Natural Product Drug Discovery in 2025: A Comprehensive Guide to Principles, AI Integration, and Future Opportunities
This article provides a comprehensive overview of the modern principles of natural product (NP) drug discovery, tailored for researchers, scientists, and drug development professionals.
Natural Product Drug Discovery in 2025: A Comprehensive Guide to Principles, AI Integration, and Future Opportunities
Abstract
This article provides a comprehensive overview of the modern principles of natural product (NP) drug discovery, tailored for researchers, scientists, and drug development professionals. It explores the renewed importance of NPs as a source of novel therapeutics, covering their historical foundations and exceptional structural diversity. The scope extends to the latest methodological advances, including the pivotal role of artificial intelligence (AI), in silico screening, and integrated workflows that are accelerating lead identification and optimization. The content also addresses key technical and regulatory challenges, offering strategies for troubleshooting and optimization. Finally, it examines contemporary validation techniques and the proven success of NPs in addressing complex diseases, particularly in oncology and antiparasitic therapy, synthesizing these insights to outline a forward-looking perspective for the field.
The Enduring Power of Nature's Pharmacy: History, Significance, and Chemical Diversity
The integration of natural products into modern therapeutic development represents a critical pathway for addressing complex medical challenges. This whitepaper examines the evolution from traditional medicine practices to evidence-based drug discovery, highlighting the continued relevance of natural products in contemporary pharmaceutical research. We present quantitative data on natural product applications, detailed experimental methodologies for their investigation, and visualization of key workflows that enable researchers to bridge traditional knowledge with modern scientific validation. Within the broader thesis of natural product drug discovery principles, this analysis demonstrates how ancient therapeutic wisdom, when investigated through rigorous scientific frameworks, continues to yield transformative treatments for malignancies, infectious diseases, and other conditions with significant unmet medical needs.
Natural products (NPs) and traditional medicinal knowledge have served as cornerstone resources in healthcare for millennia, with over 80% of the world's population relying on some form of traditional, complementary, and integrative medicine (TCIM) for primary health needs [1]. The World Health Organization recognizes the significant role of Traditional Medicine (TM) in global healthcare, noting that most member states have requested guidance on integrating evidence-based TCIM services into their healthcare systems [2]. This historical legacy continues to inform modern drug discovery, with natural products comprising approximately 41% (646/1562) of all new drug approvals between 1981 and 2014 [3].
The pharmaceutical industry's renewed interest in natural products stems from their unique structural characteristics that distinguish them from traditional small-molecule drug candidates. Natural products typically exhibit higher molecular mass, greater molecular rigidity, diverse chemical nature, and unique spatial arrangements including a greater number of sp³ carbon atoms [4]. These properties enhance their ability to target protein-protein interactions—a challenging area for conventional small molecules. Despite advances in combinatorial chemistry and computational design, natural products remain indispensable sources of novel bioactive compounds, with approximately 50% of current antibiotics derived from natural origins [4].
Historical Foundations and Contemporary Relevance
From Traditional Knowledge to Approved Therapeutics
Traditional healing systems have provided the initial observations that led to many foundational therapeutics. The journey from traditional remedy to approved pharmaceutical follows a consistent pattern: ethnobotanical observation → bioactivity confirmation → isolation of active compounds → structural modification → clinical development [5]. This pathway is exemplified by several landmark drugs:
Morphine, isolated from Papaver somniferum (opium poppy) in 1817, represents the first plant natural product to be isolated and remains a critical analgesic [4]. Paclitaxel, originally derived from Taxus brevifolia (Pacific yew), has become an essential chemotherapeutic for ovarian, breast, and lung cancers [5] [4]. The antimalarial artemisinin, discovered through investigation of traditional Chinese herbal remedies, has saved millions of lives worldwide [4].
Table 1: Historical Natural Products and Their Therapeutic Applications
| Natural Product | Natural Source | Traditional Use | Modern Therapeutic Application | Discovery Timeline |
|---|---|---|---|---|
| Morphine | Papaver somniferum (Opium poppy) | Analgesic | Severe pain management | Isolated 1817 |
| Quinine | Cinchona officinalis bark | Fever reducer | Malaria treatment | Early 19th century |
| Salicin | Salix babylonica (Willow tree) | Pain, inflammation | Aspirin (acetylsalicylic acid) | Isolated 1828 |
| Paclitaxel | Taxus brevifolia (Pacific yew) | Not documented | Chemotherapy agent | Approved 1993 |
| Artemisinin | Artemisia annua | Fever | Malaria treatment | Isolated 1972 |
Quantitative Significance in Modern Medicine
Recent analyses confirm the continued importance of natural products in addressing contemporary health challenges. The NPASS database (2023 update) now contains quantitative activity data for approximately 43,200 natural products against ~7,700 biological targets, representing a 40% and 32% increase respectively from previous versions [6]. This expanding repository reflects the growing research interest in characterizing the therapeutic potential of natural compounds.
Cancer research has particularly benefited from natural product investigation. Globally, malignancies cause one in six mortalities, creating an urgent need for innovative therapeutic approaches [5]. Natural products like Vincristine and Vinblastine (isolated from Catharanthus roseus) have demonstrated profound efficacy in managing leukemia and Hodgkin's disease, establishing the critical role of plant-derived compounds in oncology [5]. The development of Trabectedin, originally isolated from the tunicate Ecteinascidia turbinata and approved by the FDA in 2015, further exemplifies how marine natural products can yield effective anticancer medications [4].
Table 2: Quantitative Data on Natural Product Applications in Drug Discovery
| Parameter | Historical Data | Current Status (2023-2025) | Significance |
|---|---|---|---|
| NP-derived FDA approvals (1981-2014) | 41% of all new drug approvals | N/A | Demonstrates historical impact |
| NPs with documented activity data | N/A | ~43,200 compounds | 40% increase from previous database |
| Molecular targets for NPs | N/A | ~7,700 targets | 32% increase from previous database |
| Species sources documented | N/A | ~94,400 NPs with species sources | 32% increase from previous database |
| Global population using TCIM | N/A | >80% in both low and high-income countries [1] | Drives continued research interest |
Modern Methodologies: From Field Collection to Clinical Application
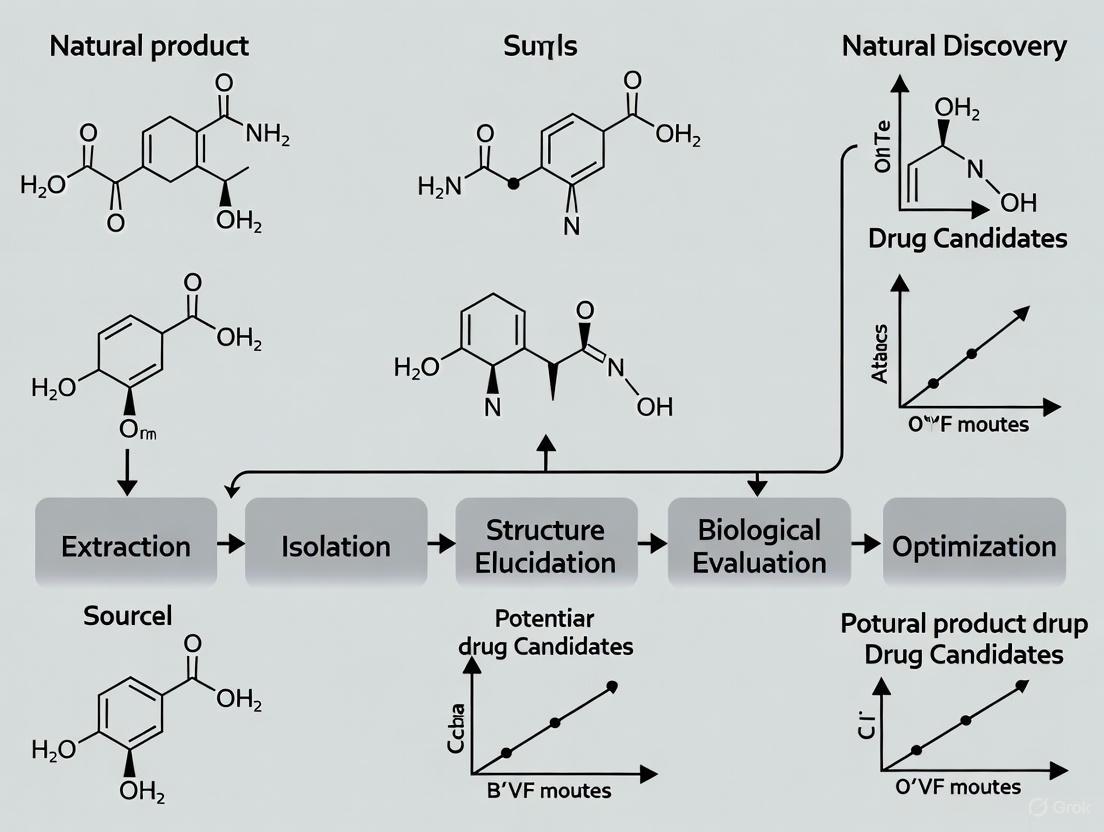
Integrated Discovery Workflow
The process of translating natural products from traditional remedies to modern therapeutics requires a multidisciplinary approach that integrates field biology, ethnopharmacology, analytical chemistry, and molecular biology. The following diagram illustrates the comprehensive workflow:
Experimental Protocols for Natural Product Research
Bioactivity-Guided Fractionation Protocol
Objective: To isolate and identify bioactive compounds from crude natural extracts through iterative fractionation and biological screening.
Materials:
- Natural source material (plant, marine organism, microbial culture)
- Extraction solvents (methanol, ethanol, ethyl acetate, water)
- Chromatography media (silica gel, C18, Sephadex LH-20)
- Bioassay systems (cellular, enzymatic, or phenotypic assays)
Methodology:
- Sample Preparation: Air-dry and pulverize source material. Perform sequential extraction with solvents of increasing polarity (hexane → ethyl acetate → methanol → water).
- Primary Screening: Test crude extracts in relevant bioassays (e.g., cytotoxicity, antimicrobial activity, enzyme inhibition). Select active extracts for further investigation.
- Fractionation: Subject active crude extract to open-column chromatography or vacuum-liquid chromatography using appropriate stationary phases. Collect fractions based on TLC or HPLC monitoring.
- Secondary Screening: Test all fractions against the same bioassay to identify active fractions.
- Compound Isolation: Purify active fractions using techniques including preparative TLC, HPLC, or counter-current chromatography. Monitor purification using analytical HPLC.
- Structural Elucidation: Employ spectroscopic methods including NMR (¹H, ¹³C, 2D experiments), mass spectrometry, and X-ray crystallography to determine complete chemical structure.
- Structure-Activity Relationship (SAR) Studies: Synthesize structural analogs to identify critical functional groups for bioactivity [5] [4].
Computational Screening Protocol for Natural Product Libraries
Objective: To prioritize natural products for experimental testing through in silico prediction of bioactivity, drug-likeness, and target engagement.
Materials:
- Natural product compound libraries (e.g., NPASS database)
- Molecular docking software (AutoDock Vina, Glide, GOLD)
- ADMET prediction tools (SwissADME, admetSAR)
- Cheminformatics platforms (RDKit, OpenBabel)
Methodology:
- Library Curation: Compile natural product structures from databases such as NPASS, which contains >94,400 compounds with species source information [6].
- Virtual Screening: Perform molecular docking against protein targets of therapeutic interest. Use consensus scoring to improve prediction reliability.
- Drug-Likeness Evaluation: Apply filters including Lipinski's Rule of Five, Veber's rules, and quantitative estimate of drug-likeness (QED).
- ADMET Profiling: Predict absorption, distribution, metabolism, excretion, and toxicity properties using in silico tools.
- Network Pharmacology Analysis: Construct compound-target-pathway networks to identify potential polypharmacology and mechanistic pathways.
- Hit Prioritization: Rank compounds based on integrated scores combining docking scores, drug-likeness, and ADMET properties [5] [7] [3].
Table 3: Essential Research Reagents and Databases for Natural Product Drug Discovery
| Resource Category | Specific Tools/Reagents | Function/Application | Key Features |
|---|---|---|---|
| Natural Product Databases | NPASS Database [6] | Quantitative activity and species source data | ~43,200 NPs with activity data, ~94,400 with species sources |
| CMAUP Database [6] | Collective molecular activities of useful plants | Plant-specific bioactivity data | |
| Analytical Instruments | High-Resolution Mass Spectrometry (HRMS) [4] | Precise molecular weight and structural information | High mass accuracy for compound identification |
| NMR Spectroscopy [4] | Structural elucidation of complex natural products | 1D and 2D experiments for complete structure determination | |
| Liquid Chromatography Systems [4] | Compound separation and purification | HPLC, UHPLC for analytical and preparative applications | |
| Bioassay Systems | High-Throughput Screening (HTS) Platforms [8] | Rapid bioactivity assessment of compound libraries | Automated screening of thousands of compounds |
| Target-Based Assays [5] | Specific molecular target engagement | Enzyme inhibition, receptor binding assays | |
| Phenotypic Screening [5] | Functional responses in cellular systems | Cell viability, antimicrobial activity, functional changes | |
| Computational Tools | Molecular Docking Software [5] | Predicting protein-ligand interactions | Virtual screening of natural product libraries |
| ADMET Prediction Tools [5] | In silico pharmacokinetic and toxicity assessment | Early elimination of problematic compounds | |
| Chemical Similarity Tools [6] | Estimating bioactivity based on structural similarity | Chemical Checker for activity prediction |
Advanced Research Applications and Case Studies
Natural Products in Targeted Cancer Therapy
Natural products have found innovative applications in advanced therapeutic modalities, particularly in targeted cancer therapy. The development of antibody-drug conjugates (ADCs) represents a paradigm shift in oncology treatment, combining the specificity of monoclonal antibodies with the potency of natural product-derived cytotoxic agents [8]. This approach minimizes systemic toxicity while maximizing tumor cell killing.
The diagram below illustrates how natural products are integrated into targeted therapeutic platforms:
Emerging Technologies and Future Perspectives
The field of natural product drug discovery is undergoing rapid transformation through the integration of artificial intelligence (AI) and machine learning. These technologies are addressing historical challenges in NP research, including structural complexity, limited supply, and unknown mechanisms of action [8] [7]. AI algorithms can now predict bioactive natural products by analyzing structural similarities to known active compounds, significantly accelerating the discovery process [7] [9].
The emerging paradigm of data-driven drug discovery leverages large-scale repositories like the NPASS database to generate therapeutic hypotheses systematically rather than relying solely on traditional bioassay-guided approaches [3]. This methodology combines cheminformatics (molecular characterization), bioinformatics (target prediction), and knowledge engineering (literature mining) to create a comprehensive framework for identifying promising natural product candidates [3].
Future directions in natural product research include the development of advanced cultivation techniques for previously unculturable microorganisms, application of synthetic biology for sustainable production of complex natural products, and implementation of blockchain technology for ensuring ethical sourcing and equitable benefit-sharing with indigenous communities [1] [2]. The WHO's establishment of the Global Centre for Traditional Medicine in Jamnagar, India, further signals the growing recognition of natural products' role in addressing global health challenges [2].
Natural products (NPs) and their structural analogues have historically served as a major source of pharmacotherapeutic agents, particularly for cancer and infectious diseases [10]. These compounds, derived from terrestrial plants, marine organisms, and microorganisms, represent a critical foundation of drug discovery, providing unique structural diversity and biological pre-validation that synthetic libraries often lack [11]. The historical use of natural products in traditional medicine systems, documented since 2600 B.C. in Mesopotamia and ancient Egypt, provided the initial leads for many scientifically validated therapeutic agents [11]. Despite a temporary decline in interest from the pharmaceutical industry from the 1990s onward due to technical challenges in screening and optimization, recent technological advancements have revitalized NP-based drug discovery, making this field more relevant than ever for addressing contemporary medical challenges [10].
This whitepaper provides a comprehensive quantitative analysis of the substantial contribution of natural products to approved drugs, framing this discussion within the core principles of natural product drug discovery research. Through systematic data compilation and methodological exposition, we aim to provide researchers, scientists, and drug development professionals with both historical context and forward-looking perspectives on NP-based drug discovery. The integration of advanced methodologies—including artificial intelligence, high-throughput screening, chemical biology, and bioinformatics—is now positioning natural products for continued impact in tackling unmet medical needs, including antimicrobial resistance and complex chronic diseases [8].
Quantitative Analysis of Natural Product-Derived Pharmaceuticals
Historical and Contemporary Impact
The quantitative contribution of natural products to the pharmaceutical landscape is substantial and enduring. Analysis of the FDA-approved drug portfolio reveals that approximately 50% of all approved drugs are natural products or natural product derivatives [12]. This figure underscores the critical role that NPs continue to play in pharmacotherapy despite the rise of combinatorial chemistry and synthetic biology approaches. Between 1981 and 2014, natural products remained significant sources of new drugs, with particularly strong representation in anti-cancer and anti-infective categories [10].
The plant kingdom accounts for the majority of known natural products, with approximately 70% of compounds recorded in the Dictionary of Natural Products originating from botanical sources [13]. Certain plant families have been exceptionally prolific; the Leguminosae family alone has contributed 44 products that have received regulatory approval or advanced to clinical development [13]. As of December 2004, the FDA approved the first marine-derived compound, ziconotide intrathecal infusion (Prialt), for severe pain management, followed by the European Union's approval of trabectedin (Yondelis) in October 2007 as the first marine-derived anticancer drug [13]. These milestones highlight the expanding diversity of NP sources with therapeutic potential.
Table 1: Natural Product Contributions to Major Therapeutic Areas
| Therapeutic Area | Percentage of NP-Derived Drugs | Representative Examples | Year of First Approval |
|---|---|---|---|
| Anti-infectives | >60% | Penicillins, Tetracyclines | 1940s |
| Anticancer Agents | ~40% | Paclitaxel, Trabectedin | 1990s |
| Immunosuppressants | ~30% | Cyclosporine, Rapamycin | 1980s |
| Central Nervous System | ~25% | Morphine, Reserpine | 1800s/1950s |
Molecular Properties and Drug-Likeness
Comparative analysis of molecular properties between natural products and FDA-approved drugs reveals both similarities and strategically important differences. The Universal Natural Products Database (UNPD), containing 197,201 unique natural products, provides a robust dataset for such comparisons [12]. Statistical analysis demonstrates that natural products exhibit larger mean values and standard deviations for key molecular descriptors compared to synthetic drugs, indicating greater structural complexity and diversity [12].
When evaluated against Lipinski's "Rule of Five" criteria for drug-likeness, 52.0% (102,605) of natural products in the UNPD comply with all four parameters, while 71.8% (141,628) satisfy at least three criteria [12]. This is comparable to the 77.17% of FDA-approved drugs that fully comply with the Rule of Five [12]. The chemical space occupied by natural products shows significant overlap with approved drugs but extends into regions associated with complex polypharmacology, providing opportunities for addressing challenging biological targets [12].
Table 2: Molecular Property Comparison: Natural Products vs. Approved Drugs
| Molecular Property | Natural Products (Mean) | FDA-Approved Drugs (Mean) | Statistical Significance |
|---|---|---|---|
| Molecular Weight | 300-350 Da interval | 250-300 Da interval | p < 0.01 |
| AlogP | Wider distribution | Narrower distribution | p < 0.01 |
| Hydrogen Bond Donors | 3.21 ± 2.34 | 2.85 ± 2.16 | p < 0.05 |
| Hydrogen Bond Acceptors | 6.02 ± 3.87 | 5.45 ± 3.24 | p < 0.05 |
| Rotatable Bonds | 4.78 ± 3.95 | 4.12 ± 3.56 | p < 0.05 |
| Aromatic Rings | 1.82 ± 1.45 | 2.15 ± 1.32 | p < 0.01 |
Methodological Framework for Natural Product Drug Discovery
Experimental Workflows and Protocols
The systematic investigation of natural products for drug discovery follows established workflows that integrate traditional knowledge with modern technological approaches. The standard methodology encompasses bioresource selection, extraction, bioactivity-guided fractionation, compound identification, and lead optimization [13].
Bioresource Selection and Authentication: The process begins with careful selection and taxonomic authentication of source material. Traditional medicinal knowledge often guides initial source selection, with the Ebers Papyrus (2900 B.C.) and Chinese Materia Medica (1100 B.C.) providing historical precedents for plant-based medicines [11]. Contemporary approaches combine this ethnobotanical knowledge with ecological considerations and biodiversity surveys.
Extraction and Fractionation: Sequential extraction using solvents of increasing polarity (hexane, ethyl acetate, methanol, water) provides preliminary fractionation based on compound polarity [12]. Advanced extraction techniques including supercritical fluid extraction, pressurized liquid extraction, and microwave-assisted extraction have improved efficiency and yield while reducing environmental impact [13]. The crude extract is typically subjected to bioactivity-guided fractionation using chromatographic methods including vacuum liquid chromatography (VLC), flash chromatography, and eventually high-performance liquid chromatography (HPLC).
Bioactivity Screening: Extracts and fractions are screened against relevant biological targets using in vitro assays. Modern approaches employ high-throughput screening (HTS) platforms, with recent technological developments enabling more efficient NP screening [10]. Phenotypic screening remains particularly valuable for NP discovery, as it detects bioactivity without requiring prior knowledge of specific molecular targets [10].
Structure Elucidation: Bioactive compounds are characterized using advanced analytical techniques, primarily hyphenated systems that combine separation with spectroscopic detection. Key methodologies include:
- LC-MS (Liquid Chromatography-Mass Spectrometry) for molecular weight and fragmentation pattern analysis
- LC-NMR (Liquid Chromatography-Nuclear Magnetic Resonance) for structural information
- HRMS (High-Resolution Mass Spectrometry) for precise molecular formula determination
- MS/MS (Tandem Mass Spectrometry) for structural elucidation through fragmentation patterns
Recent advances in analytical technologies, particularly improved NMR and mass spectrometry instrumentation, have significantly enhanced the pace and accuracy of NP structure elucidation [10].
Diagram 1: NP Drug Discovery Workflow
Dereplication Strategies
Dereplication represents a critical step in NP research to avoid redundant rediscovery of known compounds. This process combines chemical screening with database mining to rapidly identify previously characterized molecules [10]. Modern dereplication approaches utilize:
- Hyphenated Techniques: LC-UV-MS and LC-MS-SPE-NMR systems provide comprehensive chemical profiles
- Spectroscopic Databases: Mass spectral and NMR libraries enable rapid compound identification
- Cytoscape Analysis: Network visualization tools map compound-target relationships based on existing data
- In Silico Prediction: Computational models predict potential bioactivity based on structural features
The integration of these dereplication strategies has significantly improved the efficiency of NP discovery programs by focusing resources on novel chemical entities with potential therapeutic value [10].
Technological Advances Revolutionizing Natural Product Research
Analytical and Computational Innovations
Recent technological developments have addressed historical bottlenecks in NP-based drug discovery, revitalizing interest in this field [10]. Key advancements include:
Advanced Analytical Technologies: Hyphenated spectroscopic techniques have dramatically improved the pace and accuracy of NP structure elucidation. LC-HRMS-NMR systems now provide comprehensive structural information from limited quantities of material [10]. The implementation of Ultra High-Pressure Liquid Chromatography (UHPLC) has enhanced separation efficiency, while new mass spectrometry approaches like ion mobility provide additional structural dimensions [10].
Genome Mining and Engineering: The ability to sequence and analyze the genomes of NP-producing organisms has revealed numerous cryptic biosynthetic gene clusters encoding potential novel compounds [10]. Heterologous expression and pathway engineering enable production of complex NPs in more tractable host organisms, addressing supply challenges associated with rare source organisms [10].
Artificial Intelligence and Machine Learning: AI algorithms now assist in multiple aspects of NP discovery, from predicting biosynthetic pathways to identifying potential molecular targets [8] [13]. Machine learning models trained on known NP structures and activities can prioritize compounds for experimental evaluation, accelerating the discovery process [8].
Chemical Biology Tools: Advanced target identification methods, including the highly accurate non-labeling chemical proteomics approach, enable deconvolution of NP mechanisms of action [8]. These techniques are particularly valuable for NPs with complex polypharmacology.
Emerging Therapeutic Modalities
Natural products are finding new applications in emerging therapeutic modalities, most notably as payloads in antibody-drug conjugates (ADCs) [8]. The complex mechanism of action and high potency of many NPs make them ideal warheads for targeted delivery systems. Additionally, NP-derived hybrid molecules that combine natural scaffolds with synthetic elements represent a promising strategy for addressing complex diseases through multi-target engagement [8].
Diagram 2: Technological Advances in NP Research
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Essential Research Reagents and Platforms for NP Drug Discovery
| Category | Specific Tools/Reagents | Function/Application | Key Advances |
|---|---|---|---|
| Separation Media | Sephadex LH-20, C18 silica, DIAION HP-20 | Size-exclusion and reverse-phase chromatography for compound isolation | Improved resolution and recovery rates |
| Analytical Standards | Natural product libraries (e.g., UNPD, CNPD) | Dereplication and compound identification | Expanded coverage of chemical space |
| Bioassay Systems | Cell-based phenotypic assays, enzyme inhibition assays | High-throughput bioactivity screening | Miniaturization and automation |
| Spectroscopic Platforms | LC-HRMS-NMR, UPLC-QTOF-MS, MS/MS | Structural characterization and elucidation | Enhanced sensitivity and resolution |
| Bioinformatics Tools | Cytoscape, GNPS, AntiMarin | Network pharmacology and target prediction | Integration of multi-omics data |
| Biosynthetic Tools | Heterologous expression systems, CRISPR-Cas9 | Pathway engineering and production optimization | Enables production of cryptic metabolites |
| Feruloylputrescine | Feruloylputrescine, CAS:501-13-3, MF:C14H20N2O3, MW:264.32 g/mol | Chemical Reagent | Bench Chemicals |
| Lactobionic Acid | Lactobionic Acid Reagent|C12H22O12|96-82-2 | High-purity Lactobionic Acid for research. Explore applications in biochemistry, cell culture, and preservative science. For Research Use Only (RUO). Not for human use. | Bench Chemicals |
The Universal Natural Products Database (UNPD), comprising 197,201 natural products from plants, animals, and microorganisms, represents one of the largest non-commercial freely available databases for natural products research [12]. This resource, combined with commercial compound libraries, provides essential reference materials for dereplication and identification workflows. Modern NP research increasingly relies on integrated platforms that combine multiple analytical techniques with computational tools for comprehensive metabolite profiling [10].
Network pharmacology approaches have gained prominence in NP research, recognizing that many natural products exert their therapeutic effects through multi-target mechanisms rather than single-target interactions [12]. Tools like Cytoscape enable visualization and analysis of complex compound-target-disease networks, providing insights into polypharmacology and systems-level effects of NP interventions [12].
Natural products continue to make substantial contributions to approved drugs, with approximately half of FDA-approved pharmaceuticals originating from natural sources or their derivatives [12]. This quantitative impact underscores the enduring importance of NPs in addressing human disease, particularly in therapeutic areas like oncology and infectious diseases where structural complexity and specific bioactivity are paramount [10]. The historical success of NPs is now being amplified by technological innovations across the discovery pipeline, from genome-informed sourcing to AI-accelerated screening and optimization [8].
Future NP research will be characterized by increased integration of multidisciplinary approaches, with chemical biology, synthetic biology, and computational methods playing expanding roles [8]. The application of highly accurate non-labeling chemical proteomics will enhance target identification for complex NPs, while continued development of expression platforms will address supply challenges [8]. Furthermore, the growing understanding of NP biosynthetic pathways enables bioengineering approaches to expand chemical diversity and optimize therapeutic properties [10].
For researchers and drug development professionals, the principles of natural product drug discovery research remain foundational: respect for traditional knowledge, commitment to rigorous scientific validation, and openness to technological innovation. By embracing these principles while leveraging emerging tools and methodologies, the scientific community can continue to harness the substantial potential of natural products to address evolving medical needs and deliver novel therapeutics to patients worldwide.
Natural products (NPs) exhibit exceptional structural diversity that enables them to occupy chemical spaces largely inaccessible to synthetic compounds. This review examines the distinctive structural complexity, diverse biosynthetic origins, and broad bioactivity of NPs through a chemoinformatics lens. With over 1.1 million documented compounds, NPs display remarkable molecular complexity, frequent glycosylation, and unique halogenation patterns that contribute to their privileged status in drug discovery. Analysis reveals that over 50% of approved small-molecule drugs originate directly or indirectly from natural products, underscoring their profound pharmaceutical significance. This comprehensive analysis synthesizes current understanding of NP chemical space, providing researchers with methodological frameworks for exploring this diversity and highlighting emerging opportunities in pseudo-natural product design and extreme environment bioprospecting.
Natural products represent the evolved chemical defense, signaling, and regulatory systems of living organisms that have been refined through millions of years of evolutionary selection. This extensive optimization process has yielded compounds with exceptional structural diversity and biological relevance. The chemical space of NPs is distinguished by greater structural complexity compared to synthetic molecules, featuring more stereocenters, higher molecular rigidity, and increased oxygen content [14]. These characteristics enable NPs to interact with diverse biological targets through complex three-dimensional binding modes that often evade synthetically designed compounds.
The pharmaceutical significance of NPs is substantial, with analyses indicating that 39% of marketed drugs originate from natural products and their derivatives—comprising 10% unaltered natural products and 29% semi-synthetic derivatives [15]. Between 1981 and 2014, over 50% of newly developed drugs were developed from NPs, particularly in therapeutic areas including oncology, infectious diseases, and cardiovascular disorders [16] [17]. This remarkable success rate stems from the evolutionary optimization of NPs for biological interactions, positioning them as invaluable resources for drug discovery scaffolds and lead compounds.
Structural Characteristics of Natural Products
Molecular Complexity and Three-Dimensionality
Natural products exhibit substantially greater molecular complexity than their synthetic counterparts, a characteristic that directly influences their biological activity and target selectivity. Statistical analyses reveal that NPs contain a higher density of stereogenic centers, with approximately 70-91% of compounds in various databases containing defined stereochemistry [16] [17]. This stereochemical complexity enables sophisticated three-dimensional binding to biological targets, a property increasingly recognized as crucial for drug specificity.
The ring systems in NPs demonstrate exceptional diversity and complexity compared to synthetic compounds. Natural products frequently incorporate polycyclic systems with bridgehead atoms and complex ring fusions that create rigid, pre-organized three-dimensional structures [14]. This structural rigidity reduces the entropic penalty upon binding to biological targets, enhancing binding affinity and specificity. Additionally, NPs exhibit a wider variety of ring sizes and heteroatom incorporations, contributing to their distinct pharmacophoric properties.
Functional Group Diversity and Modifications
Natural products display distinctive patterns of functionalization that contribute to their biological activities and physicochemical properties. One particularly notable characteristic is their high oxygen content relative to synthetic compounds, manifested through abundant hydroxyl, carbonyl, and ether functionalities [14]. This oxygen-rich composition enhances hydrogen-bonding capacity and influences solubility profiles.
Glycosylation represents another distinctive feature of NPs, with approximately 30% of all natural products containing sugar moieties [14]. These carbohydrate modifications profoundly influence bioavailability, target recognition, and pharmacokinetic properties. Additionally, NPs exhibit unique halogenation patterns, with marine natural products particularly enriched in bromine and chlorine substituents—modifications that often enhance membrane permeability and receptor binding affinity.
Table 1: Key Structural Differentiators Between Natural Products and Synthetic Compounds
| Structural Feature | Natural Products | Synthetic Compounds | Biological Impact |
|---|---|---|---|
| Stereocenters | High density (70-91% have defined stereochemistry) | Limited | Enhanced target specificity |
| Ring Systems | Complex polycyclic frameworks | Simpler monocyclic/bicyclic | Pre-organized 3D structure |
| Oxygen Content | High (hydroxyl, carbonyl, ether) | Moderate | Improved hydrogen bonding |
| Glycosylation | ~30% of compounds | Rare | Solubility and recognition |
| Halogenation | Unique patterns (Br, Cl in marine NPs) | Standard patterns (F, Cl) | Membrane permeability |
Quantitative Analysis of Natural Product Chemical Space
Databases and Collections: Mapping the Chemical Universe
The comprehensive mapping of NP chemical space relies on extensive databases and collections that document structural and biological information. Current estimates indicate that over 1.1 million natural products have been documented across various databases [14]. However, a significant accessibility challenge exists, with only approximately 10% of known NPs being readily obtainable for experimental testing from commercial vendors and public research institutions [18] [14].
The landscape of NP databases includes over 120 different resources published since 2000, with 98 remaining accessible and only 50 providing open access [16] [17]. Among the most significant comprehensive collections is COCONUT (COlleCtion of Open NatUral prodUcTs), which contains structures and annotations for over 400,000 non-redundant NPs, representing the largest open collection available [16] [17]. Commercial databases provide additional coverage, with the Dictionary of Natural Products containing extensive curated entries, and resources like SciFinder and Reaxys encompassing over 200,000-300,000 natural compounds each [16].
Table 2: Major Natural Product Databases and Their Characteristics
| Database Name | Type | Size (Compounds) | Access | Key Features |
|---|---|---|---|---|
| COCONUT | Generalistic | >400,000 | Open | Largest open collection, non-redundant |
| Dictionary of Natural Products | Generalistic | ~250,000 | Commercial | Comprehensive, well-curated |
| SciFinder | Chemicals | >300,000 NPs | Commercial | Extensive curated content |
| Reaxys | Chemicals | >200,000 NPs | Commercial | Reaction and substance data |
| MarinLit | Marine | >30,000 | Commercial | Marine-specific, highly curated |
| AntiBase | Microbial | >40,000 | Commercial | Microbial NPs with metadata |
Comparative Chemical Space Analysis
Chemoinformatic analyses reveal that natural products occupy a broader chemical space than synthetic compounds, with distinct clustering in regions associated with biological relevance [14]. This expanded coverage is particularly evident in physicochemical property distributions, where NPs exhibit higher molecular weights, greater numbers of hydrogen bond donors and acceptors, and increased molecular rigidity compared to synthetic medicinal chemistry compounds.
The readily obtainable subset of NPs demonstrates exceptional diversity, populating regions of chemical space highly relevant to drug discovery despite representing only a fraction of known natural products [18]. Significant differences exist in the coverage of chemical space by individual databases, with specialized resources focusing on particular organismal sources (marine, microbial, plant) or structural classes (alkaloids, terpenoids, flavonoids) that collectively provide comprehensive coverage of NP chemical space.
Methodological Framework for Natural Product Chemical Space Analysis
Computational Approaches and Workflows
The systematic analysis of NP chemical space requires specialized computational approaches that address the unique challenges of natural product structures. A critical first step involves the development and application of algorithms like "SugarBuster" for the removal of sugars and sugar-like moieties from natural products, enabling focused analysis of the aglycone scaffolds that often represent the core pharmacophoric elements [18].
Rule-based automated classification systems enable the organization of NPs into natural product classes (alkaloids, steroids, flavonoids, etc.), facilitating chemotaxonomic analyses and structure-activity relationship studies [18]. These classification systems typically leverage structural fingerprints, molecular descriptors, and machine learning approaches to categorize compounds based on their biosynthetic origins and structural characteristics.
Experimental Validation and Structure Elucidation
Beyond computational analysis, comprehensive NP chemical space exploration requires sophisticated experimental approaches for structural characterization and bioactivity validation. More than 2000 natural products have been characterized through X-ray crystallography in complex with biomacromolecules, providing crucial structural insights into their molecular interactions [18].
Advanced spectroscopic techniques, particularly NMR and mass spectrometry, play essential roles in NP structure elucidation. Databases such as NIST mass spectral libraries contain over 250,000 molecules of natural origin, enabling comparative analyses and dereplication [16]. The integration of these experimental datasets with computational chemical space analysis creates powerful workflows for identifying novel scaffolds and predicting bioactivities.
Research Reagent Solutions: Essential Tools for NP Chemical Space Exploration
Table 3: Key Research Reagents and Databases for NP Chemical Space Analysis
| Resource | Type | Function | Application Context |
|---|---|---|---|
| SugarBuster Algorithm | Computational Tool | Removes sugar moieties from NPs | Focused aglycone scaffold analysis |
| COCONUT Database | Open Database | Provides 400,000+ NP structures | Virtual screening, chemoinformatics |
| Dictionary of Natural Products | Commercial Database | Authoritative NP structural data | Structure classification, validation |
| NIST Mass Spectral Library | Spectral Database | NP identification by mass spectrometry | Dereplication, structure elucidation |
| Rule-Based Classification System | Computational Method | Automates NP class assignment | Chemotaxonomy, chemical space organization |
| Crystallography Data (PDB) | Structural Data | NP-biomolecule complex structures | Interaction studies, target engagement |
Emerging Frontiers and Future Directions
The continued expansion of NP chemical space relies on the exploration of underexplored biological sources and extreme environments. Marine organisms, particularly those from deep-sea and hydrothermal vent ecosystems, produce NPs with novel scaffolds and potent bioactivities not found in terrestrial organisms [14]. These environments exert unique evolutionary pressures that shape distinct biosynthetic pathways, resulting in natural products with enhanced chemical novelty.
Microorganisms from extreme environments (extremophiles) represent another promising frontier for chemical space expansion. These organisms produce NPs adapted to function under conditions of extreme temperature, pressure, salinity, or pH, resulting in structures with exceptional stability and unique mechanisms of action [14]. Systematic bioprospecting of these environments continues to yield chemically and biologically novel compounds.
Artificial Intelligence and Novel Methodologies
Artificial intelligence approaches are revolutionizing NP chemical space exploration through enhanced pattern recognition, bioactivity prediction, and novel scaffold design. Machine learning algorithms can identify complex relationships between structural features and biological activities across vast NP datasets, enabling predictive bioactivity modeling and target identification [14].
The emerging field of pseudo-natural products (PNPs) represents a promising methodological advancement that combines NP fragments in novel arrangements not accessible through biosynthetic pathways [19]. These hybrid structures bridge distinct regions of chemical space, creating compounds that retain the biological relevance of NPs while exhibiting enhanced structural novelty. Biology-oriented synthesis (BIOS) principles guide the rational design of these compounds, creating novel chemotypes with potential for unprecedented biological activities.
The exceptional structural diversity of natural products constitutes a unique chemical space that remains indispensable for drug discovery and chemical biology. This diversity stems from evolutionary optimization across millions of years, resulting in complex molecular architectures with privileged biological activities. Despite the challenges of accessibility and redundancy, NPs continue to provide novel scaffolds and lead compounds, with emerging technologies enhancing our ability to explore and leverage this chemical universe. The integration of traditional approaches with innovative methodologies—including extreme environment bioprospecting, artificial intelligence, and pseudo-natural product design—ensures that natural products will continue to serve as essential resources for addressing unmet medical needs and expanding the frontiers of chemical space.
Natural products and their derivatives have been a cornerstone of drug discovery, providing indispensable therapeutic agents for combating infectious diseases. The journeys of artemisinin, quinine, and ivermectin from natural sources to clinical application exemplify the profound impact of this approach. These compounds, sourced from the sweet wormwood plant (Artemisia annua), the bark of the cinchona tree, and the soil-dwelling bacterium Streptomyces avermitilis, respectively, have saved millions of lives and revolutionized the treatment of parasitic diseases. This whitepaper details their discovery, mechanistic principles, and experimental methodologies, framing their development within the core principles of natural product drug discovery research to inform and guide contemporary scientific efforts.
Artemisinin: A Modern Antimalarial Powerhouse
Discovery and Clinical Impact
Artemisinin is a sesquiterpene lactone isolated from Artemisia annua, a plant used in traditional Chinese medicine for fever [20]. Its discovery and development mark a triumph of modern pharmacognosy. It and its derivatives (e.g., artesunate, artemether) form the foundation of artemisinin-based combination therapies (ACTs), which the World Health Organization recommends as the first-line treatment for Plasmodium falciparum malaria [20]. The implementation of ACTs has contributed to a significant reduction in the global malaria burden.
Beyond malaria, recent clinical research highlights the therapeutic potential of artemisinin and its derivatives in other areas, including anti-parasitic (non-malaria), anti-tumor, and anti-inflammatory applications, demonstrating a promising safety profile in clinical trials [20].
Mechanism of Action
Artemisinin's anti-malarial activity is attributed to its unique endoperoxide bridge. Upon entry into the parasite-infected red blood cell, this bridge is cleaved by intra-parasitic ferrous iron (Fe²âº), leading to the generation of cytotoxic carbon-centered radicals [21]. These radicals alkylate and damage key parasitic macromolecules, including proteins and membranes, ultimately leading to parasite death.
Key Experimental Workflow and Methodologies
A pivotal advancement in artemisinin supply was achieved through synthetic biology and metabolic engineering.
Objective: Engineer a microbial host to produce artemisinic acid, a direct precursor to artemisinin, to ensure a stable and scalable supply independent of plant cultivation [22].
Protocol:
- Gene Identification and Isolation: Identify the genes encoding the enzymes of the artemisinin biosynthetic pathway from Artemisia annua, including the amorpha-4,11-diene synthase (ADS) and cytochrome P450 monooxygenase (CYP71AV1).
- Vector Construction and Transformation: Clone these genes into expression vectors suitable for a microbial host, initially E. coli and later the yeast Saccharomyces cerevisiae.
- Host Engineering (in Yeast):
- Upstream Pathway Enhancement: Engineer the native yeast mevalonate pathway to increase the supply of isopentenyl pyrophosphate (IPP) and farnesyl pyrophosphate (FPP), the universal terpenoid precursors.
- Heterologous Pathway Expression: Introduce the genes for ADS and CYP71AV1 to convert FPP to artemisinic acid.
- Cofactor Optimization: Modify host metabolism to support the required cytochrome P450 activity.
- Fermentation and Extraction: Grow the engineered yeast in large-scale bioreactors. Extract artemisinic acid from the fermentation broth.
- Chemical Conversion: Semi-synthetically convert the microbially produced artemisinic acid to dihydroartemisinin, which can then be derivatized into final drugs like artesunate and artemether [22].
The following diagram visualizes this engineered metabolic pathway in yeast.
Diagram 1: Engineered Artemisinin Pathway in Yeast. Heterologous enzymes ADS and CYP71AV1 are introduced into yeast to convert the endogenous precursor FPP into artemisinic acid.
Research Reagent Solutions
Table: Key Reagents for Artemisinin Research and Production
| Reagent/Material | Function in Research/Production |
|---|---|
| Artemisia annua Plant Material | Source of native biosynthetic genes for pathway identification and cloning [20]. |
| Saccharomyces cerevisiae | Engineered microbial host for the heterologous production of artemisinic acid [22]. |
| Fermentation Bioreactor | System for the scalable, industrial-scale cultivation of engineered yeast [22]. |
| Artemisinin-Specific Antibodies | Key reagents for developing immunoassays (e.g., ELISA) for the quantification of artemisinin in samples. |
Ivermectin: A Broad-Spectrum Antiparasitic from the Soil
Discovery and Clinical Impact
Ivermectin, a derivative of avermectin, originates from the soil actinomycete Streptomyces avermitilis discovered by Satoshi ÅŒmura at the Kitasato Institute [23] [24]. This discovery, which earned ÅŒmura and William Campbell the 2015 Nobel Prize, yielded the first endectocide, effective against a wide range of internal and external parasites [24]. It is on the WHO's List of Essential Medicines.
Ivermectin's impact on global health is monumental, primarily through donation programs. It is the primary tool in global campaigns to eliminate onchocerciasis (river blindness) and lymphatic filariasis (elephantiasis), with hundreds of millions of people treated annually [23] [24]. It is also effective against strongyloidiasis, scabies, and ascariasis [25].
Mechanism of Action
Ivermectin exerts its potent antiparasitic effect by binding with high affinity to glutamate-gated chloride channels (GluCls) in the nerve and muscle cells of invertebrates [24]. This binding potentiates the influx of chloride ions, leading to hyperpolarization of the cell membrane. The hyperpolarization blocks neuronal signaling and causes paralytic immobilization of the pharyngeal muscle and somatic body wall, leading to parasite death [24]. Its high safety margin in mammals is due to the absence of glutamate-gated chloride channels and its poor penetration of the blood-brain barrier [24].
Key Experimental Workflow and Methodologies
The discovery of ivermectin is a paradigm for successful natural product screening.
Objective: Discover novel bioactive compounds from soil-derived microorganisms with anthelmintic properties.
Protocol:
- Sample Collection and Strain Isolation: Collect soil samples from diverse environments. Isolate pure microbial strains, primarily actinomycetes, on selective culture media.
- In vitro Bioactivity Screening: Ferment isolated strains in liquid culture and prepare crude extracts. Screen extracts using in vitro assays against target parasites (e.g., the nematode Nippostrongylus brasiliensis).
- In vivo Validation: Administer promising crude extracts to parasite-infected model animals (e.g., mice, sheep) to confirm in vivo efficacy and safety.
- Bioassay-Guided Fractionation: Fractionate the active crude extract using chromatographic techniques (e.g., HPLC). Test each fraction for bioactivity, iteratively purifying the active component until a pure compound (avermectin) is isolated.
- Structural Elucidation: Determine the chemical structure of the active compound using spectroscopic methods (NMR, MS).
- Chemical Derivatization: Chemically modify the natural compound (e.g., selective hydrogenation to create ivermectin) to improve safety and efficacy profile [23] [24].
Diagram 2: Ivermectin Drug Discovery Workflow. The process from soil sampling to the development of ivermectin, heavily reliant on bioactivity-guided purification.
Research Reagent Solutions
Table: Key Reagents for Ivermectin Research and Production
| Reagent/Material | Function in Research/Production |
|---|---|
| Streptomyces avermitilis | The producing microorganism; source of the avermectin gene cluster for fermentation [23]. |
| Model Nematodes (e.g., Nippostrongylus brasiliensis, Haemonchus contortus) | Used in in vitro and in vivo bioassays for anthelmintic activity screening [24]. |
| GluCl Channel Protein | Target protein for in vitro binding studies and mechanism of action investigations [24]. |
| Fermentation Media (Complex) | Supports the growth of S. avermitilis and the production of avermectins during fermentation [23]. |
Quinine: The Prototype Antimalarial
Discovery and Clinical Impact
Quinine, a quinoline alkaloid isolated from the bark of the cinchona tree, was the first chemical compound used for treating an infectious disease—malaria [26]. For centuries, it was the primary treatment for malaria until the emergence of resistance and newer drugs. Its use is now largely restricted to severe, chloroquine-resistant malaria when artesunate is not available [27] [26]. The FDA has banned its use for leg cramps due to the risk of serious side effects [27].
Mechanism of Action
While its precise mechanism is complex, quinine is known to accumulate in the parasite's food vacuole. It inhibits the detoxification of heme, a toxic byproduct of hemoglobin digestion. The drug is believed to bind to heme, preventing its polymerization into non-toxic hemozoin. This leads to the buildup of toxic heme, which damages the parasite's membranes and leads to its death.
Table: Comparative Analysis of Three Natural Product-Derived Drugs
| Parameter | Artemisinin | Ivermectin | Quinine |
|---|---|---|---|
| Natural Source | Artemisia annua (plant) | Streptomyces avermitilis (bacterium) | Cinchona spp. (tree bark) |
| Drug Class | Sesquiterpene lactone | Macrocyclic lactone | Quinoline alkaloid |
| Primary Indication | Malaria (especially falciparum) | Onchocerciasis, Lymphatic Filariasis | Severe/Resistant Malaria |
| Molecular Target | Fe²⺠(activates endoperoxide bridge) | Glutamate-gated Chloride Channels (GluCls) | Heme Polymerization (in food vacuole) |
| Global Health Impact | ACTs are first-line malaria treatment; millions of courses distributed annually. | >1 billion treatments donated; key to eliminating river blindness & elephantiasis [24]. | Historical prototype; now a reserve drug. |
| Supply Source | Plant extraction & Synthetic Biology (yeast) [22]. | Industrial fermentation of S. avermitilis [23]. | Primarily plant extraction & chemical synthesis. |
The stories of artemisinin, ivermectin, and quinine underscore the enduring value of natural products in drug discovery. They highlight diverse success models: quinine as the historical prototype, artemisinin as a modern plant-derived drug enhanced by synthetic biology, and ivermectin as a microbial product deployed through unprecedented philanthropic partnership. Their development required a multidisciplinary convergence of microbiology, chemistry, pharmacology, and, increasingly, genetic engineering. For researchers, these case studies validate that investigating natural compounds, coupled with innovative technologies and sustainable supply strategies, remains a powerful approach for addressing unmet medical needs and combating global health challenges.
Antimicrobial resistance (AMR) represents one of the most severe global public health threats of the 21st century, directly causing an estimated 1.27 million deaths annually and contributing to nearly 5 million more [28]. This crisis undermines the foundation of modern medicine, rendering life-saving treatments—including routine surgeries, cancer chemotherapy, and organ transplantation—increasingly risky [28]. The emergence and spread of drug-resistant pathogens are accelerated by human activities, primarily the misuse and overuse of antimicrobials in human medicine, animal agriculture, and crop production [28]. Despite increasing recognition of the problem, the antibacterial development pipeline remains inadequate, creating significant unmet medical needs, particularly for infections caused by multidrug-resistant (MDR) gram-negative bacteria [29] [30].
Within this challenging landscape, natural products (NPs) are experiencing a renaissance in drug discovery. Historically, NPs have been a vital source of therapeutic agents, and they offer unique chemical structures and biological activities that are increasingly viewed as potential solutions to combat resistant pathogens [8] [31]. This whitepaper examines the current drivers of AMR within a One Health framework, analyzes the most pressing unmet therapeutic needs, and details how modern natural product research—powered by advanced technologies like artificial intelligence (AI), high-throughput screening, and synthetic biology—is providing innovative strategies to address this escalating crisis [8] [32].
Global AMR Landscape and Key Drivers
The scale of the AMR threat is reflected in recent global surveillance data. The World Health Organization's (WHO) Global Antimicrobial Resistance and Use Surveillance System (GLASS), in its 2025 report, analyzed data from 110 countries between 2016 and 2023, encompassing over 23 million bacteriologically confirmed infections [33]. The findings present an alarming picture of resistance prevalence across common bacterial pathogens.
Table 1: Global AMR Prevalence for Key Pathogen-Antibiotic Combinations
| Pathogen | Antibiotic Class | Reported Resistance Rate | Primary Infection Types |
|---|---|---|---|
| Escherichia coli | Third-generation cephalosporins | 42% (median across 76 countries) | Urinary Tract Infections, Bloodstream Infections [28] |
| Staphylococcus aureus | Methicillin (MRSA) | 35% (median across 76 countries) | Bloodstream Infections, Skin and Soft Tissue Infections [28] |
| Klebsiella pneumoniae | Carbapenems | Increasingly observed across multiple regions | Bloodstream Infections, Pneumonia [28] |
| E. coli (Urinary) | Fluoroquinolones | 1 in 5 cases show reduced susceptibility | Urinary Tract Infections [28] |
The economic costs of AMR are equally staggering, with the World Bank estimating that AMR could result in US$1 trillion in additional healthcare costs by 2050 and US$1 trillion to US$3.4 trillion in annual GDP losses by 2030 [28]. These figures underscore that AMR is not only a health crisis but also a fundamental threat to global economic stability.
One Health Drivers of Antimicrobial Resistance
AMR is a quintessential "One Health" issue, with its drivers and consequences inextricably linked to human, animal, and environmental health.
Human Health Sector: Inappropriate and excessive use of antibiotics in human medicine remains a primary driver. This includes over-prescription, use of broad-spectrum agents when narrower alternatives are suitable, and inadequate adherence to treatment protocols [32] [28]. The COVID-19 pandemic further exacerbated this problem through increased antibiotic usage during the crisis [34].
Veterinary and Agricultural Sectors: Global veterinary antibiotic consumption was approximately 81,000 tons in 2018, with projections indicating a rise to 104,079 tons by 2030 [34]. Tetracyclines and penicillins constitute the bulk of this consumption, accounting for 40.5% and 14.1% respectively. Such massive use, particularly for non-therapeutic purposes like growth promotion, selects for resistant bacteria that can transfer through the food chain to humans [34] [32].
Environmental Sector: Environmental reservoirs, including wastewater treatment plants, surface water, and soil, are critical conduits for the dissemination of antibiotic-resistant bacteria (ARB) and antibiotic resistance genes (ARGs) [32]. Pharmaceutical manufacturing waste and agricultural runoff further contaminate ecosystems, creating hotspots for the horizontal gene transfer of resistance determinants between environmental and clinically relevant bacteria [34] [32].
The following diagram illustrates the interconnectedness of these drivers and the flow of resistance elements across the One Health spectrum.
Unmet Medical Needs and Resistance Mechanisms
Prioritized Unmet Needs in Antibacterial Therapy
From a clinical perspective, the infectious diseases community has identified clear areas of unmet need where effective therapeutic options are dwindling or non-existent. A survey of the Emerging Infections Network (EIN) found that front-line physicians view multidrug-resistant (MDR) gram-negative bacilli as the most severe unmet medical need, scoring higher (mean score 4.6/5) than MRSA, MDR tuberculosis, and MDR gonorrhea [29]. The same survey highlighted that the limited number of new antimicrobials under development was perceived as the greatest challenge (mean score 4.7/5) [29]. Clinicians are increasingly encountering infections resistant to all available antibacterial agents, with 63% of surveyed infectious disease specialists reporting caring for such a patient in the previous year [29].
Molecular Mechanisms of Resistance
At the microbial level, resistance arises through several well-characterized genetic and physiological mechanisms, which are often combined within a single bacterial cell to create MDR phenotypes.
Enzymatic Inactivation: Production of enzymes that degrade or modify antibiotics is a widespread resistance strategy. This includes extended-spectrum β-lactamases (ESBLs) and carbapenemases (e.g., KPC, NDM, OXA-48), which hydrolyze β-lactam antibiotics, rendering them ineffective [32].
Target Modification: Mutations in chromosomal genes can alter the drug target site, reducing antibiotic binding affinity. Examples include mutations in gyrA/parC genes for fluoroquinolones and rpoB for rifampicin [32].
Efflux Pumps: Overexpression of multidrug efflux systems (e.g., AcrAB–TolC in E. coli, MexAB–OprM in P. aeruginosa) actively pumps antibiotics out of the bacterial cell, decreasing intracellular drug concentration [32].
Reduced Permeability: Loss of porins (outer membrane proteins) in Gram-negative bacteria limits the entry of antibiotics into the cell, a mechanism particularly associated with carbapenem-resistant Enterobacterales [32].
Horizontal Gene Transfer (HGT): The rapid dissemination of resistance is primarily fueled by HGT via plasmids, transposons, and integrons. These mobile genetic elements can carry multiple resistance genes simultaneously, leading to the emergence of "pan-resistant" pathogens in clinical settings [32]. The co-selection of resistance genes, such as carbapenemase and colistin resistance genes on a single plasmid, compresses the available last-line treatment options [32].
The diagram below summarizes these core resistance mechanisms and their functional consequences.
Natural Product Drug Discovery: Methodologies and Workflows
The complex chemical structures of natural products, evolved for specific biological functions, make them ideal starting points for tackling sophisticated bacterial resistance mechanisms. Modern NP discovery has moved beyond traditional bioassay-guided fractionation to incorporate a suite of advanced technologies.
Key Experimental Protocols in NP Discovery
Protocol 1: High-Throughput Screening of Natural Product Libraries
- Objective: To rapidly identify novel bioactive compounds with antibacterial activity from large NP libraries.
- Methodology:
- Library Preparation: Utilize prefractionated natural product extracts or purified compound libraries in 96- or 384-well microtiter plates.
- Bacterial Strain Selection: Employ reference strains and clinically relevant MDR pathogens (e.g., MRSA, ESBL-producing E. coli, carbapenem-resistant K. pneumoniae).
- Growth Inhibition Assay: Inoculate wells with a standardized bacterial suspension (~5 x 10^5 CFU/mL) in Mueller-Hinton broth. Incubate for 16-20 hours at 35°C.
- Detection: Measure optical density (OD600) or use resazurin-based fluorescent dyes to quantify bacterial growth inhibition.
- Hit Validation: Confirm activity in dose-response assays to determine Minimum Inhibitory Concentrations (MICs) against a broader panel of resistant pathogens [8] [31].
Protocol 2: Genome Mining for Biosynthetic Gene Clusters (BGCs)
- Objective: To computationally identify the genetic potential of microorganisms to produce novel natural products.
- Methodology:
- Genome Sequencing: Perform whole-genome sequencing of microbial isolates using Illumina or PacBio platforms.
- BGC Identification: Analyze sequenced genomes with bioinformatics tools (e.g., antiSMASH, PRISM) to detect BGCs for known classes (e.g., non-ribosomal peptide synthetases (NRPS), polyketide synthases (PKS), ribosomally synthesized and post-translationally modified peptides (RiPPs)).
- Heterologous Expression: Clone silent or poorly expressed BGCs into suitable expression hosts (e.g., Streptomyces coelicolor) to activate the production of cryptic metabolites.
- Compound Isolation and Structure Elucidation: Purify expressed metabolites using HPLC and characterize structures via NMR and MS/MS spectroscopy [8].
Protocol 3: AI-Guided Prediction of NP Targets and Mechanisms
- Objective: To predict the molecular targets and mechanisms of action of novel NPs using artificial intelligence.
- Methodology:
- Data Curation: Compile training datasets from public databases (e.g., ChEMBL, DrugBank) containing NP structures, bioactivity data, and known targets.
- Model Training: Develop machine learning models (e.g., graph neural networks, random forests) to learn complex structure-activity relationships.
- Target Prediction: Input novel NP structures into trained models to predict potential protein targets (e.g., bacterial topoisomerases, cell wall synthesis enzymes).
- Experimental Validation: Confirm predicted targets through in vitro binding assays (e.g., surface plasmon resonance) and phenotypic profiling (e.g., cytological profiling) [8] [31].
The integrated workflow for modern natural product discovery, from sourcing to lead optimization, is depicted below.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagents for NP-Based AMR Research
| Reagent / Material | Function in NP Discovery | Application Example |
|---|---|---|
| Prefractionated NP Libraries | Provides diverse chemical starting points for screening; reduces complexity of crude extracts. | Identification of novel scaffolds against MDR Gram-negative pathogens [8]. |
| Genome Mining Software (e.g., antiSMASH) | Predicts Biosynthetic Gene Clusters (BGCs) in microbial genomes to prioritize strains for compound discovery. | Discovery of novel lipopeptide antibiotics with activity against vancomycin-resistant bacteria [8]. |
| CRISPR-Cas9 Systems | Enables genetic manipulation of NP-producing strains to elucidate biosynthesis pathways and engineer overproduction. | Activation of silent gene clusters in Streptomyces species [32]. |
| LC-MS/MS & NMR Platforms | Enables dereplication (identification of known compounds) and structural elucidation of novel bioactive NPs. | Determination of chemical structure and purity of a new macrocyclic antibiotic [8] [31]. |
| Artificial Intelligence (AI) Platforms | Predicts NP bioactivity, molecular targets, and mechanisms of action; optimizes pharmacokinetic properties. | In silico prediction of a natural product's ability to inhibit bacterial efflux pumps [8]. |
| Antibody-Drug Conjugate (ADC) Platforms | Facilitates targeted delivery of potent NP-derived payloads to specific bacterial or host cells. | Development of monoclonal antibodies conjugated to NP-derived cytotoxins for targeted cancer therapy related to infections [8]. |
| 7-Hydroxyguanine | 7-Hydroxyguanine, CAS:16870-91-0, MF:C5H5N5O2, MW:167.13 g/mol | Chemical Reagent |
| Dioxo(sulphato(2-)-O)uranium | Dioxo(sulphato(2-)-O)uranium, CAS:16984-59-1, MF:C2H2O6U, MW:362.08 g/mol | Chemical Reagent |
Addressing the multifaceted crisis of AMR requires a unified strategy that aligns innovative scientific discovery with robust policy and stewardship. The One Health approach provides the essential framework, recognizing that the health of humans, animals, and ecosystems is interconnected. The integration of advanced natural product research into this framework offers a promising path forward. By leveraging cutting-edge technologies—from genome mining and AI to synthetic biology and targeted delivery systems—researchers can unlock the vast, untapped potential of natural products to yield the next generation of antimicrobial agents. Sustained investment, interdisciplinary collaboration, and policies that incentivize antibacterial development are critical to translating these scientific advances into therapies that protect our present and secure our future against the relentless threat of antimicrobial resistance [8] [32] [35].
The Modern Toolkit: AI, Omics, and Integrated Workflows for Accelerated Discovery
Artificial Intelligence and Machine Learning in Target and Lead Prediction
The discovery and development of new therapeutics from natural products (NPs) represent a cornerstone of modern pharmacology, with approximately 60% of medicines approved in the last three decades originating from NPs or their derivatives [36]. However, the traditional drug discovery paradigm faces formidable challenges characterized by lengthy development cycles, prohibitive costs, and high preclinical attrition rates [37]. The process from lead compound identification to regulatory approval typically spans over 12 years with cumulative expenditures exceeding $2.5 billion, while clinical trial success probabilities decline precipitously from Phase I (52%) to Phase II (28.9%), culminating in an overall success rate of merely 8.1% [37].
Artificial intelligence (AI) and machine learning (ML) have emerged as transformative technologies poised to address these inefficiencies by systematically decoding the complex relationships between natural product structures, their protein targets, and desired biological activities [37] [38]. This technical guide provides an in-depth examination of AI-driven methodologies specifically tailored for target identification and lead optimization within natural product drug discovery, framing these computational approaches within the broader context of natural product research principles.
AI and ML Fundamentals for Drug Discovery
Artificial intelligence develops systems capable of human-like reasoning and decision-making, with contemporary AI systems integrating both machine learning (ML) and deep learning (DL) to address pharmaceutical challenges ranging from target validation to formulation optimization [37]. ML employs algorithmic frameworks to analyze high-dimensional datasets, identify latent patterns, and construct predictive models through iterative optimization processes [37]. The field has evolved into four principal paradigms, each with distinct applications in natural product research:
- Supervised Learning: Utilizes labeled datasets for classification via algorithms like support vector machines (SVMs) and for regression via algorithms like support vector regression (SVR) and random forests (RFs) [37].
- Unsupervised Learning: Identifies latent data structures through clustering and dimensionality reduction techniques (such as principal component analysis and K-means clustering) to reveal underlying pharmacological patterns and streamline chemical descriptor analysis [37].
- Semi-Supervised Learning: Boosts drug-target interaction prediction by leveraging a small set of labeled data alongside a large pool of unlabeled data, enhancing prediction reliability through model collaboration and simulated data generation [37].
- Reinforcement Learning: Optimizes molecular design via Markov decision processes, where agents iteratively refine policies to generate inhibitors and balance pharmacokinetic properties through reward-driven strategies [37].
Table 1: Machine Learning Paradigms in Natural Product Drug Discovery
| ML Paradigm | Key Algorithms | Natural Product Applications | Advantages |
|---|---|---|---|
| Supervised Learning | SVMs, Random Forests, SVR | Target classification, activity prediction, ADMET property estimation | High accuracy with quality labeled data, clear evaluation metrics |
| Unsupervised Learning | PCA, K-means, t-SNE | Compound clustering, chemical space visualization, novelty detection | No labeled data required, reveals hidden patterns in NP libraries |
| Semi-Supervised Learning | Label propagation, self-training | Target prediction with limited bioactivity data | Leverages abundant unlabeled NP data, improves generalization |
| Reinforcement Learning | Q-learning, Policy gradients | De novo molecular design, multi-parameter optimization | Discovers novel NP-inspired scaffolds, balances multiple properties |
| 1-Ethyl-3-methyl-3-phospholene 1-oxide | 1-Ethyl-3-methyl-3-phospholene 1-oxide, CAS:7529-24-0, MF:C7H13OP, MW:144.15 g/mol | Chemical Reagent | Bench Chemicals |
| Tribenuron-methyl | Tribenuron-methyl|CAS 101200-48-0|Research Compound | High-purity Tribenuron-methyl, a sulfonylurea ALS inhibitor herbicide for agricultural research. For Research Use Only. Not for human or veterinary use. | Bench Chemicals |
Deep learning, a subset of ML utilizing multi-layered neural networks, has demonstrated remarkable performance in deciphering intricate structure-activity relationships, facilitating de novo generation of bioactive compounds with optimized pharmacokinetic properties [37]. The efficacy of these algorithms is intrinsically linked to the quality and volume of training data, particularly in deciphering latent patterns within complex biological datasets [37].
AI-Driven Target Prediction for Natural Products
The Challenge of Natural Product Target Identification
Despite the significant number of NPs discovered, their interaction profiles with drug targets, primarily proteins, remain largely undefined [36]. Bioactivity data for natural products is often limited, creating a fundamental obstacle for predictive modeling [36] [39]. Furthermore, natural products exhibit distinct structural differences compared to synthetic molecules, including higher molecular weights, more complex scaffolds, and greater structural diversity, which complicates the application of conventional prediction tools trained primarily on synthetic compound libraries [39].
Similarity-Based Target Prediction Approaches
Similarity-based target prediction operates on the premise that structurally similar molecules tend to bind similar protein targets [36]. This approach represents a straightforward and efficient strategy for ligand-based target prediction due to its flexibility, low computational cost, and remarkable predictive performance [36]. The fundamental workflow involves:
- Reference Library Construction: Compounding compounds with standardized representations, precise target annotations, and significant biological activities.
- Similarity Calculation: Ranking reference compounds based on their structural similarity to query compounds using molecular fingerprints or descriptors.
- Target Assignment: Assigning targets associated with the top N most similar reference compounds to the query compound.
Table 2: Similarity-Based Target Prediction Techniques for Natural Products
| Technique | Methodology | Optimal Parameters | Advantages | Limitations |
|---|---|---|---|---|
| Top N Hits | Considers targets associated with top N compounds with highest similarities | Top 5 hits most effective [36] | Simple implementation, interpretable results | Increased false positives with larger N values |
| Mean Similarity | Ranks targets according to mean similarity scores of predefined similar compounds per target | Top 3 similar compounds [36] | Reduces bias from single compounds, more robust | Computationally more intensive |
| Statistical Significance | Transforms similarity scores into p-values or E-values | Varies by implementation | Provides statistical confidence measures | Complex implementation, may require specialized expertise |
Several specialized tools have been developed for similarity-based target prediction of natural products:
- CTAPred: An open-source command-line tool specifically designed for NP target prediction that applies fingerprinting and similarity-based search in a two-stage approach. It first generates a compound-target activity reference dataset from public data focusing on protein targets known or likely to interact with NPs, then identifies potential targets for query compounds based on similarity to this curated reference set [36].
- SEA (Similarity Ensemble Approach): Helps rationalize polypharmacology effects by relating targets based on the set-wise chemical similarity among their ligands [36].
- SwissTargetPrediction: Uses path-based fingerprints for 2D similarity and Manhattan distance between Electroshape 5D for 3D similarity [36].
- D3CARP: Provides flexibility allowing users to choose between three molecular fingerprints for 2D similarity and uses LS-align for 3D structural alignment [36].
Deep Learning with Transfer Learning for Natural Products
To address the limited bioactivity data for natural products, transfer learning has emerged as a powerful technique for building accurate target prediction models [39]. This approach involves:
- Pre-training: Training a deep learning model (such as a multilayer perceptron) on a large-scale bioactivity dataset (like ChEMBL) with natural products removed.
- Fine-tuning: Further training the pre-trained model on a limited natural product dataset with a higher learning rate and some parameters frozen, allowing the model to adapt to the specific distribution of natural products.
This methodology benefits from the knowledge embedded in the larger synthetic compound dataset while specializing for natural product applications. Research has demonstrated that transfer learning can achieve highly promising area under the receiver operating characteristic curve (AUROC) scores of 0.910, despite limited task-related training samples [39]. Embedding space analysis shows that transfer learning reduces the distribution difference between synthetic compounds and natural products, making the model's predictions for natural products more reliable [39].
Experimental Protocol: Similarity-Based Target Prediction
Objective: Identify potential protein targets for a query natural product compound using similarity-based approaches.
Materials and Reagents:
- Query natural product compound (SMILES or structure format)
- Reference database (ChEMBL, COCONUT, NPASS, or CMAUP)
- Computing environment with CTAPred or similar tool installed
Methodology:
Data Preparation:
- Obtain canonical SMILES representation of query compound
- Download and preprocess reference compound-target activity database
- Standardize molecular representations (remove salts, neutralize charges)
- Curate target annotations to ensure consistent protein identifiers
Similarity Calculation:
- Generate molecular fingerprints (e.g., ECFP4, FCFP4, MACCS) for query and reference compounds
- Calculate pairwise similarity coefficients (typically Tanimoto similarity)
- Rank reference compounds by similarity to query compound
Target Prediction:
- Select top N reference compounds (typically N=5-10)
- Extract targets associated with these reference compounds
- Apply consensus scoring across multiple fingerprints if available
- Generate ranked list of predicted targets with confidence scores
Validation:
- Compare predictions with known bioactivity data if available
- Assess chemical similarity of reference compounds to query
- Evaluate biological plausibility of predicted targets
Expected Output: Ranked list of protein targets with associated confidence scores, enabling prioritization for experimental validation.
AI-Driven Lead Optimization for Natural Products
The Lead Optimization Challenge
Lead optimization represents a critical phase in drug discovery where initial hit compounds are systematically modified to improve potency, selectivity, and pharmacokinetic properties while reducing toxicity. For natural products, this process is particularly challenging due to their structural complexity and limited synthetic accessibility.
AI Approaches for Lead Optimization
AI-driven lead optimization has demonstrated remarkable success in accelerating the traditionally lengthy hit-to-lead phase through several key approaches:
De Novo Molecular Design: AI can generate novel drug molecules from scratch, proposing natural product-inspired candidates with optimized properties rather than relying solely on screening existing libraries [38]. Reinforcement learning techniques have been used to generate 26,000+ virtual analogs, resulting in sub-nanomolar inhibitors with over 4,500-fold potency improvement over initial hits [40].
Property Prediction: Machine learning models predict key drug characteristics such as toxicity, bioactivity, solubility, and pharmacokinetics early in the pipeline [38]. This helps eliminate unsafe or ineffective candidates before costly synthesis and experimental validation.
Synthetic Accessibility Prediction: AI suggests efficient chemical synthesis routes for new natural product analogs, sometimes proposing modifications to ease manufacturing [38]. This addresses one of the key challenges in natural product-based drug discovery.
Multi-parameter Optimization: AI systems can balance multiple competing objectives simultaneously, such as optimizing binding affinity while maintaining drug-likeness and minimizing toxicity [37].
Experimental Protocol: AI-Guided Lead Optimization
Objective: Optimize a natural product-derived lead compound using AI-driven approaches to improve potency and ADMET properties.
Materials and Reagents:
- Initial lead compound structure
- Target protein structure (if available)
- ADMET prediction tools (e.g., SwissADME, pkCSM)
- Synthetic feasibility assessment tools
Methodology:
Molecular Generation:
- Define chemical space around lead scaffold
- Generate virtual analogs using deep generative models (VAE, GAN, or RNN)
- Apply structural constraints to maintain natural product-like features
Property Prediction:
- Predict binding affinity to target using QSAR models or molecular docking
- Estimate ADMET properties using machine learning models
- Assess drug-likeness using rule-based filters (Lipinski, Veber)
Multi-Objective Optimization:
- Define optimization objectives (potency, selectivity, PK properties)
- Apply Pareto optimization to identify balanced compounds
- Rank compounds by weighted scoring function
Synthetic Planning:
- Predict retrosynthetic pathways for top candidates
- Assess synthetic complexity and feasibility
- Prioritize compounds with viable synthesis routes
Expected Output: Series of optimized lead compounds with predicted improved properties, along with suggested synthesis routes for experimental validation.
Integrated Workflows and Validation
Integrating Target Prediction and Lead Optimization
The most effective AI-driven natural product discovery platforms integrate both target prediction and lead optimization into cohesive workflows. This integration enables researchers to:
- Identify novel targets for natural products of interest
- Optimize natural product scaffolds for selected targets
- Predict polypharmacology profiles to understand mechanism of action
- Assess potential toxicity and off-target effects early in discovery
Recent work has demonstrated that integrating pharmacophoric features with protein-ligand interaction data can boost hit enrichment rates by more than 50-fold compared to traditional methods [40]. These approaches not only accelerate lead discovery but improve mechanistic interpretability—an increasingly important factor for regulatory confidence and clinical translation [40].
Experimental Validation of AI Predictions
Computational predictions require experimental validation to confirm biological activity. Key validation methodologies include:
- Cellular Thermal Shift Assay (CETSA): Validates direct target engagement in intact cells and tissues, providing quantitative, system-level validation that bridges the gap between biochemical potency and cellular efficacy [40].
- High-Throughput Screening: Confirms predicted activity against intended targets
- ADMET Assays: Validates predicted pharmacokinetic and toxicity properties
- Structural Biology: Confirms predicted binding modes through X-ray crystallography or cryo-EM
Table 3: AI-Driven Drug Discovery Tools and Platforms for Natural Products
| Tool/Platform | Type | Key Features | Natural Product Applications |
|---|---|---|---|
| CTAPred | Command-line tool | Similarity-based target prediction, focused NP target database | Predicting protein targets for uncharacterized NPs [36] |
| Transfer Learning Model | Deep learning | Pre-trained on ChEMBL, fine-tuned on NPs, AUROC: 0.910 [39] | Target prediction with limited NP bioactivity data |
| SwissTargetPrediction | Web server | 2D and 3D similarity, known bioactivity data | Target prediction for NP-inspired compounds [36] |
| SEA | Web server | Set-wise chemical similarity, target family prediction | Polypharmacology prediction for NPs [36] |
| AlphaFold | AI system | Protein structure prediction | Target structure determination for NP docking [38] |
| CETSA | Experimental platform | Cellular target engagement validation | Confirming AI-predicted NP-target interactions [40] |
Table 4: Research Reagent Solutions for AI-Driven Natural Product Research
| Resource | Type | Function | Application in NP Research |
|---|---|---|---|
| ChEMBL | Database | Bioactivity data for drug-like compounds | Reference data for target prediction models [36] [39] |
| COCONUT | Database | Extensive collection of natural products | NP structures for similarity searching [36] |
| NPASS | Database | Natural product activity and species source | NP bioactivity data for model training [36] |
| CMAUP | Database | Collection of medicinal plants and constituents | Plant-derived NP structures and activities [36] |
| TensorFlow/PyTorch | Software | Deep learning frameworks | Building custom AI models for NP research [37] [41] |
| RDKit | Software | Cheminformatics toolkit | Molecular fingerprinting and descriptor calculation [36] |
| AutoDock | Software | Molecular docking | Structure-based target prediction [40] |
| SwissADME | Web tool | ADMET property prediction | Lead optimization for NP-derived compounds [40] |
Artificial intelligence and machine learning are transforming target prediction and lead optimization for natural products, addressing historical challenges through sophisticated computational approaches. Similarity-based methods and transfer learning techniques have demonstrated remarkable success in predicting protein targets for natural products, even with limited bioactivity data. Meanwhile, AI-driven lead optimization approaches are accelerating the development of natural product-derived therapeutics with improved properties.
The integration of these computational approaches with experimental validation platforms like CETSA creates powerful workflows that bridge the gap between in silico predictions and biological reality. As these technologies continue to evolve, they promise to unlock the full potential of natural products as sources of novel therapeutics, ultimately expanding the druggable genome and bringing new treatments to patients with greater speed and efficiency.
For natural product researchers, embracing these AI-driven methodologies requires developing new interdisciplinary skills that span chemistry, biology, and data science. However, the investment in these capabilities offers substantial returns through accelerated discovery timelines, reduced attrition rates, and more effective exploitation of nature's chemical diversity for therapeutic benefit.
Natural products and their structural analogues have historically been a major source of pharmacotherapeutic agents, particularly for cancer and infectious diseases [42]. Despite a decline in pursuit by the pharmaceutical industry in recent decades, technological advancements are now revitalizing interest in natural products as drug leads [42]. In silico methodologies, including virtual screening (VS), molecular docking, and molecular dynamics (MD) simulations, represent cornerstone technologies in this renaissance, offering powerful means to navigate the vast and complex chemical space of natural products efficiently. These computational approaches integrate into the early stages of the drug discovery pipeline, providing valuable insights into chemical systems in a virtual manner and productively complementing experimental analyses [43]. By applying these tools, researchers can rapidly identify promising candidate molecules from extensive digital libraries, predict their interactions with biological targets, and optimize them for enhanced efficacy and safety, thereby addressing traditional barriers such as screening, isolation, and characterization [44] [42].
This technical guide provides an in-depth examination of these core computational techniques, framed within the context of modern natural product research. It details the fundamental principles, presents structured experimental protocols, and highlights applications through contemporary case studies. The content is specifically designed for researchers, scientists, and drug development professionals seeking to leverage these in silico strategies to accelerate the discovery and development of therapeutic agents from nature's chemical treasury.
Core Methodologies and Theoretical Foundations
Virtual Screening (VS)
Virtual screening is a computational methodology designed for searching large-scale libraries of chemical structures to select a limited number of candidate molecules likely to be active against a chosen biological target [45]. It functions as a logical extension of three-dimensional (3D) pharmacophore-based database searching or molecular docking, capable of automatically evaluating vast compound databases [45]. VS approaches are broadly classified into two categories, each with distinct advantages for natural product exploration.
- Ligand-Based Virtual Screening (LBVS): LBVS methods leverage the structural and biological data of a known set of active compounds to identify potential candidates. Common techniques include quantitative structure-activity relationship (QSAR) and pharmacophore-based modeling [46]. A pharmacophore model defines the essential steric and electronic features responsible for a molecule's biological activity, providing a template for database searching [45]. This approach is particularly valuable when the 3D structure of the target protein is unavailable.
- Structure-Based Virtual Screening (SBVS): SBVS relies on the three-dimensional (3D) conformation of the target to predict compound binding within extensive libraries. Molecular docking stands as the predominant SBVS technique [46]. This method is directly applicable when a high-resolution structure of the target, often obtained from X-ray crystallography or cryo-electron microscopy, is available.
The strategic integration of both LBVS and SBVS often yields superior results. For instance, pharmacophore-based VS can serve as a pre-filter to rapidly reduce library size before applying the more computationally intensive docking-based VS, or as a post-docking filter to refine results based on key interaction features [45] [46].
Table 1: Comparison of Virtual Screening Approaches
| Feature | Ligand-Based (LBVS) | Structure-Based (SBVS) |
|---|---|---|
| Requirement | Known active ligands | 3D Structure of the target protein |
| Core Methodology | Pharmacophore modeling, QSAR, similarity search | Molecular docking |
| Primary Advantage | High efficiency, no need for protein structure | Direct insight into binding mode and interactions |
| Challenge | Limited by known chemical space | Dependent on protein structure quality and flexibility |
| Ideal Use Case | Early-stage screening when structural data is lacking; scaffold hopping | Target-focused screening; understanding structure-activity relationships |
Molecular Docking
Molecular docking is an in silico method employed to predict the binding mode and orientation of a small molecule (ligand) within a receptor's binding site [43]. The procedure aims to mimic the lock-and-key model of molecular recognition, forecasting the experimental binding pose and estimating the binding affinity through a scoring function [47]. The predictive power of docking is critically dependent on its scoring function, which is a mathematical approximation used to rank potential poses and compounds [47].
Scoring functions can be classified into four main categories:
- Physics-based methods rely on molecular mechanics force fields, including terms for van der Waals and electrostatic interactions, though solvation and entropy are often simplified [47].
- Empirical scoring functions characterize binding affinity using a set of weighted terms (e.g., hydrogen bonding, hydrophobic contact). The weights are determined by fitting experimental binding affinity data via linear regression. Prominent examples include GlideScore and AutoDock Vina [47].
- Knowledge-based potentials are derived from statistical analyses of atom-pair frequencies in experimentally determined protein-ligand complexes, such as in DrugScore and PMF [47].
- Machine Learning (ML) scoring functions represent a recent advance, using techniques like Random Forest (RF) or Deep Neural Networks (DNN) to learn the functional form of binding affinity from data. These models can capture complex patterns and have shown marked improvements in binding affinity prediction [47].
Molecular Dynamics (MD) Simulations
While molecular docking provides a static snapshot of protein-ligand interaction, it employs many approximations and often lacks receptor flexibility, casting uncertainty on the reliability of the resulting complexes [43]. Molecular dynamics (MD) simulations offer a powerful complementary approach by modeling the time-dependent behavior of the molecular system.
MD simulations calculate the movements of atoms and molecules over time, providing a dynamic view of the interactions between a protein and a ligand [43]. This allows researchers to:
- Assess complex stability: Observe whether the docked pose remains stable or undergoes significant changes.
- Calculate detailed interaction energies: Obtain more rigorous estimates of binding free energy.
- Model flexible binding sites: Capture induced-fit phenomena and allosteric effects that are missed in rigid docking.
- Investigate binding mechanisms: Uncover pathways and kinetics of ligand binding and unbinding.
The integration of docking and MD is a logical strategy to improve the drug discovery process. MD can be used a priori to generate an ensemble of flexible receptor conformations for docking, or a posteriori to optimize and validate the structures of complexes obtained from docking [43].
Integrated Workflow and Experimental Protocols
A robust in silico campaign for natural product drug discovery typically follows an integrated, multi-step workflow. The diagram below visualizes this logical pathway, from target selection to final candidate identification.
Protocol 1: Virtual Screening Implementation
This protocol outlines a hybrid VS strategy combining pharmacophore and docking methods, as demonstrated in a screening study against phosphoinositide 3-kinase delta (PI3Kδ) [46].
Step 1: Pharmacophore Model Generation.
- Objective: Construct a common feature pharmacophore hypothesis to identify key interaction elements.
- Method: Extract multiple co-crystallized ligands from target protein structures (e.g., from the Protein Data Bank). Use these ligands as a training set in software such as LigandScout or Catalyst to generate top pharmacophore hypotheses. These models typically identify features like hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), and hydrophobic (HY) regions [46].
- Validation: Validate the generated pharmacophore models using a decoy test set containing known actives and inactive compounds. Evaluate performance using enrichment factors to select the hypothesis with the best predictive capability [46].
Step 2: Pharmacophore-Based Screening.
- Objective: Rapidly filter a large compound library.
- Method: Use the validated pharmacophore model as a 3D query to screen a digital library of natural products or other compounds. This step efficiently reduces the library size by selecting only molecules that map the essential pharmacophore features [45] [46].
Step 3: Docking-Based Screening.
- Objective: Further refine the hit list by evaluating binding poses and scores.
- Method: Subject the compounds that pass the pharmacophore filter to molecular docking against the target protein. It is advisable to use multiple protein structures (a "multi-complex" approach) if available, to account for binding site flexibility. Rank the compounds based on their docking scores [46].
Protocol 2: Binding Affinity and Stability Assessment
This protocol details the steps following the initial VS to prioritize the most promising hits for a specific target, such as human hepatic ketohexokinase (KHK-C) [48].
Step 1: Multi-level Molecular Docking.
- Objective: Validate and re-rank initial VS hits with more precise docking algorithms.
- Method: Re-dock the top-ranked compounds from the VS step using standard-precision (SP) and then high-throughput or extra-precision (XP) docking protocols available in programs like Glide, GOLD, or AutoDock Vina. This step provides a more reliable ranking of binding affinities [48] [47].
Step 2: Binding Free Energy Estimation.
- Objective: Obtain a more quantitative estimate of the protein-ligand binding affinity.
- Method: Calculate the binding free energy (ΔG) for the top complexes using methods like Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) or MM/Poisson-Boltzmann Surface Area (MM/PBSA). These methods provide a more rigorous energy estimation than standard docking scores and can help differentiate very strong binders [48].
Step 3: ADMET Profiling.
- Objective: Evaluate the drug-likeness and pharmacokinetic properties of the candidates.
- Method: Perform in silico prediction of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties. Analyze parameters such as aqueous solubility, Caco-2 permeability, cytochrome P450 inhibition, and human ether-a-go-go-related gene (hERG) inhibition. This step filters out compounds with undesirable pharmacokinetic or toxicological profiles [48].
Step 4: Molecular Dynamics Simulations.
- Objective: Assess the stability and dynamics of the protein-ligand complex over time.
- Method: Solvate the top-ranked protein-ligand complex in a explicit water box, add ions to neutralize the system, and run an MD simulation for a sufficient duration (typically 50-200 nanoseconds). Analyze the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), and specific intermolecular hydrogen bonds throughout the trajectory to confirm the stability of the binding pose and the robustness of key interactions [48] [43].
Successful execution of in silico drug discovery campaigns requires a suite of computational tools and data resources. The table below details key "research reagents" essential for the field.
Table 2: Key Research Reagents and Computational Tools
| Category | Item / Software / Database | Function and Application |
|---|---|---|
| Compound Libraries | National Cancer Institute (NCI) Library, ZINC, In-house Natural Product Databases | Source of small molecules for virtual screening; provides chemical starting points. |
| Structural Databases | Protein Data Bank (PDB) | Repository for 3D structural data of biological macromolecules, essential for SBVS and docking. |
| Pharmacophore Modeling | LigandScout, Catalyst | Software for creating, validating, and applying pharmacophore models for LBVS. |
| Molecular Docking | Glide (Schrödinger), GOLD, AutoDock Vina, DOCK | Programs to predict the binding pose and affinity of a ligand in a protein's active site. |
| MD Simulation Engines | GROMACS, AMBER, NAMD | Software packages to run MD simulations for studying the dynamic behavior of protein-ligand complexes. |
| Cheminformatics & ADMET | SwissADME, admetSAR, RDKit | Online platforms and toolkits for predicting pharmacokinetic, toxicity, and drug-like properties. |
| Benchmarking Datasets | PDBbind, LIT-PCBA | Curated datasets of protein-ligand complexes with binding affinity data for developing and validating scoring functions and VS protocols [47]. |
Case Study: Discovery of KHK-C Inhibitors for Metabolic Disorders
A recent study exemplifies the integrated application of these protocols. The research aimed to discover novel human hepatic ketohexokinase (KHK-C) inhibitors for treating fructose-induced metabolic disorders like NAFLD and NASH [48].
- Workflow Implementation: The team screened 460,000 compounds from the NCI library. They first applied pharmacophore-based VS to identify hits, which were then subjected to multi-level molecular docking [48].
- Quantitative Results: The top ten compounds exhibited docking scores ranging from -7.79 to -9.10 kcal/mol and calculated binding free energies (ΔG) from -57.06 to -70.69 kcal/mol. These values were superior to clinical candidates PF-06835919 (docking score: -7.77 kcal/mol; ΔG: -56.71 kcal/mol) and LY-3522348 (docking score: -6.54 kcal/mol; ΔG: -45.15 kcal/mol) [48].
- Refinement and Validation: ADMET profiling refined the selection to five compounds. Subsequent MD simulations (e.g., 100 ns) analyzed the stability of the complexes. One candidate, "compound 2," was identified as the most stable and promising, demonstrating lower RMSD fluctuations and persistent key interactions compared to the clinical candidate, thus supporting its potential for further development [48].
The integration of virtual screening, molecular docking, and molecular dynamics simulations represents a powerful frontier in modern drug discovery, providing a robust framework for efficiently identifying and optimizing novel therapeutic agents from natural products and synthetic libraries. As computational power increases and algorithms become more sophisticated, the accuracy and predictive power of these in silico methods will continue to improve.
Key future directions include the wider adoption of machine learning and artificial intelligence to enhance scoring functions and predict ADMET properties more reliably [47]. Furthermore, the development of more efficient free energy calculation methods and the increased incorporation of protein flexibility will bring computational predictions closer to experimental reality. For the field of natural product research, these advances are particularly impactful, as they help to deconvolute complex mixtures, identify active constituents, and guide the optimization of lead compounds [44] [42]. By embracing these integrated in silico strategies, researchers can significantly accelerate the journey from traditional remedies to modern, evidence-based medicines.
The process of dereplication is a critical, early-stage methodology in natural product drug discovery aimed at the rapid identification of known compounds within complex biological extracts. Its primary purpose is to avoid the costly and time-consuming rediscovery of already documented molecules, thereby streamlining the focus toward novel bioactive entities [49] [50]. Within the broader thesis of natural product research principles, dereplication represents the essential bridge between initial bioactivity screening and the dedicated isolation of new lead compounds. Historically, natural products have been the most successful source of potential drug leads, providing unique structural diversity unmatched by standard combinatorial chemistry [11]. However, given that less than 10% of the world's biodiversity has been evaluated for biological activity, efficient strategies to access this untapped chemical diversity are paramount [11].
The evolution of analytical technologies has fundamentally transformed dereplication from a slow, isolation-heavy process to a high-throughput, informatics-driven endeavor. Modern dereplication strategies leverage advanced metabolomic approaches, which are designed to provide a comprehensive qualitative and quantitative profile of the metabolites in an organism [50]. By integrating sophisticated analytical techniques like High-Performance Liquid Chromatography coupled to High-Resolution Mass Spectrometry (HPLC-HRMS) with computational tools, researchers can now swiftly characterize the chemical repertoire of complex crude extracts [51] [52]. This paradigm shift advocates for mass spectrometry-based approaches as a powerful starting point, enabling the fast screening of secondary metabolites and aligning with the contemporary need for efficiency and innovation in drug discovery [51] [10].
Theoretical Foundations: Metabolomics and HPLC-HRMS
Metabolomics in Natural Product Research
Metabolomics is defined as the technology designed to provide a general qualitative and quantitative profile of metabolites in organisms exposed to different conditions [50]. In the context of natural product discovery, it is applied in bioactivity screening to significantly improve dereplication and identification procedures [50]. Two distinct levels of metabolomics are commonly employed:
- Metabolite Fingerprinting: This level aims for the rapid classification of samples by comparing the patterns or "fingerprints" of metabolites. It is useful for quick comparisons and grouping of extracts without immediate individual compound identification [50].
- Metabolite Profiling: This level involves a more detailed study of specific classes of compounds related to a particular biosynthetic pathway. The goal is to individually identify and quantify these metabolites, providing a deeper level of chemical insight [50].
The power of metabolomics is greatly enhanced by coupling it with multivariate data analysis. Techniques such as Principal Component Analysis (PCA) are used to classify samples into groups, identify trends, and detect outliers, thereby maximizing the information obtained from complex spectral data [50]. Furthermore, Orthogonal Projections to Latent Structures-Discriminant Analysis (OPLS-DA) can be employed to correlate the observed chemical profile with a specific tested biological activity, helping to pinpoint the active constituents in a mixture [50].
The Role of HPLC-HRMS
Liquid Chromatography coupled to High-Resolution Mass Spectrometry (HPLC-HRMS) has been recognized as a revolutionary breakthrough in the analysis and characterization of compounds from complex samples [53]. This technique synergistically combines the superior separation power of liquid chromatography with the high sensitivity and mass accuracy of high-resolution mass spectrometry.
The operational principles of HPLC-HRMS can be summarized as follows:
- Separation: HPLC efficiently separates the individual compounds present in a complex natural extract.
- Ionization: Separated analytes are then ionized, typically using soft ionization techniques like Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI), which generate ions with little fragmentation [53].
- Mass Analysis: The high-resolution mass analyzer (e.g., Quadrupole Time-of-Flight or Q-TOF) precisely determines the mass-to-charge ratio (m/z) of the molecular ions and their fragments. A key technical specification is mass accuracy, which can be less than 2 ppm RMS over 12 hours, and resolution, which can reach ≥ 42,000 FWHM for instruments like the SCIEX X500R [53].
- Fragmentation (MS/MS): Tandem mass spectrometry (MS/MS) provides supplementary information by selectively fragmenting precursor ions. The resulting MS/MS spectra are instrumental in elucidating compound structure [53].
The primary advantage of HPLC-HRMS in dereplication is its ability to generate rich, multi-dimensional data for each component in a mixture without the need for prior isolation. This data includes retention time, accurate mass, isotopic pattern, and fragmentation spectrum, which together create a unique chemical signature for rapid compound identification [51] [53].
Integrated Dereplication Strategy: A Workflow
A modern, effective dereplication strategy involves the seamless integration of metabolomic data generation with computational mining and database search. The following workflow, derived from contemporary research, outlines this process. An overview of the entire strategy is presented in Figure 1.
Stages of the Dereplication Workflow
Sample Preparation and LC-HRMS/MS Analysis: The process begins with a crude natural extract [51]. Samples are prepared (e.g., dissolved in HPLC-grade methanol) and analyzed using LC-HRMS/MS, which generates comprehensive data in both positive and negative ionization modes, including MS/MS spectra for major peaks [51] [53].
Raw Data Processing: The raw LC-HRMS data is processed using software tools like Bruker Compass DataAnalysis and MetaboScape, or open-source platforms like MZmine [51] [49] [50]. This step involves "bucketing" molecular ions, peak detection, alignment, and deconvolution to extract meaningful features from the raw data.
Dereplication via Database Mining: The processed data, particularly the accurate mass and MS/MS spectra, are used to query natural product databases. Commonly used databases include:
Molecular Networking and In-silico Annotation: MS/MS data is uploaded to the Global Natural Products Social Molecular Networking (GNPS) platform [51]. Molecular networking works by clustering MS/MS spectra based on spectral similarity, visually grouping structurally related metabolites. This facilitates the putatively annotated compound classes (Level 2). Additionally, in-silico platforms like SIRIUS 4 can be used for further annotation, providing putative formula and structure suggestions [51].
Dereplication Outcome and Prioritization: By the end of the process, metabolites are sorted into three distinct levels [50]:
- Level 1: Confidently identified compounds.
- Level 2: Putatively annotated compound classes.
- Level 3: Completely unidentified and unclassified compounds, which represent the highest priority for novel drug discovery.
Integration with Bioactivity Data: The chemical profile is correlated with biological assay results (e.g., antioxidant, anti-diabetic) using statistical models like OPLS-DA [50]. This correlation guides the subsequent targeted isolation of novel bioactive compounds [51].
Experimental Protocols and Methodologies
Protocol 1: LC-HRMS Analysis of a Plant Metabolome
This protocol is adapted from a study on the dereplication of Urtica dioica L. (stinging nettle) [51].
Sample Preparation:
- Air-dried plant material (leaves, stems, roots) is ground to a fine powder.
- The powder is extracted with a mixture of water and methanol (e.g., 10:90 ratio) [51].
- The extract is concentrated using a rotary evaporator and subjected to liquid-liquid partitioning with solvents like hexane and dichloromethane to remove non-polar and medium-polar interferences, respectively [51].
- The final extract is dissolved in HPLC-grade methanol at a concentration of 0.2 mg/mL for LC-HRMS analysis [51].
LC-HRMS Parameters (Example):
- Device Model: A high-resolution system such as a Q-TOF instrument [53].
- Column: C18 reversed-phase column.
- Mobile Phase: Gradient of water and acetonitrile, both modified with 0.1% formic acid to enhance ionization.
- Gradient Example: 5% to 100% acetonitrile over 25-30 minutes [51].
- Ionization: Electrospray Ionization (ESI) in positive and/or negative mode.
- Mass Range: Up to m/z 2000 [53].
- Data Acquisition: Full-scan MS data and data-dependent MS/MS acquisition for the most intense ions.
Protocol 2: Bioactivity-Guided Fractionation for Antioxidant Compounds
This complementary protocol details the isolation of active compounds after initial dereplication, as demonstrated in the U. dioica study [51].
Bioassay:
- The DPPH (1,1-diphenyl-2-picrylhydrazyl) free radical scavenging assay is used to track antioxidant activity.
- Briefly, test samples at various concentrations are mixed with a DPPH solution in methanol.
- After incubation at room temperature for 30 minutes, the absorbance is measured at 517 nm. A reduction in absorbance indicates free radical scavenging activity [51].
Fractionation and Isolation:
- The active crude extract is fractionated using Vacuum Liquid Chromatography (VLC) over a C18ODS stationary phase with a methanol-water gradient [51].
- All fractions are subjected to the DPPH assay to identify the most active fraction.
- The active fraction is then purified using semi-preparative or analytical HPLC with a C18 column and an acetonitrile-water gradient [51].
- The effluent is monitored with a UV detector (e.g., 280 nm), and fractions are collected automatically or manually.
- Each purified compound is re-assayed for bioactivity.
Structure Elucidation:
- The structure of the isolated active compound is elucidated using spectroscopic techniques, primarily Nuclear Magnetic Resonance (NMR) spectroscopy (1H, 13C, COSY, HSQC, HMBC) and confirmed by HRMS [51].
Essential Research Reagents and Materials
The following table details key reagents, materials, and software essential for executing the described dereplication protocols.
Table 1: Research Reagent Solutions and Essential Materials
| Item | Function/Application | Example Specifications / Notes |
|---|---|---|
| C18 Solid Phase | Stationary phase for fractionation (VLC) to simplify complex extracts. | Used for initial fractionation of crude extract [51]. |
| HPLC Solvents | Mobile phase for chromatographic separation. | HPLC-grade water, methanol, acetonitrile; often modified with 0.1% acetic or formic acid [51]. |
| DPPH Reagent | Free radical agent used in antioxidant bioassays to guide isolation. | 0.1 mM solution in methanol; used to screen for antioxidant activity [51]. |
| α-Glucosidase Enzyme | Enzyme target for anti-diabetic activity screening. | Used in an inhibition assay to detect potential anti-diabetic compounds [51]. |
| Brine Shrimp Eggs (Artemia salina) | Used in a lethality test for preliminary toxicity assessment. | A simple model to evaluate the toxicity of test samples [51]. |
| MZmine Software | Open-source software for processing raw LC-MS data. | Used for peak detection, alignment, and deconvolution [49] [50]. |
| GNPS Platform | Web-based platform for mass spectrometry data analysis and molecular networking. | Used for creating molecular networks and database searching [51]. |
| SIRIUS 4 Software | Computational tool for the annotation of compounds using tandem MS data. | Used for predicting molecular formula and structure [51]. |
| AntiBase / MarinLit | Commercial databases of natural products for dereplication. | Used for querying MS and NMR data to identify known compounds [49] [50]. |
Analytical Data Interpretation and Pathway Mapping
The data generated from HPLC-HRMS and subsequent processing requires careful interpretation to map compounds onto their biosynthetic pathways, which provides valuable biological context. The relationship between analytical data and biosynthesis is illustrated in Figure 2.
Key Biosynthetic Pathways
The interpretation of HRMS data allows researchers to link detected secondary metabolites back to their core biosynthetic origins. The most important building blocks and pathways include [11]:
- Acetyl-CoA: A key precursor for the acetate pathway, leading to polyketides and fatty acids.
- Shikimic Acid: The cornerstone of the shikimate pathway, which produces aromatic amino acids and a vast array of phenolic compounds, including flavonoids and tannins.
- Mevalonic Acid and 1-Deoxyxylulose-5-Phosphate: These are precursors for the mevalonate and non-mevalonate pathways, respectively, both of which lead to the biosynthesis of terpenoids and steroids.
The accurate mass from HRMS is used to calculate potential molecular formulae, while the MS/MS fragmentation patterns provide structural clues about the core scaffold and functional groups. This information, especially when viewed in the context of a molecular network of related analogs, enables the putative assignment of a compound to a specific chemical class and, by extension, its likely biosynthetic pathway [51] [11].
The integration of metabolomics and HPLC-HRMS has unequivocally established itself as the cornerstone of modern dereplication strategies within natural product drug discovery. This synergistic combination provides an unparalleled ability to rapidly characterize the chemical profile of complex biological extracts, efficiently distinguishing known compounds from novel chemical entities with therapeutic potential [51] [10] [50]. The workflow, which seamlessly merges advanced analytical techniques with bioinformatics and database mining, represents a powerful facilitator in the quest for new pharmacologically active compounds [50].
The broader implication for the principles of natural product research is profound. These technologies address long-standing challenges of rediscovery and inefficiency, revitalizing interest in natural products as a viable and innovative source for drug leads in the 21st century [10] [52]. By adopting these advanced metabolomic profiling and dereplication approaches, researchers can more effectively navigate the vast chemical space of nature, ensuring that the immense, untapped potential of biodiversity is explored in a rational, targeted, and productive manner [11] [50]. This positions natural products once again at the forefront of the fight against global health challenges, from antimicrobial resistance to cancer.
The escalating crisis of antibiotic resistance and the perpetual demand for novel therapeutics have revitalized interest in natural products (NPs) as indispensable resources for drug discovery. Historically, NPs and their derivatives have constituted a substantial proportion of approved pharmaceuticals, renowned for their extraordinary structural diversity and potent biological activities. However, traditional discovery and production pipelines face significant challenges, including low yields, difficulty in synthetic replication, and over-reliance on natural extraction. This technical guide delineates how engineered biosynthesis—the strategic application of synthetic biology and genetic engineering to natural product biosynthetic pathways—is revolutionizing this field. By detailing core principles, methodologies, and experimental protocols, this review provides researchers and drug development professionals with a framework for harnessing biosynthetic gene clusters (BGCs) to discover, optimize, and sustainably produce valuable bioactive compounds.
Natural products are secondary metabolites produced by living organisms—plants, microbes, and marine organisms—that exhibit a wide spectrum of biological activities. These compounds have been the cornerstone of modern pharmacopeia; approximately 50% of new therapeutic agents in the pharmaceutical market are based on natural product scaffolds or their derivatives [54] [55]. Iconic examples include the antibiotic penicillin from Penicillium fungi, the anticancer drug paclitaxel (Taxol) from the Yew tree, and the antimalarial artemisinin [54] [55]. The structural complexity of NPs, evolved for specific biological interactions, makes them superior starting points for drug development, particularly for newly discovered targets where no small molecule lead exists [54].
Despite this promise, traditional NP discovery is hampered by several bottlenecks: the low abundance of bioactive compounds in source organisms, leading to cumbersome and ecologically damaging extraction processes; the structural complexity that often defies efficient chemical synthesis; and the silencing of BGCs under standard laboratory conditions [56] [54]. Engineered biosynthesis directly confronts these challenges by using synthetic biology to manipulate the genetic blueprints of these compounds, enabling the sustainable production of complex NPs and the generation of novel "unnatural" natural products with enhanced pharmaceutical properties [57] [55].
Foundational Concepts and Strategies
Biosynthetic Pathways and Gene Clusters
The biosynthesis of major NP classes—including polyketides, nonribosomal peptides, terpenoids, and alkaloids—is governed by dedicated enzymatic pathways encoded by BGCs. These gene clusters can be extensive, containing all genes necessary for the assembly, modification, and regulation of a specific metabolite.
- Polyketides (PKs) are assembled by polyketide synthase (PKS) enzyme complexes, which iteratively condense simple acyl-CoA precursors (e.g., malonyl-CoA, methylmalonyl-CoA) in a process analogous to fatty acid biosynthesis. PKSs are categorized into types I (modular, iterative), II (iterative, dissociated), and III (chalcone synthase-like). The modular type I PKSs, such as the 6-deoxyerythronolide B synthase (DEBS) for the erythromycin precursor, are particularly amenable to engineering due to their colinear architecture, where each module is responsible for one round of chain extension and modification [57].
- Nonribosomal Peptides (NRPs) are synthesized by nonribosomal peptide synthetase (NRPS) assembly lines. These massive enzymes activate, load, and condense amino acid building blocks (including non-proteinogenic ones) into complex peptides. Like PKSs, NRPSs are modular, with each module typically comprising adenylation (A), thiolation (T), and condensation (C) domains responsible for selecting, tethering, and incorporating a single amino acid [57].
- Terpenoids are derived from five-carbon isoprene units (isopentenyl diphosphate and dimethylallyl diphosphate) produced via either the mevalonate (MVA) pathway in the cytosol or the methylerythritol phosphate (MEP) pathway in plastids. The cyclization and subsequent oxidative modifications of the linear prenyl diphosphate precursors give rise to an immense structural diversity [58] [55].
- Alkaloids are nitrogen-containing compounds synthesized from amino acids. Their biosynthetic pathways involve a variety of enzymes, including decarboxylases, transaminases, and cytochrome P450s, leading to a wide array of pharmacologically active structures [55].
Core Engineering Strategies
Combinatorial biosynthesis encompasses a suite of strategies to re-engineer these natural assembly lines, expanding chemical diversity beyond what is found in nature [57] [59]. The three primary approaches are:
- Precursor-Directed Biosynthesis: This method exploits the inherent substrate promiscuity of biosynthetic enzymes. By feeding non-native, synthetic precursors to a producing organism or engineered host, the biosynthetic machinery incorporates these analogs, yielding novel derivatives. For instance, feeding propargyl-malonyl-N-acetylcysteamine to Streptomyces cinnamonensis resulted in the production of propargyl-premonensin, a polyether antibiotic analog with potential anticancer activity [57].
- Enzyme-Level Engineering: This involves directly modifying the enzymes within the biosynthetic pathway. Techniques include:
- Domain/Module Swapping: Replacing entire catalytic domains (e.g., an acyltransferase domain in a PKS) or modules with counterparts from different systems to alter substrate specificity or processing steps [57].
- Site-Specific Mutagenesis: Rational engineering of active sites to change enzyme function or specificity.
- Directed Evolution: Using iterative rounds of random mutagenesis and screening to evolve enzymes with desired properties.
- Pathway-Level Recombination: This strategy involves refactoring and transferring entire BGCs from their native, often difficult-to-culture, hosts into genetically tractable heterologous hosts like Escherichia coli, Saccharomyces cerevisiae, or Streptomyces species. This not only facilitates the discovery of new compounds by activating silent clusters but also enables the optimization of production titers in industrial-friendly chassis [57] [55].
The following workflow visualizes the iterative process of a typical engineered biosynthesis campaign, from gene cluster discovery to compound production and analysis:
Experimental Protocols & Methodologies
Protocol 1: Precursor-Directed Biosynthesis for Novel Macrolide Antibiotics
This protocol is adapted from Harvey et al., which combined precursor-directed biosynthesis with a colony bioassay to rapidly discover new macrolide antibiotics [57].
1. Objective: To generate and screen a library of glycosylated macrolide analogs from synthetic diketide precursors for enhanced or novel antibiotic activity.
2. Materials and Reagents:
- Biological Chassis: E. coli HYL3 strain (engineered for improved polyketide production).
- Plasmids: Vectors expressing module 2–6 of the 6-deoxyerythronolide B synthase (DEBS), and genes for sugar biosynthesis and glycosyl transfer.
- Precursor Library: A suite of synthetic diketide-N-acetylcysteamine (SNAC) thioesters with varying side chains (e.g., alkynyl, alkenyl).
- Growth Media: Lysogeny Broth (LB) supplemented with appropriate antibiotics (e.g., chloramphenicol, kanamycin) for plasmid maintenance.
- Assay Strain: Bacillus subtilis for the overlay lawn bioassay.
3. Procedure: 1. Strain Preparation: Co-transform the E. coli HYL3 strain with the required plasmid set. Select positive clones on LB agar with antibiotics. 2. Fermentation and Feeding: Inoculate a single colony into liquid media with antibiotics. Grow to mid-log phase and induce gene expression with Isopropyl β-d-1-thiogalactopyranoside (IPTG). Add individual synthetic diketide-SNAC precursors to the cultures. 3. Extraction and Analysis: After 48-72 hours of incubation, extract the culture broth with an equal volume of ethyl acetate. Concentrate the organic layer under reduced air pressure and resuspend the compound for analysis (e.g., LC-MS). 4. Bioactivity Screening: Employ a colony overlay assay. Spread the production culture or the extracted compounds on an agar plate. Once dry, overlay with soft agar seeded with the indicator strain, B. subtilis. Incubate and look for zones of growth inhibition around the colonies or application spots.
4. Expected Outcome: Identification of new erythromycin analogs, such as alkynyl-substituted variants, with potent antibiotic activity against the assay strain [57].
Protocol 2: Combinatorial Biosynthesis via AT Domain Swapping in a Modular PKS
This protocol outlines the generation of a combinatorial library of polyketide analogs by swapping acyltransferase (AT) domains, which dictate the extender unit incorporated during chain elongation.
1. Objective: To create a library of 6-deoxyerythronolide B (6-DEB) analogs with altered chemical structures and potentially novel bioactivities.
2. Materials and Reagents:
- DNA Constructs: Plasmid-borne DEBS genes with targeted AT domains. Donor DNA fragments encoding heterologous AT domains from other PKSs.
- Host Strain: An appropriate Streptomyces or E. coli host strain that does not produce competing polyketides.
- Molecular Biology Reagents: Restriction enzymes, Gibson Assembly or Golden Gate Assembly master mix, PCR reagents, competent cells.
- Analysis Reagents: LC-MS solvents and columns for polyketide analysis.
3. Procedure: 1. Design and Amplification: Design primers to amplify the donor AT domain with flanking homology arms compatible with the recipient DEBS module. The swap should occur at conserved boundary sequences to maintain protein structural integrity. 2. Cloning: Use a seamless cloning strategy (e.g., Gibson Assembly) to replace the native AT domain in the DEBS module with the heterologous AT domain in the expression plasmid. 3. Library Transformation: Transform the assembled plasmids into a suitable heterologous host to create a library of mutant PKS strains. 4. Screening and Characterization: Grow individual clones in deep-well plates. Induce PKS expression and analyze the culture extracts via LC-MS to detect the production of 6-DEB and its novel analogs based on mass shifts and retention times.
4. Expected Outcome: Production of a combinatorial library of 6-DEB analogs, such as those with altered alkyl side chains, enabling structure-activity relationship (SAR) studies [57].
The Scientist's Toolkit: Essential Research Reagents
Table 1: Key Reagents for Engineered Biosynthesis Experiments
| Reagent / Tool | Function / Application | Example(s) |
|---|---|---|
| Heterologous Hosts | Genetically tractable chassis for BGC expression and pathway optimization. | Escherichia coli, Saccharomyces cerevisiae, Streptomyces coelicolor [55] |
| CRISPR/Cas9 System | Enables precise genome editing, gene knockouts, and transcriptional activation of silent BGCs. | Streptomyces genome editing, activation of cryptic clusters [55] |
| Type III PKSs | Structurally simple PKSs with remarkable substrate promiscuity, ideal for generating diverse aromatic polyketides. | HsPKS1 from Huperzia serrata [57] |
| Non-native Precursors | Synthetic building blocks fed to engineered pathways to produce "unnatural" natural products. | Diketide-SNAC esters, propargyl-malonyl-SNAC, non-proteinogenic amino acids [57] |
| Bioinformatics Tools | For in silico identification of BGCs in genomic data and prediction of their functions. | antiSMASH, PRISM |
| Myricetin 3-O-Glucoside | Myricetin 3-O-Glucoside, CAS:19833-12-6, MF:C21H20O13, MW:480.4 g/mol | Chemical Reagent |
| Illudalic Acid | Illudalic Acid, CAS:18508-77-5, MF:C15H16O5, MW:276.28 g/mol | Chemical Reagent |
Emerging Technologies and Future Directions
The field of engineered biosynthesis is being rapidly advanced by several disruptive technologies:
- CRISPR-Cas9 Genome Editing: This technology allows for precise and multiplexed genome editing in native NP producers, which are often genetically intractable. It is used to knock out competing pathways, activate silent BGCs, and seamlessly swap large PKS/NRPS modules [55].
- Artificial Intelligence and Machine Learning: AI models are being trained on genomic, metabolomic, and structural data to predict BGC function, identify novel enzyme activities, and design optimized biosynthetic pathways, drastically accelerating the discovery and design process [55].
- Sustainable Biosynthesis and Bio-refineries: There is a growing emphasis on developing environmentally friendly production processes. Engineered microbes are being developed to utilize agricultural waste products or even carbon dioxide as feedstocks for the production of high-value NPs, aligning pharmaceutical manufacturing with circular economy principles [55].
The following diagram summarizes the multi-faceted approach of modern combinatorial biosynthesis, integrating various strategies to expand chemical diversity:
Engineered biosynthesis represents a paradigm shift in natural product-based drug discovery. By moving from simple extraction to the rational design and reprogramming of biosynthetic pathways, this discipline directly addresses the critical limitations of yield, complexity, and novelty. The integration of synthetic biology, combinatorial biosynthesis, and cutting-edge tools like CRISPR and AI is creating an unprecedented capacity to access the vast, untapped reservoir of natural product diversity. As the underlying principles of BGC regulation and pathway engineering are further elucidated, the systematic and sustainable production of optimized, novel therapeutics will become increasingly feasible, ensuring that natural products continue to be a primary wellspring for the medicines of tomorrow.
The process of drug discovery and development is a vast knowledge pyramid where building blocks of information are stacked upon each other to reach ever greater heights of knowledge [60]. An extensive body of biochemistry and organic synthesis forms the basis for chemical biology, which in turn supports pharmacology and lead optimization [60]. Within the specific context of natural product (NP) research, this pyramidal structure depends critically on the seamless integration of multiple disciplines—from traditional ethnobotany to cutting-edge artificial intelligence—to transform complex natural compounds into viable therapeutic agents.
Natural products refer to chemical compounds or substances produced by living organisms including plants, animals, and microorganisms [61]. These compounds have historically been a rich source of new drugs and therapeutic agents due to their diverse chemical structures and biological activities [61]. Approximately 50% of FDA-approved medications during 1981–2006 were natural products or synthetic derivatives of natural products [61]. However, the journey from benchtop discovery to bedside application presents numerous challenges, including the limited availability of bioactive molecules, structural complexity, low yields, and obscure mechanisms of action [61].
This technical guide examines the integrated cross-disciplinary pipelines that enable researchers to navigate these challenges efficiently, with particular emphasis on how modern computational technologies are revolutionizing traditional approaches to natural product drug discovery.
Core Disciplines in the Integrated Pipeline
Chemical Informatics and Structure Representation
A foundational element of modern natural product research is the effective representation of chemical structures in formats that are computationally efficient, intuitively understandable, and scientifically meaningful [60]. Several standardized electronic formats have been developed to meet these requirements:
Character Strings: ASCII-based notations like SMILES (Simplified Molecular Input Line Entry System) and SLN provide linear string representations that encode atomic composition, connectivity, and stereochemical information unambiguously [60]. These notations enable efficient storage and processing of structural data while remaining interpretable by practicing chemists.
Bit Strings: Fixed-length binary representations, including structural keys and molecular fingerprints, enable rapid similarity searching and pattern recognition within large compound databases [60]. The Daylight and UNITY fingerprint formats employ hashing and folding techniques to compress structural information, creating representations optimized for virtual screening and similarity assessment [60].
Cartesian Coordinates: Atomic composition paired with spatial coordinates provides essential structural information for molecular modeling, docking studies, and structure-activity relationship analysis [60]. These representations bridge the gap between two-dimensional structural information and three-dimensional molecular interactions.
The integration of these representation schemes into robust chemical database systems enables the efficient storage, retrieval, and mining of natural product structural data, forming the informatics backbone of modern discovery pipelines [60].
Artificial Intelligence and Machine Learning
Artificial intelligence, particularly machine learning (ML) and deep learning (DL), has emerged as a transformative force in natural product drug discovery [61]. These technologies fundamentally enhance researchers' ability to extract meaningful patterns from complex chemical and biological datasets.
Machine Learning Techniques: Supervised learning algorithms, including support vector machines (SVMs) and random forests, enable the construction of predictive models that correlate structural features with biological activity [61] [62]. These models facilitate virtual screening of natural product libraries, identifying compounds with high probability of desired bioactivity. Unsupervised learning approaches help identify inherent patterns and clusters within natural product datasets, enabling chemical space navigation and novel scaffold identification [62].
Deep Learning Architectures: Deep neural networks, particularly convolutional neural networks (CNNs) and recurrent neural networks (RNNs), automatically extract relevant features from raw structural data without manual engineering [61] [62]. These architectures excel at processing high-dimensional data from genomics, metabolomics, and cheminformatics, enabling the identification of complex, non-linear structure-activity relationships [62].
Generative Models: Generative adversarial networks (GANs) and autoencoders represent cutting-edge applications of AI to natural product research [61]. These models learn the underlying structural and property distributions of known bioactive natural products, then generate novel compounds with optimized characteristics—a process known as de novo molecular design [61] [62].
Table 1: Artificial Intelligence Techniques in Natural Product Discovery
| Technique Category | Specific Methods | Primary Applications in NP Discovery | Key Advantages |
|---|---|---|---|
| Machine Learning | Support Vector Machines, Random Forests, Gradient Boosting Machines | Structure-activity relationship analysis, Bioactivity prediction, Compound classification | Handles diverse molecular descriptors, Works with smaller datasets, High interpretability |
| Deep Learning | Convolutional Neural Networks (CNNs), Recurrent Neural Networks (RNNs), Graph Neural Networks (GNNs) | Molecular property prediction, Virtual screening, Binding affinity prediction | Automatic feature extraction, Handles raw structural data, Identifies complex patterns |
| Generative Models | Generative Adversarial Networks (GANs), Variational Autoencoders (VAEs) | De novo molecular design, Compound optimization, Scaffold hopping | Expands chemical space, Designs novel structures, Optimizes multiple properties simultaneously |
| Natural Language Processing | Large Language Models (LLMs), Transformer Architectures | Literature mining, Patent analysis, Knowledge extraction from textual data | Processes unstructured information, Identifies hidden connections, Supports hypothesis generation |
Analytical Chemistry and Structural Elucidation
Advanced analytical techniques remain essential for the characterization and validation of natural products. Nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry (MS), and X-ray crystallography provide critical structural information that complements computational predictions [61]. These experimental methods enable researchers to determine precise molecular structures, including stereochemical configurations that profoundly influence biological activity.
The integration of these analytical techniques with computational approaches creates a powerful feedback loop: computational predictions guide analytical efforts toward promising compounds, while analytical results refine and validate computational models. This synergistic relationship accelerates the structural elucidation process and enhances the reliability of predicted structures.
Bioinformatics and Omics Technologies
Bioinformatics tools enable researchers to explore the genomic and metabolic foundations of natural product biosynthesis. The analysis of biosynthetic gene clusters (BGCs) provides insights into the metabolic pathways responsible for producing complex natural products [62]. When integrated with metabolomic profiling, these approaches facilitate the targeted discovery of novel compounds with desired structural features.
Multi-omics integration—combining genomic, transcriptomic, proteomic, and metabolomic data—represents a powerful paradigm for understanding the biological context of natural product biosynthesis and mechanism of action [62]. Deep learning models are particularly well-suited to processing and interpreting these complex, high-dimensional datasets.
Integrated Workflow: From Discovery to Development
The following diagram illustrates the core integrated pipeline that connects multidisciplinary activities in natural product drug discovery:
Stage 1: Compound Sourcing and Preparation
The initial stage involves the careful selection and preparation of natural source materials. This process begins with the collection of biological specimens from diverse ecosystems, followed by the extraction of chemical constituents using appropriate solvents and techniques [62]. Bioassay-guided fractionation then isolates individual compounds or simplified mixtures for further analysis.
Experimental Protocol: Bioassay-Guided Fractionation
- Raw Material Processing: Source materials (plant, microbial, or marine) are lyophilized and homogenized to increase surface area for extraction.
- Sequential Extraction: Employ solvents of increasing polarity (hexane, ethyl acetate, methanol, water) to extract diverse chemical constituents.
- Primary Bioactivity Screening: Screen crude extracts against target biological assays to identify active fractions.
- Chromatographic Separation: Utilize column chromatography (silica gel, Sephadex) or HPLC to separate complex mixtures into discrete fractions.
- Iterative Bioactivity Testing: Test fractions from each separation step to track bioactivity.
- Compound Isolation: Continue separation of active fractions until pure compounds are obtained.
- Purity Assessment: Verify compound purity using analytical techniques (HPLC, TLC) before structural characterization.
This systematic approach ensures that isolation efforts remain focused on compounds with relevant biological activity, conserving resources and accelerating the identification of promising leads.
Stage 2: Structural Characterization and Data Registration
Isolated compounds undergo comprehensive structural characterization using spectroscopic and spectrometric techniques. The resulting structural information is then registered in specialized chemical databases using appropriate representation schemes [60].
Experimental Protocol: Structural Elucidation of Natural Products
- High-Resolution Mass Spectrometry: Determine molecular formula and mass using HR-MS (ESI-TOF or MALDI-TOF).
- Nuclear Magnetic Resonance Spectroscopy: Conduct 1D ((^1)H, (^{13})C) and 2D (COSY, HSQC, HMBC) NMR experiments to establish atomic connectivity and stereochemistry.
- Vibrational Spectroscopy: Employ IR and Raman spectroscopy to identify functional groups and confirm structural features.
- X-ray Crystallography (when applicable): Grow single crystals and perform X-ray diffraction analysis for unambiguous structural determination.
- Computational Modeling: Utilize molecular modeling and DFT calculations to predict NMR chemical shifts and validate proposed structures.
- Stereochemical Assignment: Determine absolute configuration using circular dichroism (CD) spectroscopy or chiral derivatization agents.
This multidimensional approach to structural characterization ensures comprehensive documentation of natural product structures, including stereochemical features that are critical for biological activity.
Stage 3: AI-Enhanced Bioactivity Prediction
Registered structures become inputs for AI-driven bioactivity prediction models. These computational approaches leverage existing bioactivity data to identify potential therapeutic applications for newly characterized natural products.
Experimental Protocol: Development of AI Bioactivity Prediction Models
- Training Data Curation: Compile comprehensive dataset of known bioactive compounds with associated activity values (IC~50~, K~i~, etc.).
- Molecular Featurization: Compute molecular descriptors (topological, electronic, steric) or generate learned representations using deep learning.
- Model Selection: Choose appropriate algorithm based on dataset size and complexity (Random Forest for smaller datasets, Deep Neural Networks for large datasets).
- Model Training: Implement cross-validation protocols to optimize model parameters and prevent overfitting.
- Performance Validation: Evaluate model performance using external test sets not seen during training.
- Prospective Prediction: Apply trained model to novel natural products to predict potential bioactivities.
- Experimental Confirmation: Prioritize compounds with favorable predictions for experimental validation.
This protocol enables researchers to leverage existing knowledge systematically, increasing the efficiency of bioactivity assessment and reducing reliance on random screening approaches.
Stage 4: Experimental Validation and Lead Optimization
Computational predictions require experimental validation through in vitro and in vivo testing. Promising compounds then undergo systematic optimization to improve their drug-like properties while maintaining efficacy.
Experimental Protocol: In Vitro Bioactivity Assessment
- Compound Preparation: Prepare test compounds as 10mM DMSO stocks, with serial dilutions for dose-response studies.
- Cell-Based Assays: Implement relevant cell culture models (primary cells or cell lines) to assess functional activity.
- Target-Based Assays: Employ enzyme inhibition or receptor binding assays to measure specific target engagement.
- Cytotoxicity Screening: Include counter-screens against non-target cells to assess selectivity.
- Dose-Response Analysis: Generate concentration-response curves to determine potency (EC~50~/IC~50~ values).
- Mechanistic Studies: Investigate mode of action through pathway analysis, gene expression profiling, or proteomic approaches.
- Early ADMET Assessment: Evaluate preliminary absorption, distribution, metabolism, excretion, and toxicity properties.
Compounds that demonstrate acceptable efficacy and safety profiles in these validation assays advance to lead optimization, where medicinal chemistry approaches improve their drug-like properties.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of integrated natural product discovery pipelines requires access to specialized reagents, materials, and computational resources. The following table details essential components of the cross-disciplinary researcher's toolkit:
Table 2: Essential Research Reagents and Materials for Integrated Natural Product Discovery
| Category | Specific Items | Function/Purpose | Technical Considerations |
|---|---|---|---|
| Chromatography Media | Silica gel (various pore sizes), Sephadex LH-20, C18 reverse-phase resin, Ion-exchange media | Separation and purification of natural products from complex mixtures | Particle size affects resolution; solvent compatibility crucial; reuse potential varies |
| Spectroscopy Standards | Deuterated solvents (CDCl~3~, DMSO-d~6~), NMR reference compounds (TMS), Mass calibration standards | Enable precise structural characterization through NMR and MS | Storage conditions critical; purity affects results; proper disposal required |
| Bioassay Components | Cell culture media, Enzyme substrates, Reporter systems, Detection reagents (MTT, Resazurin) | Facilitate biological activity assessment across multiple targets | Stability varies; storage conditions important; batch-to-batch consistency critical |
| Computational Resources | Chemical databases (PubChem, ChEMBL), Cheminformatics toolkits (RDKit, OpenBabel), ML frameworks (TensorFlow, PyTorch) | Enable virtual screening, molecular modeling, and AI-driven prediction | Licensing restrictions may apply; computational requirements vary; data formats important |
| Molecular Descriptors | Topological indices, Physicochemical parameters, 3D molecular fields, Fingerprint representations | Quantify structural features for QSAR and machine learning | Information content varies; applicability domain considerations; standardization needed |
| Moronic Acid | Moronic Acid, CAS:6713-27-5, MF:C30H46O3, MW:454.7 g/mol | Chemical Reagent | Bench Chemicals |
| 3-Epiursolic acid | 3-Epiursolic Acid|Ursolic Acid Analog|Research Compound | 3-Epiursolic Acid is a ursolic acid analog for research use only (RUO). Explore its potential applications in metabolic and cancer studies. Not for human use. | Bench Chemicals |
Data Management and Knowledge Integration
Effective data management practices are essential for successful cross-disciplinary integration. The implementation of standardized data formats, metadata annotation schemes, and knowledge representation frameworks enables seamless information flow across disciplinary boundaries.
Chemical Database Management: Specialized chemical database systems employ screening approaches to accelerate structure and substructure searching [63]. These systems typically utilize linear notations (SMILES, WLN) and topological indices to enable efficient retrieval of structurally similar compounds [63]. Canonicalization algorithms ensure unique representation of molecular structures, facilitating precise structure-based queries and comparisons [63].
Knowledge Pyramid Construction: As described in the drug discovery knowledge pyramid, information progresses from "vague and voluminous" to "specialized and refined" as it moves through the discovery pipeline [60]. Each disciplinary contribution adds value to the raw data generated at previous stages, gradually building the comprehensive understanding necessary for clinical translation.
The following diagram illustrates the flow of data and knowledge through the integrated discovery pipeline:
Integrated cross-disciplinary pipelines represent the future of natural product drug discovery. By systematically combining expertise from chemistry, biology, computational science, and clinical research, these pipelines enable researchers to navigate the complexity of natural products efficiently. The strategic incorporation of artificial intelligence and machine learning technologies further enhances these pipelines, providing powerful tools for pattern recognition, prediction, and decision support.
As these integrated approaches continue to evolve, they promise to accelerate the translation of natural product discoveries from benchtop to bedside, unlocking the full therapeutic potential of nature's chemical diversity. Researchers who master these cross-disciplinary frameworks will be uniquely positioned to contribute to the next generation of natural product-derived therapeutics.
Navigating the Discovery Pipeline: Overcoming Technical and Regulatory Hurdles
The transition of a bioactive natural compound from a initial "screening hit" through a "drug lead" to a "marketed drug" is associated with increasingly challenging demands for compound amount, which often cannot be met by re-isolation from the original biological sources [64]. These supply chain complexities represent one of the most significant bottlenecks in natural product-based drug discovery programs [65]. The challenges are multifaceted, encompassing environmental, technical, and regulatory dimensions that must be systematically addressed to enable successful drug development.
The intrinsic complexity of natural product chemistry, limited availability of many natural products in their source organisms, and the critical need for sustainable sourcing and conservation create a challenging landscape for researchers [66]. Furthermore, correct species identification, taxonomic complexities, and accurate documentation of plant material represent foundational challenges that cannot be automated and require specialized expertise that is becoming increasingly rare [64]. These challenges necessitate highly integrated interdisciplinary approaches to develop effective solutions for sustainable supply and resupply of promising natural product drug candidates.
Sustainable Sourcing of Natural Products
Regulatory Frameworks and Biodiversity Access
The access and use of biological resources must be mutually agreed between the country willing to use the resource and the country of origin, which has sovereign rights over them [65]. The Convention on Biological Diversity (CBD) established by the United Nations in 1992 and the subsequent Nagoya Protocol on Access and Benefit-sharing (ABS) provide international frameworks governing genetic resource utilization [65]. These agreements aim to ensure fair and equitable sharing of benefits arising from the utilization of genetic resources while promoting conservation and sustainable use.
In Brazil, a megadiverse country, Law 13.123/15 and the SisGen registry system govern research and technological development involving genetic heritage [65]. Similar regulations exist in other biodiversity-rich nations, creating a complex regulatory landscape for researchers. Foreign researchers seeking to access native biodiversity typically must collaborate with local scientific institutions, which assume responsibility for registering the activity [65]. Negotiating appropriate benefit-sharing and access agreements can be time-consuming, requiring sophisticated knowledge of commercialization pathways, risks, and rewards from all involved parties [65].
Sustainable Harvesting and Cultivation Practices
Sustainable sourcing requires implementing practices that ensure long-term viability of natural sources while maintaining ecosystem integrity. For plant-derived natural products, this includes:
- Wildcrafting guidelines: Establishing protocols for ethical wild harvesting that minimize ecological impact
- Cultivation programs: Developing controlled cultivation of medicinal plants to reduce pressure on wild populations
- Agricultural optimization: Improving yield of target compounds through selective breeding and optimized growing conditions
The limited availability of many natural products in their source organisms creates significant supply challenges, particularly when promising compounds are present in minute quantities [66]. For instance, paclitaxel (Taxol) was originally isolated from the bark of the Pacific yew tree (Taxus brevifolia), requiring destruction of approximately three trees to obtain enough compound to treat one patient [64]. Such examples highlight the critical importance of developing sustainable and scalable sourcing strategies early in the drug discovery pipeline.
Resupply Strategies for Natural Product Drug Candidates
Total Chemical Synthesis
Total chemical synthesis represents a powerful approach for resupply of natural product drug candidates, offering potential for unlimited and reproducible production of complex molecules. This strategy involves designing and executing synthetic routes to construct target compounds from simpler, readily available starting materials [64]. Successful implementation of total synthesis can provide not only the natural product itself but also enables preparation of structural analogs for structure-activity relationship (SAR) studies.
The synthesis of complex natural products, however, presents significant challenges. Many bioactive natural products possess intricate molecular architectures featuring multiple chiral centers, complex ring systems, and sensitive functional groups that complicate synthetic efforts [66]. The development of efficient synthetic routes requires substantial investment in time and resources, with success dependent on the molecular complexity of the target compound and the current state of synthetic methodology.
Table 1: Comparison of Natural Product Resupply Strategies
| Resupply Method | Key Advantages | Major Challenges | Typical Applications |
|---|---|---|---|
| Total Synthesis | Unlimited supply; Structure-activity relationship studies possible; High purity | Often multistep, low-yielding routes; Technical expertise required; Cost-intensive at scale | Complex molecules with established synthetic routes; Production of analogs |
| Semisynthesis | Leverages natural product scaffold; Fewer steps than total synthesis; Access to analogs | Still requires natural starting material; Functionalization challenges | Natural products with synthetically accessible modification sites |
| Plant Biotechnology | Sustainable; Preserves biosynthetic machinery; Scalable | Optimization of culture conditions needed; Genetic instability; Variable yields | Plant species with established in vitro culture systems |
| Microbial Fermentation | Highly scalable; Controlled production conditions; Cost-effective | Requires host engineering; May need pathway optimization; Potential low production titers | Compounds where biosynthetic genes are characterized |
| Synthetic Biology | Sustainable production; Engineered optimization; Renewable feedstocks | Complex pathway engineering; Host compatibility issues; Regulatory considerations | High-value natural products with elucidated pathways |
Biotechnology Approaches
Biotechnology offers alternative resupply strategies that harness or mimic natural biosynthetic pathways:
Plant Cell and Tissue Culture
Plant cell and tissue culture technologies provide a controlled and sustainable platform for producing plant-derived natural products independent of geographical and seasonal constraints [64]. This approach involves establishing dedifferentiated cell cultures or organ cultures (such as roots or shoots) that can produce target compounds under optimized laboratory conditions. Success depends on numerous factors, including selection of high-producing cell lines, optimization of culture media and conditions, and application of appropriate elicitors to enhance secondary metabolite production.
Microbial Fermentation and Synthetic Biology
Microbial systems offer highly scalable and cost-effective platforms for natural product production. Advances in synthetic biology have enabled the engineering of microbial hosts (such as E. coli or S. cerevisiae) to produce complex plant natural products by introducing and optimizing heterologous biosynthetic pathways [64]. This approach requires comprehensive understanding of the biosynthetic pathway, identification and characterization of relevant enzymes, and optimization of pathway flux in the production host.
For example, semisynthetic production of the antimalarial drug artemisinin now combines extraction of the intermediate artemisinic acid from engineered yeast with subsequent chemical conversion to the final active compound, providing a more reliable and scalable supply than direct extraction from the plant Artemisia annua [64].
Analytical Methods for Compound Identification and Purity Assessment
Advanced analytical techniques are essential for characterizing natural products and ensuring quality control throughout the resupply process:
Diagram 1: Integrated Workflow for Bioactive Compound Identification and Resupply
Biochemometric approaches integrate chemical analysis with bioassay data to efficiently identify bioactive constituents within complex mixtures [67]. This methodology combines untargeted metabolomic profiling using techniques such as UPLC-HRMS (Ultra-Performance Liquid Chromatography-High Resolution Mass Spectrometry) with biological screening data to pinpoint compounds responsible for observed activities [67]. Several computational approaches facilitate this integration:
- Partial Least Squares (PLS): Decomposes spectral datasets into latent variables that maximize covariance with biological activity data [67]
- S-plots: Visualize covariance and correlation loading variables from OPLS models to identify spectral features correlating with bioactivity [67]
- Selectivity Ratio: Calculates the ratio between explained and residual variance of spectral variables, providing a quantitative measure of each variable's power to distinguish bioactive and non-bioactive samples [67]
These biochemometric methods help overcome limitations of traditional bioassay-guided fractionation, which can be biased toward abundant rather than genuinely bioactive mixture components [67].
Experimental Protocols for Sustainable Sourcing and Resupply
Biochemometric Analysis for Targeted Isolation
Biochemometric analysis enables researchers to focus isolation efforts on constituents most likely responsible for biological activity, minimizing waste of valuable source material [67].
Protocol Objectives: Identify bioactive ions in complex natural product mixtures early in fractionation to guide efficient isolation [67].
Materials and Equipment:
- UPLC-HRMS system with appropriate chromatographic columns
- Biological assay system relevant to therapeutic target
- Statistical analysis software (e.g., SIMCA, R, or MATLAB)
- Standard laboratory equipment for extraction and fractionation
Procedure:
- Sample Preparation: Prepare crude extract and sequential fractions (typically 4-6 fractions) from the source organism
- Chemical Profiling: Acquire UPLC-HRMS data for all samples in technical triplicate to ensure analytical reproducibility [67]
- Bioactivity Testing: Assess all extracts and fractions in relevant biological assays
- Data Preprocessing: Process raw UPLC-HRMS data to detect and align peaks, then normalize and scale the data
- Statistical Modeling: Apply selectivity ratio analysis to identify features (retention time-m/z pairs) with highest correlation to bioactivity [67]
- Compound Identification: Target isolation of high-priority features for structure elucidation
- Validation: Confirm bioactivity of purified compounds and verify statistical predictions
Technical Notes: The selectivity ratio method has demonstrated superior performance in identifying bioactive ions compared to PLS loading vectors or S-plots, particularly for components present in low abundance [67].
Research Reagent Solutions for Natural Product Supply Research
Table 2: Essential Research Reagents and Materials for Natural Product Supply Studies
| Reagent/Material | Function/Application | Technical Specifications | Implementation Considerations |
|---|---|---|---|
| UPLC-HRMS System | Untargeted metabolomic profiling of complex mixtures | High resolution (>50,000); MS/MS capability; Reverse-phase and HILIC options | Enables detection of major and minor mixture components; Essential for biochemometric analysis [67] |
| Bioassay Systems | Assessment of biological activity | Target-specific (e.g., antimicrobial, anticancer, anti-inflammatory) | Should be robust, reproducible, and compatible with natural product samples [64] |
| Cell Culture Systems | Plant and microbial cultivation for sustainable production | Species-optimized media; Sterile containment; Environmental control | Required for biotechnology-based resupply approaches [64] |
| Chromatography Media | Compound isolation and purification | Normal-phase, reverse-phase, and size-exclusion resins | Selection depends on compound chemistry; Impacts isolation efficiency [67] |
| Statistical Software | Biochemometric data analysis | PLS, selectivity ratio, and S-plot capabilities | Essential for integrating chemical and biological datasets [67] |
Integrated Approaches and Future Perspectives
Addressing supply and resupply challenges requires integrated strategies that combine multiple approaches tailored to specific natural products and their source organisms. The most successful programs implement sourcing triages that evaluate total synthesis, semisynthesis, and biotechnological production in parallel, selecting the most feasible approach based on molecular complexity, abundance in source organism, and available technical expertise [64].
Emerging technologies offer promising directions for overcoming current limitations. Advanced analytics and artificial intelligence are playing increasingly important roles in natural product research [13]. Genome mining and metabolic engineering continue to expand the toolbox for sustainable production [64]. Furthermore, international collaborations that respect biodiversity sovereignty and promote equitable benefit-sharing will be essential for accessing the chemical diversity needed for future drug discovery breakthroughs [65].
Diagram 2: Decision Framework for Sustainable Natural Product Supply
The declining trend in new drugs reaching the market has revitalized scientific interest in drug discovery from natural sources, despite the known challenges [64]. By developing and implementing robust solutions to supply and resupply challenges, researchers can unlock the full potential of nature's chemical diversity to address unmet medical needs. The scientific developments, technological advances, and research trends clearly indicate that natural products will be among the most important sources of new drugs also in the future, provided that sustainable sourcing and resupply strategies are successfully implemented [64].
Within the framework of natural product drug discovery research, the journey from biological source to a fully characterized bioactive compound is fraught with significant technical challenges. The initial phases of extraction, isolation, and characterization represent a critical triad of bottlenecks that can dictate the pace and success of entire discovery campaigns [68] [69]. These processes are tasked with efficiently liberating desired chemical constituents from complex biological matrices, purifying them from a vast excess of irrelevant material, and unambiguously determining their chemical structures—all while preserving the integrity of often scarce and labile molecules [70] [71]. Despite remarkable technological advancements, these foundational steps remain a major hurdle, contributing to the perception of natural product research as a slow and difficult path compared to synthetic library screening [68]. This whitepaper provides an in-depth analysis of these core technical bottlenecks, presenting both conventional and contemporary methodologies, and offers structured guidance to aid researchers in navigating these complexities.
The Extraction Bottleneck: From Raw Material to Bioactive Enrichment
Extraction is the crucial first step in the analysis of medicinal plants, as it is necessary to extract the desired chemical components from the plant materials for further separation and characterization [70]. The fundamental challenge lies in the stark chemical disparity between the native biological environment and the conditions required for analysis, necessitating a process that is both efficient and selective.
Conventional Extraction Techniques
Classical methods remain widely used due to their simplicity and minimal equipment requirements.
- Maceration: This process involves soaking the plant material in a solvent at room temperature for an extended period (3–4 days). It is a simple technique but suffers from excessive solvent usage, lengthy extraction times, and poor extraction yields [70] [71].
- Percolation: In this method, pre-wet material is packed in a percolator, and fresh solvent is continuously passed through it. While offering better extraction efficiency than maceration, it still uses large volumes of solvent and is time-consuming [71].
- Soxhlet Extraction: This technique uses a limited volume of solvent that is continuously recycled through the sample via distillation for 3–18 hours. It is efficient and cost-effective but employs elevated temperatures, which can impair Thermo-labile compounds [70] [71].
- Reflux Extraction: Similar to Soxhlet, this method is carried out at a constant heat with repeatable solvent evaporation and condensation. It is efficient but also poses a risk to heat-sensitive compounds [71].
Modern Extraction Techniques
Recently developed extraction techniques for natural products are more energy-efficient and result in shorter extraction times compared to earlier methods [72].
- Microwave-Assisted Extraction (MAE): This technique uses microwave energy to rapidly heat the solvent and plant material, reducing extraction time and solvent consumption. It has been modified into various forms, such as vacuum microwave-assisted extraction (VMAE) and ultrasonic microwave-assisted extraction (UMAE) [71].
- Ultrasound-Assisted Extraction (UAE): In UAE, the plant material is placed in an ultrasonic bath. The process reduces extraction time and improves yields owing to mechanical stress leading to cavitation and cellular disruption. A drawback is that the process can generate heat, potentially damaging Thermo-labile compounds [71].
- Accelerated Solvent Extraction (ASE): Also known as Pressurized Liquid Extraction (PLE), this technique uses solvents at elevated temperatures and pressures, which can significantly reduce extraction time and solvent volume compared to conventional methods. It also allows for on-line filtration [72] [71].
- Supercritical Fluid Extraction (SFE): This method, most commonly using supercritical COâ‚‚, is considered an environmentally friendly alternative to organic solvents. It is particularly effective for non-polar compounds, and the use of organic solvent modifiers can extend its application to more polar natural products. The technique reduces ecological complications associated with organic solvent extractions [71].
Table 1: Comparison of Common Natural Product Extraction Methods
| Extraction Method | Key Principle | Optimal Solvent | Time Required | Temperature | Key Advantages | Key Limitations |
|---|---|---|---|---|---|---|
| Maceration [71] | Passive soaking | Methanol, Ethanol, Water | 3–4 days | Room Temperature | Simple, no special equipment | Lengthy, high solvent use, low efficiency |
| Soxhlet [70] [71] | Continuous solvent recycling | Methanol, Ethanol, Hexane | 3–18 hours | Solvent Boiling Point | High efficiency, low solvent volume | High heat degrades thermolabile compounds |
| Microwave-Assisted (MAE) [72] [71] | Microwave dielectric heating | Polar solvents (e.g., Water, Ethanol) | Minutes to 1 hour | Elevated (Controllable) | Rapid, reduced solvent, high yield | Potential for localized overheating |
| Ultrasound-Assisted (UAE) [71] | Acoustic cavitation | Methanol, Ethanol, Water | ~1 hour | Can generate heat | Rapid, improved yield, simple setup | Heat generation may damage compounds |
| Accelerated Solvent (ASE) [72] [71] | Pressurized liquid at high temp | Various (Methanol to Hexane) | 10–20 minutes | Elevated (40–200°C) | Fast, automated, low solvent consumption | High initial equipment cost |
| Supercritical Fluid (SFE) [71] | Solvation with supercritical CO₂ | CO₂ (with modifiers) | 30–120 minutes | Near-ambient (31°C+) | Green, non-toxic, tunable selectivity | High pressure, cost, less effective for high-polarity compounds |
Experimental Protocol: Standardized Maceration for Bioactive Plant Extracts
The following protocol provides a baseline methodology for the initial extraction of plant material, adaptable for bioassay-guided fractionation [70] [71].
- Sample Preparation: The plant material (e.g., leaves, roots) should be air-dried in the shade at room temperature to prevent chemical degradation. The dried material is then ground to a coarse powder using a mechanical grinder to increase the surface area for solvent contact.
- Extraction: The powdered plant material (e.g., 100 g) is placed in a sealed glass container and soaked in an appropriate solvent (e.g., 500 mL of 70% ethanol in water). The container is kept at room temperature with occasional stirring or shaking for 72 hours.
- Filtration and Concentration: The mixture is first filtered through a muslin cloth to remove coarse particles, and then through filter paper (e.g., Whatman No. 1). The resulting filtrate is concentrated under reduced pressure using a rotary evaporator at a temperature not exceeding 40°C to prevent degradation of thermolabile compounds.
- Crude Extract Storage: The concentrated crude extract is transferred to a pre-weighed vial and further dried in a desiccator. The final weight is recorded, and the extract is stored at 4°C for subsequent bioactivity screening or fractionation.
The Isolation Bottleneck: Purification of Bioactive Compounds
Following extraction, the resulting crude mixture is a complex combination of various types of bioactive compounds or phytochemicals with different polarities, making their separation a significant challenge [70]. Isolation is the process of separating out the desired component(s) from this chemical mixture to obtain pure compounds for structure elucidation and biological activity testing [72].
Chromatographic Isolation Techniques
A broad range of chromatographic methods is applicable for better fractionation and finished natural product purification. The choice generally depends on the purity stage of the extract and the final goal [71].
- Thin-Layer Chromatography (TLC) and Bio-autography: TLC is a simple, quick, and inexpensive procedure that provides a rapid overview of the number of components in a mixture and can support the identity of a compound [70]. When coupled with bioautography, it becomes a powerful technique for locating antimicrobial compounds directly on the TLC plate. This combines chromatographic separation and in situ activity determination, facilitating the target-directed isolation of active constituents [70].
- Vacuum Liquid Chromatography (VLC) & Flash Chromatography (FC): These are low-pressure, open-column techniques used for the initial fractionation of crude extracts. They can handle gram quantities of material and are effective for separating compounds based on their polarity using a stationary phase like silica gel and a gradient of solvents of increasing polarity [71].
- Medium-Pressure Liquid Chromatography (MPLC): MPLC offers a superior separation efficiency to VLC and FC due to the use of a pressurized system and finer stationary phase particles. It is a workhorse for the intermediate purification of natural product fractions [71].
- High-Performance Liquid Chromatography (HPLC): HPLC is a versatile, robust, and widely used technique for the isolation of natural products, especially as the final step to obtain pure compounds [70] [71]. It provides high resolution and is readily coupled with detection methods like mass spectrometry (MS) and ultraviolet (UV) spectroscopy. Preparative HPLC is specifically designed for isolating larger quantities (milligrams to grams) of pure compounds from complex mixtures.
Table 2: Chromatographic Techniques for the Isolation of Natural Products
| Technique | Scale | Pressure/System | Key Application in NP Isolation | Throughput | Resolution |
|---|---|---|---|---|---|
| Thin-Layer Chromatography (TLC) [70] [71] | Analytical to micro-preparative | Atmospheric | Rapid profiling, bioautography, purity check | Low | Low-Medium |
| Flash Chromatography (FC) [71] | Gram-scale | Low-Pressure (Glass Column) | Initial rough fractionation of crude extracts | Medium | Low-Medium |
| Medium-Pressure LC (MPLC) [71] | Multi-gram scale | Medium-Pressure (Pump) | Intermediate fractionation and purification | Medium | Medium |
| High-Performance LC (HPLC) [70] [71] | Analytical to preparative | High-Pressure (Pump) | Final purification of compounds to high purity | Low (Prep) | High |
| Solid-Phase Extraction (SPE) [71] | Small-scale | Low-Pressure (Cartridge) | Rapid desalting, pre-cleaning, or fractionation | High | Low |
Experimental Protocol: Bioassay-Guided Fractionation using Flash Chromatography
This protocol outlines a standard approach for fractionating a crude extract based on polarity, with fractions subsequently screened for biological activity to guide further isolation efforts [71].
- Stationary Phase Packing: A glass column is dry-packed with a suitable adsorbent, typically silica gel (e.g., 40–63 µm particle size). The column is then conditioned with the initial mobile phase (e.g., 100% hexane).
- Sample Loading: The crude extract (e.g., 1 g) is adsorbed onto a small amount of silica gel (∼2 g) and dried to form a free-flowing powder. This powder is carefully layered on top of the pre-packed column.
- Gradient Elution: A stepwise gradient of solvents of increasing polarity is applied. A typical solvent series may be: 100% Hexane → Hexane/Ethyl Acetate mixtures (e.g., 90:10, 75:25, 50:50, 25:75) → 100% Ethyl Acetate → Ethyl Acetate/Methanol mixtures → 100% Methanol. The flow is maintained using gas pressure or a peristaltic pump.
- Fraction Collection: Eluate is collected in a series of pre-weighed test tubes using an automatic fraction collector. The volume per fraction is kept constant (e.g., 20 mL).
- Analysis and Pooling: Every fraction is analyzed by TLC. Fractions with similar TLC profiles are pooled together. All pooled fractions are concentrated using a rotary evaporator.
- Bioactivity Screening: The pooled fractions are subjected to relevant biological assays (e.g., antimicrobial, antioxidant). The active fraction(s) are then selected for further, higher-resolution purification (e.g., using MPLC or preparative HPLC).
The Characterization Bottleneck: Elucidating Chemical Structure
After isolation of pure compounds, the critical step is the determination of their chemical structure, including stereochemistry [70] [71]. Due to the vast structural diversity of natural products, this remains a highly complex task that relies heavily on advanced analytical techniques.
Advanced Analytical Techniques for Structure Elucidation
- Liquid Chromatography-Mass Spectrometry (LC-MS): This hybrid technique combines the physical separation of HPLC with the mass analysis capabilities of MS. It is indispensable for determining the molecular weight of a compound and obtaining fragmentation patterns that provide clues about its structure. High-resolution mass spectrometry (HRMS) can determine the exact mass, allowing for the assignment of a precise molecular formula [70] [69].
- Nuclear Magnetic Resonance (NMR) Spectroscopy: NMR is the most powerful technique for de novo structure elucidation. It provides detailed information about the carbon-hydrogen framework of a molecule. Modern multi-dimensional NMR experiments (e.g., COSY, HSQC, HMBC) are essential for determining the planar structure and, in combination, the relative stereochemistry of complex natural products [69] [71].
- Hybrid LC-MS-SPE-NMR Systems: The most advanced approach involves the hyphenation of multiple techniques. For example, LC-MS is used to separate and identify compounds of interest, which are then automatically trapped onto solid-phase extraction (SPE) cartridges. The trapped compound is subsequently eluted into an NMR flow probe for structure determination. This integrated system allows for the rapid characterization of compounds from complex mixtures, even in sub-milligram quantities, and minimizes the need for extensive isolation [69].
- Fourier Transform Infrared (FTIR) Spectroscopy: FTIR is used to identify functional groups within a molecule based on the absorption of infrared radiation. It can provide supportive data on the presence of groups such as hydroxyls, carbonyls, and alkenes [70].
Experimental Protocol: Preliminary Phytochemical Characterization of a Pure Isolate
This protocol describes the initial steps for characterizing a newly isolated natural product.
- Purity Assessment: Analyze the pure compound using analytical TLC or HPLC to confirm it is a single entity.
- Molecular Formula Determination: Acquire a high-resolution mass spectrum (HRMS) of the compound. The exact mass of the molecular ion ([M+H]⺠or [M-H]â») is used to calculate and assign a molecular formula.
- Functional Group Analysis: Obtain an FTIR spectrum (e.g., using a KBr pellet). Key absorption bands are identified (e.g., O-H stretch ∼3400 cmâ»Â¹, C=O stretch ∼1700 cmâ»Â¹, aromatic C=C ∼1600 cmâ»Â¹) to propose functional groups present.
- ¹H NMR Analysis: Dissolve the compound in a deuterated solvent (e.g., CDCl₃, DMSO-d6) and acquire a ¹H NMR spectrum. Analyze the chemical shifts (δ, ppm), integration (number of protons), and coupling constants (J, Hz) to identify the types of protons and their connectivity.
- ¹³C NMR Analysis: Acquire a ¹³C NMR spectrum of the compound. The number of distinct signals indicates the number of unique carbon environments. The chemical shifts help identify the types of carbons (e.g., carbonyl, aromatic, aliphatic).
- Advanced NMR Experiments: For complex structures, perform 2D NMR experiments such as COSY (to identify proton-proton couplings), HSQC (to identify direct C-H connections), and HMBC (to identify long-range C-H couplings) to piece together the molecular structure.
Integrated Workflow and Visualization
The journey from a natural source to a characterized bioactive compound is a multi-stage process where the output of one step becomes the input for the next. The following diagram illustrates this integrated workflow, highlighting the techniques involved at each stage and the iterative nature of bioassay-guided isolation.
Diagram 1: Integrated NP Discovery Workflow.
The Scientist's Toolkit: Essential Reagents and Materials
Successful navigation of the technical bottlenecks in natural product research requires a suite of specialized reagents and materials. The following table details key items essential for experiments in extraction, isolation, and characterization.
Table 3: Essential Research Reagents and Materials for Natural Product Research
| Item/Category | Common Examples | Primary Function in NP Research |
|---|---|---|
| Extraction Solvents [70] [72] | Hexane, Chloroform, Ethyl Acetate, Methanol, Ethanol, Water | To dissolve and release compounds from biological matrices based on polarity ("like dissolves like"). |
| Chromatography Stationary Phases [71] | Silica Gel, C18-bonded Silica, Sephadex LH-20, Alumina, Diol | The solid support for chromatographic separation, separating compounds based on adsorption, partition, or size exclusion. |
| Chromatography Mobile Phases [71] | Hexane, Ethyl Acetate, Chloroform, Methanol, Acetonitrile, Water (often with modifiers like TFA) | The liquid phase that moves through the stationary phase, eluting compounds at different rates based on their interaction with both phases. |
| Deuterated NMR Solvents [69] | CDCl₃, DMSO-d6, Methanol-d4 | To dissolve samples for NMR analysis without introducing interfering signals from protonated solvents. |
| Visualization Reagents [70] | Vanillin/Sulfuric acid, Anisaldehyde, Ceric Ammonium Molybdate | Spray reagents for TLC plates to visualize compounds that are not UV-active through color reactions. |
| Bioassay Reagents [70] | Microbial strains (S. aureus, E. coli), Cell lines, Enzyme substrates, Indicator dyes | Biological tools and chemicals used to test the pharmacological activity of extracts and compounds. |
| 3-Chloro-10H-phenothiazine | 3-Chloro-10H-phenothiazine|CAS 1207-99-4 | 3-Chloro-10H-phenothiazine is a key synthetic intermediate for phenothiazine pharmaceuticals. This product is For Research Use Only. Not for human or veterinary use. |
The path of natural product drug discovery is intrinsically linked to the successful management of the technical bottlenecks in extraction, isolation, and characterization. While these stages present significant challenges due to the complexity of natural matrices and the delicate nature of the target molecules, a robust toolkit of both conventional and modern techniques exists to address them. Strategic selection of methods—from green and efficient extraction technologies like MAE and SFE, through hyphenated chromatographic purification systems, to powerful spectroscopic platforms like LC-MS-SPE-NMR—is paramount. As technological innovations continue to emerge, particularly in automation, data processing, and miniaturization, the efficiency of overcoming these foundational hurdles will undoubtedly increase. By systematically applying the principles and protocols outlined in this whitepaper, researchers can better navigate these complexities, thereby accelerating the translation of nature's chemical diversity into novel therapeutic agents.
The discovery of new pharmaceutical drugs represents one of the preeminent challenges in biomedical research, both scientifically and economically [3]. While computational methods have increased productivity at many stages of the drug discovery pipeline, the field has experienced a significant slowdown, largely due to the extensive reliance on traditional small molecules as the primary source of novel therapeutic hypotheses [3]. Natural products (NPs) offer a promising alternative, comprising a vast and diverse source of bioactive compounds that are largely disjoint from the chemical space covered by conventional small-molecule libraries [3]. These compounds, which include plant metabolites, animal toxins, and immunological components, are supported in many cases by thousands of years of traditional medicine application [3].
However, natural products possess unique characteristics that distinguish them from traditional small molecule drug candidates, creating a significant data gap that requires new informatics methods and approaches for proper assessment of their therapeutic potential [3]. This data gap manifests primarily in two dimensions: limitations in the databases storing NP information, and fundamental challenges in accessing the compounds themselves for experimental validation. The informatics knowledge gaps and other barriers must be systematically overcome to fully leverage these compounds for drug discovery [3]. This review investigates the state-of-the-art techniques in bioinformatics, cheminformatics, and knowledge engineering that aim to bridge the divide between traditional drug discovery approaches and the unique challenges presented by diverse classes of natural products.
Fundamental Limitations of Natural Product Databases
The informatics infrastructure supporting natural product research suffers from several fundamental limitations that hinder effective data-driven discovery. These challenges span technical, structural, and methodological dimensions, creating significant bottlenecks in the research pipeline.
Structural and Compositional Heterogeneity
Natural products exhibit tremendous structural diversity that poses unique challenges for database representation and computational analysis. Unlike synthetic compounds that typically follow regular structural patterns, NPs include complex macromolecular assemblies alongside small molecules with unusual stereochemistry and ring systems [3]. This structural complexity creates representation challenges that exceed the capabilities of traditional chemical informatics frameworks designed for simpler small molecules.
The definitional ambiguity of what constitutes a natural product further complicates database organization. Some authors restrict NPs to small molecule secondary metabolites, while others adopt a broader definition that includes all classes of chemical substances produced by living organisms [3]. This definitional inconsistency leads to fragmented database architectures and inconsistent annotation practices across different resources. The resulting data fragmentation makes cross-database integration and comparative analysis particularly challenging.
Data Quality and Standardization Issues
The lack of standardized protocols for data representation, annotation, and curation represents a critical limitation in NP database infrastructure. Inconsistent metadata regarding biological source, extraction methods, and geographical origin plagues many natural product databases, limiting their utility for systematic analysis [3]. This problem is compounded by varied curation standards across different database projects, leading to significant inconsistencies in data quality and completeness.
Table 1: Key Limitations in Natural Product Databases
| Limitation Category | Specific Challenges | Impact on Research |
|---|---|---|
| Structural Representation | Complex stereochemistry, macromolecular assemblies, unusual ring systems | Hinders computational analysis and similarity searching |
| Data Completeness | Inconsistent annotation, partial spectroscopic data, missing source organism metadata | Limits utility for data-driven discovery and meta-analysis |
| Standardization | Varied curation standards, incompatible data formats, inconsistent taxonomy | Creates integration barriers between different databases |
| Accessibility | Proprietary data, licensing restrictions, limited programmatic access | Restricts widespread utilization and collaborative research |
The absence of community-wide standardization protocols for data representation creates significant integration barriers between different NP databases. This problem is particularly acute for spectroscopic data, where inconsistent formatting and annotation limit the utility of valuable experimental data for computational analysis and machine learning applications [3]. Without improved standardization, the natural product research community cannot fully leverage its collective data assets for transformative discovery.
Technical Challenges in Compound Accessibility
Beyond database limitations, natural product research faces substantial technical challenges in actually accessing compounds for experimental validation. These accessibility constraints create a critical bottleneck in the transition from computational prediction to experimental confirmation.
Sourcing and Supply Chain Limitations
The procurement challenges associated with natural products are multifaceted and significantly more complex than those for synthetic compounds. Many source organisms are difficult to cultivate, have limited geographical distribution, or are subject to environmental protection regulations [3]. The sustainable sourcing of natural products presents additional complications, particularly for compounds derived from endangered species or slow-growing organisms.
Supply chain complexity for natural products far exceeds that of synthetic compounds, involving multiple steps from original sourcing through extraction and purification. Each stage introduces potential bottlenecks that can delay or prevent researchers from obtaining sufficient quantities of material for comprehensive biological evaluation. These supply constraints particularly impact the study of animal toxins and marine natural products, where collection challenges and limited biomass availability create significant accessibility barriers.
Analytical and Characterization Challenges
The structural complexity of natural products creates significant analytical challenges that impact both database quality and experimental research. Spectroscopic characterization of complex natural products requires specialized expertise and instrumentation, creating a technical barrier that limits the pace of compound identification and validation [3]. This problem is particularly acute for compounds existing in complex mixtures that resist straightforward separation and purification.
Table 2: Technical Barriers to Compound Accessibility
| Barrier Category | Specific Challenges | Potential Mitigation Strategies |
|---|---|---|
| Physical Sourcing | Limited source availability, environmental regulations, cultivation difficulties | Synthetic biology approaches, improved cultivation methods, conservation programs |
| Supply Chain | Multi-step extraction and purification, limited biomass, geographic constraints | Development of efficient synthetic routes, analog screening, miniaturized assays |
| Structural Characterization | Complex stereochemistry, mixture analysis, unusual functional groups | Advanced spectroscopic techniques, computational prediction tools, collaborative networks |
| Quantity and Purity | Limited amounts of pure compound, batch-to-batch variability, stability issues | Microscale screening technologies, sensitive analytical methods, stabilization approaches |
The isolation and purification of natural products from complex biological matrices presents significant technical hurdles that impact both the quality and quantity of material available for biological screening. These challenges are compounded by the frequent occurrence of natural products as mixtures of closely related analogs that are difficult to separate and characterize individually [3]. Without improved analytical methodologies and miniaturized screening technologies, these accessibility barriers will continue to constrain the systematic evaluation of natural products for drug discovery.
Computational and Experimental Protocols
Bridging the data gap in natural product research requires integrated computational and experimental approaches that address both database limitations and compound accessibility challenges. This section outlines key methodologies for advancing NP-based drug discovery.
Cheminformatics Approaches for NP Characterization
Cheminformatics methods provide powerful tools for characterizing natural products and predicting their properties despite data limitations. Molecular descriptor calculations form the foundation of these approaches, enabling the quantitative representation of NP structures for subsequent analysis [73]. These calculations typically involve generating thousands of descriptors capturing structural, topological, and physicochemical properties that can be used to build predictive models.
Quantitative Structure-Activity Relationship (QSAR) modeling represents a critical methodology for connecting NP structures to biological activity [73]. The standard QSAR protocol involves: (1) calculating molecular descriptors for a training set of compounds with known activities; (2) selecting the most relevant descriptors using statistical or machine learning methods; (3) building a mathematical model relating descriptor values to biological activity; and (4) rigorously validating the model using external test sets. For natural products, this approach must be adapted to account for structural complexity and data sparsity through specialized descriptor sets and regularization techniques.
Virtual screening protocols enable the computational prioritization of natural products for experimental testing [73]. These protocols typically employ molecular docking to predict the binding poses and affinities of NPs against specific therapeutic targets, followed by more sophisticated molecular dynamics simulations to assess binding stability and interaction patterns. The application of these methods to natural products requires special consideration of NP flexibility and the potential for unexpected binding modes due to their structural complexity.
Bioinformatics and Knowledge-Based Methods
Bioinformatics approaches provide essential context for understanding the mechanisms of action of natural products by connecting compound structures to biological systems. Target prediction protocols use computational methods to identify potential protein targets for natural products based on chemical similarity, machine learning, or network-based approaches [3]. These methods help bridge the gap between compound identification and biological evaluation by generating testable hypotheses about mechanism of action.
Pathway analysis methodologies enable researchers to place natural products within their broader biological context by identifying the pathways and processes they impact [3]. Standard protocols involve: (1) identifying differentially expressed genes or proteins following NP treatment; (2) performing enrichment analysis to identify affected pathways; and (3) constructing interaction networks to visualize the systems-level impact of NP exposure. For natural products with complex mechanisms, these approaches can help disentangle polypharmacology and identify primary versus secondary effects.
Knowledge engineering approaches address the data integration challenges in NP research by developing formal ontologies and semantic frameworks that enable more sophisticated data mining and reasoning [3]. These methods include the development of specialized ontologies for natural product sources, traditional medicine applications, and mechanism-of-action annotations that facilitate the integration of disparate data types and enable more powerful knowledge discovery.
Research Reagent Solutions for NP Drug Discovery
Advancing natural product research requires specialized reagents and computational tools that address the unique challenges of working with these compounds. The table below outlines key resources that enable researchers to bridge the data and accessibility gaps in NP drug discovery.
Table 3: Essential Research Reagents and Resources for NP Drug Discovery
| Resource Category | Specific Tools/Reagents | Function and Application |
|---|---|---|
| Computational Tools | Molecular descriptors, QSAR models, docking software | Enable virtual screening and property prediction for NP prioritization |
| Database Resources | NP-specific databases, spectral libraries, traditional medicine repositories | Provide structural, spectral, and bioactivity data for data-driven discovery |
| Analytical Standards | Reference compounds, isotopic labels, fragment libraries | Facilitate compound identification, quantification, and mechanism studies |
| Screening Resources | Targeted assay panels, phenotypic screening platforms, protein production systems | Enable biological evaluation of NP activity and selectivity |
| Synthetic Biology Tools | Heterologous expression systems, pathway engineering kits, CRISPR-Cas9 components | Support alternative production strategies for scarce NPs |
The effective application of these research reagents requires integrated workflows that connect computational prediction with experimental validation. Commercial fragment libraries based on natural product scaffolds provide valuable starting points for medicinal chemistry optimization, while heterologous expression systems enable alternative production routes for compounds that are difficult to source from their native organisms [3]. As the field advances, the development of specialized reagents tailored to the unique challenges of natural product research will be essential for overcoming current limitations.
Integrated Roadmap for Overcoming NP Data Challenges
Addressing the data gap in natural product research requires a systematic approach that integrates computational innovation with experimental advancement. This section outlines key priorities for bridging the current limitations in NP databases and compound accessibility.
Data Management and Integration Strategies
Developing standardized data formats specifically designed for natural product information represents a critical priority for the research community [3]. These standards should encompass structural representations, spectral data, biological activity measurements, and source organism metadata in a unified framework that enables seamless data exchange and integration. The adoption of community-wide data standards will dramatically improve the interoperability of different NP databases and enable more powerful meta-analyses.
Implementing advanced data integration platforms that combine natural product information with broader biomedical knowledge bases can help contextualize NP activity within biological systems [3]. These platforms should incorporate protein-protein interaction networks, gene expression data, and disease association information to enable systems-level analysis of NP mechanisms and potential therapeutic applications. Such integration is essential for translating NP chemical diversity into actionable therapeutic hypotheses.
Establishing data federation protocols that maintain the autonomy of individual NP databases while enabling unified querying across resources can help overcome the current fragmentation of NP information [3]. These protocols should include standardized application programming interfaces (APIs), common query languages, and cross-database identity resolution services that allow researchers to access distributed NP data resources as a unified knowledge graph.
Technological Innovation for Compound Access
Accelerating natural product discovery requires new technologies that overcome current bottlenecks in compound access and evaluation. Miniaturized screening platforms that enable high-content biological evaluation of limited NP quantities can dramatically reduce the material requirements for biological testing [3]. These platforms should incorporate advanced detection technologies that maximize information content from minimal compound amounts, thereby expanding the range of NPs that can be systematically evaluated.
Synergistic public-private partnerships that address supply chain challenges for promising natural products can help bridge the gap between initial discovery and clinical development [3]. These partnerships should develop sustainable sourcing strategies, efficient synthetic routes, and bioproduction approaches that ensure a reliable supply of NPs for thorough pharmacological evaluation. Such collaborations are particularly important for compounds derived from sensitive ecosystems or difficult-to-cultivate organisms.
Advanced analytical methodologies that accelerate the dereplication and characterization of natural products can dramatically increase the throughput of NP discovery pipelines [3]. These methodologies should combine sophisticated separation techniques with high-resolution mass spectrometry and NMR technologies to enable rapid structural elucidation directly from complex mixtures, thereby reducing the purification burden associated with traditional natural product research.
The data gap characterizing natural product research—manifested through limitations in databases and accessibility of compounds—represents both a formidable challenge and a significant opportunity for advancing drug discovery. While natural products offer tremendous chemical diversity with proven therapeutic potential, fully leveraging this resource requires overcoming substantial informatics and accessibility barriers [3]. The structural complexity of NPs, coupled with challenges in sourcing and characterization, has created a disconnect between the theoretical promise of natural products and their practical application in modern drug discovery pipelines.
Addressing these limitations requires integrated computational and experimental strategies that span from improved data management to innovative compound access technologies. Computational methods including cheminformatics, bioinformatics, and knowledge engineering provide powerful approaches for extracting maximum value from existing NP data, while experimental innovations in microscreening and analytical characterization can expand the range of compounds accessible for biological evaluation [3] [73]. The continued development of specialized research reagents and databases tailored to the unique challenges of natural products will be essential for advancing this field.
As these technical challenges are addressed, natural products are poised to make increasingly significant contributions to pharmaceutical discovery, particularly for complex diseases that have proven resistant to conventional small-molecule approaches [3]. By systematically bridging the data gap through collaborative, multidisciplinary efforts, the research community can unlock the full potential of natural chemical diversity to address unmet medical needs. The roadmap outlined in this review provides a pathway toward realizing this goal, transforming natural products from challenging research subjects into productive sources of therapeutic innovation.
Natural products are an indispensable resource in drug discovery, accounting for nearly half of all FDA-approved drugs over the past several decades, including a significant majority of anticancer agents [74]. Despite their structural diversity and potent biological activities, identifying their precise molecular targets remains a major obstacle in the drug development pipeline. Traditional target identification methods, such as affinity-based protein profiling (AfBPP) and activity-based protein profiling (ABPP), require chemical modification of the natural compound—a process that can alter its bioactivity and lead to misleading results [74]. The emergence of label-free techniques, particularly the Cellular Thermal Shift Assay (CETSA), provides a powerful alternative for studying drug-target engagement in a physiological context, enabling researchers to directly monitor interactions between unmodified natural products and their protein targets within native cellular environments [75] [74].
CETSA, introduced in 2013, represents a transformative biophysical method that exploits the fundamental principle of ligand-induced thermal stabilization [76]. When a small molecule binds to a protein, it often reduces the protein's conformational flexibility, thereby enhancing its thermal stability and resistance to heat-induced denaturation [74]. Unlike traditional approaches that measure downstream cellular effects, CETSA directly quantifies this stabilization event, providing a critical link between the observed phenotypic response to a drug and its specific molecular interaction [76]. This capability is particularly valuable for natural product research, where complex chemical structures often defy straightforward modification and where multi-target effects may contribute to therapeutic efficacy.
CETSA Core Principles and Methodological Evolution
Fundamental Mechanism
The underlying principle of all thermal shift assays is that ligand binding alters the thermal stability of proteins. In its cellular application, CETSA measures how drug binding affects the thermal denaturation profile of proteins in a physiologically relevant context [77]. The assay is predicated on the biophysical phenomenon that a protein-ligand complex typically requires more thermal energy to unfold than the apo-protein [76]. When cells are heated, unbound proteins denature and aggregate, while ligand-bound proteins remain soluble and functionally folded. This differential solubility allows for separation and quantification, providing a direct readout of target engagement [76].
The basic CETSA protocol consists of four key steps: (1) compound incubation with live cells or lysates, (2) heat treatment at different temperatures, (3) separation of folded proteins from denatured aggregates, and (4) detection and quantification of remaining soluble protein [77]. The resulting data generates thermal melting curves for proteins in both the presence and absence of a ligand, with the difference between these curves (ΔTm) serving as a robust indicator of direct drug-target interaction [76] [74].
Format Evolution and Applications
Since its initial development, CETSA has evolved into multiple specialized formats tailored to different stages of drug discovery, from target identification to lead optimization.
Table 1: Key CETSA Formats and Their Applications in Drug Discovery
| Format | Detection Method | Throughput | Primary Applications | Advantages | Limitations |
|---|---|---|---|---|---|
| Western Blot CETSA | Antibody-based detection | Low | Target validation for hypothesis-driven studies [75] [74] | Easy implementation; requires only standard lab equipment [74] | Limited throughput; requires specific antibodies [77] [74] |
| High-Throughput CETSA | Dual-antibody proximity assays or split reporter systems [77] | High | Primary screening, hit confirmation, lead optimization [77] [75] | Amenable to 384- and 1536-well formats; requires small sample volumes [78] | May require specialized reagents or engineered cell lines [77] |
| MS-CETSA (TPP) | Mass spectrometry | Low to medium | Proteome-wide target identification, selectivity profiling, mode-of-action studies [79] [77] [75] | Unbiased detection of thousands of proteins simultaneously [77] [74] | Resource-intensive; requires advanced instrumentation and expertise [74] |
| Isothermal Dose-Response (ITDR) | Various (WB, MS, etc.) | Varies | Quantifying drug-binding affinity and potency (EC50) [79] [74] | Provides quantitative assessment of binding affinity [74] | Fixed temperature must be carefully selected [74] |
The Split Nano Luciferase (SplitLuc) CETSA represents a particularly significant advancement for screening applications. This homogeneous, standardized platform uses a 15-amino acid tag appended to target proteins, enabling rapid assay development and compatibility with both 384- and 1536-well formats without the need for affinity purification or centrifugation steps [78]. The broad applicability of this approach has been demonstrated for diverse targets across multiple subcellular compartments, providing a versatile platform for medium to ultra-high-throughput applications [78].
Experimental Protocols for CETSA Implementation
Basic Workflow for Live-Cell CETSA
The following protocol outlines a standardized approach for live-cell CETSA, adaptable to various detection modalities:
Cell Preparation and Compound Treatment: Culture cells under appropriate conditions. For adherent cells, harvest using standard methods. Treat cell suspensions with the compound of interest dissolved in suitable vehicle, with control samples receiving vehicle alone. Incubation time and temperature should reflect physiological relevance to the biological question [76] [74].
Heat Challenge: Aliquot compound-treated and control cells into PCR tubes or microplates. Subject samples to a temperature gradient (typically 37-73°C) using a thermal cycler with precise temperature control. The specific range should be determined empirically for each system, as some proteomes contain highly thermostable proteins requiring extended gradients [79].
Cell Lysis and Soluble Protein Extraction: Lyse heated cells using multiple freeze-thaw cycles (rapid freezing in liquid nitrogen followed by thawing at 37°C) or detergent-based lysis (e.g., 1% NP-40). Centrifuge lysates at high speed (e.g., 20,000 × g) to separate soluble proteins from denatured aggregates [76] [74]. For high-throughput formats, centrifugation may be omitted if validation confirms minimal impact on results [78].
Protein Detection and Quantification:
- For WB-CETSA: Separate soluble proteins by SDS-PAGE, transfer to membranes, and probe with target-specific antibodies. Quantify band intensities to generate melting curves [74].
- For MS-CETSA: Digest soluble proteins with trypsin, label peptides with tandem mass tags (TMT), and analyze by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [79].
- For HT-CETSA: Use compatible detection methods such as AlphaLISA, proximity extension assay, or split luciferase systems in plate-based formats [77] [78].
Specialized Methodological Considerations
Sample Preparation Challenges for Intracellular Pathogens: When applying CETSA to complex biological systems such as Plasmodium falciparum-infected erythrocytes, specialized sample preparation is required. The high abundance of hemoglobin (>90% of soluble RBC proteins) poses substantial challenges for mass spectrometry analysis. Effective hemoglobin depletion strategies, such as HemogloBind-based methods, can increase peptide spectrum matches for parasite and non-hemoglobin human proteins by 40-50% [79].
Isothermal Dose-Response (ITDR) Protocol: Instead of varying temperature, this approach applies a concentration gradient of the compound at a fixed temperature near the protein's Tm [74]:
- Treat cells with serially diluted compound concentrations.
- Heat all samples at a single predetermined temperature.
- Process samples through standard lysis and separation steps.
- Quantify remaining soluble target protein to generate dose-response curves and calculate EC50 values for target engagement potency [74].
Two-Dimensional Thermal Proteome Profiling (2D-TPP): This comprehensive approach combines both temperature and compound concentration gradients to provide a multidimensional view of drug-target interactions [74]:
- Treat cells with multiple compound concentrations across a temperature gradient.
- Process samples for MS-based proteomic analysis.
- Analyze data to identify proteins showing concentration-dependent thermal shifts.
- Generate 2D heat maps visualizing stability changes across both dimensions.
CETSA Applications in Natural Product Research
Target Identification and Validation
For natural products with unknown mechanisms of action, MS-CETSA enables unbiased proteome-wide screening for direct molecular targets. The method has successfully identified targets for various natural product classes, resolving previously ambiguous mechanisms of action [75] [74]. In proof-of-concept studies, CETSA has confirmed known interactions, such as the engagement of dihydrofolate reductase-thymidylate synthase by the antifolate pyrimethamine and falcipain 2/3 by the cysteine protease inhibitor E64d [79]. Furthermore, it has revealed novel targets, including the identification of PfPNP as a putative target for quinine and mefloquine [79].
Lead Optimization and Selectivity Profiling
During lead optimization, CETSA provides critical structure-activity relationship (SAR) data by quantifying target engagement potency (EC50) across compound series [77]. The method enables selectivity profiling by comparing thermal shifts across the proteome, identifying off-target interactions that may contribute to efficacy or toxicity [77]. For natural product derivatives, this application helps guide synthetic efforts toward improved selectivity and reduced off-target effects.
Mechanism of Action Studies
Beyond simple target identification, CETSA can illuminate complex mechanisms of action by detecting downstream effects of target engagement [79]. Varying drug exposure times enables characterization of the sequence of downstream events following initial target binding, providing insights into contextual relationships between target stabilization and phenotypic outcomes [79]. This is particularly valuable for natural products with pleiotropic effects, where multiple protein stabilizations may be observed.
Table 2: Key Reagent Solutions for CETSA Implementation
| Reagent/Category | Function/Purpose | Examples/Specifications |
|---|---|---|
| Cell Culture Systems | Provide physiologically relevant environment for target engagement | Primary cells, cell lines, engineered lines (e.g., SplitLuc-tagged) [78] |
| Lysis Buffers | Release soluble proteins while maintaining ligand-bound complexes | NP-40 detergent (1%), freeze-thaw cycles [78] [74] |
| Hemoglobin Depletion Reagents | Reduce hemoglobin interference in erythrocyte-based studies | HemogloBind resin for Plasmodium studies [79] |
| Proteomics Materials | Enable MS-based protein detection and quantification | Trypsin for digestion, Tandem Mass Tags (TMT) for multiplexing [79] |
| Detection Antibodies | Target-specific detection for WB-CETSA | Validated primary antibodies for proteins of interest [77] |
| SplitLuc Components | Enable high-throughput luciferase-based detection | 86b peptide tag, 11S large fragment, luciferase substrate [78] |
Integration with Complementary Methods and Future Perspectives
While CETSA provides robust evidence of direct target engagement, its integration with complementary approaches strengthens target validation. Combining CETSA with functional assays establishes links between binding events and phenotypic outcomes [77]. Correlation with downstream efficacy data helps confirm the therapeutic relevance of observed interactions. Other label-free techniques, including Drug Affinity Responsive Target Stability (DARTS) and Stability of Proteins from Rates of Oxidation (SPROX), can provide orthogonal validation through different biophysical principles [74].
Future developments in CETSA methodology continue to enhance its applications for natural product research. Compressed formats like the protein integral stability assay (PISA) reduce sample requirements and analysis time while maintaining statistical power [77]. Advances in mass spectrometry sensitivity progressively improve coverage of low-abundance proteins. Furthermore, the application of CETSA to map global protein-metabolite interactions and monitor protein complex dynamics opens new possibilities for fundamental research into natural product mechanisms [79].
CETSA represents a paradigm shift in target engagement assessment for natural product drug discovery. Its capacity to directly monitor drug-target interactions in physiologically relevant environments, without requiring compound modification, addresses a critical bottleneck in the development of natural product-based therapeutics. Through its various implementations—from simple Western blot formats to sophisticated proteome-wide profiling—CETSA provides a versatile toolkit for identifying and validating molecular targets, optimizing lead compounds, and elucidating complex mechanisms of action. As the methodology continues to evolve through technical improvements and integration with complementary approaches, its impact on natural product research will undoubtedly grow, accelerating the transformation of bioactive natural compounds into effective therapeutics with well-defined mechanisms of action.
The Nagoya Protocol on Access and Benefit-Sharing (ABS) represents a critical international framework that directly impacts natural product drug discovery. This whitepaper provides researchers, scientists, and drug development professionals with a comprehensive technical guide to navigating the regulatory complexities of the Protocol. Within the context of natural product research, we examine the core pillars of ABS—access obligations, benefit-sharing mechanisms, and compliance requirements—and their implications for drug discovery workflows. By integrating detailed methodologies, compliance checklists, and visual frameworks, this document aims to equip researchers with practical tools to ethically and legally leverage genetic resources while fostering equitable collaborations and promoting biodiversity conservation.
Natural products have historically been a cornerstone of pharmacotherapy, particularly for cancer and infectious diseases, with between 25-50% of currently marketed drugs owing their origins to natural compounds [80]. These compounds offer special features including enormous scaffold diversity, structural complexity, and molecular rigidity that make them particularly valuable for drug discovery, especially in tackling challenging target classes like protein-protein interactions [81]. The structural optimization of natural products through evolution has enriched this resource pool with bioactive compounds covering a wider area of chemical space compared with typical synthetic small-molecule libraries [81].
The Convention on Biological Diversity (CBD), which came into force in 1993, established a framework for the conservation and sustainable use of biodiversity, with the Nagoya Protocol on Access and Benefit-Sharing supplementing its third objective by providing a legal framework for the fair and equitable sharing of benefits arising from the utilization of genetic resources [82]. Adopted in 2010 and entering into force in October 2014, the Protocol emerged partly in response to historical practices in which commercial entities exploited natural and indigenous resources without fair compensation [83]. This international agreement plays a fundamental role in protecting plant genetic resources while presenting specific challenges and opportunities for research and industry [83].
For drug discovery professionals, understanding the Nagoya Protocol is no longer merely a legal consideration but an essential component of sustainable research practice. The Protocol aims to provide more predictable conditions for access to genetic resources and associated traditional knowledge, help ensure benefit-sharing, and promote initiatives to conserve biodiversity [82]. In the pharmaceutical sector, this creates both challenges and opportunities for researchers working with natural products.
Core Principles of the Nagoya Protocol
Definition and Scope
The Nagoya Protocol defines genetic resources as any material of plant, animal, microbial, or other non-human origin containing functional units of heredity with actual or potential value, including derivatives such as enzymes, proteins, and metabolites [84]. The key concept of utilization refers to conducting research and/or development on the genetic and/or biochemical composition of these resources or their derivatives [84].
The Protocol's temporal scope is significant; the CBD established a critical threshold in 1992, distinguishing actions and acquisitions of biological resources before and after this date under the new international legal framework [83]. For practical purposes, this means genetic resources collected before 1992 generally fall outside the Protocol's requirements, though institutions may choose to manage them ethically regardless of legal obligations [83].
The Protocol covers all genetic resources within national jurisdiction, including exclusive economic zones, while specifically excluding human DNA [84]. However, human pathogens and human-associated microorganisms might be covered under certain conditions and countries [84]. The Protocol also excludes organisms covered by specialized international treaties such as the International Treaty on Plant Genetic Resources for Food and Agriculture and the Pandemic Influenza Preparedness Framework when used under those agreements' terms [84].
The Three Pillars: ABC of ABS
The Nagoya Protocol operates through three interconnected pillars that form the foundation of its implementation:
Access Obligations (Pillar 1: Prior Informed Consent)
Prior Informed Consent (PIC) represents the initial step in the ABS process, requiring that access to genetic resources be granted by the provider country through a permit issued to the researcher before acquisition of the resource [84]. The providing country refers to where the genetic resource is found in situ and where it will be initially accessed [84]. Each country maintains sovereignty over its genetic resources and can establish its own specific PIC requirements and procedures.
Benefit-Sharing (Pillar 2: Mutually Agreed Terms)
Mutually Agreed Terms (MAT) establish the contractual obligations between the providing country and the user of the genetic resource [84]. These terms define monetary and/or non-monetary benefit-sharing measures and establish permitted uses (commercial or non-commercial research), authorized users, duration of use, and conditions for transfer to other researchers [84]. Once a user obtains PIC and MAT, the providing country may issue an Internationally Recognized Certificate of Compliance (IRCC) to demonstrate legal access and established MATs [84].
Compliance Obligations (Pillar 3: Regulatory Implementation)
Compliance obligations are implemented at national levels, requiring that researchers utilizing genetic resources within their jurisdiction comply with ABS rules of the country where the resource originated [84]. In the European Union, these are implemented through EU Regulation No. 511/2014, which requires due diligence from researchers and includes monitoring measures at designated checkpoints during research, development, and commercialization phases [84]. In Germany, violations can constitute administrative offenses with fines up to €50,000 [84].
Table 1: Core Concepts and Definitions of the Nagoya Protocol
| Term | Definition | Practical Significance for Researchers |
|---|---|---|
| Genetic Resources | Any material of plant, animal, microbial or other non-human origin containing functional units of heredity with actual or potential value [84] | Determines whether research materials fall under the Protocol's scope |
| Utilization | Conducting research and/or development on the genetic and/or biochemical composition of genetic resources, including their derivatives [84] | Defines which research activities trigger compliance requirements |
| Prior Informed Consent (PIC) | Permission granted by the provider country prior to accessing genetic resources [84] | First formal step in legal access to genetic resources |
| Mutually Agreed Terms (MAT) | Contractual agreements establishing benefit-sharing conditions between user and provider [84] | Defines specific obligations, including benefit-sharing and transfer conditions |
| Internationally Recognized Certificate of Compliance (IRCC) | Document issued by provider country demonstrating legal access and established MATs [84] | Provides proof of compliance throughout the research chain |
Implementation in Natural Product Research
Determining Protocol Applicability
For natural product researchers, a critical first step is determining whether specific genetic resources fall under the Nagoya Protocol's scope. The applicability often depends on how 'known', 'widespread', and 'familiar' a genetic resource is [83]. Traditional and widespread varieties, such as many common cultivars of major crops, are typically not covered due to their long-standing global dissemination [83]. For example, an assessment from Brazil indicates that the country would not have to share benefits from coffee genetic resources introduced before the Protocol came into force [83].
However, specific wild accessions or locally domesticated cultivars and landraces with unique genetic patterns, particularly those conserved by indigenous communities, generally fall under the Protocol [83]. The case of Coffea arabica illustrates these distinctions well. While widespread cultivars like Bourbon or Typica are excluded, unique landraces from regions like Hararghe in Ethiopia, Yemen, or Timor Leste with distinctive genetic patterns would be covered [83].
Researchers must also consider the ethical dimension beyond strict legal requirements. Some institutions, such as the Centro Agronómico Tropical de Investigación y Enseñanza (CATIE) in Costa Rica, choose to manage certain genetic resources as if they were under the Nagoya Protocol, acknowledging their origin and promoting fair benefit-sharing practices despite not being legally bound to do so [83].
ABS Workflow for Natural Product Drug Discovery
The following diagram illustrates the complete ABS workflow for natural product research, from resource identification to benefit-sharing implementation:
ABS Workflow for Natural Product Research
The Researcher's Compliance Toolkit
Successful navigation of the Nagoya Protocol requires researchers to utilize specific tools and resources throughout the drug discovery process:
Table 2: Essential Research Tools for Nagoya Protocol Compliance
| Tool/Resource | Function | Access Point |
|---|---|---|
| ABS Clearing-House | Platform for exchanging ABS information, providing legal certainty on procedures, and monitoring utilization [85] | https://absch.cbd.int/ |
| National Focal Points (NFPs) | Country-specific contacts providing official information on national ABS requirements and procedures [82] | Listed on ABS Clearing-House |
| Due Diligence Declarations | Formal declarations of compliance required under EU Regulation 511/2014 and other implementing measures [84] | EU DECLARE portal or national systems |
| Material Transfer Agreements (MTAs) | Contracts ensuring subsequent users continue utilizing resources under initial terms and conditions [84] | Institutional technology transfer offices |
| Internationally Recognized Certificates of Compliance (IRCCs) | Documents demonstrating legal access and established MATs [84] | ABS Clearing-House or provider country |
Methodologies for Compliant Natural Product Research
Pre-Access Assessment Protocol
Before initiating research on genetic resources, researchers must conduct thorough due diligence to determine regulatory obligations:
Resource Characterization: Document the taxonomic identification, geographic origin, and collection date of the genetic resource. For coffee research, this includes determining whether materials are widespread cultivars (e.g., Bourbon, Typica) or unique landraces with restricted distribution [83].
Temporal Assessment: Verify whether the resource was acquired before or after the 1992 CBD cutoff or the 2014 Nagoya Protocol implementation, as this determines applicability [83].
Jurisdictional Analysis: Identify the provider country where the resource was originally accessed and determine its status as a Party or non-Party to the Nagoya Protocol [84].
Legislative Review: Consult the ABS Clearing-House to research specific ABS legislation in the provider country, including any requirements for Prior Informed Consent [85] [82].
Traditional Knowledge Screening: Determine if the resource is associated with traditional knowledge held by indigenous or local communities, which triggers additional protections and requirements under the Protocol [83].
Access and Benefit-Sharing Negotiation Framework
The successful establishment of legally sound ABS agreements requires systematic approach:
Initial Contact: Reach out to the identified National Focal Point in the provider country to initiate the access process [82].
PIC Application: Submit a comprehensive application for Prior Informed Consent, including detailed research objectives, methodology, anticipated outcomes, and potential commercial applications [84].
MAT Negotiation: Negotiate Mutually Agreed Terms that clearly define:
- Research scope and limitations
- Rights to intellectual property arising from utilization
- Monetary and non-monetary benefit-sharing mechanisms
- Conditions for transfer of resources to third parties
- Reporting and monitoring obligations [84]
Documentation Management: Secure and maintain all ABS documentation (PIC, MAT, IRCC) for at least 20 years after the end of utilization, as required by EU regulations [84].
Benefit-Sharing Implementation Models
The Nagoya Protocol allows for flexible benefit-sharing arrangements tailored to specific research contexts:
Table 3: Benefit-Sharing Models in Natural Product Research
| Benefit Type | Research Phase | Examples | Considerations |
|---|---|---|---|
| Up-front Payments | Pre-access | Access fees, sample acquisition payments | Provides immediate benefits but may create barriers for academic researchers |
| Milestone Payments | Research & Development | Payments upon patent filing, clinical trial initiation, regulatory approval | Aligns benefits with research progression; requires long-term tracking |
| Royalty Sharing | Commercialization | Percentage of sales (typically 1-5% in India's framework [86]) | Potentially high returns but dependent on successful product development |
| Non-Monetary Benefits | All phases | Research collaboration, capacity building, technology transfer | Often more accessible for academic researchers and developing country institutions |
| IP Sharing | Intellectual Property Development | Joint patent ownership, licensing preferences | Ensures ongoing involvement and benefits for provider countries |
Current Challenges and Emerging Solutions
Implementation Barriers in Research Practice
Despite the established legal framework, researchers face significant practical challenges in implementing Nagoya Protocol requirements:
Transboundary Resources: Genetic resources and traditional knowledge often exist across political borders, making it difficult to obtain PIC or fairly share benefits among multiple stakeholders [87].
Academic-Commercial Distinction: The difficulty in distinguishing between purely academic research and commercially motivated studies can lead to potential misuse of exemptions and bypassing of benefit-sharing requirements [87].
Capacity Limitations: Many biodiversity-rich countries have insufficient human and institutional capacity to implement the CBD and Nagoya Protocol effectively, creating administrative bottlenecks [87].
Documentation Complexity: Documenting biodiversity and associated knowledge through tools like People's Biodiversity Registers is time-intensive and technically complex, though essential for legal protection and benefit-sharing [87].
Digital Sequence Information (DSI): The regulation of DSI—digital representations of genetic sequences—represents an emerging challenge, with recent developments at CBD COP16 (2024) working to establish multilateral benefit-sharing mechanisms for these resources [86].
Innovative Approaches and Future Directions
The field of ABS is evolving with new approaches to address existing challenges:
Multilateral Benefit-Sharing: Development of global mechanisms for fair sharing across borders, particularly relevant for transboundary resources and DSI, in line with decisions from recent CBD COPs [87] [86].
Streamlined Documentation: Digital platforms for creating and maintaining biodiversity documentation can speed up processes like People's Biodiversity Register creation while ensuring protection and transparency [87].
Standardized MAT Elements: Development of model contractual clauses for specific research contexts (e.g., academic non-commercial research, pharmaceutical screening) can reduce negotiation burdens.
Integrated Compliance Systems: Establishment of centralized digital platforms linking national and institutional authorities for real-time compliance tracking, as envisioned in India's updated ABS framework [87].
The integration of artificial intelligence and machine learning in cheminformatics and natural product research also presents new opportunities for navigating the ABS landscape, potentially enabling more efficient identification of promising compounds while ensuring compliance with regulatory requirements [88].
The Nagoya Protocol on Access and Benefit-Sharing represents a fundamental shift in how researchers approach natural product drug discovery, moving from a model of unrestricted access to one of equitable collaboration. For drug discovery professionals, understanding and effectively implementing the Protocol's requirements is no longer optional but essential for ethically and legally sound research practice. While the regulatory landscape presents significant challenges, from determining applicability to negotiating benefit-sharing agreements, the frameworks and methodologies outlined in this whitepaper provide a roadmap for compliance. By embracing these principles, researchers can not only avoid legal pitfalls but also contribute to a more sustainable and equitable model of natural product research that values both biological diversity and the knowledge of indigenous and local communities that have conserved it. As the field continues to evolve with new challenges like digital sequence information, ongoing engagement with the ABS framework will remain critical to the future of natural product drug discovery.
From Hit to Drug: Validation Strategies and Comparative Success in the Clinic
In natural product drug discovery, establishing a compound's biological activity is only the first step. The subsequent and more formidable challenge is functional validation—definitively confirming its mechanism of action (MoA) within complex biological systems. This process is the critical bridge linking phenotypic observation to therapeutic development, ensuring that a natural product's efficacy is grounded in a specific and understood interaction with its molecular target[s]. [89] [90]
The historical difficulty of identifying targets for bioactive natural products has been a major bottleneck in their development. [90] However, the field is rapidly evolving. Emerging technologies in chemical biology, proteomics, and computational analysis are now providing the tools to deconvolute these complex interactions with unprecedented precision, offering new pathways to revitalize and innovate within natural product research. [8] [89]
The Imperative for Validation in Natural Product Research
Unlike synthetic compounds designed for a specific target, many natural products are discovered through phenotype-based screens in complex biological systems (e.g., cell-based assays or whole-organism models). While this captures therapeutically relevant bioactivity, it leaves a "black box" regarding the specific molecular target(s) and signaling pathways involved. [90] Functional validation is the process of opening this black box. It moves beyond merely identifying a binding partner to demonstrating that the observed phenotypic outcome is a direct consequence of that specific interaction.
This confirmation is strategically essential for:
- De-risking Drug Development: Understanding the MoA allows for the rational optimization of lead compounds and helps anticipate potential toxicity or off-target effects. [40]
- Providing a Basis for Regulatory Approval: A clearly defined MoA is a cornerstone of Investigational New Drug (IND) and New Drug Application (NDA) submissions.
- Enabling New Discoveries: Identifying a novel target for a natural product can reveal new biology and uncover previously unknown pathways involved in disease. [89]
Methodologies for Target Identification and Validation
A robust functional validation strategy employs a hierarchical approach, integrating initial, system-wide target identification with subsequent, rigorous confirmation of the target's functional role.
Initial Target Identification and Engagement
This phase focuses on discovering potential molecular partners of a natural product from within a native cellular environment.
A. Chemical Proteomics
Chemical proteomics uses modified, yet bioactive, natural product analogs as molecular bait to fish out interacting proteins directly from cell lysates or tissue samples.
Detailed Protocol:
- Probe Design and Synthesis: A functional group (e.g., an alkyne or azide) is chemically incorporated into the natural product structure. This moiety must be placed such that it does not interfere with the compound's biological activity.
- Cell/Tissue Treatment: Live cells or tissue homogenates are treated with the probe. The bioactive probe engages with its endogenous targets in their native state.
- "Click Chemistry" Reaction: After incubation, a biotin reporter tag is attached to the probe via a copper-catalyzed azide-alkyne cycloaddition ("click reaction").
- Affinity Purification: The biotin-tagged complex is captured using streptavidin-coated beads.
- Protein Elution and Identification: Bound proteins are eluted and digested into peptides. The resulting peptides are analyzed by high-resolution liquid chromatography-mass spectrometry (LC-MS/MS) to identify the natural product's protein interaction partners. [89]
B. Cellular Thermal Shift Assay (CETSA)
CETSA and its quantitative variant, thermal proteome profiling (TPP), measure the thermal stabilization of a protein upon ligand binding. When a natural product binds to its target, it often makes the protein more resistant to heat-induced denaturation.
Detailed Protocol:
- Sample Preparation: Live cells or cell lysates are divided into two groups: one treated with the natural product, and a vehicle-controlled group.
- Heat Challenge: Aliquots from each group are heated to a range of different temperatures (e.g., from 37°C to 65°C).
- Soluble Protein Isolation: The heated samples are centrifuged to remove denatured, aggregated proteins. The remaining soluble protein fraction is collected.
- Protein Quantification:
- CETSA (for single targets): The soluble target protein is quantified by Western blot.
- TPP (for proteome-wide screening): The entire soluble proteome is digested and labeled with tandem mass tags (TMT). The peptides are analyzed by LC-MS/MS to measure the melting curves of thousands of proteins simultaneously, identifying those stabilized by the natural product. [40]
- A rightward shift in the melting curve (( T_m ) shift) of a protein in the treated sample indicates direct target engagement.
Functional Validation of Identified Targets
After candidate targets are identified, their functional role in the observed phenotype must be confirmed.
A. Gene Silencing and Editing
This approach tests whether the biological effect of the natural product is abolished when the putative target is removed or knocked down.
Detailed Protocol (using CRISPR-Cas9):
- Design gRNAs: Design guide RNAs (gRNAs) specific to the gene encoding the candidate target protein.
- Transduction: Transduce cells with a lentiviral vector expressing Cas9 and the target-specific gRNA.
- Selection and Validation: Select for successfully transduced cells (e.g., using puromycin) and validate gene knockout via Western blot or sequencing.
- Phenotypic Re-assessment: Treat the knockout cell line with the natural product and re-evaluate the original phenotypic readout (e.g., cell death, inhibition of inflammation). If the natural product's effect is lost, it strongly validates the target's functional role. [89]
B. Resistance Mutation Analysis
This classical genetic method involves selecting for mutant cells that are resistant to the natural product and then identifying the mutated gene, which often encodes the compound's direct target or a protein in the same pathway.
Detailed Protocol:
- Mutagenesis and Selection: Cells are exposed to a mutagen and then grown in the presence of a lethal concentration of the natural product.
- Clone Isolation: Surviving, resistant clones are isolated and expanded.
- Genetic Analysis: The genomes of resistant clones are sequenced (e.g., via whole-exome or whole-genome sequencing) and compared to the parental, sensitive cell line to identify causative mutations.
- Validation: The identified mutation is introduced into a naive cell line via gene editing to confirm it confers resistance.
C. Network Pharmacology and Bioinformatics
For natural products with polypharmacological effects, such as many from Traditional Chinese Medicine, network pharmacology provides a framework for validating multi-target mechanisms.
Detailed Protocol:
- Data Integration: Construct a network by integrating data on the natural product's identified protein targets, the associated disease-related genes, and the known biological pathways.
- Network Analysis: Use bioinformatics tools to analyze the network topology, identifying key hubs and nodes that are critical to the network's function.
- Experimental Perturbation: Experimentally inhibit or modulate the key nodes identified in the network and measure the effect on the natural product's overall activity. This helps validate that the predicted network is functionally relevant. [89]
A Scientist's Toolkit: Essential Reagents and Technologies
The following table details key reagents and technologies essential for conducting functional validation studies.
Table 1: Key Research Reagent Solutions for Functional Validation
| Research Reagent / Technology | Function in Functional Validation |
|---|---|
| Bioactive Chemical Probes (e.g., alkyne-tagged natural products) | Serve as molecular bait for affinity purification of direct protein targets in chemical proteomics. [89] |
| Tandem Mass Tag (TMT) Reagents | Enable multiplexed, quantitative proteomics in Thermal Proteome Profiling (TPP) by allowing simultaneous MS analysis of samples from multiple temperatures. [40] |
| CRISPR-Cas9 Gene Editing Systems | Used to create knockout cell lines for validating the functional necessity of a putative target protein. [89] |
| Single-Cell Multiomics Platforms | Allow for the simultaneous analysis of transcriptomic, proteomic, and epigenomic changes in individual cells, revealing MoA and heterogeneity of response in complex tissues. [89] |
| CETSA Kits | Provide standardized protocols and reagents for implementing the Cellular Thermal Shift Assay to confirm target engagement in a physiologically relevant cellular context. [40] |
Quantitative Data and Analysis
Rigorous functional validation is supported by quantitative data that demonstrates the strength and specificity of the compound-target interaction.
Table 2: Quantitative Metrics for Key Functional Validation Assays
| Assay Method | Key Quantitative Metric | Interpretation & Benchmark |
|---|---|---|
| CETSA/TPP | Thermal Shift (( \Delta T_m )) | A significant positive shift (e.g., >2°C) indicates stabilization due to target engagement. Dose-dependency strengthens the evidence. [40] |
| Chemical Proteomics | Fold-Enrichment (vs. control) | Proteins significantly enriched in the probe pull-down (e.g., >5-fold, p-value < 0.05) compared to a control are considered high-confidence hits. |
| Gene Silencing | Loss of Efficacy (IC50 shift) | In a dose-response assay, a significant rightward shift in the IC50 curve (e.g., >10-fold) in knockout vs. wild-type cells validates functional importance. |
| Cellular Potency Assay | Half-Maximal Inhibitory Concentration (( IC_{50} )) | Measures the functional potency of the natural product in a cell-based model (e.g., nM to µM range). [40] |
Visualizing Workflows and Pathways
Effective visualization of experimental workflows and the resulting mechanistic insights is crucial for communication and understanding.
Diagram 1: Hierarchical Validation Workflow
This diagram outlines the strategic flow from initial, unbiased target discovery to conclusive functional validation.
Diagram 2: CETSA Experimental Protocol
This diagram details the step-by-step protocol for the Cellular Thermal Shift Assay (CETSA).
Diagram 3: Multi-Target Network Action
This diagram illustrates how a single natural product, like Berberine, can engage multiple targets to produce a coordinated therapeutic effect, a common principle in natural product pharmacology.
The paradigm of natural product drug discovery is shifting from serendipitous finding to systematic, mechanism-driven science. Functional validation is the cornerstone of this transformation. By integrating advanced methodologies for target identification, engagement, and validation—powered by artificial intelligence and multiomics technologies—researchers can confidently elucidate the complex mechanisms of natural products. [8] [89] [40] This rigorous approach unlocks the full potential of these ancient therapeutic treasures, paving the way for their revitalization and the development of innovative, effective, and safe medicines to address unmet medical needs.
Parasitic diseases continue to inflict significant social and economic consequences globally, particularly in developing regions. Natural products (NPs) and their derivatives, historically used in traditional medicines, have provided some of the most impactful antiparasitic therapeutics, underscoring their enduring value in modern drug discovery pipelines. This case study examines the central role of NPs in combating parasitic infections, highlighting the historical successes, the application of contemporary revolutionized technologies, and the persistent challenges. The discussion is framed within the broader thesis that NPs, with their exceptional structural diversity and marked bioactivities, remain an indispensable source of chemical agents for addressing unmet medical needs, especially when integrated with innovative methodological approaches [91].
The drug discovery process is notoriously resource-intensive, often requiring over a decade and costing upwards of $2 billion per approved drug, with a very high failure rate [92] [93]. For neglected tropical diseases (NTDs), which affect approximately one billion people, the need for new therapeutics is acute, as existing drugs often suffer from limitations in cost, safety, administration, and efficacy due to drug resistance [94]. Historically, drug discovery was rooted in natural products, and NPs continue to be a major contributor to the pharmacopeia. From 1981 to 2019, NPs, their derivatives, and NP-inspired synthetic compounds constituted a significant proportion of all approved small-molecule drugs [95]. This is particularly true for antiparasitic drugs, where renowned NPs like artemisinin, quinine, and ivermectin have served as foundational treatments [91]. The pseudo-natural product (PNP) concept, which involves combining NP fragments in novel, non-biogenetic ways, is a modern evolution of NP-inspired discovery, demonstrating increased likelihood of clinical success and highlighting the untapped potential of nature's building blocks [95]. This case study explores how the unique structural complexity of NPs is being harnessed, in conjunction with cutting-edge technologies, to address the ongoing challenge of parasitic diseases.
Historical and Clinical Impact of Antiparasitic NPs
Natural products have provided the cornerstone for antiparasitic therapy for centuries. Their clinical significance is underscored by several landmark compounds derived from traditional medicines and microbial sources [91]. Artemisinin, isolated from the plant Artemisia annua (sweet wormwood), remains the most critical treatment for malaria, a disease caused by Plasmodium parasites. Quinine, and its derivatives, originating from the bark of the Cinchona tree, were the primary antimalarials for centuries. Ivermectin, a semi-synthetic derivative of the bacterial product avermectin, is a broad-spectrum antiparasitic agent essential for the control and elimination of onchocerciasis (river blindness) and lymphatic filariasis [91] [94]. These examples exemplify the "natural selection" in drug discovery, where NP-derived structures have proven to be privileged scaffolds for therapeutic development.
Table 1: Clinically Significant Natural Products in Antiparasitic Therapy
| Natural Product | Source | Parasitic Disease Target | Key Clinical Significance |
|---|---|---|---|
| Artemisinin | Plant (Artemisia annua) | Malaria (Plasmodium spp.) | Foundation of first-line combination therapies (ACTs) for malaria; fast-acting. |
| Quinine | Plant (Cinchona bark) | Malaria (Plasmodium spp.) | Historical and continued-use antimalarial; prototype for synthetic analogues. |
| Ivermectin | Bacterium (Streptomyces avermitilis) | Onchocerciasis, Lymphatic Filariasis | Instrumental in global disease elimination programs; broad-spectrum. |
A Framework for Target Assessment in Antiparasitic Discovery
Advancing a putative molecular target into a drug discovery program requires rigorous assessment to maximize the chances of success. This process is guided by a pre-defined Target Product Profile (TPP), which outlines the desired attributes of the new drug, such as route of administration, cost, spectrum of activity, and safety profile [94]. For parasitic diseases like human African trypanosomiasis (HAT), a TPP would specify the need for an oral drug that is curative, safe in pregnancy, and stable under tropical conditions. Once a TPP is established, potential targets can be evaluated using a "traffic light" scoring system across several key criteria [94]:
Table 2: Target Assessment Criteria for Antiparasitic Drug Discovery [94]
| Criterion | Red (Stop/High Risk) | Amber (Medium Risk/Caution) | Green (Go/Low Risk) |
|---|---|---|---|
| Target Validation | No/weak evidence of essentiality for parasite survival. | Genetic or chemical evidence of essentiality. | Strong genetic and chemical evidence of essentiality. |
| Druggability | No known drug-like inhibitors; active site not druggable. | Drug-like inhibitors known or active site potentially druggable. | Drug-like inhibitors known and druggable active site (clinical precedent). |
| Assay Feasibility | No assay; significant reagent cost/supply problems. | In vitro assay exists; development into HTS format feasible. | Assay ready in plate format; protein supply assured. |
| Toxicity/Specificity | Essential human homologue present; no evidence of selective inhibition. | Human homologue present, but structural/chemical evidence for selectivity. | No human homologue or human homologue is non-essential. |
| Resistance Potential | Multiple gene copies/isoforms; high potential for escape. | Target has isoforms or may be subject to escape. | No known isoforms; low potential for resistance. |
Modern Strategies and Technologies in NP-Based Discovery
The field of NP drug discovery has moved beyond simple bioassay-guided fractionation. Modern, integrated approaches are significantly enhancing the efficiency and success rate of identifying and developing new antiparasitic agents.
Network Pharmacology and the Multi-Target Paradigm
Network pharmacology (NP) is a paradigm shift from the traditional "one drug-one target" model. It integrates systems biology, pharmacology, and computational techniques to understand drug actions within complex biological networks. This approach is particularly suited to NPs, which often exert their effects through polypharmacology—simultaneously modulating multiple targets. The core of NP involves constructing and analysing various networks, such as:
- Drug-Target Networks: Map the interactions between drugs and their protein targets, revealing polypharmacology and opportunities for drug repurposing [93].
- Disease-Gene Networks (Human Disease Networks): Connect diseases that share associated genes, providing insights into common pathological mechanisms and potential new therapeutic targets [93].
- Protein-Protein Interaction (PPI) Networks: Elucidate the functional relationships between proteins, helping to identify key nodes in parasitic pathways that can be targeted for intervention [93].
NMR-Based Metabolomics for Target Identification and Validation
Nuclear Magnetic Resonance (NMR) metabolomics is a powerful tool for understanding the metabolic dysregulation caused by parasitic diseases and for identifying the mechanism of action of NP-based drugs. Parasites like cancer cells often undergo specific metabolic reprogramming, such as the Warburg effect (aerobic glycolysis) and glutaminolysis [92]. By profiling the global metabolic changes in parasites treated with NP leads, researchers can identify the inhibited pathways and proteins, thereby validating the target. Furthermore, this approach can reveal off-target effects early in the discovery process [92].
Experimental Protocol: NMR Metabolomics for MoA Studies [92]
- Sample Collection & Preparation: Treat parasitic cell cultures with the NP-based compound and appropriate controls (e.g., DMSO vehicle). Use proper quenching methods to instantly halt metabolism and extract intracellular metabolites and extracellular media.
- NMR Data Acquisition: Acquire 1D ¹H NMR spectra (e.g., using the CPMG pulse sequence to suppress protein signals) and/or 2D ¹H J-resolved (JRES) spectra from the prepared samples.
- Data Pre-processing: Process the raw NMR data, including Fourier transformation, phasing, baseline correction, and spectral alignment. Segment the spectra into bins (e.g., 0.01 ppm) for multivariate analysis.
- Multivariate Statistical Analysis: Apply unsupervised (e.g., Principal Component Analysis, PCA) and supervised (e.g., Partial Least Squares-Discriminant Analysis, PLS-DA) methods to the processed spectral data.
- Biomarker Identification & Pathway Analysis: Statistically significant metabolites (biomarkers) are identified from the loadings plots of the multivariate models. These metabolites are then mapped onto biochemical pathways using databases like KEGG or MetaboAnalyst to deduce the affected biological processes and propose the mechanism of action.
The Scientist's Toolkit: Essential Reagents and Materials
Successful NP-based antiparasitic drug discovery relies on a suite of specialized reagents and tools.
Table 3: Key Research Reagent Solutions for NP Drug Discovery
| Reagent / Material | Function and Application in Discovery |
|---|---|
| ChEMBL Database | A manually curated database of bioactive molecules with drug-like properties, used for target validation and cheminformatics analysis [95]. |
| NP Fragment Libraries | Defined collections of low molecular-weight scaffolds derived from natural products, used for the design and synthesis of pseudo-natural products (PNPs) [95]. |
| Derwent Innovations Index | A comprehensive database used for patent literature searches to track the first disclosure and novelty of clinical compounds [8]. |
| NMR Metabolomics Kits | Commercial kits for standardized metabolite extraction from cell cultures or tissues, ensuring reproducibility in metabolomics studies [92]. |
| High-Throughput Screening (HTS) Assays | Miniaturized, automated in vitro assays (e.g., target-based or whole-parasite phenotypic) used to screen large NP extract or compound libraries [94]. |
Natural products have proven their profound and central role in the fight against parasitic diseases through historical triumphs. The future of NP-based antiparasitic discovery is not a return to traditional methods but an evolution into an integrated, technology-driven discipline. By leveraging frameworks for target assessment, adopting holistic approaches like network pharmacology, utilizing advanced analytical techniques such as NMR metabolomics, and embracing novel design concepts like pseudo-natural products, researchers can systematically unlock the vast potential of the NP chemical space. This integrated approach, framed within the enduring principles of NP drug discovery, promises to deliver the next generation of safe, effective, and accessible antiparasitic therapies.
The landscape of cancer therapy has been revolutionized by the advent of Antibody-Drug Conjugates (ADCs), which represent a paradigm shift in targeted treatment approaches. These sophisticated "biological missiles" integrate the precision of monoclonal antibodies with the potent cytotoxicity of natural product (NP)-derived payloads, enabling direct lethal agent delivery to tumor cells while minimizing damage to healthy tissues [96]. Within the broader context of natural product drug discovery research, ADCs exemplify how traditional NP scaffolds can be synthetically modified and strategically deployed to address modern therapeutic challenges, particularly in oncology [8].
The fundamental architecture of ADCs comprises three critical components: a monoclonal antibody for target-specific recognition, a chemical linker system for stability and controlled release, and a highly potent cytotoxic payload typically derived from natural products [96] [97]. This case study examines the central role of NP-derived payloads in ADC development, exploring their mechanisms, experimental characterization, and integration into the drug discovery pipeline.
Core Components and Mechanism of ADCs
Structural Architecture
ADCs constitute a targeted delivery system where each structural element plays a indispensable role in determining overall efficacy and safety [96]:
Monoclonal Antibody (mAb): Serves as the targeting component, enabling specific recognition and binding to antigens expressed on cancer cell surfaces. Humanized or fully human mAbs are preferred due to their high specificity, extended circulation half-life, and diminished immunogenicity. Immunoglobulin G (IgG) has emerged as the preferred antibody class due to its sustained efficacy, engineering adaptability, and well-established conjugation methodologies [96].
Chemical Linker: Connects the antibody to the cytotoxic payload through stable bonds designed to maintain integrity during circulation while allowing efficient payload release upon internalization into target cells. Linker stability is crucial for minimizing systemic toxicity and maximizing tumor-specific drug delivery [96] [97].
Cytotoxic Payload: The therapeutic warhead, typically a natural product-derived cytotoxin with high potency (100-1000 times stronger than conventional chemotherapeutic agents). These payloads are selected for their ability to kill cancer cells at minimal concentrations once delivered intracellularly [96].
Mechanism of Action
The therapeutic activity of ADCs follows a multi-step process [96]:
Target Binding: The monoclonal antibody component specifically binds to tumor-associated antigens expressed on the surface of cancer cells.
Internalization: The ADC-antigen complex undergoes endocytosis and is internalized into the cell within endosomal vesicles.
Payload Release: The linker is cleaved within the acidic environment of endosomes or lysosomes, or by specific proteolytic enzymes, freeing the cytotoxic payload.
Cell Killing: The released payload exerts its mechanism of action (microtubule disruption or DNA damage), ultimately triggering apoptotic cell death.
Table: Chronological Development of Approved ADCs
| Approval Era | Time Period | Representative ADC | Target Antigen | Payload Type | Key Advancement |
|---|---|---|---|---|---|
| First Generation | 2000 | Gemtuzumab Ozogamicin | CD33 | Calicheamicin (DNA damaging) | Pioneering proof-of-concept |
| Second Generation | 2011-2013 | Brentuximab Vedotin, Trastuzumab Emtansine | CD30, HER2 | MMAE, DM1 (Tubulin inhibitors) | Improved linker technology |
| Third Generation | 2019-Present | Trastuzumab Deruxtecan, Sacituzumab Govitecan | HER2, TROP2 | Deruxtecan, Govitecan (DNA topoisomerase I inhibitors) | Enhanced bystander effect |
NP-Derived Payload Classes and Mechanisms
Natural products serve as the foundation for most ADC payloads due to their inherently high potency and diverse mechanisms of action. These compounds are typically derived from microbial sources or plants and synthetically modified to optimize their therapeutic properties for ADC incorporation [8].
Tubulin Inhibitors
Tubulin inhibitors represent the most extensively utilized class of ADC payloads, particularly in second-generation ADCs. These agents target the dynamic assembly and disassembly of microtubules, essential components of the cytoskeleton that play critical roles in intracellular transport, cell division, and morphology maintenance [96].
Mechanism of Action: During cell division, microtubules reorganize to form the mitotic spindle apparatus, which provides the structural framework for chromosomal segregation. Tubulin inhibitors disrupt the dynamic equilibrium of microtubule assembly, arresting cells in the G2/M phase of the cell cycle and ultimately triggering apoptosis through mitochondrial-dependent pathways [96].
Table: Major Tubulin Inhibitor Classes in ADCs
| Payload Class | Representative Compounds | Mechanism | Approved ADCs | Key Characteristics |
|---|---|---|---|---|
| Auristatins | MMAE, MMAF | Suppress tubulin polymerization, accelerate disassembly | Brentuximab vedotin, Polatuzumab vedotin | MMAF more hydrophilic with lower systemic toxicity |
| Maytansinoids | DM1, DM4 | Promote tubulin polymerization, inhibit depolymerization | Trastuzumab emtansine, Mirvetuximab soravtansine | 20% of ADCs in development use maytansinoid derivatives |
The following diagram illustrates the differential mechanisms of tubulin inhibitors:
DNA-Damaging Agents
While tubulin inhibitors demonstrate high efficacy against rapidly proliferating tumor cells, their activity is significantly diminished in quiescent cancer cell populations. To overcome this limitation, third-generation ADCs increasingly utilize DNA-damaging agents capable of targeting all phases of the cell cycle as cytotoxic payloads [96].
Mechanism of Action: DNA-damaging agents disrupt DNA structure through various mechanisms, including double-strand breaks, alkylation, intercalation, and cross-linking. These damages overwhelm the cellular repair mechanisms, leading to irreversible genomic instability and cell death [96].
Table: DNA-Damaging Agent Classes in ADCs
| Payload Class | Representative Compounds | Molecular Target | Approved ADCs | Advantages |
|---|---|---|---|---|
| Topoisomerase I Inhibitors | Exatecan derivatives (DXd), Govitecan | Topoisomerase I-DNA complex | Trastuzumab deruxtecan, Sacituzumab govitecan | Cell cycle-independent activity |
| Calicheamicin | N-acetyl gamma calicheamicin | Minor DNA groove | Gemtuzumab ozogamicin | Potent double-strand breaks |
| Pyrrolobenzodiazepines (PBDs) | PBD dimers | DNA minor groove | Loncastuximab tesirine | Interstrand cross-linking |
The following diagram illustrates the mechanisms of DNA-damaging agents:
Experimental Methodologies for ADC Payload Characterization
Pharmacokinetic and Efficacy Evaluation
Comprehensive characterization of ADC pharmacokinetics and efficacy requires sophisticated experimental designs that evaluate multiple parameters simultaneously. The PRINT (Particle Replication in Non-wetting Templates) fabrication technique enables formulation of nanoparticles with identical size, shape, and surface chemistry while systematically varying drug loading capacity [98].
Key Experimental Protocol:
- Formulation Design: Fabricate NP formulations with identical physical parameters (size: 100-200 nm, shape: spherical, surface chemistry: PEGylated) but differential drug loading (e.g., 9% vs. 20% docetaxel loading) using soft-lithography techniques [98].
- Pharmacokinetic Profiling: Administer formulations intravenously to murine cancer models (e.g., SKOV-3 human ovarian carcinoma xenografts). Collect plasma and tissue samples at predetermined intervals (0.5, 2, 8, 24, 48, 72 hours post-administration). Quantify drug concentrations using LC-MS/MS analysis [98].
- Tissue Distribution Analysis: Measure payload accumulation in target (tumor) and off-target tissues (liver, spleen, lung) using radiolabeled compounds or fluorescent tags. Calculate area under the curve (AUC) and clearance rates for comparative analysis [98].
- Efficacy Assessment: Monitor tumor volume changes over 21-28 days using caliper measurements or bioluminescent imaging. Compare treatment groups using RECIST criteria [98].
Critical Finding: Lower drug loading (9%-NP docetaxel) demonstrated superior pharmacokinetic profiles and enhanced efficacy compared to higher loading (20%-NP) formulations, with increased plasma and tumor exposure and reduced liver, spleen, and lung accumulation [98].
Next-Generation Risk Assessment
Advanced risk assessment methodologies integrate in vitro data with physiologically-based pharmacokinetic (PBPK) modeling to establish safety profiles for ADC payloads [99].
PBPK Modeling Protocol:
- Model Development: Construct species-specific PBPK models for target payloads using specialized software (e.g., PK-Sim v.11.3). Incorporate critical parameters including tissue permeability, plasma protein binding, and metabolic clearance pathways [99].
- Model Verification: Validate predictive performance by comparing simulated pharmacokinetic profiles with observed clinical or preclinical data. Apply statistical measures (e.g., fold error <2) to confirm model adequacy [99].
- Reverse Dosimetry: Translate in vitro concentration-response data into in vivo dose-response estimates using PBPK-facilitated reverse dosimetry. Calculate benchmark dose lower confidence limits (BMDL) as points of departure for permitted daily exposure (PDE) derivation [99].
- Uncertainty Factor Application: Apply appropriate uncertainty factors (interspecies, interindividual, database adequacy) to establish health-based exposure limits. Compare traditionally calculated PDE values with PBPK-derived values for assessment refinement [99].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagents for ADC Payload Development
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Humanized monoclonal antibodies (IgG1) | Targeting component | Low immunogenicity, high specificity and affinity, extended half-life [96] |
| Cleavable linkers (Val-Cit, SEaA) | Payload attachment and release | pH-sensitive or enzyme-cleavable; balance plasma stability with intracellular release [96] |
| Tubulin inhibitor payloads (MMAE, DM1) | Cytotoxic warheads | 100-1000x more potent than conventional chemotherapy; require synthetic modification [96] |
| DNA-damaging payloads (exatecan, calicheamicin) | Cytotoxic warheads | Cell cycle-independent activity; effective against quiescent cells [96] |
| PRINT nanoparticle system | Formulation platform | Controls size, shape, and drug loading independently; evaluates formulation parameters [98] |
| PBPK modeling software (PK-Sim) | Predictive toxicology | Integrates in vitro data with physiological parameters; supports reverse dosimetry [99] |
| Patient-derived xenograft (PDX) models | Efficacy evaluation | Maintains tumor heterogeneity and microenvironment; predicts clinical response [96] |
Emerging Trends and Future Perspectives
The ADC landscape continues to evolve with several promising directions focused on enhancing the therapeutic potential of NP-derived payloads:
Next-Generation ADC Constructs: Emerging platforms include bispecific ADCs targeting multiple tumor antigens, dual-payload ADCs delivering complementary mechanisms, and radionuclide drug conjugates (RDCs) combining cytotoxic and radiation approaches [96].
Pipeline Expansion: As of June 2025, 19 ADCs have received global regulatory approval, with numerous candidates in advanced development targeting novel antigens including TROP2, HER3, c-Met, BCMA, PSMA, MUC16, DLL3, and CLDN18.2 [96] [97].
Methodological Innovation: Future advancements will leverage artificial intelligence for target identification, high-throughput screening platforms, chemical biology approaches, bioinformatics, gene regulation technologies, and highly accurate non-labeling chemical proteomics to explore novel NP targets and optimize payload properties [8].
The integration of NP-derived payloads into targeted delivery systems represents a compelling case study within natural product drug discovery research, demonstrating how traditional natural products can be re-engineered through modern chemical and biological approaches to address contemporary therapeutic challenges in oncology.
The pursuit of novel therapeutic agents remains a cornerstone of pharmaceutical research, relying heavily on diverse chemical libraries for screening and lead compound identification. Within this landscape, two principal sources of chemical diversity stand out: Natural Products (NPs) and Combinatorial Compound Libraries. This analysis provides a comparative examination of these two approaches, contextualized within the broader principles of natural product drug discovery research. NPs, derived from biological sources such as plants, microorganisms, and marine organisms, have a long and successful history in drug discovery, contributing to a significant proportion of approved small-molecule drugs [8]. In contrast, combinatorial chemistry emerged in the mid-1980s as a synthetic approach to systematically generate vast arrays of compounds by covalently linking diverse "building blocks" [100]. While both strategies aim to explore chemical space for bioactive molecules, their underlying philosophies, methodologies, and applications present distinct advantages and challenges. This review synthesizes current advances, detailing experimental protocols, computational design tools, and the evolving synergy between these approaches in addressing complex medical needs.
Core Characteristics and Historical Context
Natural Products (NPs) are chemical compounds produced by living organisms through secondary metabolism. Their historical significance is profound, serving as the foundation for many early drugs. NPs are characterized by their immense structural complexity, diverse carbon skeletons, and high density of stereocenters, features honed by evolution for specific biological functions. The exploration of NPs involves the extraction, purification, and identification of compounds from nature, often guided by ethnobotany or ecological observations [8]. The landscape of NP-based discovery is rapidly evolving, with recent advances focusing on innovative target identification and harnessing NP-derived payloads for advanced therapeutic modalities like antibody-drug conjugates (ADCs) [8].
Combinatorial Compound Libraries, conversely, are a product of human ingenuity. The field was pioneered in the mid-1980s with Geysen's multi-pin and Houghten's "tea-bag" methods for parallel peptide synthesis [100]. The core concept involves the systematic and repetitive covalent linkage of a set of building blocks to generate a large collection of compounds, known as a library. These libraries can be composed of peptides, non-peptide oligomers, peptidomimetics, or small molecules [100]. Key methodologies include the One-Bead-One-Compound (OBOC) library, parallel synthesis, and DNA-encoded chemical libraries (DECLs), each with unique synthesis, screening, and decoding strategies [100]. The initial focus was on generating immense diversity, but the field has matured to emphasize the design of smarter, more focused libraries.
Table 1: Fundamental Comparison of NPs and Combinatorial Libraries
| Attribute | Natural Products (NPs) | Combinatorial Libraries |
|---|---|---|
| Origin | Biological organisms (plants, fungi, bacteria, marine life) | Synthetic chemical synthesis |
| Chemical Space | Explores biologically relevant, evolutionarily refined chemical space | Explores vast, theoretically possible chemical space |
| Structural Complexity | High; rich in stereocenters and complex ring systems | Typically lower; often designed for synthetic feasibility |
| Molecular Diversity | Broad scaffold diversity but limited by nature's biosynthetic pathways | Can be immense, but often clustered around similar scaffolds ("library noise") |
| Historical Success | High; foundation of a large portion of approved small-molecule drugs | More significant in lead optimization; growing number of clinical candidates from DECLs |
| Primary Challenge | Supply, purification, dereplication, and low yields | Achieving drug-like properties and biological relevance in designed libraries |
Library Design and Construction Methodologies
Designing and Building Natural Product Libraries
The success of NP discovery is predicated on constructing high-quality, chemically diverse libraries. A modern, rational approach moves beyond simple collection to incorporate genetic and metabolomic tools for maximizing chemical diversity.
Key Experimental Protocol: Clade-Based Metabolomics for Fungal NP Libraries A representative protocol for building a chemically diverse NP library from fungal isolates involves a bifunctional analysis of genetic and chemical data [101].
- Sample Collection and Isolation: Environmental samples (e.g., soil) are collected, and fungal strains are isolated. For instance, the University of Oklahoma's Citizen Science Soil Collection Program yielded 78,581 fungal isolates [101].
- Genetic Barcoding: Each isolate is identified by sequencing the Internal Transcribed Spacer (ITS) region, a standard genetic barcode for fungi. Phylogenetic analysis groups isolates into sequence-based clades (e.g., Clades U, V, W, X, Y) [101].
- Metabolome Profiling: Liquid Chromatography-Mass Spectrometry (LC-MS) is used to profile the secondary metabolome of each isolate. The data are processed to identify "chemical features" based on retention time and mass-to-charge ratio [101].
- Diversity Analysis: Principal Coordinate Analysis (PCoA) is performed on the metabolomics data to group isolates into "chemical clusters" based on their metabolite profiles. The relationship between genetic clades and chemical clusters is then analyzed [101].
- Feature Accumulation Curves: The data is used to generate feature accumulation curves, which predict how chemical diversity increases with the number of isolates screened. This allows for rational library sizing. A study on Alternaria fungi found that a modest 195 isolates captured nearly 99% of the chemical features in the dataset, though 17.9% of features were unique to single isolates, highlighting the value of deep sampling [101].
The Scientist's Toolkit: Key Reagents for NP Library Construction Table 2: Essential Materials for Natural Product Library Construction
| Item | Function |
|---|---|
| Internal Transcribed Spacer (ITS) Primers | For PCR amplification and sequencing of the fungal ITS barcode region for phylogenetic identification. |
| Liquid Chromatography-Mass Spectrometry (LC-MS) | For high-throughput metabolome profiling to detect and quantify chemical features from microbial extracts. |
| Culture Media | Various growth media (e.g., potato dextrose broth, ISP media) to support the growth of diverse microbial isolates and potentially trigger silent biosynthetic gene clusters. |
| Solid Support for Extraction | Resins (e.g., XAD) for in-situ capture of metabolites from liquid fermentation broths. |
The following workflow diagram illustrates the integrated process of building a rationally designed natural product library:
Designing and Synthesizing Combinatorial Libraries
Combinatorial library construction is defined by systematic synthesis. The two primary approaches are Combinatorial Synthesis (e.g., OBOC, DECL using split-pool method) and Parallel Synthesis [102].
Key Experimental Protocol: One-Bead-One-Compound (OBOC) Combinatorial Library Synthesis The OBOC method, introduced by Lam et al. in 1991, is a classic combinatorial approach for generating vast libraries of peptides or small molecules [100].
- Solid Support and Split-Pool Synthesis: Synthesis begins with microbeads as the solid support. The key is the "split-pool" strategy:
- Split: The starting beads are divided into several equal portions.
- Couple: Each portion is coupled with a different building block (e.g., amino acid Fmoc-AAâ‚, Fmoc-AAâ‚‚, etc.).
- Pool: All portions of beads are mixed together.
- Deprotect: The protecting group (e.g., Fmoc) is removed, readying the beads for the next cycle.
- Repetition: This split-pool process is repeated for each additional building block in the sequence. After 'n' cycles, a library of Bâ¿ compounds is generated (where B is the number of building blocks per step). Crucially, each individual bead carries many copies of a single, unique compound [100].
- Screening and Decoding: The bead-bound library can be screened against a biological target. Positive beads are isolated, and the structure of the active compound is determined through chemical decoding (e.g., Edman degradation for peptides) or via physical barcodes incorporated during synthesis [100].
Computational Design Tools Modern combinatorial chemistry heavily relies on computational tools for in silico library design prior to synthesis, saving time and resources.
- CoLiDe (Combinatorial Library Design): A Python-based tool for designing combinatorial protein libraries with precise control over amino acid composition, length, and diversity. It uses an evolutionary algorithm to find optimal degenerate or spiked codon solutions to approximate a desired amino acid distribution [103].
- Structure-Based Design: A protocol exists for optimizing combinatorial protein libraries beyond simple saturation mutagenesis. It uses cluster expansion and regression to design "smarter" degenerate codon libraries highly enriched for favorable, low-energy sequences, as assessed by an all-atom scoring function [104].
- Target-Focused Design: Computational chemistry is used to generate virtual libraries, perform analogue docking, and conduct in silico screening. ADMET (absorption, distribution, metabolism, excretion, and toxicity) filters are often incorporated to improve the drug-likelihood of library members [100].
Table 3: Comparison of Combinatorial Synthesis vs. Parallel Synthesis
| Attribute | Combinatorial Synthesis | Parallel Synthesis |
|---|---|---|
| Definition | Creates large libraries by systematically combining building blocks using a split-pool method. | Simultaneously synthesizes multiple compounds in separate, addressable reaction vessels. |
| Approach | Sequential addition of building blocks in a non-addressable manner. | Simultaneous synthesis using multiple reaction vessels or microarrays. |
| Library Size & Diversity | Very high (thousands to millions); excellent for generating diverse libraries. | Smaller (hundreds to thousands); ideal for focused libraries and SAR studies. |
| Throughput | High for generating diversity, but lower for screening/decoding individual compounds. | High throughput in synthesis; each compound is addressable and easier to handle. |
| Compound Identification | Requires decoding (chemical or barcode) after positive hit identification. | The structure of each compound is known based on its synthesis location. |
| Primary Application | Lead discovery and exploration of vast chemical space. | Lead optimization and Structure-Activity Relationship (SAR) studies [102]. |
Screening, Identification, and Optimization
From Hit to Lead in Natural Product Discovery
Screening NP libraries often involves bioassay-guided fractionation. A crude extract showing desired biological activity is sequentially fractionated (e.g., using chromatography), with each fraction re-tested until the single active NP is isolated and identified structurally (e.g., via NMR, MS) [8]. A significant challenge is dereplication—the early identification of known compounds to avoid rediscovery—which is now accelerated by databases and LC-MS/MS techniques.
Modern innovations are expanding the scope of NP discovery:
- NP-Derived Payloads in ADCs: NPs serve as potent cytotoxic "warheads" in Antibody-Drug Conjugates (ADCs). The antibody provides targeted delivery to cancer cells, minimizing systemic toxicity while leveraging the evolved bioactivity of NPs [8].
- Hybrid NP Molecules: Creating hybrid molecules by combining NP scaffolds with other pharmacophores is a promising strategy to address complex diseases and overcome resistance [8].
- Gene Regulation and Metabolomics: Techniques like the "simulated fermentation" method use ketoacid ligation in water to generate complex organic molecules from small building blocks, bypassing the need for living organisms and enabling the discovery of new bioactive scaffolds [100].
Screening and Optimization in Combinatorial Libraries
Screening combinatorial libraries requires high-throughput methods compatible with the library format.
- OBOC Libraries: Screened on-bead using binding assays or functional assays. Positive beads are physically picked for decoding [100].
- DNA-Encoded Libraries (DECLs): Screened as a mixture by incubating the entire library with a purified protein target. Binding members are isolated, and the attached DNA tag is PCR-amplified and sequenced to identify the hit compound [100].
- Parallel Synthesis Libraries: Each compound is screened individually in a high-throughput screening (HTS) platform, as the identity and location of each compound are known.
Once a hit is identified, the optimization process begins. Combinatorial chemistry, particularly parallel synthesis, is exceptionally powerful for lead optimization and establishing Structure-Activity Relationships (SAR). By synthesizing focused libraries around the initial hit with systematic variations at specific positions, researchers can rapidly determine which structural features are critical for potency, selectivity, and other drug-like properties [102].
Comparative Analysis and Future Outlook
The strengths and limitations of NPs and combinatorial libraries are complementary, and the future lies in their strategic integration.
Table 4: Strategic Comparison and Application
| Aspect | Natural Products | Combinatorial Libraries |
|---|---|---|
| Key Strengths | High success rate in drug discovery; evolutionarily optimized for bioactivity; unparalleled structural complexity and scaffold diversity. | Total synthetic control; rapid generation of vast numbers of compounds; amenable to precise computational design and optimization. |
| Inherent Limitations | Supply, (re)discovery of known compounds, low yields, complex purification. | Can lack structural complexity and biological relevance; "library noise"; limited by available coupling chemistries (especially DECLs). |
| Ideal Application | Primary lead discovery for novel targets and therapeutic areas, especially when natural bioactivity is suspected. | Lead optimization, SAR studies, and targeting well-defined binding pockets or enzymes. |
| Trends & Synergies | Used as inspiration for combinatorial library design (natural product-like libraries); source of payloads for targeted therapies (ADCs). | Used to create analogs and hybrids of NP scaffolds; computational tools are applied to predict NP biosynthesis and targets. |
Expert Opinion and Future Directions: According to a 2025 update, NPs remain "vital to drug discovery, demonstrating adaptability in tackling complex medical challenges" [8]. Future efforts in NP research will focus on integrating advanced methodologies such as artificial intelligence (AI), high-throughput screening, chemical biology, bioinformatics, and gene regulation to explore novel NP targets and overcome traditional bottlenecks [8]. The convergence of NP inspiration with combinatorial design and synthesis, powered by AI and machine learning, represents the next frontier. This synergy will enable the creation of "smarter" libraries that capture the best of both worlds: the biological relevance and complexity of NPs with the synthetic tractability and focus of combinatorial chemistry, ultimately accelerating the discovery of new therapeutics for unmet medical needs.
Abstract The high attrition rate of drug candidates due to unfavorable pharmacokinetic or toxicity profiles remains a significant challenge in pharmaceutical development. This is particularly pertinent in the field of natural product drug discovery, where promising bioactive compounds often fail due to poor Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties. This whitepaper provides an in-depth technical guide to the principles and practices of ADMET profiling, framing them within the context of modern natural product research. We detail a multidisciplinary framework that integrates in silico computational predictions with subsequent in vitro and in vivo experimental validation. By presenting structured protocols, quantitative data comparisons, and essential research tools, this document serves as a resource for researchers and drug development professionals aiming to de-risk the development of natural product-based therapeutics.
Natural products (NPs) and their derivatives have historically been a vital source of new therapeutic agents, contributing significantly to the treatment of diseases such as cancer, infectious diseases, and metabolic disorders [8] [44]. However, the intrinsic complexity of NPs, including their unique chemical structures and often unknown mechanisms of action, presents specific challenges for modern drug discovery campaigns. A major hurdle is the late-stage attrition of pharmacophores with good predictive power but undesirable ADMET properties [105].
Consequently, the early-stage integration of ADMET profiling has become a cornerstone of efficient drug development. This paradigm shift involves evaluating ADMET properties before lead optimization, thereby saving substantial time and resources. While several high-throughput in vitro models are available, in silico methods are gaining prominence due to their economic and faster prediction ability without the requirement for expensive laboratory resources [105]. Nonetheless, in silico ADMET tools alone are not sufficiently accurate; they are ideally adopted in an integrated manner with in vitro and, ultimately, in vivo models to enhance predictive power and build a robust dataset for decision-making [105]. This guide elaborates on this integrated approach, providing a roadmap for its application in natural product research.
Computational (In Silico) ADMET Predictions
Computational methods provide the first line of assessment in modern ADMET profiling, enabling the screening of vast virtual libraries of natural products and their analogues.
Key Methodologies and Tools
In silico ADMET prediction relies on quantitative structure-activity relationship (QSAR) models and machine learning (ML) algorithms trained on experimentally verified datasets [106]. These tools use molecular descriptors to predict a wide range of properties.
Table 1: Prominent In Silico ADMET Prediction Tools and Their Applications
| Tool Name | Type | Key Predictions | Application in NP Research |
|---|---|---|---|
| SwissADME [107] | Web Server | Drug-likeness (Lipinski's Rule of 5), pharmacokinetic parameters, solubility. | Initial rapid filtering of NP libraries for oral bioavailability potential. |
| admetSAR [107] [108] | Web Server | >20 ADMET endpoints, including human intestinal absorption, Ames mutagenicity, and acute toxicity. | Broad-spectrum toxicity and absorption profiling of phytoconstituents. |
| ADMET Predictor [109] | Standalone AI/ML Platform | >175 properties, including solubility vs. pH, pKa, CYP metabolism, DILI, and PBPK parameters. | High-accuracy, enterprise-level screening for lead NP optimization. |
| ADMETlab 2.0 [110] | Integrated Web Server | Comprehensive ADMETox properties, molecular properties, and drug-likeness. | Used in integrated workflows for identifying promising anti-TB NP hits. |
A critical concept in computational screening is the "ADMET Risk" score, an extension of Lipinski's Rule of 5. This score uses "soft" thresholds to assign a risk value for poor absorption, high CYP metabolism, and toxicity. The overall risk is the sum of these individual risks, providing a more nuanced assessment than a simple pass/fail filter [109].
A Standard Protocol for Computational Screening
The following workflow, adapted from several studies [107] [111] [110], details the steps for computationally profiling natural products.
- Ligand Preparation: Obtain 3D structures of natural product compounds from databases like PubChem or ZINC. Use software like LigPrep (Schrödinger) or Spartan to perform energy minimization and conformational analysis. Generate possible ionization states at physiological pH (e.g., 7.0 ± 2.0) [108] [110].
- Molecular Descriptor Calculation: Calculate key physicochemical descriptors, such as molecular weight (MW), partition coefficient (Log P), number of hydrogen bond donors (HBD) and acceptors (HBA), and topological polar surface area (TPSA).
- Drug-Likeness Screening: Apply filters like Lipinski's Rule of 5 or the more advanced ADMET Risk rules to assess the potential for oral bioavailability [109].
- ADMET Property Prediction: Input the prepared structures into web servers like SwissADME and admetSAR or a platform like ADMET Predictor to obtain predictions for:
- Absorption: Human intestinal absorption (HIA), Caco-2 permeability.
- Distribution: Plasma protein binding (PPB), blood-brain barrier (BBB) penetration.
- Metabolism: Interactions with Cytochrome P450 (CYP) enzymes (e.g., inhibition of 2D6, 3A4).
- Excretion: Clearance mechanisms.
- Toxicity: Ames mutagenicity, drug-induced liver injury (DILI), acute toxicity, and environmental toxicity [107].
- Analysis and Prioritization: Rank compounds based on their favorable predicted ADMET profiles and low risk scores for progression to experimental validation.
Figure 1: A standard workflow for the computational ADMET screening of natural products.
Experimental Validation of ADMET Properties
Computational predictions are probabilistic and must be confirmed experimentally. This section outlines key in vitro and in vivo protocols for validating critical ADMET parameters.
In Vitro Methodologies
In vitro models provide a controlled, high-throughput means of assessing ADMET properties before moving to complex in vivo studies.
Table 2: Key Experimental Models for ADMET Validation
| ADMET Property | Experimental Model | Protocol Summary | Key Output |
|---|---|---|---|
| Absorption | Caco-2 Cell Monolayer [107] [105] | Grow human colon adenocarcinoma cells on a permeable filter for 21 days. Apply test compound and measure apparent permeability (Papp) from apical to basolateral side. | Papp > 1 x 10-6 cm/s suggests high absorption. |
| Metabolism | Liver Microsomal Stability [109] [105] | Incubate test compound with liver microsomes (human or species-specific) and NADPH cofactor. Sample at time points (0, 5, 15, 30, 60 min) and analyze by LC-MS/MS. | % parent compound remaining. Calculates half-life (t1/2) and intrinsic clearance (CLint). |
| Toxicity | MTT Cytotoxicity Assay [112] | Seed cells (e.g., HepG2 human liver cells). Treat with serially diluted test compound for 24-72 hrs. Add MTT reagent; viable cells reduce it to purple formazan. Measure absorbance. | IC50 value (concentration that inhibits 50% of cell growth). |
| Toxicity | Ames Test [109] | Incubate test compound with specific strains of Salmonella typhimurium that cannot synthesize histidine, in the presence/absence of metabolic activation (S9 fraction). Count revertant colonies. | A significant increase in revertants indicates mutagenicity. |
An Integrated Validation Protocol: Cytotoxicity and Metabolic Stability
The following combined protocol illustrates how to gather critical toxicity and metabolism data in parallel.
A. MTT Cytotoxicity Assay on HepG2 Cells [112]
- Cell Line: Human hepatocellular carcinoma (HepG2).
- Reagents: DMEM culture medium, Fetal Bovine Serum (FBS), Penicillin-Streptomycin, MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), Dimethyl Sulfoxide (DMSO).
- Procedure:
- Culture HepG2 cells in DMEM supplemented with 10% FBS and 1% Penicillin-Streptomycin at 37°C in a 5% CO2 incubator.
- Seed cells in a 96-well plate at a density of 1 x 104 cells/well and incubate for 24 hours to allow attachment.
- Treat cells with a serial dilution of the natural product (typically from 0.1 µM to 100 µM). Include a negative control (vehicle, e.g., DMSO) and a positive control (e.g., a known cytotoxic agent).
- After 48 hours of incubation, carefully remove the medium and add fresh medium containing MTT (0.5 mg/mL final concentration). Incubate for 4 hours.
- Carefully remove the MTT-containing medium and dissolve the formed formazan crystals in DMSO.
- Measure the absorbance at 570 nm using a microplate reader. Calculate the percentage of cell viability and determine the IC50 value using non-linear regression analysis.
B. Liver Microsomal Stability Assay [109] [105]
- Reagents: Human or rat liver microsomes, NADPH regenerating system, Phosphate Buffered Saline (PBS), stop solution (e.g., acetonitrile with internal standard).
- Procedure:
- Prepare the incubation mixture containing liver microsomes (0.5 mg/mL protein) and the test compound (1 µM) in PBS.
- Pre-incubate the mixture for 5 minutes at 37°C.
- Initiate the reaction by adding the NADPH regenerating system.
- At predetermined time points (0, 5, 15, 30, and 60 minutes), withdraw an aliquot and mix it with an ice-cold stop solution to terminate the reaction.
- Centrifuge the samples to precipitate proteins and analyze the supernatant using LC-MS/MS to quantify the amount of parent compound remaining.
- Plot the natural logarithm of the percent remaining versus time. The slope of the linear regression is used to calculate the in vitro half-life (t1/2) and intrinsic clearance (CLint).
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful ADMET profiling relies on specific, high-quality reagents and computational tools. The following table details essential components of the researcher's toolkit.
Table 3: Key Research Reagent Solutions for ADMET Profiling
| Tool/Reagent | Function | Application Context |
|---|---|---|
| Caco-2 Cell Line | A human colon cancer cell line that differentiates into a monolayer mimicking the intestinal epithelium. Used for predicting human intestinal absorption. | In vitro absorption studies [107] [105]. |
| Cryopreserved Hepatocytes | Primary liver cells that contain the full complement of drug-metabolizing enzymes and transporters. Used for more physiologically relevant metabolism and toxicity studies. | Hepatic clearance prediction, metabolite identification, and DILI assessment [109]. |
| NADPH Regenerating System | Provides a constant supply of NADPH, a crucial cofactor for Cytochrome P450 enzymes. Essential for all microsomal stability assays. | In vitro metabolic stability and reaction phenotyping studies [109] [105]. |
| admetSAR Web Server | A freely available online tool for predicting multiple ADMET endpoints using QSAR models. | Initial computational screening and toxicity risk assessment of natural products [107] [108]. |
| ADMET Predictor Software | A comprehensive AI/ML platform for predicting over 175 ADMET properties, including PBPK parameters. | High-fidelity prediction and de-risking in late-stage lead optimization [109]. |
Case Studies in Integrated ADMET Profiling
The synergy between computational predictions and experimental validation is best illustrated through case studies from recent literature.
Case Study 1: Anti-Tuberculosis Chromene Glycoside [110] A study aimed at discovering novel inhibitors of Mycobacterium tuberculosis protein kinase G (PknG) screened a library of 460,000 compounds.
- Computational Phase: Virtual screening and molecular docking identified seven top hits. Subsequent in silico ADMET profiling using ADMETlab 2.0 was performed to evaluate drug-likeness and toxicity risks. A chromene glycoside (Hit 1) was prioritized not only for its high binding affinity but also for its predicted favorable pharmacokinetic and low toxicity profile.
- Experimental Phase: The stability of the Hit 1-PknG complex and the compound's properties were further validated through molecular dynamics simulations and density functional theory (DFT) calculations, confirming the stability of the interaction and the electronic properties underlying its reactivity. This integrated approach highlighted Hit 1 as a promising lead candidate for further development.
Case Study 2: Antidiabetic Natural Products [111] In a study to discover novel antidiabetic agents, 24,316 natural compounds from the ZINC database were screened against human pancreatic amylase.
- Computational Phase: The top three compounds (e.g., ZINC85593620) demonstrated higher binding scores than the standard drugs acarbose and ranirestat. Their drug-likeness and ADMET properties were profiled computationally to ensure they had characteristics suitable for drug development.
- Experimental Validation: Molecular dynamics (MD) simulations were used as an advanced in silico validation step, revealing that the complexes of these compounds with amylase showed minimal fluctuations, indicating strong and stable binding interactions. This provided a high level of confidence in the computational predictions before committing to costly wet-lab experiments.
The following diagram conceptualizes the decision-making process in this integrated ADMET risk assessment.
Figure 2: The iterative cycle of computational prediction and experimental validation in ADMET risk assessment.
The landscape of ADMET profiling is continuously evolving. The integration of in silico tools at the earliest stages of natural product drug discovery is no longer optional but a necessity for improving efficiency and success rates [106] [105]. The future lies in the deeper integration of artificial intelligence and machine learning to build more accurate predictive models, fueled by high-quality experimental data [109] [8]. Furthermore, the adoption of more sophisticated in vitro models, such as 3D organoids and organs-on-chips, will provide even more physiologically relevant data for validation, bridging the gap between traditional in vitro assays and human clinical outcomes [105]. For researchers in natural product drug discovery, embracing this integrated, multi-faceted approach to ADMET profiling is paramount for translating the immense potential of natural compounds into safe and effective therapeutic agents of the future.
Conclusion
Natural product drug discovery is experiencing a powerful revitalization, driven by a synergy of traditional knowledge and cutting-edge technologies. The foundational principles of exploring nature's chemical diversity remain as relevant as ever, providing a unique and privileged source of scaffolds for new therapeutics. The integration of AI, in silico methods, and advanced omics technologies is decisively overcoming historical challenges, compressing discovery timelines, and enabling a more targeted approach. Looking forward, future success will depend on a continued commitment to interdisciplinary collaboration, the development of high-quality datasets for machine learning, and the sustainable and equitable management of genetic resources. By embracing these advanced methodologies and navigating the associated challenges, natural products will continue to be a major force in delivering novel drugs to address pressing global health concerns, from antimicrobial resistance to complex chronic diseases.
References
- 1. A review of the WHO strategy on traditional, complementary, and integrative medicine from the perspective of academic consortia for integrative medicine and health - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11201178/]
- 2. Evidence-based traditional medicine for transforming global health & wellbeing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10645034/]
- 3. Informatics and Computational Methods in Natural Product Drug Discovery: A Review and Perspectives - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6503039/]
- 4. The Role of Natural in Products Modern Drug Discovery [https://www.azolifesciences.com/article/The-Role-of-Natural-Products-in-Modern-Drug-Discovery.aspx]
- 5. Anticancer Drug Discovery Based on Natural Products: From Computational Approaches to Clinical Studies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10813144/]
- 6. NPASS database update 2023: quantitative natural product activity and species source database for biomedical research - PubMed [https://pubmed.ncbi.nlm.nih.gov/36624664/]
- 7. Frontiers | Exploring chemical space for “druglike†small molecules in the age of AI [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1553667/full]
- 8. The latest advances with natural in products and... drug discovery [https://pubmed.ncbi.nlm.nih.gov/40391763/]
- 9. The latest trends in drug | CAS discovery [https://www.cas.org/resources/cas-insights/2025-drug-discovery-trends]
- 10. Natural products in drug discovery: advances and opportunities | Nature Reviews Drug Discovery [https://www.nature.com/articles/s41573-020-00114-z]
- 11. A Historical Overview of Natural Products in Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901206/]
- 12. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0062839]
- 13. Frontiers | Plant and marine-derived natural : sustainable... products [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1497668/full]
- 14. Navigating the chemical of space from diverse... natural products [https://link.springer.com/article/10.1007/s11101-025-10198-3]
- 15. (PDF) Characteristics of known drug space . Natural , their... products [https://www.academia.edu/32450924/Characteristics_of_known_drug_space_Natural_products_their_derivatives_and_synthetic_drugs]
- 16. Review on natural products databases: where to find data in 2020 | Journal of Cheminformatics | Full Text [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-00424-9]
- 17. Review on natural products databases: where to find data in 2020 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7118820/]
- 18. Characterization of the Chemical of Known and Readily... Space [https://pubmed.ncbi.nlm.nih.gov/30010333/]
- 19. CIMB | Special Issue : Applications of Natural and Pseudo- Natural ... [https://www.mdpi.com/journal/cimb/topical_collections/1V6D29J00P]
- 20. New clinical application prospects of artemisinin and its derivatives... [https://idpjournal.biomedcentral.com/articles/10.1186/s40249-023-01152-6]
- 21. Frontiers | Artemisinin and its derivatives: all-rounders that may... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1602581/full]
- 22. : A Synthetic Biology Artemisinin | Bio 2.0 | Learn... Success Story [https://www.nature.com/scitable/blog/bio2.0/artemisinin_a_synthetic_biology_success/?error=cookies_not_supported&code=2efbac4d-3a8c-41d7-b30c-6b07811803c5]
- 23. Ivermectin, ‘Wonder drug’ from Japan: the human use perspective - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3043740/]
- 24. Ivermectin: enigmatic multifaceted ‘wonder’ drug continues to surprise and exceed expectations | The Journal of Antibiotics [https://www.nature.com/articles/ja201711]
- 25. Ivermectin - Wikipedia [https://en.wikipedia.org/wiki/Ivermectin]
- 26. - Wikipedia Quinine [https://en.wikipedia.org/wiki/Quinine]
- 27. Uses, Side Effects & Warnings Quinine [https://www.drugs.com/mtm/quinine.html]
- 28. Antimicrobial resistance [https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance]
- 29. Unmet Medical Need in Infectious Diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3404719/]
- 30. Prioritized current unmet needs for antibacterial therapies - PubMed [https://pubmed.ncbi.nlm.nih.gov/25056396/]
- 31. in Modern Natural : Challenges, Advances... Products Drug Discovery [https://zenodo.org/records/14652293]
- 32. Frontiers | Antimicrobial at a turning point: resistance ... microbial [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1698809/full]
- 33. Global antibiotic resistance surveillance report 2025 [https://www.who.int/publications/i/item/9789240116337]
- 34. Frontiers | Addressing the global challenge of bacterial drug resistance: insights, strategies, and future directions [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1517772/full]
- 35. World Antimicrobial Awareness Week Resistance ... - ESICM 2025 [https://www.esicm.org/esicm-waaw-2025/]
- 36. Exploring natural products potential: A similarity-based target prediction tool for natural products - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0010482524014367]
- 37. Progress of AI -Driven Drug –Target Interaction Prediction and Lead ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563295/]
- 38. How AI is Transforming Drug : Innovations, Benefits, and... Discovery [https://www.onedaymd.com/2025/07/how-is-ai-used-in-drug-discovery.html]
- 39. Target Prediction Model for Natural Products Using Transfer Learning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8124298/]
- 40. Top 5 Drug Trends Discovery Driving Breakthroughs 2025 [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 41. How Machine In Learning And Development Works... Drug Discovery [https://www.linkedin.com/pulse/how-machine-learning-drug-discovery-development-works-a119f]
- 42. in Natural ... | products Reviews drug Nature Drug Discovery [https://www.nature.com/articles/s41573-020-00114-z?error=cookies_not_supported&code=10bcfbb8-4943-4dc3-b75b-49d0ccf37d51]
- 43. Integrating Molecular and Docking Simulations Molecular Dynamics [https://pubmed.ncbi.nlm.nih.gov/31452096/]
- 44. Modern Approaches in the Discovery and Development of... [https://pubmed.ncbi.nlm.nih.gov/35056662/]
- 45. Pharmacophore-based virtual screening versus docking-based virtual screening: a benchmark comparison against eight targets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4007494/]
- 46. Optimization of virtual screening against phosphoinositide 3-kinase delta: Integration of common feature pharmacophore and multicomplex-based molecular docking - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1476927123002025]
- 47. Protein–Ligand Docking in the Machine-Learning Era - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9323102/]
- 48. | Pharmacophore-based Frontiers and virtual ... screening in silico [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1531512/full]
- 49. and Metabolomics strategies in dereplication natural products [https://pubmed.ncbi.nlm.nih.gov/23963915/]
- 50. and Metabolomics strategies in the dereplication of... discovery [https://pureportal.strath.ac.uk/en/publications/metabolomics-and-dereplication-strategies-in-the-discovery-of-nat]
- 51. Bioprospecting the Metabolome of Plant Urtica dioica L.: A Fast... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9050285/]
- 52. Natural Products for Drug Discovery in the 21st Century: Innovations for Novel Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6032166/]
- 53. Liquid Chromatography High Resolution Mass Spectrometry (LC-HR-MS) | Nawah Scientific [https://nawah-scientific.com/all-services/tests/chemical-characterization/hr-ms/]
- 54. , Development, and Regulation of Discovery Natural Products [https://www.intechopen.com/chapters/44805]
- 55. Journal of Nutrition Science Research - Biosynthesis of Natural ... [https://www.omicsonline.org/open-access/biosynthesis-of-natural-products-and-bioactive-compounds-current-trends-and-advances-134405.html]
- 56. Enhancement of Natural in Plant in the... | CoLab Products [https://colab.ws/articles/10.1007%2F978-981-97-2166-5_3]
- 57. Recent advances in combinatorial biosynthesis for drug discovery [https://www.dovepress.com/recent-advances-in-combinatorial-biosynthesis-fornbspdrug-discovery-peer-reviewed-fulltext-article-DDDT]
- 58. , Discovery , Regulation, Transport, Release and... Biosynthesis [https://www.frontiersin.org/research-topics/26414/discovery-biosynthesis-regulation-transport-release-and-engineering-of-plant-natural-products/magazine]
- 59. Recent advances in combinatorial biosynthesis for drug discovery [https://experts.illinois.edu/en/publications/recent-advances-in-combinatorial-biosynthesis-for-drug-discovery]
- 60. Chemical Informatics and the Drug Discovery Knowledge Pyramid - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3796186/]
- 61. Artificial Intelligence in Natural : Current... Product Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC11874025/]
- 62. Application of artificial intelligence in bioprospecting for natural ... [https://link.springer.com/article/10.1186/s44398-025-00004-7]
- 63. Chemoinformatics and Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6146447/]
- 64. Discovery and resupply of pharmacologically active plant-derived... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4748402/]
- 65. Building Natural Product–Based Libraries for Drug Discovery... [https://link.springer.com/article/10.1007/s43450-024-00540-9]
- 66. Unlocking Nature 's Pharmacy [https://www.numberanalytics.com/blog/ultimate-guide-pharmaceuticals-chemistry-natural-products]
- 67. Biochemometrics for Natural Products Research: Comparison of Data Analysis Approaches and Application to Identification of Bioactive Compounds - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5135737/]
- 68. and Natural : a survey of stakeholders in... products drug discovery [https://pubmed.ncbi.nlm.nih.gov/26578954/]
- 69. in Natural ... | products Reviews drug Nature Drug Discovery [https://www.nature.com/articles/s41573-020-00114-z?error=cookies_not_supported&code=b84b6eed-98b2-49dd-a762-b41579fb39d8]
- 70. , Extraction and Isolation of Bioactive Compounds... Characterization [https://pmc.ncbi.nlm.nih.gov/articles/PMC3218439/]
- 71. Frontiers | Natural Products’ Extraction and Isolation-Between Conventional and Modern Techniques [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2022.873808/full]
- 72. Contemporary methods for the extraction and isolation of natural products - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10314546/]
- 73. Concepts and Experimental Protocols of Modelling and Informatics in Drug Design | ScienceDirect [https://www.sciencedirect.com/book/9780128205464/concepts-and-experimental-protocols-of-modelling-and-informatics-in-drug-design]
- 74. Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review [https://www.mdpi.com/1422-0067/26/9/3940]
- 75. CETSA and thermal proteome profiling strategies for target identification and drug discovery of natural products - PubMed [https://pubmed.ncbi.nlm.nih.gov/37216761/]
- 76. Cellular Thermal Shift Assay ( CETSA ) [https://www.news-medical.net/life-sciences/Cellular-Thermal-Shift-Assay-(CETSA).aspx]
- 77. A Shift in Thinking: Cellular Thermal Shift Assay-Enabled Drug... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10108388/]
- 78. A widely-applicable high-throughput cellular thermal shift assay... [https://www.nature.com/articles/s41598-018-27834-y?error=cookies_not_supported&code=ec946a81-96a6-4669-a15a-3c7000dfcaf7]
- 79. Cellular thermal shift assay for the identification of... | Nature Protocols [https://www.nature.com/articles/s41596-020-0310-z?error=cookies_not_supported&code=2437c29c-77a2-44fe-aba6-f866f16eb8c9]
- 80. Modern Natural Products Drug Discovery and its Relevance to Biodiversity Conservation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3061248/]
- 81. Natural products in drug discovery: advances and opportunities - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7841765/]
- 82. Nagoya Protocol: sharing pharma products and protecting biodiversity [https://www.europeanpharmaceuticalreview.com/article/96377/the-nagoya-protocol-sharing-the-benefits-of-pharmaceutical-products-and-protecting-biodiversity/]
- 83. Convention on Biological Diversity (CBD) and the Nagoya ... Protocol [https://pmc.ncbi.nlm.nih.gov/articles/PMC10814485/]
- 84. | Uni Kiel Nagoya protocol [https://www.uni-kiel.de/en/research/integrity-ethics/nagoya-protocol]
- 85. The Nagoya on Access and Protocol - Benefit sharing [https://www.cbd.int/abs?ref=moodfuel-news]
- 86. Biological Diversity (Access and Benefit ) Regulation, Sharing 2025 [https://www.drishtiias.com/daily-updates/daily-news-analysis/biological-diversity-access-and-benefit-sharing-regulation-2025]
- 87. Biological Diversity (Access And Benefit ) Regulation Sharing 2025 [https://visionias.in/current-affairs/monthly-magazine/2025-06-17/environment/biological-diversity-access-and-benefit-sharing-regulation-2025]
- 88. Frontiers | Core publications in drug discovery and natural product research [https://www.frontiersin.org/journals/natural-products/articles/10.3389/fntpr.2024.1493720/full]
- 89. New opportunities and challenges of natural products research: When target identification meets single-cell multiomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9643300/]
- 90. Recent advances in target identification of bioactive natural products - PubMed [https://pubmed.ncbi.nlm.nih.gov/30343630/]
- 91. in Natural : advances... products antiparasitic drug discovery [https://pubmed.ncbi.nlm.nih.gov/40501439/]
- 92. NMR Metabolomics Protocols for Drug - PMC Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC7025395/]
- 93. Harnessing network pharmacology in drug : an integrated... discovery [https://link.springer.com/article/10.1007/s00210-024-03625-3]
- 94. Target assessment for antiparasitic drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2979298/]
- 95. Occurrence of “Natural Selection†in Successful Small Molecule Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11247505/]
- 96. Frontiers | Antibody-drug conjugates in cancer : therapy ... current [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1669592/full]
- 97. New ADC Drugs 2025 : Targeted is Coming – A New... Cancer Therapy [https://www.creative-biolabs.com/blog/adc/new-adc-drugs-2025/]
- 98. Nanoparticle drug loading as a design parameter to improve docetaxel... [https://pubmed.ncbi.nlm.nih.gov/23899444/]
- 99. Next-generation risk assessment: integrating in vitro data and... [https://repositorio.bc.ufg.br/items/ce6e3022-743f-4970-8a8f-7c4a598e54a8]
- 100. Combinatorial Chemistry in Drug Discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5645069/]
- 101. Building Natural Using Product Clade-Based... Libraries Quantitative [https://pmc.ncbi.nlm.nih.gov/articles/PMC8547436/]
- 102. Synthesis Combinatorial . Parallel Synthesis... | This vs . That vs [https://thisvsthat.io/combinatorial-synthesis-vs-parallel-synthesis]
- 103. CoLiDe: Combinatorial Library Design tool for probing protein sequence space - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8088326/]
- 104. A Structure-Based Design Protocol for Optimizing Combinatorial Protein Libraries - PubMed [https://pubmed.ncbi.nlm.nih.gov/27094288/]
- 105. ADMET Profiling in Drug Discovery and Development: Perspectives of In Silico, In Vitro and Integrated Approaches - PubMed [https://pubmed.ncbi.nlm.nih.gov/34225615/]
- 106. tools in the digital era: Applications and limitations | CoLab ADMET [https://colab.ws/articles/10.1016%2Fbs.apha.2025.01.004]
- 107. Journal of Higher Education and Science » Submission »... [https://dergipark.org.tr/en/pub/higheredusci/issue/94453/1697221]
- 108. Target-based drug discovery, ADMET profiling and bioactivity studies of antibiotics as potential inhibitors of SARS-CoV-2 main protease (Mpro) - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8246438/]
- 109. Predictor® - Simulations Plus - Machine Learning- ADMET ... ADMET [https://www.simulations-plus.com/software/admetpredictor/]
- 110. Frontiers | Molecular docking, free energy calculations, ADMETox... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1531152/full]
- 111. De novo in silico screening of natural products for antidiabetic drug... [https://pubmed.ncbi.nlm.nih.gov/39974370/]
- 112. Tri-metallic (Au–Pt–Ag) nanofluids: unveiling synergistic anti-malarial... [https://pubs.rsc.org/en/content/articlehtml/2025/ra/d5ra02037a]