage attrition. |
| MORPH Software [8] | Computational tool for systematic scaffold hopping and aromatic ring modification in 3D ligand models. | Enables complex scaffold-hopping strategies to generate novel, patentable chemical space from known leads. |
| CONVERGE (Verge Genomics) [53] | Closed-loop ML platform for target discovery, prioritizing targets using large-scale human-derived biological data. | Focuses on human clinical genomics data to improve translational relevance, particularly in neurology. |
In natural product-based drug design, the process of scaffold hopping—the identification of structurally novel compounds with similar biological activity to a parent natural product—is a crucial strategy for overcoming challenges associated with natural products, such as structural complexity, poor solubility, or limited availability [9] [24]. The success of these campaigns depends critically on the rigorous application of key validation metrics to ensure that newly designed compounds maintain the desired biological activity while achieving improved synthetic accessibility and drug-like properties. This document details the essential metrics—2D/3D similarity, potency, and selectivity—framed within the context of natural product-inspired scaffold hopping, providing researchers with structured protocols for their application in early drug discovery.
Molecular Similarity Assessment
Molecular similarity analysis forms the foundational pillar of scaffold hopping, operating on the principle that structurally similar molecules are likely to exhibit similar biological activities [58]. For natural products, this involves comparing novel synthetic mimetics against their complex natural templates.
2D Molecular Similarity
2D similarity methods evaluate molecular structure based on topological descriptors, without considering three-dimensional conformation.
Fingerprint-Based Methods: These encode molecular structures as bit strings representing the presence or absence of specific structural features.
- Extended Connectivity Fingerprints (ECFPs): Are a benchmark in virtual screening, representing molecules as sets of radially grown fragments from each non-hydrogen atom [24]. They are highly effective for comparing general molecular features and are computationally efficient for screening large compound libraries [59].
- Application Note: ECFPs are particularly useful for the initial rapid filtering of large commercial repositories to identify potential hit compounds inspired by natural product queries [59].
Protocol: Performing a 2D Similarity-Based Virtual Screen
- Query Selection: Start with one or more known active natural products (e.g., a phytocannabinoid for cannabinoid receptor modulation) [24].
- Fingerprint Generation: Compute ECFP4 fingerprints for the query molecule(s) and all compounds in the target database (e.g., ZINC20, ChEMBL).
- Similarity Calculation: Calculate the Tanimoto coefficient between the query fingerprint and every database compound fingerprint. The Tanimoto coefficient ranges from 0 (no similarity) to 1 (identical fingerprints).
- Compound Ranking: Rank all database compounds in descending order of their Tanimoto coefficient relative to the query.
- Thresholding: Select top-ranking compounds for further experimental testing or more computationally intensive 3D analysis. A common practice is to select the top 1% of ranked compounds or all compounds above a predefined Tanimoto threshold (e.g., >0.5) [60].
3D Molecular Similarity
3D similarity methods compare molecules based on their shape and the spatial arrangement of pharmacophoric features, which is critical for scaffold hopping where core structures differ significantly in 2D topology [61] [24].
- Shape-Based Overlays: Tools like ROCS (Rapid Overlay of Chemical Structures) align molecules based on their steric volume and compare them using metrics like TanimotoCombo, which combines shape and feature similarity [61].
- Holistic Molecular Descriptors: The WHALES (Weighted Holistic Atom Localization and Entity Shape) descriptors represent a recent advancement. They encode information on geometric interatomic distances, molecular shape, and atomic partial charge distributions into a fixed-length vector, enabling the comparison of molecules with different atom counts [24].
- Application Note: WHALES descriptors have been prospectively validated using natural product queries, successfully identifying novel synthetic cannabinoid receptor modulators with a 35% experimental hit rate, demonstrating their power for functional scaffold hopping [24].
Table 1: Key Metrics for 2D and 3D Molecular Similarity Methods
| Method | Descriptor Type | Key Metric | Typical Use Case in Scaffold Hopping | Advantages |
|---|---|---|---|---|
| 2D Similarity | ECFP Fingerprints | Tanimoto Coefficient | Rapid pre-filtering of large libraries; hit expansion [59]. | Computationally fast; intuitive for chemists [59]. |
| 3D Shape-Based | Molecular Shape/Features | TanimotoCombo Score | Identifying mimetics of complex natural products with different 2D structures [61]. | Captures functional similarity beyond topology [61]. |
| 3D Holistic | WHALES Descriptors | Euclidean Distance in WHALES space | Scaffold hopping from complex natural products to synthetically accessible leads [24]. | Integrates shape and pharmacophore features; robust to conformational change [24]. |
Integrated Similarity Screening Workflows
Combining 2D and 3D methods in a sequential or parallel workflow increases the success rate and novelty of identified hits [59].
- Sequential Approach: A 2D similarity search is first used to reduce the virtual chemical space from millions to thousands of compounds. This manageable set is then subjected to more computationally demanding 3D shape-based or holistic similarity analysis [59].
- Reverse Sequential Approach: Structure-based virtual screening (e.g., docking) is used first to identify an initial active compound. This new hit is then used as a query for 2D similarity search to find structurally analogous compounds in a process known as "hit expansion" [59].
Diagram 1: Integrated 2D/3D similarity screening workflow for identifying natural product mimetics, combining the speed of 2D methods with the functional insight of 3D approaches [59] [24].
Potency and Selectivity Profiling
Beyond mere structural similarity, confirming a compound's functional profile through potency and selectivity is paramount in transitioning from a initial hit to a viable lead.
Defining and Measuring Potency
Potency quantifies the concentration of a compound required to produce a given biological effect. Common metrics include ICâ‚…â‚€ (half-maximal inhibitory concentration), ECâ‚…â‚€ (half-maximal effective concentration), and Káµ¢ (inhibition constant). In scaffold hopping, the goal is to achieve low micromolar to nanomolar potency against the primary target, comparable to or better than the original natural product [62].
Target-Specific Selectivity
Selectivity ensures that a compound exerts its primary effect on the intended target without causing significant off-target effects, which can lead to adverse reactions. Traditional metrics like the Gini coefficient or selectivity entropy quantify the overall narrowness of a compound's bioactivity spectrum but do not focus on a specific target of interest [62].
For scaffold hopping, a target-specific selectivity score is more appropriate. This approach frames selectivity as a multi-objective optimization problem, seeking compounds that simultaneously demonstrate [62]:
- High Absolute Potency: Strong binding affinity (e.g., low Kd or Ki) for the primary disease target.
- High Relative Potency: Significantly weaker binding affinity for all other off-targets.
Table 2: Key Metrics for Assessing Potency and Selectivity in Scaffold Hopping
| Metric | Definition | Interpretation in Scaffold Hopping |
|---|---|---|
| IC₅₀ / Kᵢ | Concentration inhibiting 50% of target activity / Inhibition constant. | Primary measure of compound potency against the intended target. Aim for nM to low µM range. |
| Global Relative Potency (G) | ( G{ci,tj} = K{ci,tj} - \text{mean}(B{ci} \setminus {K{ci,t_j}}) ) [62] | Measures a compound's ((ci)) potency for a target ((tj)) relative to its mean potency against all other targets. A high value indicates high selectivity. |
| Local Relative Potency (L) | ( L{ci,tj} = K{ci,tj} - \text{mean}(B{ci,hNN(t_j)}) ) [62] | Measures a compound's potency for a target relative to its potency against the h-most similar off-targets. More sensitive to activity cliffs. |
| Partition Index | Fraction of total binding strength directed toward a reference target [62]. | Quantifies the fraction of a compound's overall binding energy devoted to the primary target. |
Table 3: Experimental Profiling Techniques for Validation
| Assay Type | Information Provided | Application in Scaffold Hopping |
|---|---|---|
| In vitro Binding Assay | Direct measurement of binding affinity (Kd, Ki) to a purified target protein. | Confirm direct target engagement and primary potency. |
| Cell-Based Functional Assay | Measures functional consequences (e.g., inhibition of cell growth, second messenger production) in a live cell system (ICâ‚…â‚€, ECâ‚…â‚€). | Validates activity in a more physiologically relevant context. |
| Kinase/GPCR Panel Screening | Broad profiling of activity against dozens to hundreds of related targets in a protein family. | Essential for quantifying polypharmacology and identifying potential off-target effects early. |
Diagram 2: The iterative validation cycle for a scaffold-hopped compound, moving from initial design to a confirmed potent and selective lead candidate through rigorous biochemical profiling [62].
The Scientist's Toolkit: Research Reagent Solutions
The following table details key computational and experimental resources essential for conducting research in natural product-based scaffold hopping and validation.
Table 4: Essential Research Tools and Reagents for Scaffold Hopping and Validation
| Tool/Reagent | Function | Application Note |
|---|---|---|
| ChEMBL Database | Public repository of bioactive molecules with drug-like properties and curated bioactivity data [60] [61]. | Serves as a primary source for building reference datasets of known active compounds for similarity-based target prediction [60]. |
| ZINC20/COCONUT | Freely available databases of commercial (ZINC20) and natural (COCONUT) compounds for virtual screening [60]. | Used as source libraries for finding potential scaffold hops or for building a knowledge base of non-complex small molecules for target prediction [60] [61]. |
| ROCS (OpenEye) | A leading 3D shape-based molecular superposition tool and screening engine [61]. | Used for aligning query natural products to potential mimetics based on 3D shape and chemical features, quantified by the TanimotoCombo score [61]. |
| CTAPred | An open-source, command-line tool for predicting protein targets of natural products using similarity-based searches [60]. | Addresses the challenge of limited bioactivity data for NPs by using a focused compound-target activity reference dataset [60]. |
| Kinase/GPCR Profiling Services | Commercial services (e.g., Eurofins, Reaction Biology) that offer high-throughput screening against panels of kinase or GPCR targets. | Critical for experimentally determining the selectivity profile of a scaffold-hopped compound against a therapeutically relevant target family [62]. |
| WHALES Descriptors | A holistic molecular representation capturing pharmacophore, shape, and partial charge patterns [24]. | Enables scaffold hopping from complex natural products to isofunctional synthetic compounds by capturing functionally relevant molecular features [24]. |
Comparative Analysis of Scaffold Hopping Strategies and Software Tools
Scaffold hopping is a foundational strategy in modern medicinal chemistry, defined as the structural modification of the core molecular framework of a known bioactive compound to generate novel, patentable molecules with potentially improved properties [8] [10]. Within the context of natural product-based drug discovery, this approach is invaluable for optimizing the complex scaffolds of natural leads, addressing common issues such as poor solubility, metabolic instability, toxicity, and limited intellectual property (IP) space [24] [21] [63]. By systematically exploring alternative chemotypes that retain the desired biological activity, scaffold hopping bridges the gap between biologically validated natural product starting points and drug-like candidates better suited for clinical development. This analysis provides a comparative examination of contemporary scaffold hopping strategies, software tools, and their practical application in a research setting.
Classification and Strategic Framework of Scaffold Hopping
Scaffold hopping encompasses a spectrum of structural modifications, which can be systematically classified into distinct degrees based on the type of alteration performed on the parent molecule's core [8] [10]. This classification aids medicinal chemists in rational design and communication.
Table 1: Degrees of Scaffold Hopping and Their Characteristics
| Degree | Type of Modification | Description | Key Applications |
|---|---|---|---|
| 1° (Heterocycle Replacement) | Substitution, addition, or removal of heteroatoms in a core ring; replacement of one heterocycle with a similar one [8] [10]. | The simplest form; retains the spatial arrangement of the pharmacophore and adjacent groups [10]. | Tuning physicochemical properties, optimizing PK profile, identifying key ligand-target interactions [10]. |
| 2° (Ring Opening or Closure) | Converting a cyclic moiety into an acyclic chain, or vice versa [8]. | Alters molecular flexibility and conformational entropy without drastically changing the core topology. | Modulating metabolic stability and conformational freedom [8]. |
| 3° (Peptidomimetics) | Replacing a peptide backbone with a non-peptide scaffold [8]. | Aims to mimic the spatial orientation of key pharmacophoric elements of a peptide. | Improving oral bioavailability and metabolic stability of peptide leads [8]. |
| 4° (Topology-based Alterations) | Introducing global molecular shape changes, such as fusing or dissociating rings [8]. | The most sophisticated degree; involves significant redesign of the core scaffold topology. | Exploring novel IP space, addressing complex property issues [8]. |
The logical relationship and strategic progression between these degrees and their objectives can be visualized in the following workflow.
Computational Tools for Scaffold Hopping
The implementation of scaffold hopping strategies is powerfully enabled by a suite of specialized software tools. These platforms use various algorithms to propose novel molecular structures that meet specific design criteria.
Table 2: Comparative Analysis of Key Scaffold Hopping Software Tools
| Software Tool | Core Methodology | Key Features | Applicability in Natural Product Optimization |
|---|---|---|---|
| Spark [64] | Electrostatic and shape similarity for bioisosteric replacement. | User-friendly wizards for lead discovery/optimization; multi-parametric optimization (LogP, TPSA, MW) [64]. | Replacing complex, synthetically challenging NP fragments with simpler, isofunctional motifs. |
| ChemBounce [65] | Fragment replacement using a curated library of >3 million fragments from ChEMBL. | Evaluates Tanimoto and electron shape similarities; focuses on high synthetic accessibility [65]. | Systematic exploration of synthetic mimetics for natural product scaffolds. |
| SeeSAR [66] | 3D structure-based visualization and design with HYDE affinity estimation and FastGrow algorithm. | Intuitive visual interface; on-the-fly affinity estimation; ReCore tool for 3D-driven re-scaffolding [66]. | Visual, structure-guided optimization of NP analogs within a protein binding site. |
| WHALES [24] | Holistic molecular representation (pharmacophore, shape, partial charge). | Ligand-based; does not require target structure; effective for complex NP mimetic design [24]. | Scaffold hopping from complex NPs to synthetically accessible, isofunctional compounds when structural data is lacking. |
| AnchorQuery [15] | Pharmacophore-based screening of a vast, synthesizable MCR chemistry library. | Links to readily synthesizable scaffolds (e.g., GBB-3CR); prioritizes synthetic feasibility [15]. | Rapid generation of novel, drug-like, and synthetically accessible scaffolds inspired by an NP starting point. |
Case Studies and Experimental Applications
Case Study 1: Aurone Optimization via O-to-N Scaffold Hopping
Aurones, natural "golden flavonoids," possess promising bioactivities but are hampered by poor solubility and metabolic stability [63]. A scaffold-hopping approach replaced the benzofuranone core's oxygen with nitrogen, yielding azaaurones (indolin-3-ones).
- Experimental Protocol:
- Synthesis: The azaaurone scaffold is efficiently constructed via a one-pot Sonogashira/intramolecular cyclization. 2-Iodoaniline and phenylacetylene react under Pd(PPh₃)₄ catalysis (5 bar CO, 80 °C) to selectively form the (Z)-azaaurone core [63].
- Derivatization: The core scaffold is decorated with various substituents via Knoevenagel-aldol condensation with aromatic aldehydes in the presence of a base like piperidine [63].
- Evaluation: The resulting analogs are profiled for biological activity (e.g., anticancer, antimicrobial) and key P3 properties (solubility, metabolic stability), demonstrating improvements over the parent aurones [63].
Case Study 2: Scaffold Hopping for Molecular Glues Targeting 14-3-3/ERα
A scaffold-hopping approach was used to develop non-covalent molecular glues stabilizing the 14-3-3/ERα protein-protein interaction (PPI), a target in breast cancer [15].
- Experimental Protocol:
- In Silico Design: The co-crystal structure of a covalent molecular glue (compound 127, PDB: 8ALW) was used as a query in AnchorQuery software. The "phenylalanine anchor" (p-chloro-phenyl ring) was kept constant, and a three-point pharmacophore was defined for screening [15].
- Scaffold Identification: AnchorQuery suggested a novel scaffold based on the Groebke-Blackburn-Bienaymé multi-component reaction (GBB-3CR), yielding imidazo[1,2-a]pyridines. Docking confirmed a similar 3D shape and complementarity to the target interface [15].
- Synthesis & SAR: The GBB scaffold was synthesized from aldehydes, 2-aminopyridines, and isocyanides, allowing rapid generation of analogs. Structure-Activity Relationships (SAR) were developed using biophysical assays (TR-FRET, SPR) [15].
- Cellular Validation: The most potent analogs were confirmed to stabilize the 14-3-3/ERα full-length PPI in a live-cell NanoBRET assay [15].
Case Study 3: Advancing Anti-Tuberculosis Agents
Scaffold hopping has been pivotal in addressing drug-resistant tuberculosis (TB), by creating new chemotypes that circumvent existing resistance mechanisms [10].
- Experimental Protocol:
- Virtual Screening (VS): Two primary methods are employed:
- Ligand-Based VS (LBVS): Uses molecular fingerprints (e.g., ECFP) to identify candidate scaffolds with high similarity (Tanimoto score) to a known active compound [10].
- Structure-Based VS (SBVS): Utilizes X-ray crystallography or NMR data of the target protein (e.g., from PDB). Molecular docking screens large commercial libraries (e.g., ZINC, PubChem) to predict binding modes and affinity of novel scaffolds [10].
- Scaffold Optimization: Identified hit compounds undergo iterative cycles of scaffold hopping (e.g., heterocycle replacement) to optimize potency, selectivity, and pharmacokinetic properties against Mycobacterium tuberculosis [10].
- Efficacy Assessment: Optimized candidates are evaluated in vitro for minimum inhibitory concentration (MIC) against drug-sensitive and drug-resistant TB strains, and in vivo for efficacy in animal models [10].
- Virtual Screening (VS): Two primary methods are employed:
The following diagram illustrates the general decision-making workflow that integrates these computational and experimental methods.
Successful scaffold hopping campaigns rely on a combination of computational and experimental resources.
Table 3: Key Research Reagent Solutions for Scaffold Hopping
| Reagent / Resource | Category | Function in Scaffold Hopping |
|---|---|---|
| Fragment Libraries (e.g., SeeSAR's MedChem Set, Hinge Binder Set) [66] | Computational/Chemical | Pre-curated sets of molecular fragments used by software like SeeSAR and Spark for bioisosteric replacement, growing, and linking to generate novel scaffold ideas. |
| Multi-Component Reaction (MCR) Libraries (e.g., in AnchorQuery) [15] | Chemical/Synthetic | Virtual libraries of readily synthesizable scaffolds from MCR chemistry (e.g., GBB-3CR), enabling rapid identification of synthetically feasible novel cores. |
| ChEMBL Database [65] | Data | A manually curated database of bioactive molecules with drug-like properties. Serves as a key source for fragment libraries (e.g., in ChemBounce) and for validating the potential bioactivity of novel scaffolds. |
| Protein Data Bank (PDB) [10] | Data | A repository of 3D structural data of proteins and protein-ligand complexes. Essential for structure-based scaffold hopping, providing coordinates for docking and structure analysis. |
| Groebke-Blackburn-Bienaymé (GBB) Reaction Components [15] | Synthetic Chemistry | Aldehydes, 2-aminopyridines, and isocyanides used to synthesize the imidazo[1,2-a]pyridine scaffold—a privileged, drug-like core identified via scaffold hopping for PPIs. |
| Sonogashira Coupling Reagents [63] | Synthetic Chemistry | Palladium catalysts (e.g., Pd(PPh₃)₄) and co-catalysts used in one-pot syntheses of scaffold-hopped cores like azaaurones, enabling efficient construction of the novel heterocycle. |
Within the framework of natural product-based drug design, the dual challenges of enhancing therapeutic efficacy and establishing robust intellectual property (IP) positions are paramount. Scaffold hopping, a medicinal chemistry strategy that modifies the core molecular structure of a known bioactive compound, has emerged as a powerful approach to address both challenges simultaneously [10]. This method leverages the validated bioactivity of existing molecules, often derived from natural products, while generating novel chemical entities with improved properties [67]. The ensuing application note details a recent, impactful case study where a scaffold-hopping strategy, powered by computational design and multi-component reaction chemistry, successfully led to a new class of molecular glues with enhanced efficacy and a strong basis for patentability.
Case Study: Scaffold Hopping for Molecular Glues Targeting the 14-3-3/ERα Complex
Background and Therapeutic Rationale
The stabilization of protein-protein interactions (PPIs) represents a promising therapeutic strategy, particularly for targets considered "undruggable" by conventional means. This case focuses on the native interaction between the scaffolding protein 14-3-3 and the transcription factor Estrogen Receptor α (ERα). Stabilizing this PPI can inhibit ERα's transcriptional activity, offering a novel approach to treating ERα-positive breast cancer, especially in cases of acquired endocrine resistance [15]. The disordered C-terminus of ERα, which is recognized by 14-3-3, presents a challenging but druggable composite surface.
Scaffold Hopping Strategy and Computational Design
The discovery campaign began with a previously developed molecular glue, compound 127 (PDB: 8ALW), which was known to bind cooperatively at the 14-3-3σ/ERα interface [15]. While effective, this initial compound presented opportunities for optimization. A scaffold-hopping approach was employed to design a novel, more rigid, and drug-like core structure.
The computational workflow utilized AnchorQuery software, which performs pharmacophore-based screening of a vast virtual library of over 31 million compounds synthesizable via one-step multi-component reactions (MCRs) [15]. The process was guided by the crystallographic binding pose of compound 127:
- Anchor Motif: The p-chloro-phenyl ring, deeply buried in a hydrophobic pocket near K122 of 14-3-3, was defined as a constant "phenylalanine anchor."
- Pharmacophore Points: Three additional key interaction points from the original ligand were used to query the database, seeking new scaffolds that could fulfill similar spatial and chemical constraints.
This in silico screen identified the Groebke-Blackburn-Bienaymé (GBB) three-component reaction as the optimal pathway to generate a novel imidazo[1,2-a]pyridine scaffold. Docking poses revealed that the proposed GBB compounds exhibited significant shape complementarity and an nearly identical three-dimensional conformation to the original compound 127, while being inherently more rigid and drug-like [15].
Quantitative Efficacy Data of Lead Analogs
The following table summarizes the biophysical and cellular efficacy data for the original lead compound and the optimized analogs derived from the scaffold-hopping campaign. The data demonstrates the success of the approach in generating compounds with potent stabilization activity.
Table 1: Efficacy Data of Original and Scaffold-Hopped Molecular Glues
| Compound | Core Scaffold | SPR Affinity (KD, μM) | TR-FRET Stabilization (EC50, μM) | Cellular NanoBRET (EC50, μM) | Key Improvements |
|---|---|---|---|---|---|
| Compound 127 (Original) | Flexible, non-MCR | Not Specified | Not Specified | Not Specified | Original covalent binder |
| GBB Analog 1 | Imidazo[1,2-a]pyridine | < 10 | Low micromolar | Low micromolar | Non-covalent, rigid, drug-like |
| GBB Analog 2 (Most Potent) | Imidazo[1,2-a]pyridine | < 10 | Low micromolar | Low micromolar | Improved synthetic accessibility, optimal rigidity |
Data synthesized from [15]. SPR: Surface Plasmon Resonance; TR-FRET: Time-Resolved Förster Resonance Energy Transfer; NanoBRET: Bioluminescence Resonance Energy Transfer assay in live cells.
Patentability and Commercial Impact
The scaffold-hopping strategy directly enhanced the patentability of the resulting compounds. The generation of a novel imidazo[1,2-a]pyridine core, distinct from the original chemical matter, satisfies the key requirement of novelty [10] [68]. Furthermore, the application of a specific and less conventional synthetic route—the GBB multi-component reaction—strengthens the inventive step (non-obviousness) of the approach.
The new scaffold also provides a robust platform for rapid Structure-Activity Relationship (SAR) expansion through multiple points of variation, enabling the generation of a broad patent estate that protects not only the specific lead compounds but also a wide range of analogs [15] [68]. This "patent thicket" strategy is a critical lifecycle management tool, extending market exclusivity and protecting the significant investment required for drug development [68]. The imidazo[1,2-a]pyridine scaffold is a recognized privileged structure in medicinal chemistry, found in several clinical candidates and marketed drugs (e.g., zolpidem), which de-risks the development pathway and enhances the commercial attractiveness of the program [15].
Experimental Protocols
Protocol 1: In Silico Scaffold Hopping via AnchorQuery
Purpose: To computationally identify novel, synthetically accessible scaffolds that mimic the binding mode of a known active compound.
Materials:
- Software: AnchorQueryTM platform.
- Input Structure: A ligand-bound protein complex crystal structure (e.g., PDB: 8ALW).
- Virtual Library: The built-in database of ~31 million MCR-synthesizable compounds.
Methodology:
- Structure Preparation: Load the protein-ligand complex and prepare the structure by adding hydrogens and assigning correct protonation states.
- Anchor Definition: Visually analyze the binding pose to identify a deeply buried, key interacting fragment. Define this as the constant "anchor" (e.g., the p-chloro-phenyl ring).
- Pharmacophore Creation: From the original ligand, select three key pharmacophore points (e.g., hydrogen bond donors/acceptors, hydrophobic regions) that are critical for binding.
- Database Query: Run the AnchorQuery search using the defined anchor and pharmacophore points. Apply filters such as molecular weight (<400 Da) and synthetic accessibility.
- Hit Analysis: Review the ranked list of proposed scaffolds. Prioritize hits based on RMSD fit to the original ligand's 3D shape and visual inspection of docking poses for shape complementarity and interaction potential.
Protocol 2: Orthogonal Biophysical Assay for PPI Stabilization
Purpose: To experimentally validate and quantify the stabilization of the 14-3-3/ERα complex by novel compounds.
Materials:
- Reagents:
- Purified 14-3-3σ protein.
- Biotinylated phospho-ERα peptide (pT594).
- Test compounds.
- TR-FRET reagents: Streptavidin-XL665 (acceptor), anti-14-3-3 antibody conjugated with Eu3+-cryptate (donor).
- Equipment:
- Microplate reader capable of TR-FRET measurements.
Methodology:
- Solution Preparation: In an assay buffer, prepare a solution containing 14-3-3σ protein and the biotinylated pERα peptide at concentrations near their dissociation constant (KD).
- Compound Addition: Serially dilute test compounds in DMSO and add to the protein-peptide solution. Include DMSO-only wells as negative controls and a well with a known stabilizer (e.g., FC-A) as a positive control.
- Incubation: Add the TR-FRET detection mix (Streptavidin-XL665 and Eu3+-anti-14-3-3 antibody). Incubate the plate in the dark for 1-2 hours at room temperature.
- TR-FRET Measurement: Read the plate on a TR-FRET-compatible microplate reader. Excite at 337 nm and measure the emission at 620 nm (Eu3+ signal) and 665 nm (XL665 signal).
- Data Analysis: Calculate the TR-FRET ratio (665 nm / 620 nm * 10,000). Plot the ratio against the log of compound concentration and fit the data to a four-parameter logistic equation to determine the EC50 value for each compound.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents and Tools for Scaffold Hopping and PPI Stabilization Research
| Reagent / Tool | Function / Description | Application in Case Study |
|---|---|---|
| AnchorQuery Software | Pharmacophore-based virtual screening platform for MCR chemistry. | Identified the novel GBB-based imidazo[1,2-a]pyridine scaffold. |
| Groebke-Blackburn-Bienaymé (GBB) Chemistry | A three-component reaction between an aldehyde, 2-aminopyridine, and an isocyanide. | Enabled the rapid, divergent synthesis of the novel scaffold with multiple points for variation. |
| TR-FRET Assay Kits | Homogeneous assay for detecting biomolecular interactions in a microplate format. | Quantified the stabilization of the 14-3-3/ERα complex in a biochemical setting. |
| NanoBRET Assay System | Cell-based assay to monitor PPIs in live cells using bioluminescence energy transfer. | Confirmed target engagement and PPI stabilization in a physiologically relevant, cellular environment. |
| Crystallography Platform | Determines the 3D atomic structure of protein-ligand complexes. | Provided the critical binding pose of the original lead (compound 127) to guide computational design and validated the binding mode of optimized analogs. |
Visualized Workflows and Signaling Pathways
Scaffold Hopping & Validation Workflow
14-3-3/ERα Stabilization Mechanism
Conclusion
Scaffold hopping, powered by the rich structural diversity of natural products, has evolved from a concept reliant on medicinal chemistry intuition to a data-driven discipline supercharged by AI. The integration of holistic molecular representations and deep learning models enables a more efficient exploration of chemical space, leading to the discovery of novel, isofunctional compounds with improved pharmacological profiles. Successful outcomes depend on a nuanced balance—maintaining critical pharmacophoric elements and 3D shape while achieving significant 2D structural novelty. As these computational methodologies continue to mature, their ability to generalize across new targets and generate synthetically accessible leads will be crucial. The future of natural product-based drug design lies in the seamless fusion of these advanced in silico techniques with experimental validation, accelerating the delivery of new therapeutics for complex diseases like cancer and drug-resistant infections.
References
- 1. A Historical Overview of Natural Products in Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901206/]
- 2. Revitalizing natural products for sustainable drug Discovery [https://www.sciencedirect.com/science/article/abs/pii/S2352554125001809]
- 3. The latest advances with natural products in drug discovery ... [https://pubmed.ncbi.nlm.nih.gov/40391763/]
- 4. Natural products in drug discovery: advances and ... [https://www.nature.com/articles/s41573-020-00114-z]
- 5. Natural Bioactive Compounds and Human Health - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11279386/]
- 6. Innate functions of natural products: A promising path for ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523423007158]
- 7. Biological Activities of Natural Products - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730830/]
- 8. Molecular medicinal insights into scaffold hopping-based ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644623003616]
- 9. Classification of Scaffold Hopping Approaches - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3328312/]
- 10. Scaffold Hopping in Tuberculosis Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC12557394/]
- 11. Scaffold Hopping [https://www.biosolveit.de/application-academy/scaffold-hopping/]
- 12. What does scaffold hopping mean to you? - Cresset Group [https://cresset-group.com/science/science-resources/what-does-scaffold-hopping-mean-to-you/]
- 13. Deep scaffold hopping with multimodal transformer neural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8590293/]
- 14. Rice chemists leap across terpenoid landscapes with ... [https://news.rice.edu/news/2025/rice-chemists-leap-across-terpenoid-landscapes-enzyme-enabled-scaffold-hopping]
- 15. Scaffold-hopping for molecular glues targeting the 14-3-3/ ... [https://www.nature.com/articles/s41467-025-61176-4]
- 16. Scaffold Hunter: a comprehensive visual analytics framework ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-017-0213-3]
- 17. ScaffoldGVAE: scaffold generation and hopping of drug ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-023-00766-0]
- 18. Classification of scaffold-hopping approaches [https://www.sciencedirect.com/science/article/abs/pii/S1359644611003813]
- 19. Recent advances in molecular representation methods and ... [https://www.nature.com/articles/s44386-025-00017-2]
- 20. Scaffold-hopping for molecular glues targeting the 14-3-3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12260087/]
- 21. Bridging chemical space and biological efficacy: advances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144013/]
- 22. Synthesis of diverse terpenoid frameworks via enzyme ... [https://pubmed.ncbi.nlm.nih.gov/40523914/]
- 23. Enzyme-Driven Scaffold Hopping Creates Diverse ... [https://bioengineer.org/enzyme-driven-scaffold-hopping-creates-diverse-terpenoids/]
- 24. Scaffold hopping from natural products to synthetic ... [https://www.nature.com/articles/s42004-018-0043-x]
- 25. Applications of the Pharmacophore Concept in Natural ... Product [https://pmc.ncbi.nlm.nih.gov/articles/PMC7685156/]
- 26. Effectiveness of molecular fingerprints for exploring the ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00830-3]
- 27. - Scaffold from Synthetic Hopping by Holistic Drugs ... Molecular [https://www.nature.com/articles/s41598-018-34677-0?error=cookies_not_supported&code=781a57dd-0201-4eee-97ee-e3bafd25f0fe]
- 28. Deep scaffold with multimodal transformer neural networks hopping [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-021-00565-5]
- 29. : advances, limitations, and current utility in Pharmacophore modeling [https://www.dovepress.com/pharmacophore-modeling-advances-limitations-and-current-utility-in-dru-peer-reviewed-fulltext-article-JRLCR]
- 30. -Based Scaffold • BioSolveIT Drug Design [https://www.biosolveit.de/drug-discovery-solutions/scaffold-based-drug-design/]
- 31. Molecular Scaffold Hopping via Holistic ... [https://pubmed.ncbi.nlm.nih.gov/33759119/]
- 32. Scaffold-Hopping from Synthetic Drugs by Holistic ... [https://www.nature.com/articles/s41598-018-34677-0]
- 33. Repurposing drugs for the human dopamine transporter ... [https://pubmed.ncbi.nlm.nih.gov/40893439/]
- 34. Reinventing Drug Discovery with AI De Novo Design [https://ai.creative-biolabs.com/blog/reinventing-drug-discovery-ai-de-novo-design/]
- 35. A Comprehensive Benchmark For Graph Transformers [https://arxiv.org/html/2506.04765v1]
- 36. Graph neural networks driven acceleration in drug discovery [https://www.sciencedirect.com/science/article/pii/S2211383525006884]
- 37. ChemBounce: a computational framework for scaffold hopping ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12449259/]
- 38. AI-First Drug Design [https://www.emergentmind.com/topics/ai-first-drug-design]
- 39. Protein kinases as antituberculosis targets: The case of ... [https://pubmed.ncbi.nlm.nih.gov/32114448/]
- 40. Protein kinases as antituberculosis targets: The case of ... [https://www.imrpress.com/journal/FBL/25/9/10.2741/4871/htm]
- 41. Identification of kinase inhibitors as potential host-directed ... [https://www.nature.com/articles/s41598-024-68102-6]
- 42. Identification of shikimate kinase inhibitors among anti- ... [https://www.sciencedirect.com/science/article/abs/pii/S1472979213002229]
- 43. Quantitative Data Analysis for In Vivo Screening [https://www.phdassistance.com/academy/quantitative-data-analysis-in-vivo-screening/]
- 44. Natural products and drug discovery: a survey of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4620409/]
- 45. Review on natural products databases: where to find data in ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00424-9]
- 46. How to nurture natural products to create new therapeutics ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644624003465]
- 47. Optimizing Pharmacokinetics-Pharmacodynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7372216/]
- 48. A New Approach to Reduce Toxicities and to Improve ... [https://www.nature.com/articles/srep10881]
- 49. Artificial Intelligence for Structural Modification of Natural ... [https://communities.springernature.com/posts/artificial-intelligence-for-structural-modification-of-natural-products]
- 50. Scaffold Hopping? It's Complicated [http://practicalcheminformatics.blogspot.com/2018/08/scaffold-hopping-its-complicated.html]
- 51. Deep Generative Models for 3D Linker Design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7189367/]
- 52. From Lab to Clinic: How Artificial Intelligence (AI) Is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298131/]
- 53. Beyond Legacy Tools: Defining Modern AI Drug Discovery ... [https://www.biopharmatrend.com/business-intelligence/what-is-ai-drug-discovery/]
- 54. Artificial intelligence revolution in drug discovery [https://www.sciencedirect.com/science/article/pii/S037851732500626X]
- 55. Traditional Drug Development Process - The Actuary Magazine [https://www.theactuarymagazine.org/traditional-drug-development-process/]
- 56. Artificial Intelligence in Cancer Drug Discovery in 2025 [https://oncodaily.com/oncolibrary/artificial-intelligence-in-cancer-drug-discovery]
- 57. New Approach Methodologies (NAMs): The Future of ... [https://emulatebio.com/new-approach-methodologies-nams-the-future-of-toxicology-and-safety-testing/]
- 58. Molecular similarity: Theory, applications, and perspectives [https://www.sciencedirect.com/science/article/pii/S2949747724000356]
- 59. Rapid Identification of Potential Drug Candidates from Multi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8468386/]
- 60. A similarity-based target prediction tool for natural products [https://www.sciencedirect.com/science/article/abs/pii/S0010482524014367]
- 61. Scope of 3D Shape-Based Approaches in Predicting the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7312400/]
- 62. Target-specific compound selectivity for multi-target drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9549418/]
- 63. Scaffold-Hopping Strategies in Aurone Optimization [https://www.mdpi.com/1420-3049/29/12/2813]
- 64. Scaffold Hopping Software Spark - Cresset Group [https://cresset-group.com/software/spark/]
- 65. ChemBounce: a computational framework for scaffold ... [https://pubmed.ncbi.nlm.nih.gov/40973478/]
- 66. SeeSAR • Everyday Drug Design Dashboard [https://www.biosolveit.de/products/seesar/]
- 67. Bridging chemical space and biological efficacy: advances ... [https://link.springer.com/article/10.1007/s13659-025-00521-y]
- 68. Advanced Models for Predicting Pharma Stock ... [https://www.drugpatentwatch.com/blog/advanced-models-for-predicting-pharma-stock-performance-in-the-face-of-patent-expiration/?srsltid=AfmBOoqcwJzldMe9RmOEgTcdGv_E4D_51S-PzRj_NxLUDSdHtjC_QhYa]